

Abstract
Introduction
To elucidate the mechanism of late-phase 3 early after depolarization (EAD) in ventricular arrhythmogenesis, we hypothesized that intracellular calcium (Cai) overloading and action potential duration (APD) shortening may promote late phase 3 EAD and triggered activity, leading to development of ventricular fibrillation (VF).
Methods and Results
In isolated rabbit hearts, we performed microelectrode recording and simultaneous dual optical mapping of transmembrane potential (Vm) and Cai transient on left ventricular endocardium. An IKATP channel opener, pinacidil, was used to abbreviate action potential duration (APD). Rapid-pacing was then performed. Upon abrupt cessation of rapid pacing with cycle lengths of 60–200 ms, there were APD90 prolongation and the corresponding Cai overloading in the first post-pacing beats. The duration of Cai transient recovered to 50% (DCaT50) and 90% (DCaT90) in the first post-pacing beats was significantly longer than baseline. Abnormal Cai elevation coupled with shortened APD produced late-phase 3 EAD induced triggered activity and VF. In additional 6 preparations, the heart tissues were treated with BAPTA-AM, a calcium chelator. BAPTA-AM significantly reduced the maximal Cai amplitude (26.4±3.5% of the control; p<0.001) and the duration of Cai transients in the mapped region, preventing the development of EAD and triggered activity that initiated VF.
Conclusions
IKATP channel activation along with Cai overloading are associated with the development of late phase 3 EAD and VF. Because acute myocardial ischemia activates the IKATP channel, late phase 3 EADs may be a mechanism for VF initiation during acute myocardial ischemia.
Keywords: ventricular fibrillation, early afterdepolarization, triggered activity, calcium, APD shortening
INTRODUCTION
Abbreviation of repolarization by ATP-regulated potassium channel openers (IKATP), as occurs in acute myocardial ischemia, has been shown to increase ventricular vulnerability to reentry and fibrillation.1–3 Pinacidil is known to augment IKATP channel in cardiac tissues, leading to a shortening of action potential duration (APD).4–7 Although previous studies have demonstrated that pinacidil facilitates the induction of ventricular fibrillation (VF) by promoting reentrant activity due to refractoriness dispersion,8;9 information regarding the mechanism of its arrhythmogenesis in response to the corresponding intracellular calcium (Cai) dynamics is largely unknown.
Cai exerts significant influence on many membrane ion channels and plays an important role in arrhythmogenesis.10–14 Elevated Cai may activate Na-Ca exchanger current (INCX), causing diastolic elevation of transmembrane potential (Vm). During VF, the levels of diastolic Cai may increase to that of systolic Cai, reaching levels that could trigger spontaneous sarcoplasmic reticulum (SR) calcium release.15 This raises the possibility that VF might be maintained in part by effects of Cai-sensitive membrane currents on action potential propagation. Burashnikov and Antzelevitch showed that Cai overloading contributes to the development of late phase 3 early afterdepolarization (EAD) induced triggered activity, and this mechanism may be responsible for the extrasystolic activity that reinitiates fibrillation in canine atrial tissues.16 Whether or not late phase 3 EAD is also important in the generation of ventricular arrhythmias, such as VF, is not clear. In addition, VF itself induces Cai overloading.17 Whether the Cai elevation is responsible for late phase 3 EAD and VF reinitiation remains unknown. We hypothesized that Cai overloading with abbreviation of APD by pinacidil may promote late phase 3 EAD and triggered activity, leading to development of spontaneous VF. To test this hypothesis, we performed simultaneous Vm and Cai dual optical mapping of left ventricular endocardium in isolated Langendorff-perfused rabbit hearts.
MATERIALS AND METHODS
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996). The study protocol was approved by the Institutional Animal Care and Use Committee (IACUC) and followed the guidelines of the American Heart Association.
Isolated Rabbit Heart with Endocardial Exposure Preparations
New Zealand White rabbits (weight of 4 to 5 kg) of either sex were intravenously injected with 1000 units of heparin and anesthetized with ketamine (20 mg/kg) and xylazine (5 mg/kg). After a median sternotomy, the whole heart was rapidly excised and retrogradely perfused through ascending aorta with 37°C Tyrode solution (pH 7.3–7.4) equilibrated with 5% CO2 and 95% O2. The coronary perfusion pressure was regulated between 80 and 95 mmHg. Each isolated heart was allowed to stabilize with perfusion for about 15 minutes (equilibration period) before the following dissection and mapping protocols.
Since most previous studies were performed on the epicardium of left ventricle (LV) without elucidation of the calcium dynamics in the endocardium, we performed a dual optical mapping on the LV endocardium. We first cut open the right ventricle (RV) following procedures detailed elsewhere.18 Briefly, we cut open the right atrial (RA) free wall toward atrial septum above the right coronary artery (RCA). The distal end of the RCA was tied off to ensure continuous perfusion of the RV free wall without significant leakage. Then, a base-to-apex cut along the posterior descending artery separated the free wall from the septum on one side of the ventricle, forming an RV flap. Once the RV flap was created, the middle portion of septum was cut off to expose LV endocardium (Fig. 1A). This preparation ensured a well-perfused endocardium with normal anatomic structures exposed in the isolated rabbit heart. There were no observations of dark areas throughout the fluorescently imaged areas, indicating nonischemic tissue preparations. ECG was recorded by two widely spaced RA-LV bipoles (RA-LV) and/or RV-LV bipoles.
Figure 1.
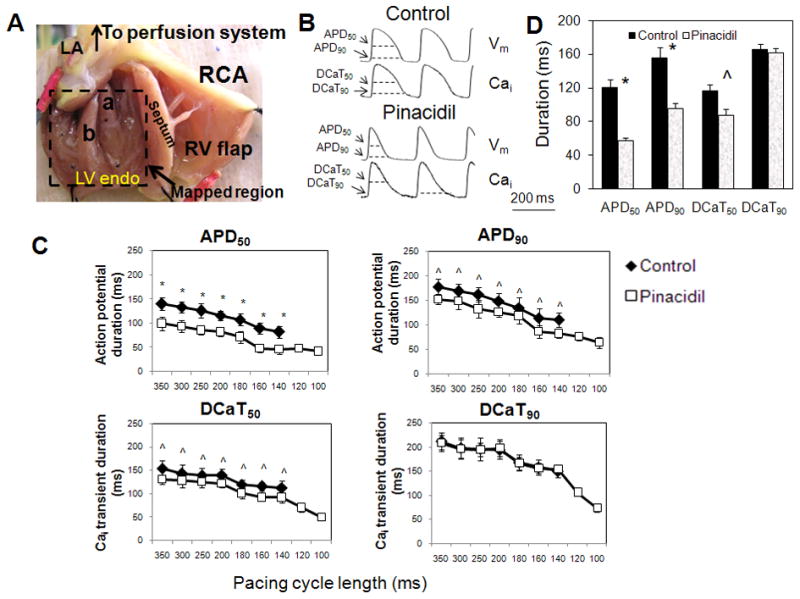
A: Cut-open LV preparation in Langendorff-perfused isolated rabbit hearts. LA: left atrium; RCA: right coronary artery; LV endo: left ventricle endocardium; RV: right ventricle; a: anterior papillary muscle; b: posterior papillary muscle. B: Optical recording traces of Vm and Cai of LV endocardium at pacing cycle length (PCL) of 250 ms. APD50: action potential duration measured to 50% repolarization; APD90: action potential duration measured to 90% repolarization; DCaT50: duration of Cai transient measured to 50% repolarization; DCaT90: duration of Cai transient measured to 90% repolarization. C: Effects of pinacidil on APD50, APD90, DCaT50, and DCaT90 as a function of pacing cycle length between 100 and 350 ms. D: Comparison of the APD and DCaT in control and pinacidil at PCL of 180 ms. *p<0.0001; ^p<0.001
Experimental Protocol
We studied 22 Langendorff-perfused isolated normal rabbit hearts with and without IKATP channels opener, pinacidil (Sigma-Aldrich). We performed a dose ranging study of pinacidil level at 5, 20 40, and 80 μM. At a low level such as 5 μM, pinacidil abbreviated APD90 in the endocardium by 6.3±1.6%. Neither EADs nor arrhythmias were inducible in the endocardial mapped region. With the increase of pinacidil concentrations to 20 and 40 μM, higher degree of APD abbreviations and some triggered activity were observed in the endocardium. Based on the results of dose ranging study, 80 μM of pinacidil was found to be the most effective in shortening APD and inducing EADs and triggered arrhythmias. In group I (n=10), we performed simultaneous dual optical mapping of transmembrane potential (Vm) and Cai on LV endocardium. In group II (n=6), glass microelectrode single cell recording of transmembrane potential (TMP) was performed. In group III (n=6), we examined the effects of BAPTA-AM (20 μM; Sigma), a calcium chelator, on late-phase 3 EAD using dual Vm and Cai optical mapping. For the BAPTA-AM study, the protocol was started with control, then pinancidil, followed by BAPTA-AM perfusion in the preparations. Optical and glass microelectrode measurements were made in each step to assess the effect of BAPTA-AM on arrhythmias. The ventricular endocardium was paced at a pacing cycle length (PCL) between 60 ms and 350 ms (2-ms pulse duration, 2x diastolic threshold) for 29 beats.
High Resolution, Dual Optical Mapping of Vm and Cai Transient
After each isolated heart was allowed to stabilize, heart tissues were double stained with Vm (RH237; Invitrogen) and Cai sensitive fluorescent dyes (Rhod-2 AM, 1.48 μM; Invitrogen). After illumination by excitation laser light at 532 nm, the fluorescence was collected using two cameras (MiCAM Ultima) at 2 ms/frame with spatial resolution of 0.35×0.35 mm2 per pixel. The fluorescence induced by the laser illumination was obtained through a common lens, separated with a dichroic mirror (650 nm cut-off wavelength), and directed to the respective camera with additional filtering (715 nm long-pass for Vm and 580 ± 20 nm band-pass for Cai). Electromechanical uncoupler blebbistatin (Tocris; Ellisville, MO) was added to the perfusate at a concentration of 15 μM to eliminate motion artifacts during optical recording.19
Single Cell TMP Recordings
To confirm the optical mapping data in single myocyte, transmembrane action potential was recorded from LV endocardium using standard glass microelectrodes filled with 3M KCl, as our laboratory previously described.20;21 The TMP data were continuously acquired and stored for analysis using Axoscope software and an Axon Instruments Digidata 1440A acquisition system (Molecular Devices).
Statistical Analysis
Data are presented as mean ± SD. Paired and unpaired Student’s t-tests were used to compared the means between two groups. A p value of < 0.05 was considered significant.
RESULTS
Effect of Pinacidil on the Duration of Action Potential and Cai Transient
Fig. 1B shows optical recordings of Vm and Cai signals in LV endocardium with and without pinacidil. Pinacidil significantly reduced the action potential duration (APD) at 50% (APD50), 90% repolarization (APD90), and the corresponding duration of calcium transient (DCaT) at 50% (DCaT50) in the mapped region. Fig. 1C shows the effect of pinacidil on APD and DCaT as a function of pacing cycle length (PCL) between 100 ms and 350 ms. At all PCLs, pinacidil (80 μM) reduced APD50, APD90, and DCaT50 of LV endocardium by 34.8±7.4%, 17.4±5.7%, 14.7±3.7%, respectively, as compared to control. However, DCaT90 was almost unchanged (reduced by 1.2±0.3%). For example, at PCL of 180 ms (Panel D), the APD50 was reduced from 120±10 ms at baseline to 58±3 ms with pinacidil, while the APD90 from 156±12 ms at baseline to 96±6 ms (p<0.0001). The corresponding DCaT50 repolarization was shortened (116±8 ms at baseline to 88±7 ms; p<0.001). However, DCaT90 was very similar (166±6 ms) compared to control (162±5 ms; p=NS). Due to the APD shortening by pinacidil, rapid pacing was able to capture the myocardium at PCL < 140 ms, with the minimum PCL for 1:1 capture at 60 ms.
Vm and Cai Dynamics of First Beat after the Cessation of Rapid Pacing
The heart preparations with and without pinacidil were paced at a PCL range from 100 to 350 ms. At the cessation of a pacing train, there was a post-pacing pause followed by spontaneous beats (Fig. 2A). At the shortest PCL (140 ms) achievable for both control and experimental groups, the pause period was significantly longer in preparations with pinacidil perfusion (795±122 ms) than control (488±27 ms; p<0.001, Fig. 2B). In the first beat following the pacing trains, both DCaT50 and DCaT90 were found to be lengthened by pinacidil as compared to their last paced beats (Table 1). Additionally, the amplitudes of Cai in the first postpacing beats were found to be increased under pinacidil (Fig. 2A, red lines). To evaluate the Cai amplitude change following rapid pacing, the Cai amplitude of first post-pacing beat was normalized to the Cai signal of the last paced beat. With the perfusion of pinacidil, the Cai level of the first post-pacing beat was increased by 35.2 ± 6.6% as compared to baseline.
Figure 2.
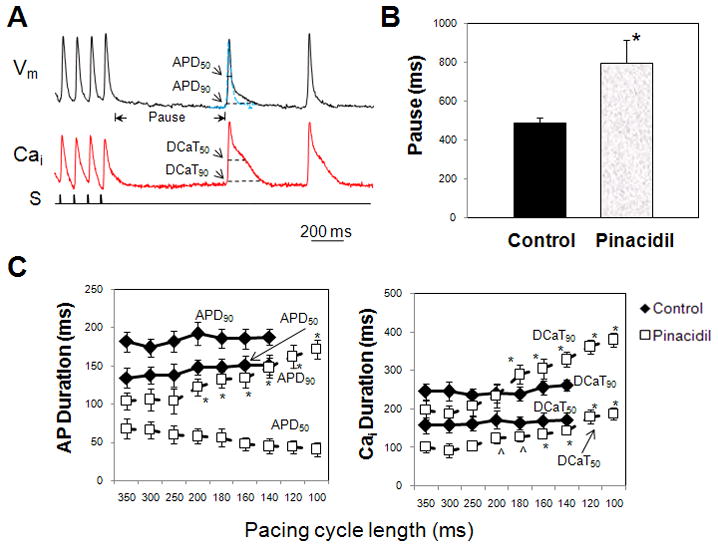
A: Optical signals of Vm and Cai of beats following pause after pacing with pinacidil. Rapid pacing with pinacidil produced an APD prolongation of first post-pacing beat. Blue traces indicate the morphology of action potentials of the last beat in the pacing train. S, pacing stimulus. B: Pinacidil induced a longer pause following pacing train. *p<0.001. C: Effects of pinacidil on APD50, APD90, DCaT50, and DCaT90 of first post-pacing beat when PCL decreased from 350 to 100 ms. Rapid pacing under pinacidil produced a prolongation of APD90 and increase of DCaT50 and DCaT90. *p<0.001; ^p<0.01 as compared to pacing cycle length greater than 250 ms.
Table 1.
Comparison of post-pacing beats’ action potential duration (APD), Cai duration (DCaT) and amplitude to those of the last paced beats in heart preparation of control (n=10), under pinacidil (n=10), and after BAPTA-AM infusion (n=6).
| Control | Pinacidil | BAPTA-AM | |
|---|---|---|---|
| Post-pacing Pause | 488±27 ms | 795±122 ms * | 320±22 ms § |
| Post-pause APD50 prolongation | 5.7±1.1% | 2.6±0.3% | 1.6±0.5% |
| Post-pause APD90 prolongation | 3.2±0.8% | 102±18% * | 2.1±0.4% § |
| Post-pause DCaT50 prolongation | 8±1.4% | 125±19% * | 22±2% § |
| Post-pause Cai Amplitude Increase | -- | 35.2±6.6% * | 9.6±3.9% § |
| EAD | None | In all 10 preparations | None |
| VF induction | None | In all 10 preparations | None |
p<0.001 between control and pinacidil group;
p<0.001 between pinacidil and BAPTA-AM group
The APD90 of first post-pacing beat was also significantly lengthened by pinacidil (Table 1). At baseline, the APD50 and APD90 were not significantly increased as compared to those before pacing (APD50: 140±11 ms at baseline and 148±10 ms in first post-pacing beat, p=NS; APD90: 186±12 ms at baseline and 192±15 ms in first post-pacing beat, p=NS). Under pinacidil at rapid pacing (e.g. 100 ms), the APD90 was increased from 92±16 ms at baseline to 186±21 ms (p<0.001) in the first post-pacing beat. The APD50, however, did not show significant prolongation (38±6 ms at baseline to 39±6 ms; p=NS).
Fig. 2C shows the APD50, APD90, DCaT50 and DCaT90 values of the first post-pacing beats as function of the pacing cycle length between 100 ms and 350 ms. At baseline without pinacidil, the duration of calcium transients did not significantly increase with shortening of PCL. In contrast, with pinacidil, the DCaT50 and DCaT90 of first post-pacing beat were significantly (p<0.01) increased when PCL was shortened to be < 200 ms with a longer postpacing pause. It should be noted that abbreviated repolarization caused by pinacidil allowed very rapid pacing of the hearts. Thus, while the shortest possible PCL was 140 ms at baseline, evaluation of APD and DCaT at PCL < 140 ms was enabled after pinacidil perfusion.
Late-phase 3 EAD Induced by Cai Overloading
While there was no EADs or arrhythmias induced in control, abbreviation of APD by pinacidil perfusion and rapid pacing can lead to arrhythmia inductions. Figure 3A shows an episode of VF induced by late-phase 3 EAD. After the pause at the cessation of a pacing train at PCL of 100 ms, the first spontaneous beat was followed by a late phase 3 EAD (indicated by dashed line) and onset of VF. The EAD was the delayed repolarization that occurred before the dashed line on the Vm tracing. At the EAD initiation, the corresponding Cai signals remained elevated as shown in the simultaneous Vm and Cai optical recordings. When the Vm was depolarized (blue color in the Vm imaging), the same recording site (cross) in the Cai map showed high Cai signal (red color), indicating a persistently elevated Cai level. Fig. 3B shows another example of VF inductions in which Cai level was significantly elevated compared to that of the last paced beat (PCL: 70 ms) and remained high at the onset of late-phase 3 EAD and arrhythmia initiation. Approximately 30% of EADs in the first post-pacing beats led to VF induction where post-pacing Cai amplitude was at least 1.5 folds higher than that of the last paced beat. The incidence of VF induction increased at faster pacing rate. Approximately 62% EAD induced VF occurred after a pacing train at 70–100 ms, 35% at PCL of 110–130 ms, and 3% for PCL > 140 ms. This distribution is consistent with the fact that rapid pacing of the myocardium is known to induce Cai loading.
Figure 3.
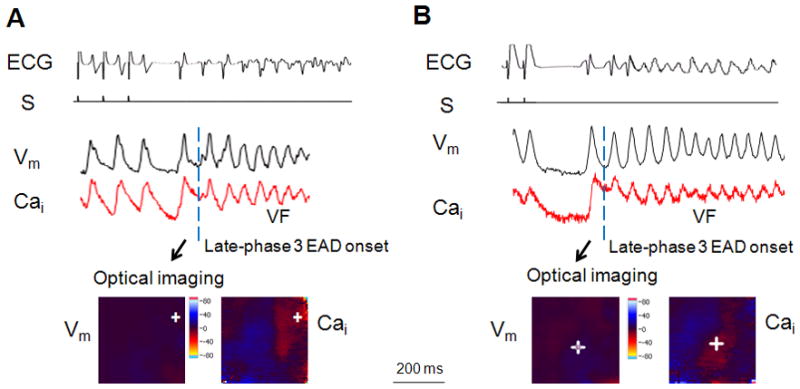
A: Typical Vm and Cai signals of late-phase 3 EAD induced triggered activity and ventricular fibrillation. The late phase 3 EAD is the delayed repolarization that occurred before the dashed line on the Vm tracing. PCL: 90 ms. S: pacing stimulus. B: Vm and Cai recordings for VF onset induced by EADs after post-pacing pause. PCL at 70 ms. Crosse indicates the origin of the EAD in the mapped region.
Transmembrane Potential Recordings Using Standard Glass Microelectrodes
We performed single cell TMP recording using glass microelectrodes in LV endocardium of six isolated rabbit hearts. Table 2 summarizes the experimental results in the single cell TMP study. The APD90 of the first post-pacing beat with pinacidil was increased by 56±5.4% as compared to the last beat of the pacing train, demonstrating a repolarization prolongation. In control, the APD90 was only increased by 2.3±0.7% (p < 0.0001). Fig. 4 shows the TMP recordings with and without pinacidil at PCL of 120 ms. To better compare the TMP morphology before and after pacing, we superimposed the last paced beat (blue traces) with the first post-pacing beat. At baseline, there was no EAD or repolarization prolongation in the first post-pacing beat (Panel A). Nor arrhythmias were induced. With pinacidil infusion, the abbreviated APD led to a prolonged post-pacing pause followed by a delayed repolarization in the first post-pacing beat (Panel B). A total of 41 episodes of repolarization prolongation were recorded in the six preparations, among which about 30% resulted in triggered activities and VF induction as shown in Panel C. The post-pacing pause duration appeared to positively correlate with the incidence of EAD and VF inductions (Panel D). At PCL of 120 ms, the pause was 182±13 ms in control. Pinacidil significantly prolonged the pause duration. When EAD was induced without VF, the pause was 356±21 ms (p < 0.001). When the pause was increased to 495±18 ms which might facilitate Cai overloading,16;22 EADs resulted in triggered activities and VF inductions.
Table 2.
Summary of the post-pacing pause, post-pacing APD, early afterdepolarization (EAD) and ventricular fibrillation (VF) inductions in glass microelectrode study (n=6; PCL: 120 ms).
| # | Post-pacing Pause (ms) | Post-pause APD90 prolongation (%) | No. of EAD episodes | No. of VF episodes | |||||
|---|---|---|---|---|---|---|---|---|---|
| Control | Pinacidil + EAD | Pinacidil + VF | Control | Pinacidil | Control | Pinacidil | Control | Pinacidil | |
| 1 | 160 | 340 | 480 | 2.2 | 53.1 | -- | 8 | -- | 2 |
| 2 | 180 | 330 | 520 | 1.5 | 47.3 | -- | 5 | -- | 1 |
| 3 | 176 | 345 | 473 | 2.6 | 61.5 | -- | 7 | -- | 3 |
| 4 | 193 | 367 | 498 | 3.2 | 54.6 | -- | 6 | -- | 1 |
| 5 | 182 | 380 | 486 | 2.8 | 59.2 | -- | 8 | -- | 3 |
| 6 | 198 | 376 | 510 | 1.8 | 60.4 | -- | 7 | -- | 2 |
| Mean | 182±13 | 356±21 § | 495±18 § | 2.3±0.7 | 56±5.4 § | None | Total: 41 | None | Total: 12 |
p<0.001
Figure 4.
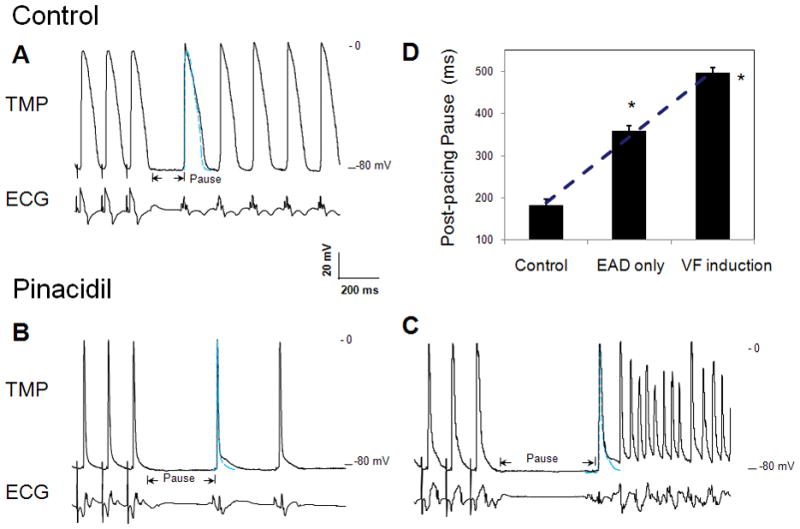
Glass microelectrode recordings of transmembrane potential (TMP) of single myocyte in LV endocardium of isolated rabbit hearts. Blue traces indicate the TMP recording of the last paced beat in the train. A: control study. B and C: pinacidil infusion to produce APD90 prolongation in first post-pacing beat (B) and late-phase 3 EAD induced triggered activities and VF (C). PCL at 120 ms. D: comparison of the post-pacing pause duration in control, EAD, and VF induction. *p<0.0001
BAPTA-AM
In additional six LV endocardial preparations, BAPTA-AM (20 μM), a calcium chelator,23 was added to the Tyrode perfusate and infused for 60 minutes. The maximal Cai amplitude was reduced to 26.4±3.5% (p < 0.001) of the baseline amplitude (Fig. 5A). Neither EAD nor APD prolongation was observed in the first post-pacing beats in any of the six preparations. This is true for data obtained both from optical mapping techniques (Fig. 5B) and from single cell TMP recordings (Fig. 5C). Additionally, in none of the preparations was ventricular arrhythmia inducible after BAPTA-AM. Fig. 5D shows the effect of BAPTA-AM on the APD90 prolongation of the first post-pause beat. With pinacidil only, the APD90 of the first post-pacing beat was increased from 82±9 ms of last pacing beat to 148±13 ms (p<0.0001), showing a late phase repolarization prolongation due to Cai overloading. After BAPTA-AM infusion of the same heart preparation, the APD90 of the first post-pacing beat was only slightly increased from 82±9 ms of last pacing beat to 96±10 ms (p=NS). To evaluate the Cai level, we normalized the Cai signal amplitude of the first post-pacing beat to that of the last paced beat (Fig. 5E). Before BAPTA-AM, the Cai level of the first post-pacing beat was increased by 35.2 ±6.6% as compared to baseline. In contrast, after BAPTA-AM, the Cai increased only by 9.6±3.9% (p<0.001 when compared to Cai increase before BAPTA-AM perfusion). The duration of Cai transient, DCaT50, of the first post-pacing beating was lengthened by approximately 125±19% normalized to the last paced beat during pinacidil infusion. In contrast, the DCaT50 change was almost negligible after BAPTA-AM (22±2%; p<0.0001).
Figure 5.
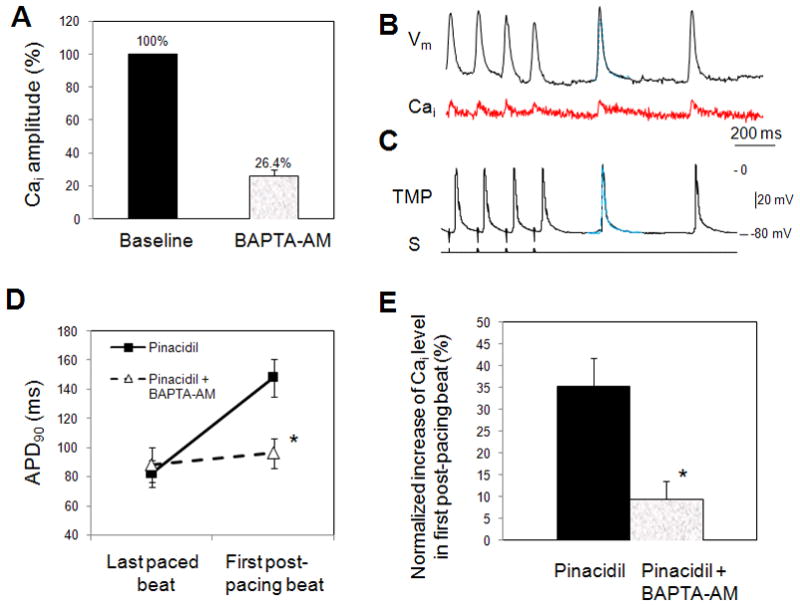
A: BAPTA-AM significantly reduced the maximal Cai amplitudes normalized to baseline. B: Vm and Cai optical signal recordings after BAPTA-AM perfusion showed no APD prolongation in first post-pacing beats. Blue traces indicate the action potential recording of the last paced beats in the train. C: Glass microelectrode TMP recording in BAPTA-AM study. D: Effect of BAPTA-AM on post-rapid pacing APD90. Optical imaging data (n=6). E: BAPTA-AM significantly reduced the corresponding Cai increase in first post-pacing beat. The Cai increase magnitude was normalized to the Cai level in last pacing beat. * p<0.001.
DISCUSSION
We performed simultaneous Vm and Cai optical mapping study of LV endocardium in isolated Langendorff-perfused rabbit hearts. Activating IKATP by infusion of pinacidil at high concentration of 80 μM markedly shortened the APD in the mapped region. Abbreviation of APD allowed very rapid pacing rates of the myocardium, known to induce Cai loading as occurs during VF. Our study provides evidence, for the first time, in support of the hypothesis that very rapid rates of excitation with APD shortened by activation of IKATP can lead to an elevation of Cai and prolongation of APD after pause. These changes in turn contribute to the development of late-phase 3 EAD, triggered activity, and VF induction. To the best of our knowledge, the present study is the first that not only demonstrates late-phase 3 EAD in ventricles, but also elucidates the Cai dynamics during late-phase 3 EADs and arrhythmia inductions by simultaneous Vm and Cai dual-mapping.
Late-phase 3 EAD and VF
Studies suggested that a relatively small increase in the intensity of the IKATP can contribute importantly to the development of electrical inhomogeneity in the ventricles and thus to the genesis of cardiac arrhythmias.24–26 Pinacidil activates IKATP, leading to an abbreviation of APD.4 The present study demonstrated a direct evidence that abbreviation of repolarization coupled with the Cai overload following post-pacing pause can contribute to VF induction. Our findings provided insights into a novel mechanism of ventricular arrhythmogenesis caused by late-phase 3 EAD.
The results indicated a positive relationship between the duration of post-pacing pause and the VF incidence. Approximately 30% of EADs resulted in VF inductions. The pause was significantly longer in those episodes where VFs were induced following EAD in the beats after train. Faster pacing increased the incidence of delayed repolarization and VF initiation. No endocardial reentry was observed during the onset of VF induced by late-phase 3 EADs. This is mainly because the mapping area in this study was focused on the endocardium. Phase 2 reentry is an epicardial phenomenon, for the appearance of which a prominent phase 2 must be present. A Prominent phase 2 can be seen in epicardium but not in endocardium. Although no reentry was present in the mapping area, these two mechanisms are not mutually exclusive, and may both be important in the induction of VF during acute ischemia. Previous studies showed that phase 2 reentry originated on epicardial surface of the right ventricle was a trigger for VF initiation during acute ischemia in canine model.27
Role of Cai Elevation
Our observations in the present Vm and Cai dual mapping study demonstrated, as discussed by Burashnikov and Antzelevitch16, that levels of Cai would peak during the plateau of action potential under control condition, but during the late phase of repolarization in the presence of pinacidil. The relationship between Cai and APD is complex and bidirectional. Cai determines the shape of Vm and APD through various calcium mediated ionic channels, such as the ICaiL and the INCX.13;14 The elevated Cai level are restored during repolarization by the rapid reuptake of Cai into the SR. When the APD is shortened by pinacidil, the duration of Cai transient did not proportionally shorten because the Cai reuptake is not entirely a voltage dependent process. As a result, there was a persistent and heterogeneous Cai elevation after the end of action potential. The elevated Cai level in the first post-pacing beat could activate the electrogenic INCX which is most likely an underlying mechanisms for late-phase 3 EAD development. Additionally, the hypothesis was supported by the suppression of EADs and arrhythmia inductions after infusion of BAPTA-AM over a 60-min period. BAPTA-AM, a calcium chelator, significantly reduced the Cai level and prevented calcium accumulation by rapid pacing rates of excitation (Fig. 5). Consequently, no VF was inducible after BAPTA-AM infusion.
Cai plays an important role in controlling cardiac excitation, implying a direct coupling between Cai and membrane currents. Although cellular Cai overload plays a dominant role, it may not be the only factor responsible for EAD and arrhythmias development. Indeed, rapid pacing and pauses could lead to some degree of Cai overload. Our control study in the absence of pinacidil did not demonstrate EADs and arrhythmias after pacing, although strong post-pacing contractions were recorded both in controls and in the presence of pinacidil. The present study suggested that dramatic APD abbreviation and apparently strong SR Cai release are needed to elicit EADs and arrhythmias in the initial period after rapid pacing train.
Role of APD Shortening and Hyperpolarization
Pinacidil is known to dramatically shorten APD. In addition to APD shortening, pinacidil induced hyperpolarization, which may be responsible for longer pauses that enhanced Cai accumulation. Subsequent large SR calcium release in the first post-rapid-pacing beats played an important role in causing late phase 3 EADs and triggered activity. As discussed elsewhere, APD abbreviation is commonly thought to be an arrhythmogenic substrate by reducing the wavelength for reentry and promoting dispersion of repolarization and refractoriness. Our data clearly demonstrated no reentrant activities in endocardium, thereby we believe for the first time that dramatic APD shortening coupled with a pause-induced strong Cai overload may contribute to ventricular arrhythmogenesis by allowing for the development of late-phase 3 EAD induced triggered activity.
Clinical Implications
The results of the present study have significant clinical implications for recurrent spontaneous VF during cardiopulmonary resuscitation and in short-QT syndrome. In approximately 50% of the patients who receive defibrillation shocks during cardiopulmonary resuscitation, initial successful defibrillation is followed by recurrent spontaneous VF.28 Recurrent spontaneous VF is associated with high morbidity and mortality, and may lead to arrhythmic death in spite of an ICD.29–32 A possible mechanism of spontaneous VF is that the cessation of coronary circulation during VF may result in acute myocardial ischemia, which activates IKATP and shortens the APD. In addition to causing ischemia, VF also results in Cai accumulation. A combination of short APD and Cai accumulation may promote late phase 3 EAD, triggered activities and VF. Short-QT syndrome33 is a rare genetic disorder characterized by short-QT interval but normal duration of mechanical systole,34 suggesting short APD and normal Cai transient. The present study shows that a combination of short APD and prolonged DCaT can lead to spontaneous VF through late-phase 3 EAD. A clinical implication of this present study is that late phase 3 EAD is responsible for recurrent spontaneous VF during cardiopulmonary resuscitation and patients with short-QT syndrome. Prevention of late phase 3 EAD may reduce the incidence of recurrent VF in these patients.
Acknowledgments
This work was supported in part by National Institutes of Health [P01 HL78931, R01s HL78932, 66389, 71140], a Medtronic-Zipes Endowment and an AHA Established Investigator Award [#0540093N].
Footnotes
Dr. Chen reports donation of equipment for animal research from Medtronic, Inc, St. Jude Medical, Cryocath & Cyberonics. Other authors: No disclosures.
Reference List
- 1.Padrini R, Bova S, Cargnelli G, Piovan D, Ferrari M. Effects of pinacidil on guinea-pig isolated perfused heart with particular reference to the proarrhythmic effect. Br J Pharmacol. 1992;105:715–719. doi: 10.1111/j.1476-5381.1992.tb09044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uchida T, Yashima M, Gotoh M, Qu Z, Garfinkel A, Weiss JN, Fishbein MC, Mandel WJ, Chen P-S, Karagueuzian HS. Mechanism of acceleration of functional reentry in the ventricle: effects of ATP-sensitive potassium channel opener. Circulation. 1999;99:704–712. doi: 10.1161/01.cir.99.5.704. [DOI] [PubMed] [Google Scholar]
- 3.Coromilas J, Costeas C, Deruyter B, Dillon SM, Peters NS, Wit AL. Effects of pinacidil on electrophysiological properties of epicardial border zone of healing canine infarcts: possible effects of K(ATP) channel activation. Circulation. 2002;105:2309–2317. doi: 10.1161/01.cir.0000016292.14390.16. [DOI] [PubMed] [Google Scholar]
- 4.Arena JP, Kass RS. Enhancement of potassium-sensitive current in heart cells by pinacidil. Evidence for modulation of the ATP-sensitive potassium channel. Circ Res. 1989;65:436–445. doi: 10.1161/01.res.65.2.436. [DOI] [PubMed] [Google Scholar]
- 5.Traverse JH, Chen Y, Hou M, Li Y, Bache RJ. Effect of K+ATP channel and adenosine receptor blockade during rest and exercise in congestive heart failure. Circ Res. 2007;100:1643–1649. doi: 10.1161/CIRCRESAHA.107.150219. [DOI] [PubMed] [Google Scholar]
- 6.Lee TM, Lin MS, Chang NC. Effect of ATP-sensitive potassium channel agonists on ventricular remodeling in healed rat infarcts. J Am Coll Cardiol. 2008;51:1309–1318. doi: 10.1016/j.jacc.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 7.Wu S, Hayashi H, Lin SF, Chen PS. Action Potential Duration and QT Interval During Pinacidil Infusion in Isolated Rabbit Hearts. J Cardiovasc Electrophysiol. 2005;16:872–878. doi: 10.1111/j.1540-8167.2005.40811.x. [DOI] [PubMed] [Google Scholar]
- 8.Chi L, Uprichard ACG, Lucchesi BR. Profibrillatory actions of pinacidil in a conscious canine model of sudden coronary death. J Cardiovasc Pharmacol. 1990;15:452–464. doi: 10.1097/00005344-199003000-00016. [DOI] [PubMed] [Google Scholar]
- 9.Di Diego JM, Antzelevitch C. Pinacidil-induced electrical heterogeneity and extrasystolic activity in canine ventricular tissues. Does activation of ATP-regulated potassium current promote phase 2 reentry? Circulation. 1993;88:1177–1189. doi: 10.1161/01.cir.88.3.1177. [DOI] [PubMed] [Google Scholar]
- 10.Merillat JC, Lakatta EG, Hano O, Guarnieri T. Role of calcium and the calcium channel in the initiation and maintenance of ventricular fibrillation. Circ Res. 1990;67:1115–1123. doi: 10.1161/01.res.67.5.1115. [DOI] [PubMed] [Google Scholar]
- 11.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res. 2005;96:459–466. doi: 10.1161/01.RES.0000156891.66893.83. [DOI] [PubMed] [Google Scholar]
- 12.Omichi C, Lamp ST, Lin SF, Yang J, Baher A, Zhou S, Attin M, Lee MH, Karagueuzian HS, Kogan B, Qu Z, Garfinkel A, Chen PS, Weiss JN. Intracellular Ca dynamics in ventricular fibrillation. Am J Physiol Heart Circ Physiol. 2004;286:H1836–H1844. doi: 10.1152/ajpheart.00123.2003. [DOI] [PubMed] [Google Scholar]
- 13.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 14.Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea-pig hearts: mechanisms underlying concordant alternans. J Physiol. 2000;529 (Pt 1):171–188. doi: 10.1111/j.1469-7793.2000.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stefenelli T, Wikman-Coffelt J, Wu ST, Parmley WW. Intracellular calcium during pacing-induced ventricular fibrillation. Effects of lidocaine. J Electrocardiol. 1992;25:221–228. doi: 10.1016/0022-0736(92)90007-m. [DOI] [PubMed] [Google Scholar]
- 16.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 17.Zaugg CE, Wu ST, Barbosa V, Buser PT, Wikman-Coffelt J, Parmley WW, Lee RJ. Ventricular fibrillation-induced intracellular Ca2+ overload causes failed electrical defibrillation and post-shock reinitiation of fibrillation. J Moll Cell Cardiol. 1998;30:2183–2192. doi: 10.1006/jmcc.1998.0777. [DOI] [PubMed] [Google Scholar]
- 18.Tang L, Hwang GS, Hayashi H, Song J, Ogawa M, Kobayashi K, Joung B, Karagueuzian HS, Chen PS, Lin SF. Intracellular calcium dynamics at the core of endocardial stationary spiral waves in Langendorff-perfused rabbit hearts. Am J Physiol Heart Circ Physiol. 2008;295:H297–H304. doi: 10.1152/ajpheart.00137.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fedorov VV, Lozinsky IT, Sosunov EA, Anyukhovsky EP, Rosen MR, Balke CW, Efimov IR. Application of blebbistatin as an excitation-contraction uncoupler for electrophysiologic study of rat and rabbit hearts. Heart Rhythm. 2007;4:619–626. doi: 10.1016/j.hrthm.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 20.Ikeda T, Yashima M, Uchida T, Hough D, Fishbein MC, Mandel WJ, Chen P-S, Karagueuzian HS. Attachment of meandering reentrant wave fronts to anatomic obstacles in the atrium. Role of the obstacle size. Circ Res. 1997;81:753–764. doi: 10.1161/01.res.81.5.753. [DOI] [PubMed] [Google Scholar]
- 21.Kim Y-H, Yashima M, Wu T-J, Doshi R, Chen P-S, Karagueuzian HS. Mechanism of procainamide-induced prevention of spontaneous wave break during ventricular fibrillation. Insight into the maintenance of fibrillation wave fronts. Circulation. 1999;100:666–674. doi: 10.1161/01.cir.100.6.666. [DOI] [PubMed] [Google Scholar]
- 22.Patterson E, Lazzara R, Szabo B, Liu H, Tang D, Li YH, Scherlag BJ, Po SS. Sodiumcalcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins. J Am Coll Cardiol. 2006;47:1196–1206. doi: 10.1016/j.jacc.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 23.Tsien RY. A non-disruptive technique for loading calcium buffers and indicators into cells. Nature. 1981;290:527–528. doi: 10.1038/290527a0. [DOI] [PubMed] [Google Scholar]
- 24.Antzelevitch C, Sicouri S, Litovsky SH, Lukas A, Krishnan SC, Di Diego JM, Gintant GA, Liu D-W. Heterogeneity within the ventricular wall. Electrophysiology and pharmacology of epicardial, endocardial, and M cells. Circ Res. 1991;69:1427–1449. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- 25.Smeets JL, Allessie MA, Lammers WJ, Bonke FI, Hollen J. The wavelength of the cardiac impulse and reentrant arrhythmias in isolated rabbit atrium. The role of heart rate, autonomic transmitters, temperature, and potassium. Circ Res. 1986;58:96–108. doi: 10.1161/01.res.58.1.96. [DOI] [PubMed] [Google Scholar]
- 26.Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 27.Yan GX, Joshi A, Guo D, Hlaing T, Martin J, Xu X, Kowey PR. Phase 2 reentry as a trigger to initiate ventricular fibrillation during early acute myocardial ischemia. Circulation. 2004;110:1036–1041. doi: 10.1161/01.CIR.0000140258.09964.19. [DOI] [PubMed] [Google Scholar]
- 28.Hess EP, White RD. Out-of-hospital cardiac arrest in patients with cardiac amyloidosis: presenting rhythms, management and outcomes in four patients. Resuscitation. 2004;60:105–111. doi: 10.1016/j.resuscitation.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Ohgo T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, Aihara N, Kamakura S, Ohe T, Shimizu W. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm. 2007;4:695–700. doi: 10.1016/j.hrthm.2007.02.014. [DOI] [PubMed] [Google Scholar]
- 30.Exner DV, Pinski SL, Wyse DG, Renfroe EG, Follmann D, Gold M, Beckman KJ, Coromilas J, Lancaster S, Hallstrom AP. Electrical storm presages nonsudden death: the antiarrhythmics versus implantable defibrillators (AVID) trial. Circulation. 2001;103:2066–2071. doi: 10.1161/01.cir.103.16.2066. [DOI] [PubMed] [Google Scholar]
- 31.Maury P, Couderc P, Delay M, Boveda S, Brugada J. Electrical storm in Brugada syndrome successfully treated using isoprenaline. Europace. 2004;6:130–133. doi: 10.1016/j.eupc.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 32.Nademanee K, Taylor R, Bailey WE, Rieders DE, Kosar EM. Treating electrical storm: sympathetic blockade versus advanced cardiac life support-guided therapy. Circulation. 2000;102:742–747. doi: 10.1161/01.cir.102.7.742. [DOI] [PubMed] [Google Scholar]
- 33.Gaita F, Giustetto C, Bianchi F, Wolpert C, Schimpf R, Riccardi R, Grossi S, Richiardi E, Borggrefe M. Short QT Syndrome: a familial cause of sudden death. Circulation. 2003;108:965–970. doi: 10.1161/01.CIR.0000085071.28695.C4. [DOI] [PubMed] [Google Scholar]
- 34.Schimpf R, Antzelevitch C, Haghi D, Giustetto C, Pizzuti A, Gaita F, Veltmann C, Wolpert C, Borggrefe M. Electromechanical coupling in patients with the short QT syndrome: further insights into the mechanoelectrical hypothesis of the U wave. Heart Rhythm. 2008;5:241–245. doi: 10.1016/j.hrthm.2007.100.015. [DOI] [PMC free article] [PubMed] [Google Scholar]