

Conspectus
Interactions between ions and aromatic rings are now a mainstay in the field of supramolecular chemistry. The prototypical cation-π interaction, first characterized in the gas phase, is now well known as an important contributor to protein structure, enzyme function, and as a noncovalent force found in many synthetic systems. The complementary “anion-π interaction” – defined as an electrostatic attraction between an anion positioned over the centroid of an aromatic ring – has recently emerged as another reversible ion-π interaction in supramolecular systems. This type of interaction could offer new selectivity in binding poorly basic, strongly solvated anions and may also affect structure, biological function, and anion transport.
This Account describes our group’s efforts in ion-π interactions in two areas. We first describe a series of self-assembled Group 15 (pnictogen)-thiolate complexes, all featuring prominent cation-π interactions between the trivalent pnictogen and an aromatic ring of the ligand. This structural feature appears to stabilize a variety of self-assembled dinuclear macrocycles, dinuclear M2L3 cryptand-analogs, and a tetranuclear As4L2 metallocyclophane. These complexes are all remarkably robust and feature intramolecular cation-π interactions, which suggest that these interactions could be an important feature in ligand design for the Group 15 elements. We also highlight our efforts to characterize the interaction between anions and electron-deficient aromatic rings in solution. Complementary crystallographic and computational studies suggest that off-center weak-σ interactions play the dominant role in stabilizing the anion-arene adducts unless an acidic CH bond is present to participate in favorable CH···anion hydrogen bonds. In solution the weak-σ complexes show downfield shifts of the proton resonances in their NMR spectra. With more polarizable anions such as bromide and iodide, we also observe anion binding by UV/Vis spectroscopy. Initial solution studies suggest these reversible interactions are weak in organic solvents, but the Hofmeister bias in anion binding could be mitigated, if not reversed, in the halides using these anion-π type interactions.
Introduction
The use of reversible interactions between ions and aromatic rings as a directing force for self-assembly and/or ion binding continues to be an active area of investigation.1 This Special Issue on Aromatic Interactions points to the high-level of research activity in these areas. Specifically, cation-π, anion-π/weak-σ, and a variety of secondary bonding interactions between main group cations and aromatic rings comprise a number of such ion-π interactions used in supramolecular chemistry. Cation-π interactions are now well-recognized as important structural features in proteins,1a,b and it is becoming increasingly apparent that anions may serve as complementary attractive partners to electron-deficient aromatic rings.2 Such weak, reversible interactions are conceptually related to their covalent congeners, the more well-established metallocene, and Meisenheimer complexes.
We entered this area with a program to design specific chelators for toxic ions, primarily As(III), and discovered a preponderance of Group15 cation-π interactions in our supramolecular assemblies. This became a key component of our ligand design for toxic ions (vide infra). At the same time we were investigating these well-known main group ion-π interactions,1d several papers also rekindled interest in understanding interactions between anions and aromatic rings.3 We therefore sought to design receptors that could take advantage of these so-called anion-π interactions. This review is a comprehensive summary of our work in the area of ion-π interactions, in the context of two research directions: i) the development of a supramolecular ligand design strategy for main group cations that features cation-π interactions as an important design component, and ii) the design of receptors that bind anions in solution through interactions with electron-deficient aromatic rings. This Accounts article is not meant to be a review of other work in these quickly moving fields; rather, the reader is referred to other articles in this Special Issue, and we provide references in the subsequent sections to representative reviews with more in-depth surveys of these ion-π interactions.
Main Group Supramolecular Coordination Chemistry – Group 15
Over the last two decades, a diverse compilation of supramolecular coordination complexes emerged from self-assembly between a d-block, or less commonly an f-block metal, and a multidentate ligand. The resulting metal-ligand self-assemblies typically have square-planar, tetrahedral, or octahedral coordination geometries. Development of self-assembled complexes featuring main group metalloid ions has received less attention, perhaps due to the unusual and sometimes unpredictable coordination geometries of main group elements. The result is a lack of highly specific chelators for toxic ions such as arsenic (As). With this in mind, we launched a program in 2004 to investigate a “supramolecular approach” to toxic metal coordination chemistry and initially targeted arsenic by employing stabilizing secondary bonding contacts, more specifically, an ion-π interaction, to direct self-assembly.
Brief Overview of Interactions Between Main Group Cations and Aromatic Rings
Recently the role of weak intermolecular forces for complex formation between arenes and heavier metals of Group 13–16 was highlighted.1d In low-oxidation states, post-transition metals with stereochemically active lone pairs, such as As, Sb and Bi, produce stable arene complexes. Recent measurements of binding enthalpies between SbCl3 and neutral arenes (ca. 5–10 kcal mol−1) are in the typical range for other “weak interactions”1d, suggesting these interactions might serve as a motif for directing the formation of supramolecular self-assemblies. This weak, predominately electrostatic interaction has been both described as a traditional Lewis acid (main group metalloid) to π-base (arene ring) interaction1d (Figure 1) and as electron transfer between As (lone pair) to the π-aryl ring.4 In the latter description, calculations show the intrinsic molecular dipole of AsCl3 induces polarization in the arene ring upon complexation, revealing a weak sharing of electrons between the As center and each C atom of the arene ring (Aslone-pair-πaryl interactions). In the Lewis acid to π-base interaction, sometimes referred to as “Menshutkin complexes”, donor orbitals on the arene interact with acceptor orbitals on the metal center. Such a description explains the shortening of the metalloid-π distances in ECl3·arene complexes (E = As, Sb, Bi), as the metalloids become more Lewis acidic going down the group. Along with this, it is important to note dispersion forces may become dominant for heavier congeners of the Group.5
Figure 1.

Illustrative depiction of pnictogen-π interactions in supramolecular assemblies. (a) View down the As-As axis of As2(L)3 showing the possibility of interactions (red arrows) between the π-systems of the arene spacers with the σ* orbitals of As. (b) Locations of σ* orbitals in the primary coordination sphere about an As(III) center.
The evidence illustrating the significance of πaryl interactions upon complexation with Group 15 metal cations sparked our interest in applying this weak, and reversible force as part of a design strategy for forming self-assembled supramolecular main group coordination complexes. The self-assembly of the macrocycles, cryptands, and metallocycles described below all proceed with the apparent assistance of stabilizing As-πaryl interactions.
Pnictogen-π Interactions and “Supramolecular” Ligand Design
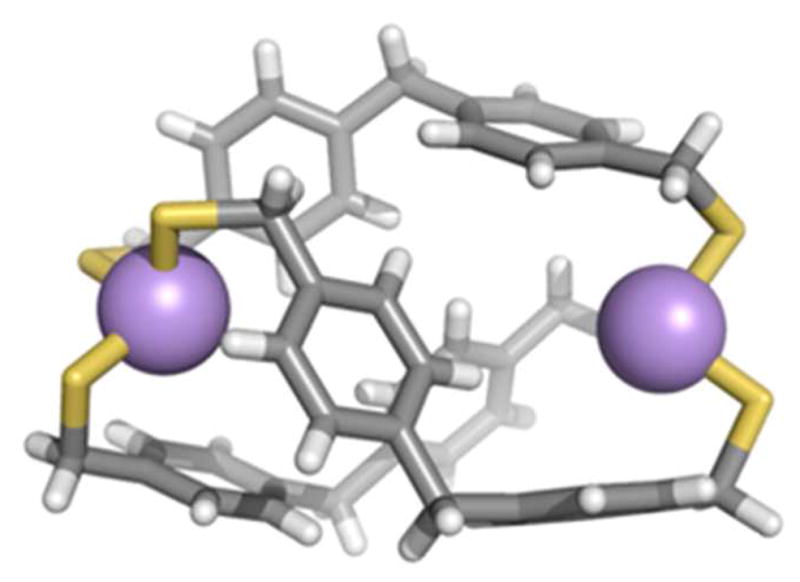
Our supramolecular design strategy focuses on the self-assembly of an As(III) source and an organothiol ligand (Chart 1). The As-thiolate bond is attractive due to its lability and reversibility, as well as the predictable trigonal-pyramidal coordination geometry of As(III)-thiolates. Our first approach for self-assembled As(III)-organothiolate complexes exploiting a stabilizing As-π interaction utilized a rigid, multidendate 1,4-bis(mercaptomethyl)benzene (H2L) ligand.6 When treated with stoichiometric amounts of AsCl3 and KOH in THF/methanol, a discrete dinuclear As2L3 complex emerges, with As positioned endo relative to the ligands, allowing for the stereochemically active lone pair to be directed inside the small cavity of the cryptand (Scheme 1). The crystal structure revealed six short As-Caryl contacts with distances between 3.18 and 3.33 Å, indicating a stabilizing metal-π interaction between the arene and the As lone pair (Figure 2). DFT calculations of this weak force in an AsCl3-benzene dimer suggest the As-π interaction is strong (7.3 kcal mol−1), with a shallow potential for its preferred geometry.
Chart 1.

Ligands used to prepare pnictogen-containing supramolecular assemblies.
Scheme 1.

Synthesis of As2L3 assemblies.
Figure 2.

(a) ORTEP representation of the X-ray crystal structure of As2L3. (b) Wireframe representations with arsenic atoms shown as space-filling spheres.
In the absence of base, a macrocyclic As2L2Cl2 complex forms as a mixture of syn and anti diastereomers as seen by 1H NMR spectroscopy.7 However, either stereoisomer can be selectively isolated by altering crystallization conditions (Figure 3). Structural analysis of the anti conformer displays two As-Cl bonds pointing in opposite directions. In the anti conformation, the aromatic rings are parallel and the As(III) centers twist allowing each As(III) center, providing a short As1-As1a separation (4.65 Å) and decreased As-Caryl distances (3.16 Å, far smaller than the sum of the van der Waals radii of 3.70 Å). In the syn-As2L2Cl2 diastereomer both As-Cl bonds point in the same direction. In contrast to its diastereomeric partner, the two As(III) centers in the syn conformation align along the ligand axis (As1-As1a = 5.02 Å), and correspondingly longer As-π interactions were observed (avg. As-Caryl = 3.25 Å). When molecular mechanics modeling was examined, both syn-As2L2Cl2 and anti-As2L2Cl2 macrocycles were shown to have more torsional freedom to shift the As ion away from the aryl rings than the As2L3 cryptand.8 Nevertheless, both macrocycles show closer As-π contacts than the cryptand, indicating As-π interactions might play a more significant role in the self-assembly of the macrocycle than the apparently more thermodynamically stable cryptand. Interestingly, 1H NMR spectroscopy reveals a slow equilibrium between syn and anti, slightly favoring the formation of the anti macrocycle (diastereomeric ration ca. 5:4). This suggests the shorter As-π interaction observed in the solid state might persist in solution to provide a stronger stabilization to the anti-macrocycle.
Figure 3.

ORTEP representation of the single crystal X-ray structure of syn-As2L2Cl2 and anti-As2L2Cl2 macrocycle with 50% probability ellipsoids and hydrogen atoms omitted.
The directing role of As-π interactions was further investigated by expanding the choice of organothiol ligand to achiral, isomeric bis(mercaptomethyl)naphthalene ligands (H2Lnap1,4, H2Lnap1,5, H2Lnap2,6).9 As before, a secondary As-π interaction directs the formation of each syn and anti macrocycle; however, the choice of naphthalene ligand dictates the syn-to-anti ratio. The variation in substitution pattern alters the environment of the As-Cl bond upon complexation, and directs formation of the major diastereomer: the isomer that places the chlorine ligands farthest from the electron-rich arene ring is favored. Given this repulsive interaction, crystallization gave the following expected isomers as the preferred products: a nearly equal mixture of anti-As2(Lnap2,6)2Cl2 and syn-As2(Lnap2,6)2Cl2, primarily anti-As2(Lnap1,5)2Cl2, and lastly, syn-As2(Lnap1,4)2Cl2, all of which minimize the unfavorable steric interactions (Figure 4). The As-As distances vary (7.45, 5.64, 4.66 Å) for each macrocycle, but the As-Caryl distances remain comparable (3.30, 3.22, 3.14 Å).
Figure 4.

ORTEP (30% probability ellipsoids) and wireframe representations of single-crystal X-ray structures for (a) anti-As2(Lnap2,6)2Cl2, (b) anti-As2(Lnap1,5)2Cl2, and (c) syn-As2(Lnap1,4)2Cl2·CHCl3 macrocycles.
We also sought to explore the host-guest chemistry of macrocycles featuring larger interior cavities assembled from extended ligands (e.g., biphenyl, stilbene, anthracene linkers). The assembly expansion revealed no effect on the As-π interaction, although the presence of the interaction itself may inhibit guests from fitting entirely within the cavity.10 In each case, the macrocycles were too small to completely surround a guest molecule, but were large enough to accommodate one or two guests just on the periphery of the macrocyclic cavity (Figure 5). Each macrocycle displays multiple intramolecular As-π interactions. Self-assembly of H2LOMe1,4 and AsCl3 provided a macrocycle wide enough to host a guest due to the protruding methoxy substituents (Figure 6). When crystallized from a solvent that serves as a suitable guest, such as p-xylene, the macrocycle forms a homodimer [(syn-As2(LOMe1,4)2Cl2)2·p-xylene], where the ligand walls of one macrocycle tilt out of parallel allowing one end of the cavity to open and share a guest molecule with the cavity of a second macrocycle (Figure 7). A slight tilt of one ligand from parallel with the other clearly indicates guest inclusion in the macrocyclic dimeric “capsule” does not destroy the As-π interaction, and therefore the guest alone may control the diastereoselectivity during the crystallization. When no suitable guest molecules are present, the anti macrocycle is crystallized exclusively and the As-π interaction is shorter than in any other macrocycles reported to date (As-Caryl = 3.11 Å), presumably due to the increased π-basicity of H2LOMe1,4.
Figure 5.

(a) The packing diagram of [anti-As2(Lstilbene)2Cl2·benzene] along the macrocyclic axis. (b) Space-filling representation of inclusion complex.
Figure 6.

ORTEP (30% probability ellipsoids) representation of single-crystal X-ray structure of (a) anti-As2(LOMe1,4)2Cl2. Stick representation showing the packing from (b) side and (c) top views.
Figure 7.

ORTEP (30% probability ellipsoids) and space-filling representations of single-crystal X-ray structure of (a,b) [(syn-As2(LOMe1,4)2Cl2)2·p-xylene] and (c,d) [(syn-As2(LOMe1,4)2Cl2)2·toluene].
To expand preliminary work probing the role of steric repulsions in self-assembly of the macrocycles, two types of dimethyl-substituted phenyl ligands were synthesized: H2LMe2,5 and H2LMe2,3.11 Each showed facile formation of the anti macrocycle indicating the bulky methyl groups do not disrupt the apparently strong directing effect of the As-π interaction. In the crystal structure of anti-As2(LMe2,5)2Cl2 the two chloride ligands point away from the methyl groups, avoiding unfavorable steric repulsions while maintaining favorable As-π interactions (Figure 8a, c). Preferential formation of the sterically disfavored anti-As2(LMe2,3)2Cl2 macrocycle was surprising since one of the anti-conformers has one chloride ligand folded into the cavity of the macrocycle to avoid steric repulsions with the methyl groups (Figure 9b, d). Rather than participating in potential weak anion-π interactions, this “folded in” chloride ligand participates in secondary bonding interactions (SBIs) with an As center in an adjacent macrocycle. As anticipated, this conformation does not disrupt intramolecular As-π interactions, maintaining As-Caryl distances of 3.27 and 3.32 Å with the thiol ligand (Figure 9).
Figure 8.

Molecular structure in stick and space-filling representations of (a, c) As2(LMe2,5)2Cl2 and (b, d) As2(LMe2,3)2Cl2.
Figure 9.

A fragment of the crystal packing structure of As2(LMe2,3)2Cl2 showing As-Cl, As-S, and S-S SBIs (dash lines).
Another example where this interaction imparts stereochemical direction upon self-assembly of organo-arsenic coordination complexes is in the structure of a tetranuclear arsenic(III) As4(Ltet)2Cl4 S4 metallacyclophane (Chart 1, Figure 10).12 This unusual S4-symmetric coordination complex co-crystallizes with two cis- and trans-As2LtetCl2 intermediates in a single crystal. The S4-symmetry complex houses four As-π interactions per aryl ring (As-Caryl range: 3.24 and 3.45 Å) with the four As(III) metalloids pointing their lone pairs directly into the cavity, in an endo configuration. Surprisingly, no intramolecular As-π contacts were observed in the cis and trans As2LtetCl2 complexes (Figure 9).
Figure 10.

Wireframe representations of the three discrete complexes observed in a single crystal: (a) trans-[As2LtetCl2], (b) cis-[As2LtetCl2], and (c) the [As4(Ltet)2Cl4] metallacyclophane.
These supramolecular approaches to coordination chemistry helped us develop generalized rules for designing ligands for specific arsenic(III) coordination.13 However, it was uncertain if the ligands always needed to be as rigid as those previously described. To probe this constraint, diphenylmethane ligand H2Ldpm (4,4′-dimercaptomethyldiphenylmethane) was utilized,13b where the methyl spacer between the two phenyl rings would possibly lengthen the cavity of the cryptand and the added flexibility might also widen it. The solid state structure of As2(Ldpm)3 reveals a distorted mesocate in which the three ligands are unevenly distributed around the metalloid centers. The diphenylmethane aryl rings are twisted to direct their edges toward the inner cavity, resulting in an “imploded” cryptand that fills its own cavity interior (Figure 11). Despite the distorted geometry, the structure still maintains multiple intramolecular As-π interactions, with one ligand folded into the cavity shortening the metal-metal distance to 9.19 Å.
Figure 11.
Molecular structure of As2(Ldpm)3 shown as wireframe; spheres represent arsenic atoms.
Successful incorporation of arsenic into supramolecular self-assemblies led us to explore other Group 15 metalloids. Early work in our lab revealed two dinuclear complexes, Sb2L3 cryptand and Sb2L2Cl2 macrocycle, can be formed upon treatment of H2L with SbCl3, featuring intramolecular Sb-π contacts (Sb-Caryl contact distances range from 3.20 to 3.61 Å.14 Self-assembly of related bismuth- and antimony-containing cryptands, E2(Lnap1,4)3 was achieved using H2Lnap1,4, DIPEA, and ECl3 (E = Bi, Sb).15 The metalloid-metalloid (E-E) distances decrease from As2(Lnap1,4)3 (5.11 Å) to Sb2(Lnap1,4)3 (4.83 Å) to Bi2(Lnap1,4)3 (4.68Å) to compensate for longer E-S bonds and smaller S-E-S angles, thus allowing the ligands to retain near identical positions in each cryptand. Previous research1 reports E-π interactions to be stronger for larger pnictogens, typically resulting in a decrease in the E-Caryl distance. Due to the constraints of the cryptand structure, the trend is reversed; the shortest observed E-Caryl distance increases slightly from As (3.30 Å) to Sb (3.34 Å) to Bi (3.36 Å) complexes. This does not necessarily indicate a weakening of the E-π interactions, but an increasing ionic radius of E giving the longer E-S bond lengths and constricted metal coordination inside the cavity. In addition to E-π interactions, all of the cryptands formed from H2Lnap1,4 maintain favorable intramolecular edge-to-face aromatic interactions giving the complex a propeller twist (Figure 12).
Figure 12.

Stick and space-filling representations of the molecular structures of symmetric cryptands (a, d) As2(Lnap1,4)3, (b, e) Sb2(Lnap1,4)3, and (e, f) Bi2(Lnap1,4)3.
The dynamic solution behavior of these supramolecular complexes allowed us to experimentally measure the relative stability of each complex by subjecting each pnictogen cryptand to a different trivalent Group 15 metalloid. Upon addition of AsCl3 to dissolved crystals of the Sb2(Lnap1,4)3 cryptand, a transmetalation reaction was observed to provide the As2(Lnap1,4)3 cryptand within a week. The weaker Sb-Cl bonds likely help drive the formation of the more thermodynamically stable As cryptand. This “transmetalation” reaction has since allowed us to synthesize a new phosphorus-containing supramolecular complex, which was unattainable by direct methods (Figure 13).15 The P2(Lnap1,4)3 crystal structure reveals the same endohedral coordination as the As, Sb and Bi cryptands with the phosphorous lone pairs directed inside the small cryptand cavity. Despite close P-Caryl contacts (3.54 Å, sum of the van der Waals radii: 3.50 Å), the presence of P-π interactions is not apparent. Such interactions are typically only observed in more Lewis acidic cationic phosphorus complexes.16
Figure 13.

(a) Transmetalation of Sb2(Lnap1,4)3. (b, c) Stick and space-filling representations of the molecular structures of symmetric cryptands P2(Lnap1,4)3.
Surprisingly, closer inspection of the homometallic cryptands E2(Lnap1,4)3 (E = As, Sb, Bi) in solution identified the presence of a second, less symmetric species, asym-E2(Lnap1,4)3, in which one ligand has “flipped” perturbing the C3 symmetry of the complex (Figure 14).17 Each conformer maintains numerous intramolecular E-π interactions. The asymmetric species is in equilibrium with the C3h-symmetric cryptand is solution and is increasingly favored in the heavier pnictogens (<5% component for As, 47% for Sb, 60% for Bi).
Figure 14.

Cartoon representations of (a) symmetric E2(Lnap1,4)3 (left) and asym-E2(Lnap1,4)3 (right) in which the ligands are represented by arrows. The DFT-calculated structure of asym-Bi2(Lnap1,4)3 is shown in (b) a stick and (c) space-filling representation. The “flipped” ligand is indicated in red, blue arrows indicate the protons correlated by their NOEs in solution.
We also have pursued a systematic study of E2L3 cryptands formed from a variety of different organic ligand backbones, each with small structural alterations to change the electronics, cavity size, and flexibility of the aromatic framework (H2Lanth, H2Ltolane, H2Lstilbene, H2Ldpm, H2Ldpe, Chart 1).18 Despite the diversity of the ligands, each of the cryptands synthesized showed clear As-π interactions, with varied metal-metal distances depending on the distance between the two thiolate functional groups (5.03 – 12.28 Å). Again, the same somewhat surprising trend appears in comparing these As2L3 cryptands to their Sb2L3 congeners: the Caryl-E contacts are approximately the same distance in the arsenic complexes (3.25 – 3.31 Å) as in the antimony cryptands (3.31 – 3.36 Å), despite the higher Lewis acidity of Sb. As a comparison, while the S-S distances in H2L and H2Lnap1,4 are identical, the Sb-Sb distances in the corresponding cryptands vary dramatically (Figure 15b, d): 4.30 Å for Sb2L3 compared to 4.83 Å for Sb2(Lnap1,4)3. This greater than 0.5 Å difference is even more remarkable when one considers that the As-As distances in As2L3 and As2(Lnap1,4)3 vary by less than 0.1 Å. The result is a profound helical twist in the Sb2L3 cryptand (Figure 15).
Figure 15.

Stick representations of the molecular structures of previously reported E2L3 cryptands: (a) As2L3, (b) Sb2L3, (c) As2(Lnap1,4)3, (d) Sb2(Lnap1,4)3.
The observed endohedral directionality of the As centers in our complexes can perhaps be attributed to the intramolecular As-π interaction, meaning its stability (whether endo or exo at the metal center for similar systems with stereochemically active lone pairs) depends on the macrocycle’s ring size and the steric strain imposed. The presence of such short interactions in the pnictogen macrocycles and cryptands could result from strong mechanical coupling in the self-assembled complex due to the constraints imposed by the rigid ligand framework. Interestingly, the endo conformation also appears to be the more stable geometry in de novo designed and native proteins that bind As(III) as well, suggesting another mechanism for outer sphere metal coordination may explain some modes of metal ion toxicity in vivo.19
Anion-Arene Interactions – Historical Overview
Multiple binding modes of anions with arenes have been demonstrated through experimental and theoretical studies. The ‘Meisenheimer’ or strong-σ complex, known since its isolation in 1902, results from the nucleophilic addition of an anion to an electron-deficient aromatic ring.20 In a gas phase mass spectrometry study, binding energies were determined for a series of arenes hydrogen bonded to Cl−, including hexafluorobenzene, for which a binding energy was surprisingly still found despite the lack of aryl hydrogens.21 Initial calculations assigned this binding to an interaction of the halide anion over the center of the arene.22 Further advances on these studies, at the time deemed “anion-π” interactions, were not made until 2002, when several papers inspired renewed interest and demonstrated favorable interactions between select anions and aromatic rings through theoretical calculations.3 These four theoretical papers spawned new investigations probing anion-arene interactions, in particular anion-π interactions and sparked a lively discussion on the nature and definition of the interaction.2 For the purposes of this review, anion-π will be used to define an interaction of an anion over the centroid of an aromatic ring.
A large effort has focused on observing anion-arene interactions in solution, particularly anion-π. The nature of the interaction itself makes this a difficult task because binding of anions to the π-system is typically too weak for detection via NMR spectroscopy. As a result, systems have been investigated which allow observation of such interactions through other techniques. More recently, a perfluorobenzene ditopic calixarene-based system showed Cl− ion transport across a lipid membrane whereas no detectable Cl− binding was seen via 19F NMR spectroscopy.23 For more examples, we direct the reader to the recent review listed as reference 2c. Our studies probing anion-arene interactions were performed primarily with the goal of quantifying the strength and selectivity of anion-π interactions in solution and are highlighted below.
Interactions Between Anions and Electron-Deficient Arenes: Our Investigations Using Combined Crystallographic, Computational, and Solution Phase Studies
Neutral electron-deficient sulfonamide receptor 1 and control receptor 2 were synthesized to investigate anion-π interactions in solution (Figure 16a).24 1H NMR spectroscopic titrations were used to measure the association constants of receptors 1 and 2 with chloride, bromide, and iodide tetra-n-butylammonium salts. The electron-deficient ring of 1 encourages a small, but measurable association constant for each of the halides, determined by monitoring the change in chemical shift of the sulfonamide hydrogen. Receptor 2, lacking an electron-deficient ring, shows no measurable binding with the halides. Receptor 1 binds Cl−, Br−, and I− in a 1:1 fashion with binding constants of 30 ± 3, 20 ± 2, and 36 ±6 M−1, respectively. In comparison to receptor 2, the electron-deficient arene clearly encourages or assists in the binding of the halides, therefore the interaction between a halide and pentafluorobenzene is less repulsive than with benzene. While Hartree-Fock calculations found a centered anion-π interaction as one of two local minima (Figure 16b), no direct experimental evidence classifies the interaction as anion-π. Our future studies served as a reminder for the existence of other binding modes between anions and arene rings. Even with this in mind, neither charge transfer nor weak-σ complexes have been demonstrated for perfluoroarenes thus far. A more systematic change of the substituents may provide more insight on the nature of the anion-arene interaction present in this system.
Figure 16.

(a) Structures of neutral sulfonamide receptors. (b) One of two minimum geometries of 1 with chloride optimized at HF/6-31+G*.
Further investigation into anion-π interactions led to the study of 1,2,4,6-tetracyanobenzene (TCB) in the presence of halides.25 Crystal structures of the halide salts KBr, KI, and NaI with 18-crown-6 binding TCB demonstrated three of the four binding modes possible between anions and electron-deficient arenes. Two charge transfer, or weak-σ, complexes were formed above the arene plane and along the periphery of the ring; situated nearest the C-CN bond (3) and the C-H bond (4). The third interaction was an aryl C-H hydrogen bond within the plane of the arene (5, Figure 17). The local environment around the anion in the crystal structure for each of the halides was surprisingly similar. Interestingly, no centered anion-π interaction was found as part of the crystal structure. Calculations of 1:1 complexes between TCB and F−, Cl−, and Br− at the MP2/aug-cc-pVDZ level of theory were used to evaluate the relative energy differences between the three geometries listed above in addition to the anion-π complex (6). 3-5 were found as minima for each of the halides, with the exception of 3 for Br−. Configuration 6 was not found as a minimum on the potential energy surface for any of the three halides, but the binding energy of each halide was calculated by imposing C2ν symmetry. These results were the first example in which non-covalent anion-π complexes with Cl− and Br− were not found as minima on the potential energy surface. Global minima corresponded to complex 4 for each of the halides. Bond distances of less than 2 Å and a binding strength of −53.06 kcal mol−1 for F− in complex 4 indicated a strong σ or Meisenheimer complex. Cl− binds more strongly than Br− by 1.12 kcal/mol with binding energies of −29.80 and −28.68 kcal mol−1, respectively. This study demonstrated the weak nature of anion-π interactions, as well as their less favorable formation, in comparison to other anion-arene modes of binding, at least for extremely electron-deficient arenes.
Figure 17.

MP2/aug-cc-pVDZ optimized geometries for chloride complexes with TCB demonstrating the four modes of binding between anions and arenes.
Equipped with this knowledge, our next studies sought to reduce the weak-σ interaction to practice in solution, in a designed synthetic receptor. Neutral tripodal receptors utilizing only electron-deficient aromatic rings were shown to bind halides in a 1:1 stoichiometry by 1H NMR spectroscopic titrations and DFT calculations (Figure 18).26 Unsubstituted receptor 9, designed as a negative control, failed to bind halides. While 7 and 8 are structural isomers, the substitution pattern of 8 does not allow for aryl C-H hydrogen bonding interactions due to the steric hindrance the nitro groups provide. Titration experiments were performed with tetra-n-heptylammonium halide salts in C6D6 at 27 °C and the determined 1:1 association constants are shown in Table 1. Titration of 7 vs 8 with Cl−, Br−, and I− yielded larger changes in chemical shifts (as well as color changes) for 7, despite having association constants of the same magnitude as 8. The difference in Δδ between receptors 7 and 8 is indicative of the modes by which each receptor binds the anion. Since 7 is able to bind anions through a stronger aryl C-H interaction, a larger overall chemical shift is observed. DFT calculations of the two receptors binding Br− confirm this finding (Figure 18b). These results demonstrate the binding of anions to receptors in solution by interaction with only electron-deficient arenes through either weak-σ or C-H···X− hydrogen bonds, as well as provide a quantitative measure and comparison of the relative stabilities for such interactions in solution.
Figure 18.

(a) Structures of tripodal anion receptors 7-9 and optimized geometries of 7 and 8 complexed with bromide at the B3LYP/DZVP level of theory.
Table 1.
Average Ka is reported from two or three titration experiments, except for 9.
All titrations were performed in C6D6; initial receptor concentrations at ~2 mM; errors are estimated at ± 10%.
Tetra-n-heptylammonium halide salts were used; titrations were performed at 27 °C to account for the insolubility of NHep4+I− at room temperature.
Changes in chemical shift for control receptor 9 were too small to determine Ka values.
While we have been unable to quantify or definitively display purely centered (electrostatic) anion-π interactions, we have learned much about the interaction of anions with electron-deficient arenes. Our efforts have served as a reminder of the various types of anion-arene interactions possible, of which pure anion-π seems to be the least favored with arenes we have studied. Nevertheless, these remain an important component in ligand design for anions, and understanding the preferred geometry of the various interactions for receptor optimization is critical. We continue to design new receptors to test the selectivity of these interactions in solution for a variety of anions.
Conclusions
Ion-π interactions continue to play a prominent role in supramolecular chemistry. This Accounts article highlighted our group’s efforts on the design of ligands and receptors featuring prominent ion-π interactions in their complexes. We discussed a number of dinuclear self-assembled pnictogen-thiolate complexes and one tetranuclear assembly. In every example, pnictogen-π interactions appear to provide additional stability to these surprisingly robust complexes. We also described efforts to understand and to quantify anion-arene interactions in solution. Our work has revealed that a variety of interactions are favorable between anions and electron-deficient arenes, including weak-σ and C-H···anion hydrogen bonds; purely centered anion-π interactions have remained elusive in the systems we have studied, although they presumably comprise a portion of the attractive forces in these complexes.
These ion-arene interactions are quite weak in solution, perhaps even non-existent for only weakly polarized aromatic rings. In addition, the nature of the interactions is a topic of current debate and interest, stimulating new scientific discovery. Nevertheless, our studies in these two areas have taught us important lessons: these weak interactions may play a supporting role in ligand design for toxic main group elements, and interactions between aromatic rings and anions may provide materials with selectivity for more weakly basic anions and help alter the Hofmeister bias in anion binding.
Acknowledgments
The work presented on anion-arene interactions has evolved into a collaboration with UO colleague Prof. Michael M. Haley investigating molecular probes and sensors for ions with a continued interest in investigating the role of electron-deficient aromatic rings in anion binding.27 We are grateful to the National Institutes of Health for generous support of this work (R01-GM087398). Our studies on supramolecular main group coordination chemistry were primarily been funded by UO, the National Science Foundation (CAREER award CHE-0545206) and the Research Corporation for Science Advancement through a Cottrell Scholar award. We are indebted to our numerous coworkers who contributed to the research described in this review; their creativity and hard work continue to serve as inspiration.
Biographies
Michelle M. Watt received a B.S. in biochemistry in 2007 from St. Louis University. She continued her studies at SLU and received a M.S.(R) in 2009 working under Professor M. Lewis, investigating cation-arene and arene-arene interactions. She is currently a Ph.D. student in the groups of Professors D. W. Johnson and M. M. Haley, where she works on the synthesis and analysis of receptors for binding anions.
Mary S. Collins obtained her B.S. from the University of Texas, Austin in 2010 under the supervision of Dr. Christopher Bielawski. She is currently a graduate researcher in Prof. Darren W. Johnson’s laboratory at the University of Oregon. Her research focuses on the coordination chemistry of pnictogen-containing self-assemblies.
Darren W. Johnson joined the University of Oregon in 2003, where he is currently an Associate Professor of Chemistry. Born in Columbus, Ohio in 1974 and raised in College Station, Texas, Darren received his B.S. in Chemistry at the University of Texas at Austin in 1996. After his studies in Jonathan Sessler’s laboratories at Texas, he earned his Ph. D. in Chemistry in 2000 from the University of California at Berkeley working with Kenneth N. Raymond. He then spent two years at the Scripps Research Institute as a National Institutes of Health post-doctoral fellow with Julius Rebek, Jr. Research in his group uses supramolecular chemistry as a tool to explore a variety of problems in coordination chemistry, molecule/ion recognition and inorganic cluster synthesis, much of which is investigated within the NSF Phase II CCI Center for Sustainable Materials Chemistry (uoregon.edu/grnchem).
References
- 1.For a selection of the numerous relevant reviews, see: Ma JC, Dougherty DA. The cation-π interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744.Gokel GW. The aromatic sidechains of amino acids as neutral donor groups for alkali metal cations. Chem Commun. 2003:2847–2852. doi: 10.1039/b305483g.Meyer EA, Castellano RK, Diederich F. Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319.Schier A, Schmidbauer H. π-Complexation of post-transition metals by neutral aromatic hydrocarbons: the road from observations in the 19th century to new aspects of supramolecular chemistry. Organometallics. 2008;27:2361–2395.
- 2.Reviews, and references within: Hay BP, Bryantsev VS. Chem Commun. 2008:2417–2428. doi: 10.1039/b800055g.Berryman OB, Johnson DW. Experimental evidence for interactions between anions and aromatic rings. Chem Commun. 2009:3143–3153. doi: 10.1039/b823236a.Frontera A, Gamez P, Mascal M, Mooibroek TJ, Reedijk J. Putting anion-π interactions into perspective. Angew Chem Int Ed. 2011;50:9564–9583. doi: 10.1002/anie.201100208.
- 3.(a) Mascal M, Armstrong A, Bartberger MD. Anion-aromatic bonding: a case for anion recognition by pi-acidic rings. J Am Chem Soc. 2002;124:6247–6276. doi: 10.1021/ja017449s. [DOI] [PubMed] [Google Scholar]; (b) Quinoñero D, Garau C, Frontera A, Ballester P, Costa A, Deyà PM. Counterintuitive interaction of anions with benzene derivatives. Chem Phys Lett. 2002;359:486–492. [Google Scholar]; (c) Quinoñero D, Garau C, Rotger C, Frontera A, Ballester P, Costa A, Deyà PM. Anion-pi interactions: do they exist? Angew Chem, Int Ed. 2002;41:3389–3392. doi: 10.1002/1521-3773(20020916)41:18<3389::AID-ANIE3389>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]; (d) Alkorta I, Rozas I, Elguero J. Interaction of anions with perfluoro aromatic compounds. J Am Chem Soc. 2002;124:8593–8598. doi: 10.1021/ja025693t. [DOI] [PubMed] [Google Scholar]
- 4.Zukerman-Schpector J, Otero-de-la-Roza A, Lauña V, Tiekink ERT. Supramolecular architectures based on As(lone pair)-π(aryl) interactions. Chem Commun. 2011;47:7608–7610. doi: 10.1039/c1cc11412c. [DOI] [PubMed] [Google Scholar]
- 5.Auer AA, Mansfeld D, Nolde C, Schneider W, Schurmann M, Mehring M. Bismuth-arene π-interaction: a combined experimental and theoretical approach. Organometallics. 2009;28:5405–5411. [Google Scholar]
- 6.Vickaryous WJ, Herges R, Johnson DW. Arsenic-π interactions stabilize a self-assembled As2L3 supramolecular complex. Angew Chem Int Ed. 2004;43:5831–5833. doi: 10.1002/anie.200461011. [DOI] [PubMed] [Google Scholar]
- 7.Vickaryous WJ, Rather Healy E, Berryman OB, Johnson DW. Synthesis and characterization of two isomeric, self-Assembled arsenic-thiolate macrocycles. Inorg Chem. 2005;44:9247–9252. doi: 10.1021/ic050954w. [DOI] [PubMed] [Google Scholar]
- 8.Molecular mechanics minimization were performed using the CAChe software program with MM3 and MM2 force fields: CAChe, version 5.0. Fujitsu, Ltd; Kawasaki, Japan: 2000–2001. [Google Scholar]
- 9.Cangelosi VM, Sather AC, Zakharov ZN, Berryman OB, Johnson DW. Diastereoselectivity in the self-assembly of As2L2Cl2 macrocycles is directed by the As-π interaction. Inorg Chem. 2007;46:9278–9284. doi: 10.1021/ic701290h. [DOI] [PubMed] [Google Scholar]
- 10.Cangelosi VM, Zakharov LN, Fontenot SA, Pitt MA, Johnson DW. Host-guest interactions in a series of self-assembled As2L2Cl2 macrocycles. Dalton Trans. 2008:3447–3453. doi: 10.1039/b802178c. [DOI] [PubMed] [Google Scholar]
- 11.Cangelosi VM, Zakharov LN, Crossland JL, Franklin BC, Johnson DW. A surprising ‘folded-in’ conformation of a self-assembled arsenic-thiolate macrocycle. Cryst Growth Des. 2010;10:1471–1473. [Google Scholar]
- 12.Lindquist NR, Carter TG, Cangelosi VM, Zakharov LN, Johnson DW. Three’s company: co-crystallization of a self-assembled S4 metallacyclophane with two diastereomeric metallacycle intermediates. Chem Commun. 2010;46:3505–3507. doi: 10.1039/b927034e. [DOI] [PubMed] [Google Scholar]
- 13.(a) Pitt MA, Zakharov LN, Vanka K, Thompson WH, Laird BB, Johnson DW. Multiple weak supramolecular interactions stabilize a surprisingly ‘twisted’ As2L3 assembly. Chem Commun. 2008:1–3. doi: 10.1039/b806958a. [DOI] [PubMed] [Google Scholar]; (b) Cangelosi VM, Pitt MA, Vickaryous WJ, Allen CA, Zakharov LN, Johnson DW. Design considerations for the group 15 elements: the pnictogen-π interaction as a complementary component in supramolecular assembly design. Cryst Growth Des. 2010;10:3531–3536. [Google Scholar]
- 14.Vickaryous WJ, Zakharov LN, Johnson DW. Self-assembled antimony-thiolate Sb2L3 and Sb2L2Cl2 complexes. Main Group Chem. 2006;5:51–59. [Google Scholar]
- 15.Cangelosi VM, Zakharov LN, Johnson DW. Supramolecular transmetalation leads to an unusual self-assembled P2L3 cryptand. Angew Chem Int Ed. 2010;49:1248–1251. doi: 10.1002/anie.200906460. [DOI] [PubMed] [Google Scholar]
- 16.Burford N, Clyburne JAC, Bakshi PK, Cameron TS. η6-Arene complexation to a phosphenium cation. J Am Chem Soc. 1993;115:8829–8830. [Google Scholar]
- 17.Cangelosi VM, Carter TG, Crossland JL, Zakharov LN, Johnson DW. Self-assembled E2L3 cryptands (E = P, As, Sb, Bi): transmetalation, homo- and heterometallic assemblies, and conformational isomerism. Inorg Chem. 2010;49:9985–9992. doi: 10.1021/ic1012186. [DOI] [PubMed] [Google Scholar]
- 18.Fontenot SA, Cangelosi VM, Pitt MAW, Sather AC, Zakharov LN, Berryman OB, Johnson DW. Design, synthesis and characterization of self-Assembled As2L3 and Sb2L3 cryptands. Dalton Trans. 2011;40:12125–12131. doi: 10.1039/c1dt10817d. [DOI] [PubMed] [Google Scholar]
- 19.Zampella G, Neupane KP, De Gioia L, Pecoraro VL. The importance of stereochemically active lone pairs for influencing Pb(II) and As(III) protein binding. Chem Eur J. 2012;18:2040–2050. doi: 10.1002/chem.201102786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meisenheimer J. Reactions of aromatic nitro structures. Justus Liebigs Ann Chem. 1902;323:205–246. [Google Scholar]
- 21.Chowdhury S, Kebarle P. Role of binding energies in A−•B and A•B− complexes in the kinetics of gas phase electron transfer reactions: A− + B = A + B− involving perfluoro compounds: SF6, C6F11CF3, C6F6. J Chem Phys. 1986;85:4989–4994. [Google Scholar]
- 22.Hiraoka K, Mizuse S, Yamabe S. High-symmetric structure of the gas-phase cluster ions X−•••C6F6 (X = CI, Br, and I) J Phys Chem. 1987;91:5294–5297. [Google Scholar]
- 23.Vargas Jentzsch A, Emery D, Mareda J, Metrangolo P, Resnati G, Matile S. Ditopic ion transport systems: anion-π interactions and halogen bonds at work. Angew Chem Int Ed. 2011;50:11675–11678. doi: 10.1002/anie.201104966. [DOI] [PubMed] [Google Scholar]
- 24.Berryman OB, Hof F, Hynes MJ, Johnson DW. Anion-π interaction augments halide binding in solution. Chem Commun. 2006:506–508. doi: 10.1039/b511570a. [DOI] [PubMed] [Google Scholar]
- 25.Berryman OB, Bryantsev VS, Stay DP, Johnson DW, Hay BP. Structural criteria for the design of anion receptors: the interaction of halides with electron-deficient arenes. J Am Chem Soc. 2007;129:48–58. doi: 10.1021/ja063460m. [DOI] [PubMed] [Google Scholar]
- 26.Berryman OB, Sather AC, Hay BP, Meisner JS, Johnson DW. Solution phase measurement of both weak-σ and C-H•••X− hydrogen bonding interactions in synthetic anion receptors. J Am Chem Soc. 2008;130:10895–10897. doi: 10.1021/ja8035652. [DOI] [PubMed] [Google Scholar]
- 27.(a) Carroll CN, Naleway JJ, Haley MM, Johnson DW. Arylethynyl receptors for anions and emerging applications in cellular imaging. Chem Soc Rev. 2010;39:3875–3888. doi: 10.1039/b926231h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Engle JM, Carroll CN, Johnson DW, Haley MM. Molecular self-assembly: solvent guests tune the conformation of a series of versatile 2,6-bis(2-anilinoethynyl)pyridine-based ureas. Chem Sci. 2012;3:1105–1110. [Google Scholar]