

Abstract
Aims
We aimed to investigate the role of the nuclear carrier and binding proteins, transportin-1 (TRN1) and transportin-2 (TRN2), TATA-binding protein-associated factor 15 (TAF15) and Ewing’s Sarcoma protein (EWS) in inclusion body formation in cases of Frontotemporal Lobar Degeneration (FTLD) associated with Fused in Sarcoma protein (FTLD-FUS).
Methods
Eight cases of FTLD-FUS (5 cases of atypical FTLD-U (aFTLD-U), 2 of Neuronal Intermediate Filament Inclusion Body Disease (NIFID) and 1 of Basophilic Inclusion Body Disease (BIBD)) were immunostained for FUS, TRN1, TRN2, TAF15 and EWS. 10 cases of FTLD associated with TDP-43 inclusions served as reference cases.
Results
The inclusion bodies in FTLD-FUS contained TRN1 and TAF15 and, to a lesser extent, EWS, but not TRN2. The patterns of immunostaining for TRN1 and TAF15 were very similar to that of FUS. None of these proteins was associated with tau or TDP-43 aggregations in FTLD.
Conclusion
Data suggest that FUS, TRN1 and TAF15 may participate in a functional pathway in an interdependent way, and imply that the function of TDP-43 may not necessarily be in parallel with, or complementary to, that of FUS, despite each protein sharing many similar structural elements.
Keywords: Frontotemporal Lobar degeneration, Fused in Sarcoma, TDP-43, transportins, TATA-binding protein-associated factor 15, Ewing’s sarcoma protein
Introduction
Frontotemporal lobar degeneration (FTLD) is clinically, pathologically and genetically heterogeneous. The prototypical clinical syndromes are behavioural variant frontotemporal dementia (BvFTD), a disorder of behaviour, progressive non-fluent aphasia (PNFA), a disorder of expressive language and semantic dementia (SD), a disorder of conceptual knowledge [1]. Moreover, the clinical signs of motor neurone disease (MND) occur in some patients with FTLD [2,3]. Pathologically, approximately half of FTLD cases show an accumulation of hyperphosphorylated tau proteins in neurones and glia (and are known as FTLD-tau), whereas others are tau-negative and show immunoreactivity for ubiquitin (FTLD-U) [4]. The pathological protein in the majority of cases of FTLD-U, as well as in around 85% of MND cases, has been identified as TDP [5,6]. Four subtypes of TDP-43 pathology have been delineated [7,8], reflecting the predominance of pathological change within nerve cell bodies and/or neurites. Most of the tau and TDP-43 negative FTLD cases show a pathology characterised by accumulations of fused in sarcoma (FUS) protein, this being present in cases labelled as atypical FTLD-U (aFTLD-U) [9-13], neuronal intermediate filament inclusion disease (NIFID) [14] and basophilic inclusion body disease (BIBD) [15]. Current nomenclature includes aFTLD-U, NIFID and BIBD as three forms of FUS pathology [16].
FUS is a 526-amino acid long protein with a predicted molecular mass of 53kDa which is ubiquitously expressed and has multiple cellular functions [17,18]. It is normally present more in the nucleus than in the cytoplasm, and more is present in neurones than glial cells [19]. It binds both DNA and RNA [17], with its C-terminus being involved in RNA-protein interactions and its N-terminus having a role in transcription. Under normal physiological conditions, FUS shuttles between nucleus and cytoplasm through the nuclear pore [20]. Nuclear import of FUS is effected by transportin 1 (TRN1) – a 890 amino acid long protein, also known as M9-interacting protein or karyopherin β1 (karβ1). Karβ1 is part of a family of proteins (karyopherinβ-s, also termed importins or exportins) which are responsible for performing most of the cell’s nucleocytoplasmic transport traffic. TRN1 is responsible for the import of particular RNA processing proteins which include FUS [21]. Binding of FUS to TRN1 is dependent on the PY motif, which is located at the C-terminus of FUS. A recent study [22] has suggested that TRN1 is incorporated into the FUS-positive inclusions in patients with FTLD associated with aFTLD-U and NIFID. Other studies have reported that the inclusion bodies in FTLD-FUS may contain other members of the FET protein family such as TATA-binding protein-associated factor 15 (TAF15) and Ewing’s sarcoma protein (EWS) [23].
In the present report we have investigated patients with all 3 forms of FTLD-FUS (ie aFTLD-U, NIFID and BIBD), and others with FTLD-tau and FTLD-TDP using antibodies against both TRN1 and Transportin 2 (TRN2) (also known as karyopherinβ2). We further immunostained the same cases for TAF15 and EWS proteins.
Materials and Methods
Eight patients with FTLD-FUS were drawn from those cases with FTLD collected across 4 Academic Centres: 3 patients (patients#1-3, all with aFTLD-U) were from Manchester Brain Bank, 3 patients (patients #4 and 6 with aFTLD-U and patient #8 with BIBD) were from Northwestern University, Chicago, patient #6 (with NIFID) was from Washington University, and patient #7 (also with NIFID) was from the Newcastle Brain Tissue Resource (NBTR) at the Institute for Ageing and Health, Newcastle University, Campus for Ageing and Vitality (Table 1). Full clinical and pathological descriptions for patients #1-3 with aFTLD-U, and patients #6 and 7 with NIFID, have been presented elsewhere [see refs 12 and 24, respectively]. Ten other patients with other histological forms of FTLD (1 with MAPT exon 10 +16 mutation, 2 with FTLD-tau with Pick bodies, 3 with GRN mutation, 1 with C9ORF72 hexanucleotide repeat expansion with FTLD-TDP type A, 1 other patient with FTLD-TDP type A, 1 with FTLD-TDP type B and 1 with FTLD-type C: these latter 3 patients were known not to bear mutation in any of the aforementioned genes) were drawn from the Manchester Brain Bank and served as reference cases. All FTLD cases selected fulfilled Lund-Manchester clinical diagnostic criteria for FTLD [1]. In all instances, brains had been obtained with full ethical permission and appropriate consent/declaration procedures.
Table 1.
Demographic details of cases.
| case ID | pathology | gender | Age at onset (yr) | Duration (yr) | Brain weight (g) |
|---|---|---|---|---|---|
| 1 | aFTLD-U | M | 37 | 5 | 1201 |
| 2 | aFTLD-U | M | 45 | 4 | 1290 |
| 3 | aFTLD-U | M | 46 | 4 | 1160 |
| 4 | aFTLD-U | M | 58 | 6 | 1210 |
| 5* | aFTLD-U | F | 34 | 7 | 910 |
| 6 | NIFID | F | 38 | 3 | 904 |
| 7 | NIFID | F | 52 | 3 | 813 |
| 8 | BIBD | M | 48 | 7 | 1240 |
also has Adult Polyglucosan Body Disease with haploinsufficiency of branching enzyme
Brains had been fixed for variable periods up to 12 months before documentation of external appearances and cutting into coronal sections for the reporting of macroscopic changes and preparation of tissue blocks for histological inspection. Representative fixed tissue blocks were cut from the temporal cortex (to include the hippocampus) and processed routinely into paraffin wax. Only the motor cortex was studied in the BIBD case as this was the cortical region showing maximal FUS pathology. Sections were cut at a thickness of 6μm and immunostained for non phosphorylated TDP-43 (rabbit polyclonal antibody (10782-2-AP (ProteinTech, Manchester, UK) 1:1000), FUS protein (rabbit polyclonal antibody HPA-008784 (Sigma, Poole, UK) 1:50 to 1:200), TRN1 (also known as importin beta-1 sub unit or karyopherin β1 (KPNB1)) (mouse monoclonal antibody ab10303 (Abcam, Cambridge, UK), and rabbit polyclonal antibody sc-11367 (Santa Cruz Biotechnology Inc), both used at 1:200), TRN2 (also known as karyopherin β2/2B or importin 3) (goat polyclonal antibody sc-6914 (Santa Cruz Biotechnology Inc), TAF15 (also known as TAF1168) (rabbit polyclonal antibody A300-308A (Bethyl Laboratories Inc), 1:200)) and EWS (mouse monoclonal antibody EWS-G5, sc-28327 (Santa Cruz Biotechnology Inc) 1:200), employing a standard ABC Elite kit (Vector, Peterborough, UK) with DAB as chromogen.
The number of NCI immunostaining for each protein was assessed semiquantitatively. For FUS, Abcam TRN1, TAF15 and EWS, the ratings refer to the relative frequencies of nerve cells bearing NCI, where 0 = none; + = few cells with NCI; ++ = moderate number; of cells with NCI; +++ = many cells with NCI. For Santa Cruz TRN1, the ratings refer to the number of cells and the intensity of staining of the cytoplasmic granules and nuclear membrane in each cell, and for TRN2 the ratings refer to the number of cells and intensity of the cytoplasmic granular staining, where 0=none; + = few/weak staining; ++ = moderate number/moderate staining; +++ = many/most/strong staining.
Results
All 8 cases were characterised by FUS-immunoreactive neuronal cytoplasmic inclusions (NCI) widely distributed throughout the temporal cortex and hippocampus in aFTLD-U and NIFID cases, and motor cortex in the BIBD case (Table 2). Employing the FUS antibody (Sigma), a mix of rounded, more solid or granular NCI and ring-like NCI were present in the majority of granule cells of the dentate gyrus of the hippocampus, and ‘vermiform’ NII were commonly seen in the aFTLD-U (Figure 1a) and NIFID cases (Figure 2a). In the temporal cortex, the FUS-immunopositive NCI in the aFTLD-U and NIFID cases appeared rounded, either solid or resembling clumps of granules; ‘vermiform’ NII were occasionally seen in the aFTLD-U and NIFID cases, but no glial cell inclusions were present (not shown). In the motor cortex in the BIBD case, NCI had a ring-like, perinuclear distribution (Figure 3a), but no NII were seen.
Table 2.
Rating of immunohistochemical staining of the various nuclear transport proteins in hippocampus and cerebral cortex in 5 cases of atypical FTLD-U (aFTLD-U), 2 of Neuronal Intermediate Filament Inclusion Body Disease (NIFID) and 1 of Basophilic Inclusion Body Disease (BIBD). For FUS, Abcam TRN1, TAF15 and EWS the ratings refer to the relative frequencies of the pathological cytoplasmic inclusion bodies, for Santa Cruz TRN1 the ratings refer to the staining of the cytoplasmic granules and nuclear membrane, and for TRN2 the ratings refer to the intensity of the cytoplasmic granular staining (see text for detail).
| case ID | pathology | region | FUS | TRN1 (Abcam) | TRN1 (Santa-Cruz) | TRN2 | TAF15 | EWS |
|---|---|---|---|---|---|---|---|---|
| 1 | aFTLD-U | Hippocampus | +++ | +++ | ++ | + | +++ | + |
| 2 | aFTLD-U | Hippocampus | + | + | + | + | + | 0 |
| 3 | aFTLD-U | Hippocampus | + | + | + | + | + | 0 |
| 4 | aFTLD-U | Hippocampus | +++ | +++ | +++ | ++ | +++ | + |
| 5 | aFTLD-U | Hippocampus | +++ | +++ | +++ | ++ | +++ | + |
| 6 | NIFID | Hippocampus | +++ | +++ | +++ | ++ | +++ | + |
| 7 | NIFID | Hippocampus | + | + | + | + | + | 0 |
| 1 | aFTLD-U | Temporal cx | +++ | +++ | ++ | ++ | +++ | + |
| 2 | aFTLD-U | Temporal cx | + | + | + | + | + | 0 |
| 3 | aFTLD-U | Temporal cx | + | + | + | + | + | 0 |
| 4 | aFTLD-U | Temporal cx | +++ | +++ | +++ | ++ | +++ | + |
| 5 | aFTLD-U | Temporal cx | +++ | +++ | +++ | ++ | +++ | + |
| 6 | NIFID | Temporal cx | +++ | +++ | ++ | ++ | +++ | + |
| 7 | NIFID | Temporal cx | ++ | ++ | + | + | ++ | 0 |
| 8 | BIBD | Motor cx | +++ | +++ | +++ | ++ | +++ | + |
FUS = Fused in sarcoma; TRN = transportin; TAF15 = TATA-binding protein-associated factor 15; EWS = Ewing’s Sarcoma protein
Figure 1.
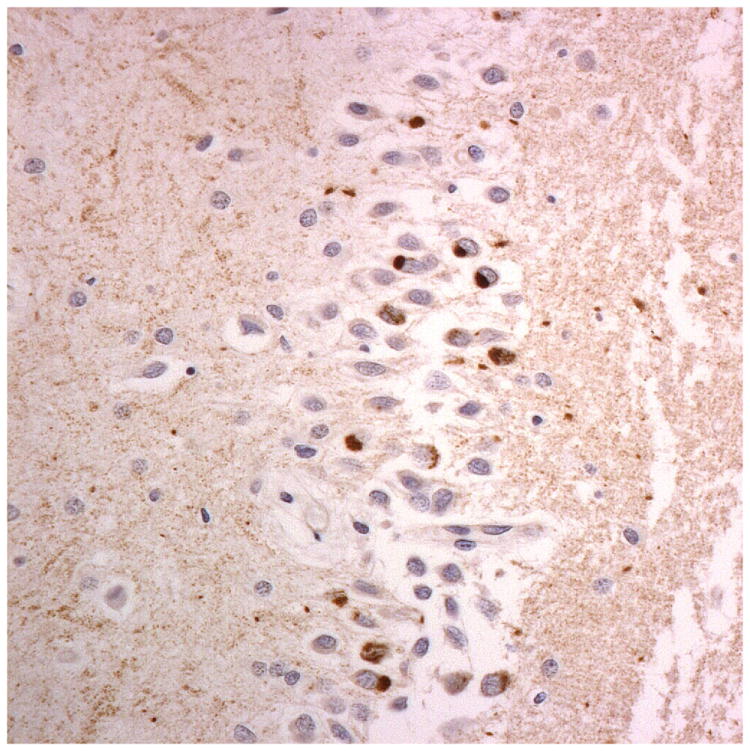
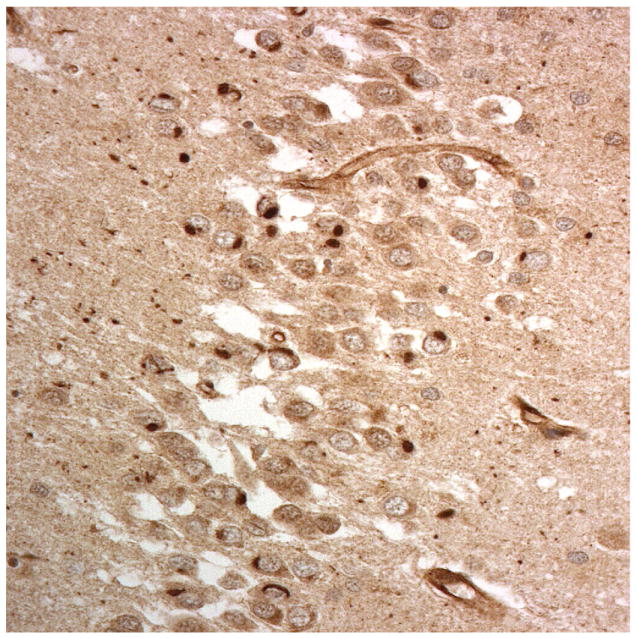
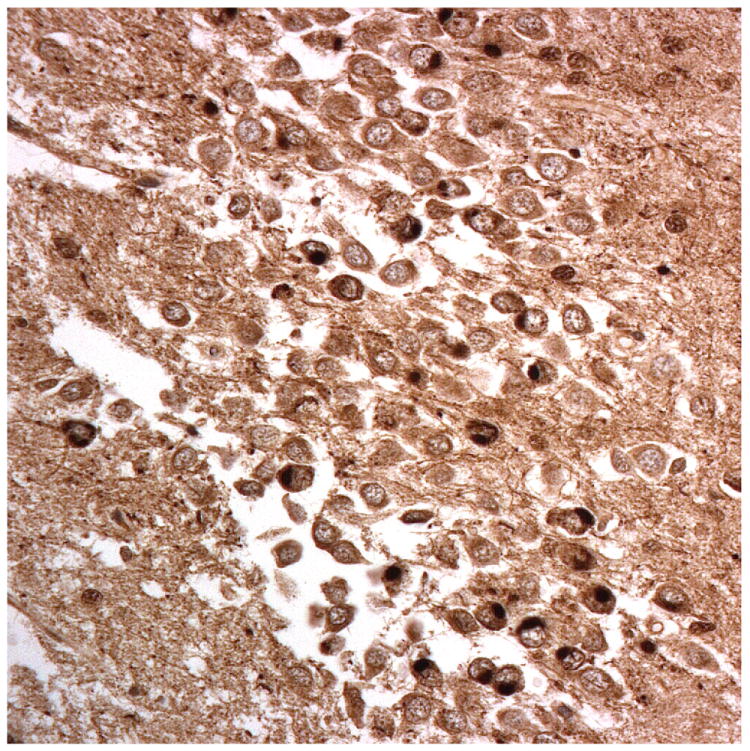
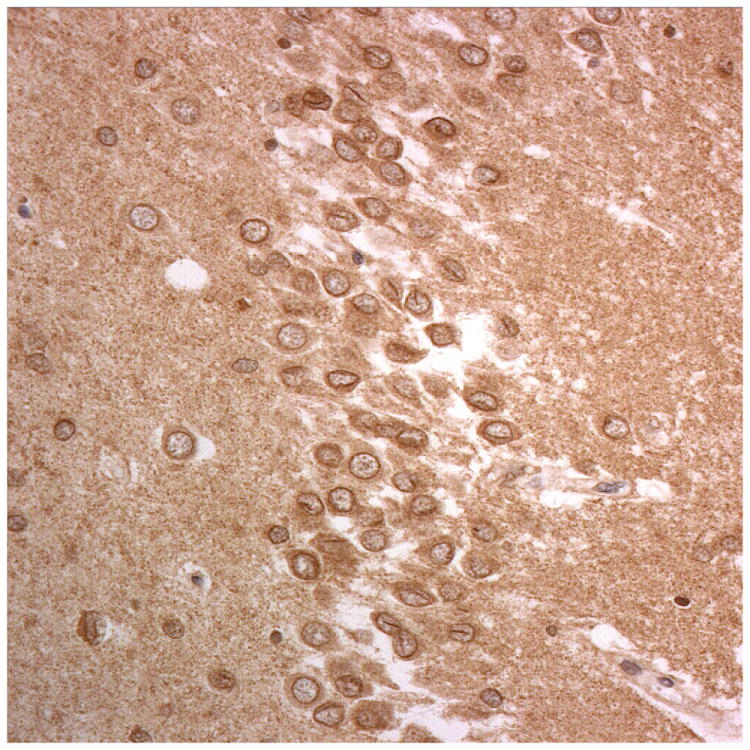
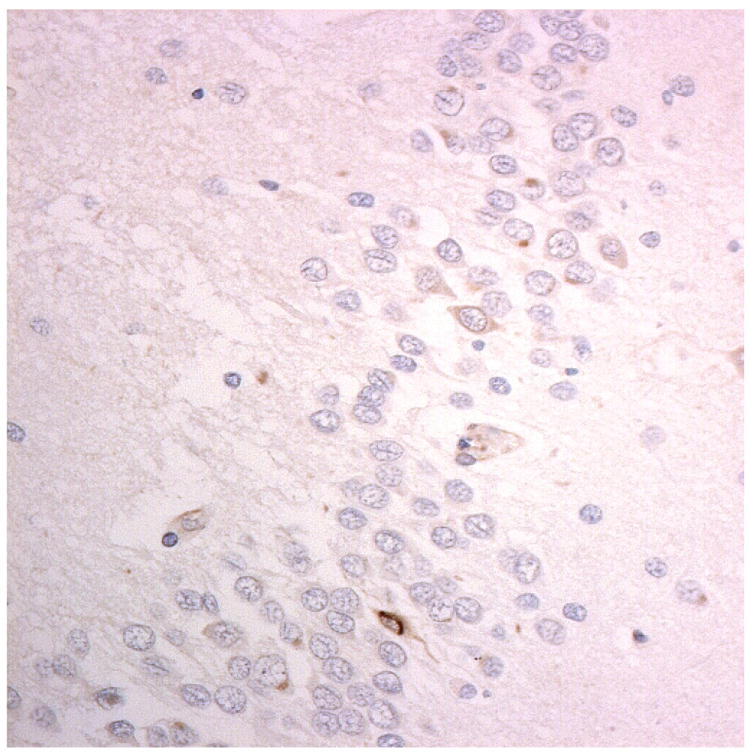
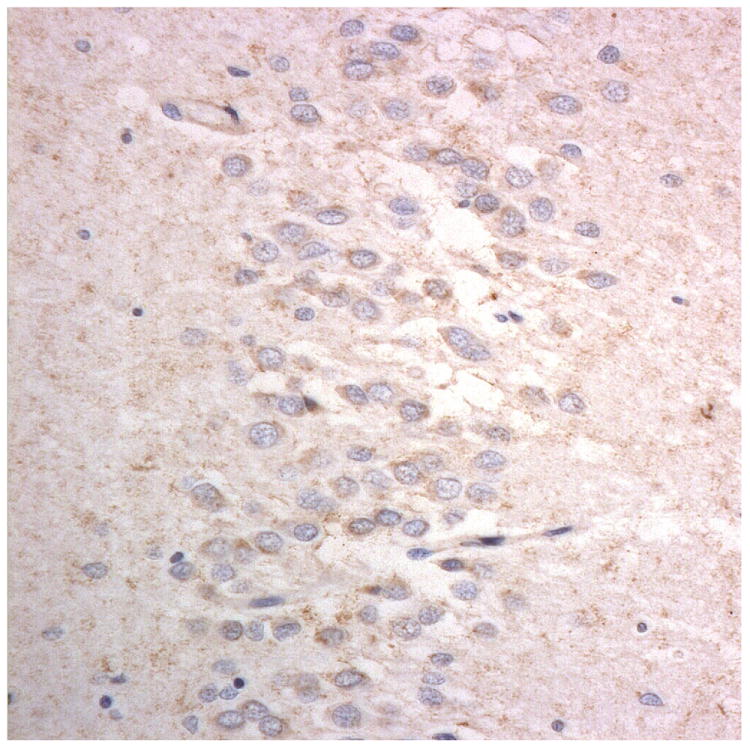
Immunostaining for nuclear transport associated proteins in consecutive sections from aFTLD-U case #4. There is strong immunostaining of neuronal cytoplasmic inclusions (NCI) and intranuclear inclusions (NII) in dentate gyrus granule cells, in a similar distribution, for FUS (a), Abcam TRN1 (b) and TAF15 (c). The Sigma TRN1 antibody immunostains cell cytoplasm and nuclear membrane, but not NCI or NII (d). The EWS antibody only weakly immunostains a proportion of those NCI and NII immunolabelled by FUS, TRN1 and TAF15 (e). The TRN2 antibody immunolabels small amounts of granular material in the cytoplasm of both neurones and astrocytes (f).
Immunoperoxidase – haematoxylin, all x40 microscope objective magnification.
Figure 2.
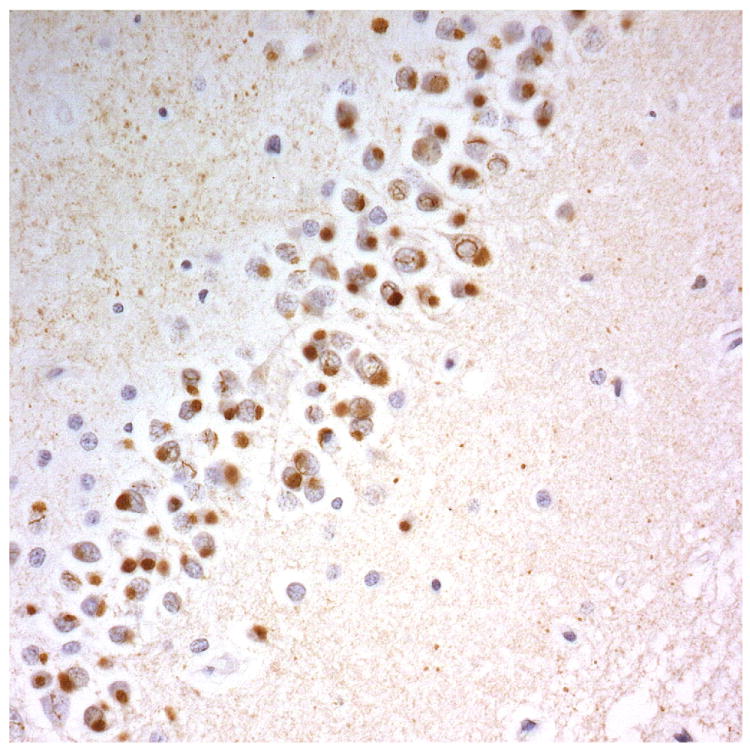
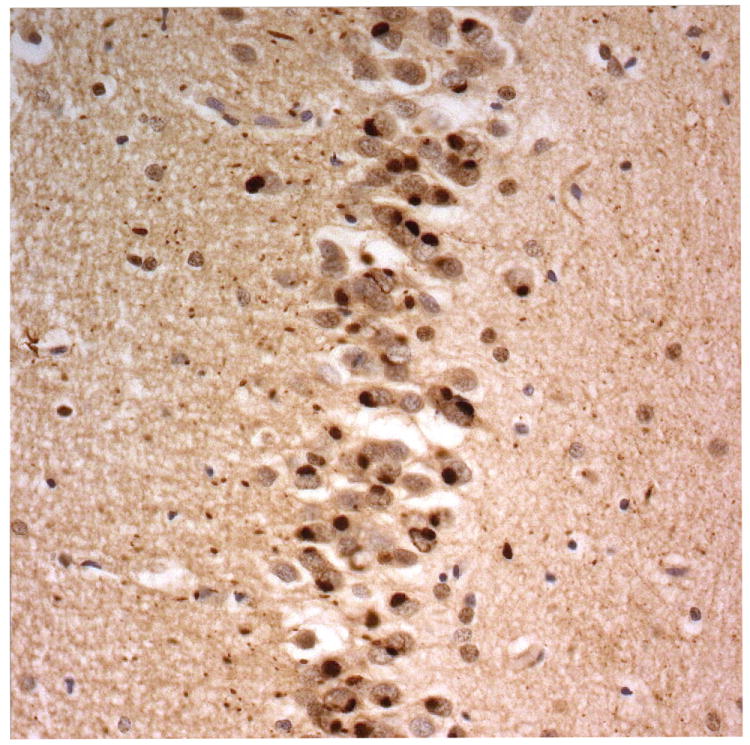
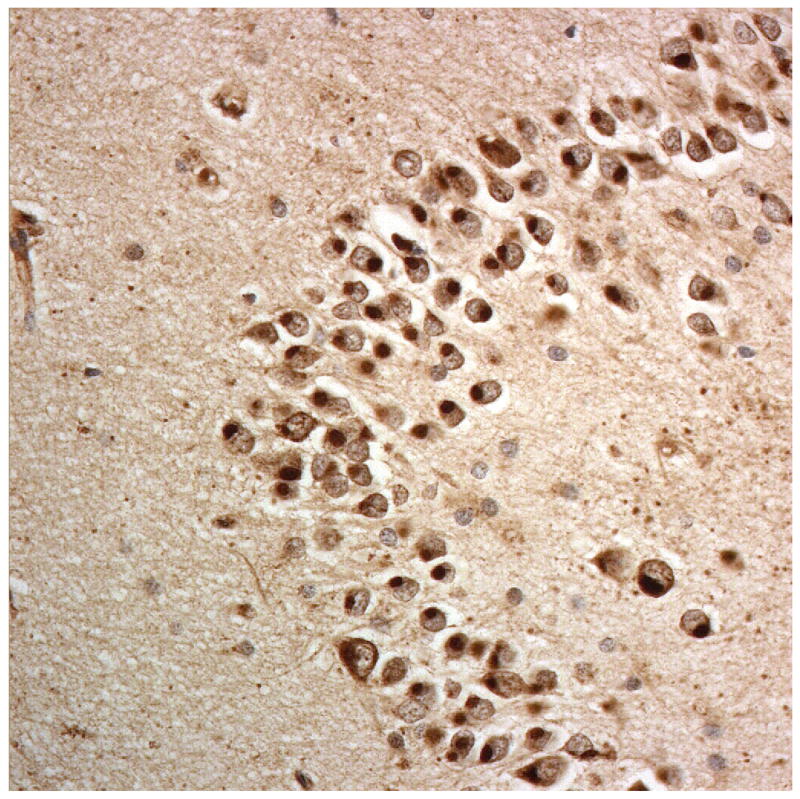
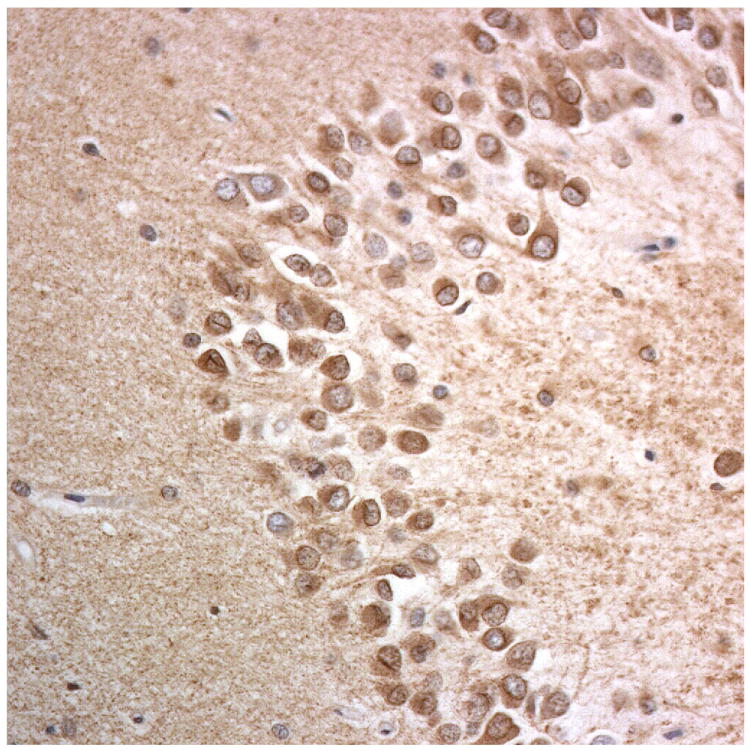
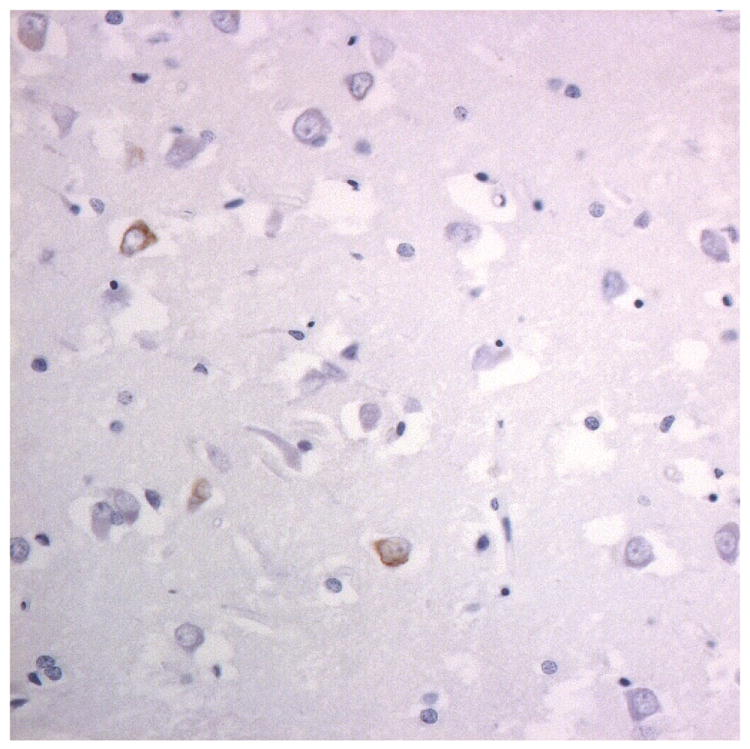
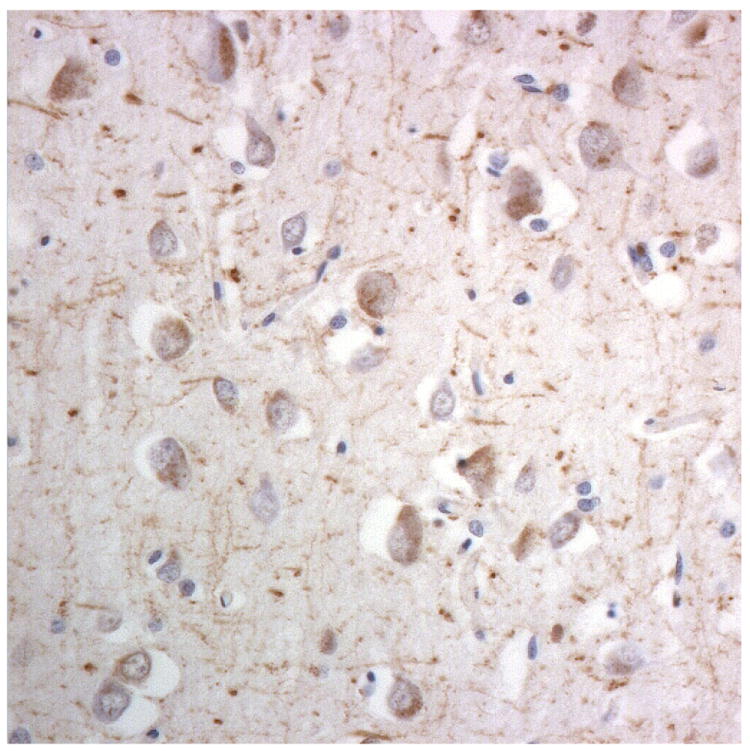
Immunostaining for nuclear transport associated proteins in consecutive sections from NIFID case #6. There is strong immunostaining of neuronal cytoplasmic inclusions (NCI) and intranuclear inclusions (NII) in dentate gyrus granule cells, in a similar distribution, for FUS (a), Abcam TRN1 (b) and TAF15 (c). The Sigma TRN1 antibody immunostains cell cytoplasm and nuclear membrane, but not NCI or NII (d). In the temporal cortex (and dentate gyrus – not shown), the EWS antibody only weakly immunostains a small proportion of NCI immunolabelled by FUS, TRN1 and TAF15 (e). The TRN2 antibody immunolabels small amounts of granular material in the cytoplasm of both neurones and astrocytes (f).
Immunoperoxidase – haematoxylin, all x40 microscope objective magnification.
Figure 3.
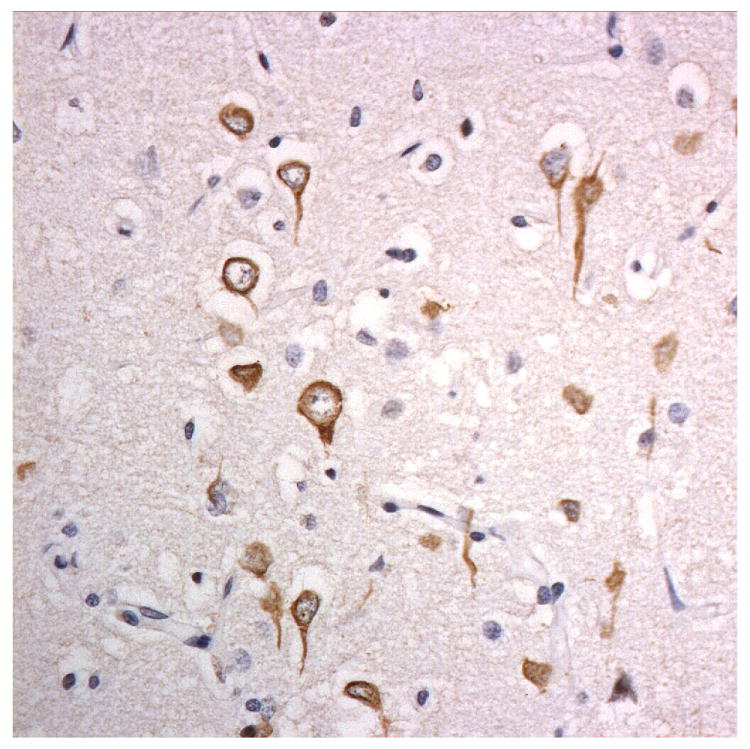
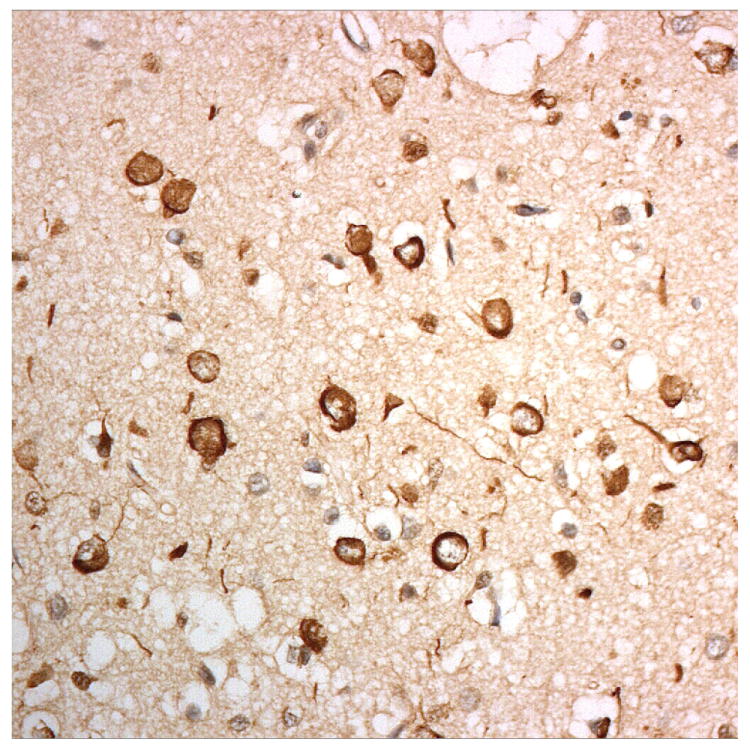
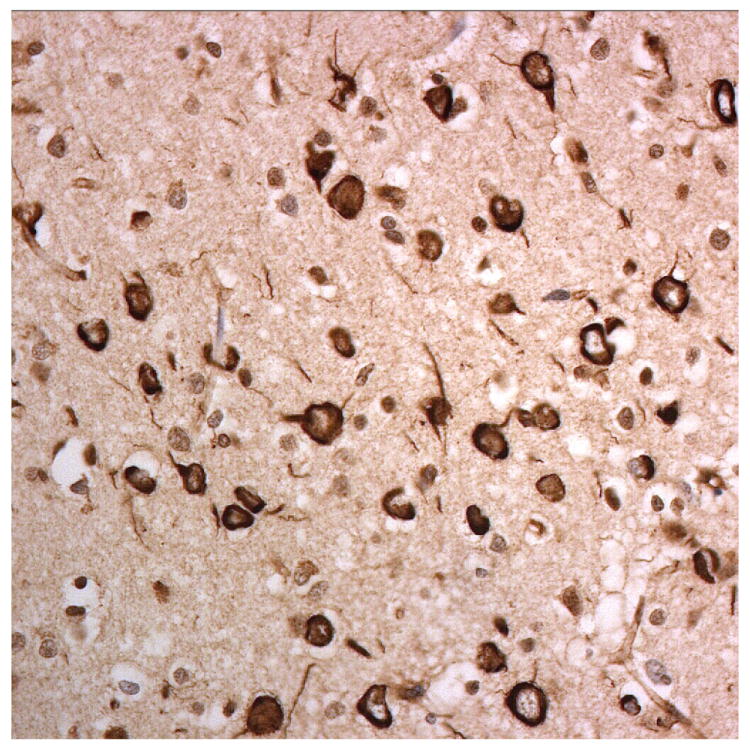
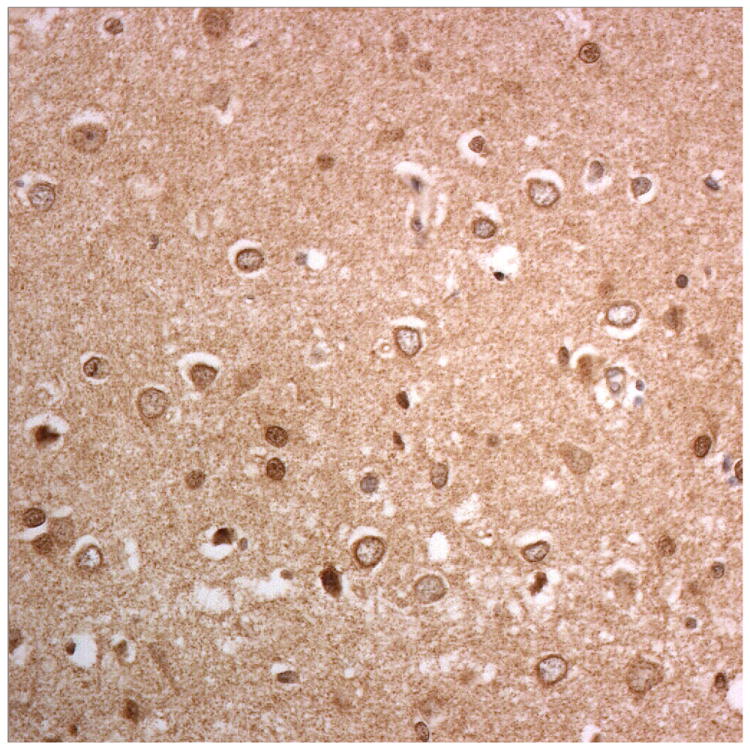
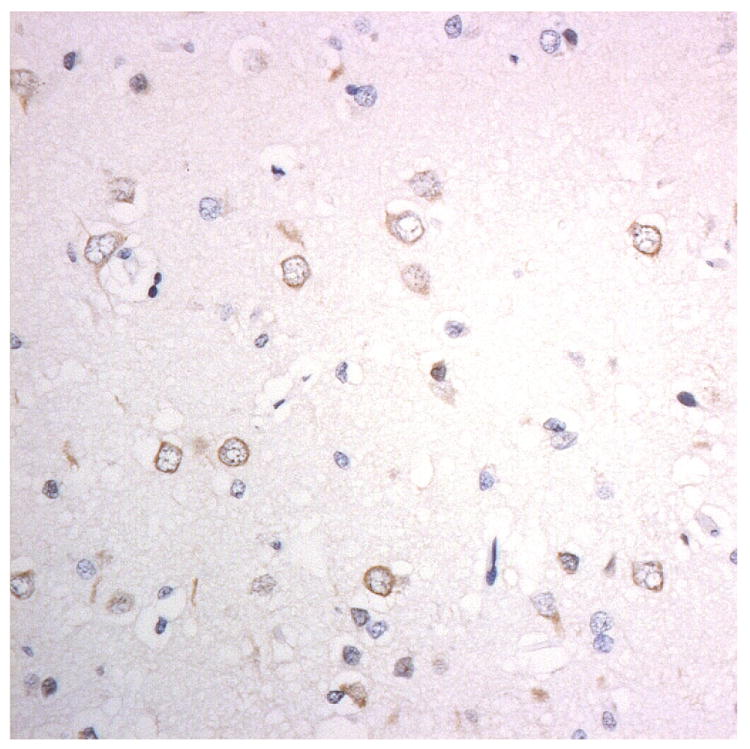
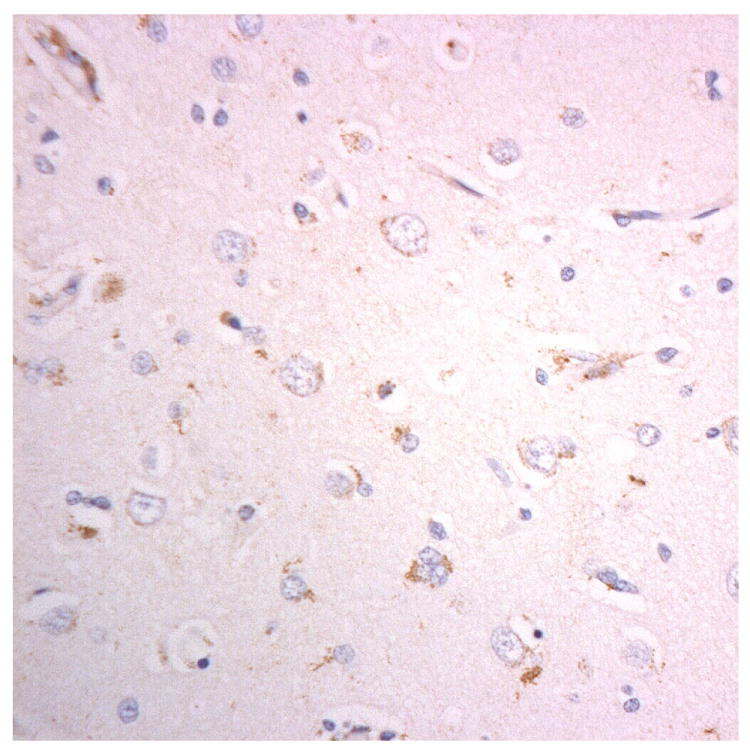
Immunostaining for nuclear transport associated proteins in consecutive sections from BIBD case #8. There is strong immunostaining of neuronal cytoplasmic inclusions (NCI) in pyramidal cells of the motor cortex, in a similar distribution, for FUS (a), Abcam TRN1 (b) and TAF15 (c). The Sigma TRN1 antibody immunostains cell cytoplasm and nuclear membrane, but not NCI (d). The EWS antibody immunostains, less strongly, only a proportion of those NCI immunolabelled by FUS, TRN1 and TAF15 antibodies (e). The TRN2 antibody immunolabels small amounts of granular material in the cytoplasm of both neurones and astrocytes (f).
Immunoperoxidase – haematoxylin, all x40 microscope objective magnification.
The patterns of immunostaining using the Abcam antibody, ab10303, for TRN1 (Figures 1b, 2b and 3b, Table 1) and Bethyl antibody A300-308A for TAF15 (Figures 1c, 2c and 3c) were similar to that of FUS. However, immunostaining for TRN1 with Santa Cruz antibody, sc-11367, did not detect the FUS-positive NCI, but demonstrated a fine punctuate immunostaining within neurones and neuropil with distinct nuclear membrane immunostaining (Figures 1d, 2d and 3d). Immunostaining for EWS protein was generally weak with, only a (small) proportion of all TRN1/TAF15 positive structures being immunostained (Figures 1e, 2e and 3e).
None of the FTLD-TDP or FTLD-tau cases showed any immunopositivity with TRN1, TAF15 or EWS antibodies.
Immunostaining for TRN2 revealed small granules within dentate gyrus granule cells and cortical and hippocampal pyramidal cells, as well as cortical astrocytes (Figures 1f, 2f and 3f). The FTLD-FUS cases all showed similar staining patterns for TRN2, with no apparent qualitative or quantitative differences in immunostaining for this between these and the FTLD-tau or FTLD-TDP cases.
Discussion
In the present report we have shown that the inclusion bodies in cases of FTLD-FUS, whether these be ones in cases of aFTLD-U, NIFID or BIBD, contain other proteins besides FUS, notably TRN1 and TAF15 (and to a lesser extent apparently, EWS), whereas none of these proteins or other the transportin proteins TRN1 and TRN2 seem to be associated with tau or TDP-43 aggregations in FTLD. These data imply FUS, TRN1 and TAF15 participate in a functional pathway in an interdependent way. Present data agree with those recently reported by Brelstaff et al [22] who also showed there was good colocalisation of FUS and TRN1 protein in the inclusion bodies of the hippocampus and cerebral cortex in 7 cases of aFTLD-U and 6 cases of NIFID. In the present study, comparison between FUS and TRN1 immunostained sections indicated close anatomical overlaps. Similar to Neumann et al [23], we find that the inclusion bodies in all forms of FTLD-FUS consistently immunostain for TAF15, and that EWS protein immunostaining not only appears to be much weaker in intensity in aFTLD-U, NIFID and BIBD cases, but is also localised to a much lesser proportion of the inclusions. However, the commercial EWS antibody employed in the present study (Santa Cruz) may not always behave optimally in its immunoreactivity, and therefore the results obtained need to be interpreted with caution. Similarly, although we did not test the specificity of the commercial TRN1 and TRN2 antibodies from Santa Cruz, ourselves, both of these have been employed in siRNA knockdown and other immunohistochemical studies [25,26], and their specificity validated. According to manufacturer’s datasheets, on western blots, the Santa Cruz TRN1 antibody recognises a single band (of the correct molecular mass) at 97kDa, whereas TRN2 antibody recognises a single band at around 90kDa. It is curious, therefore, that the NCI immunostained by Abcam TRN1 antibody were not immunostained by the Santa Cruz TRN1 antibody, and therefore the results obtained with this antibody likewise need to be viewed with caution.
Present data therefore imply that FUS, TRN1, TAF15, and possibly to a lesser extent EWS, are all implicated in the pathogenesis of inclusion body formation in FTLD-FUS, and indicate that disruption of FUS transport between nucleus and cytoplasm may underpin the pathogenesis of FTLD-FUS. This may not be an entirely unexpected conclusion when it is considered that all 4 proteins form part of the FET family of proteins whose predicted roles include RNA transcription, processing and transport, microRNA processing and DNA repair [27,28], and that protein interaction studies have suggested that they can interact with each other to form protein complexes [27,29]. All of these proteins are normally located within the nucleus, but can continuously shuttle between nucleus and cytoplasm [20,30]. In the present study, we observed nuclear staining to be weak or absent in many cases of FTLD-FUS. While this might on face value be taken as evidence of a cytoplasmic translocation or sequestration (in inclusion bearing cells at least) its absence, in non-inclusion bearing cells in FTLD-FUS, and in cases of FTLD-tau and FTLD-TDP may be greater testament to poor preservation of antigenicity following death or fixation, and not misinterpreted as a specific phenomenon in long fixation cases.
It has also been noted that the FUS inclusions associated with cases of ALS due to mutations in FUS do not apparently contain either TAF15 or EWS proteins [23]. Whether they contain TRN1 or not is presently unknown, though in view of the previously noted observations [23] it might be surmised they would not. Finally, mutations in FUS are associated with ALS and not with FTLD [31-33]. Hence, there may be further mechanistic heterogeneities within the FUSopathies with inherited forms of disease progressing along different pathogenic routes to those in sporadic disease.
It is interesting, as shown here and elsewhere [22,23], that other histological forms of FTLD, particular FTLD-TDP, were not associated with TAF15, EWS or transportins including TRN1. This implies that the function of TDP-43 may not necessarily parallel, or even be complementary to, that of FUS, despite a commonality of many structural elements within each protein. It is becoming increasingly clear that carrier proteins such as TDP-43 and FUS (and their binding partners) play a critical and primary role in the pathogenesis of disorders such as FTLD and Motor Neurone Disease, but they may also contribute secondarily to the pathological outcomes in other disorders such as Alzheimer’s disease, where a substantial minority of cases also display TDP-43 tissue changes. Future research should be directed towards the roles of carrier proteins in normal neurobiology and in neurodegenerative disease.
Acknowledgments
We thank all staff at the Neuropathology Department, Newcastle General Hospital, and Histopathology Department, Salford Royal Hospitals NHS Foundation Trust for technical assistance, particularly Andrew Brown and Janet Thompson at Newcastle for cutting and staining of the sections and Lynne Ramsay for the TDP43 staining. Dr E. Jaros, Andrew Brown and Lynne Ramsay have been supported by the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Hospitals NHS Foundation Trust. Quan Hu is supported through the Program for Changjiang Scholars and Innovative Research Team in University (IRT0810) and the “Project on Absorption of Intellects by Institutions of Higher Education for Academic Disciplinary Innovations” (the “111 Project”) (No: B08006) to Professor Jinzhou Tian, funded by Ministry of Education of People’s Republic of China. We also acknowledge the support of Alzheimers Research UK and Alzheimer’s Society through their funding of the Manchester Brain Bank under the Brains for Dementia Research (BDR) initiative, and the Newcastle Brain Tissue Resource which manages the brain tissue from the Newcastle FTLD series. Also, Newcastle Brain Tissue Resource is funded in part by a grant from the UK Medical Research Council (G0400074, G1100540). DMAM also receives funding from MRC and Wellcome Trust which supported this study in part. EHB receives funding from the Northwestern ADC, AG13854. Support for this work was also provided by grants from the National Institute on Aging of the National Institutes of Health (P50 AG05681 and P01 AG03991), and the Friedman Award to NJC.
Yvonne S Davidson, Andrew C Robinson, Quan Hu, and Manjari Mishra performed all histological and technical work.
Atik Baborie, Evelyn Jaros, Robert H Perry, Nigel J Cairns, Eileen H Bigio supplied case material and assisted with the writing and editing of the manuscript.
Anna Richardson, Alex Gerhard, David Neary and Julie S Snowden classified cases clinically and assisted with the writing and editing of the manuscript.
David M A Mann was responsible for study design, case analysis and principal author
References
- 1.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 2.Neary D, Snowden JS, Mann DMA, Northen B, Goulding PJ, MacDermott N. Frontal lobe dementia and motor neurone disease. J Neurol Neurosurg Psychiatry. 1990;53:23–32. doi: 10.1136/jnnp.53.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strong MJ, Grace GM, Freedman M, Lomen-Hoerth C, Woolley S, Goldstein LH, Murphy J, Shoesmith C, Rosenfeld J, Leigh PN, Bruijn L, Ince P, Figlewicz D. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:131–46. doi: 10.1080/17482960802654364. [DOI] [PubMed] [Google Scholar]
- 4.Cairns NJ, Bigio EH, Mackenzie IRA, Neumann M, Lee VMY, Hatanpaa KJ, White CL, III, Schneider JA, Tenenholz Grinberg L, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DMA. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:2–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 6.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar H, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 7.Mackenzie IRA, Baborie A, Pickering-Brown S, Du Plessis D, Jaros E, Perry RH, Neary D, Snowden JS, Mann DMA. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol. 2006;112:539–49. doi: 10.1007/s00401-006-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, Bruce J, Grossman M, Trojanowski JQ, Lee VM. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–52. doi: 10.2353/ajpath.2006.060438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baborie A, Griffiths TD, Jaros E, Richardson A, Ferrari R, Moreno J, Momeni P, McKeith IG, Burn DJ, Duplessis D, Pal P, Rollinson S, Pickering-Brown SM, Thompson JC, Neary D, Snowden JS, Perry R, Mann DMA. Pathological correlates of frontotemporal lobar degeneration in the elderly. Acta Neuropathol. 2011;121:365–71. doi: 10.1007/s00401-010-0765-z. [DOI] [PubMed] [Google Scholar]
- 10.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–31. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seelaar H, Klijnsma KY, de Koning I, van der Lugt A, Zheng Chiu W, Azmani A, Rozemuller AJM, Van Swieten JC. Frequency of ubiquitin and FUS-positive, TDP-43-negative frontotemporal lobar degeneration. J Neurol. 2010;257:747–53. doi: 10.1007/s00415-009-5404-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snowden JS, Hu Q, Rollinson S, Halliwell N, Robinson A, Davidson YS, Momeni P, Baborie A, Griffiths TD, Jaros E, Perry RH, Richardson A, Neary D, Pickering-Brown SM, Mann DMA. The most common form of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia, but not related to mutations in the FUS gene. Acta Neuropathol. 2011;122:99–110. doi: 10.1007/s00401-011-0816-0. [DOI] [PubMed] [Google Scholar]
- 13.Urwin H, Josephs KA, Rohrer JD, Mackenzie IR, Neumann M, Authier A, Seelaar H, Van Swieten JC, Brown JM, Johannsen P, Nielsen JE, Holm IE, Dickson DW, Rademakers R, Graff-Radford NR, Parisi JE, Petersen RC, Hatanpaa KJ, White Iii CL, Weiner MF, Geser F, Van Deerlin VM, Trojanowski JQ, Miller BL, Seeley WW, van der Zee J, Kumar-Singh S, Engelborghs S, De Deyn PP, Van Broeckhoven C, Bigio EH, Deng HX, Halliday GM, Kril JJ, Munoz DG, Mann DM, Pickering-Brown SM, Doodeman V, Adamson G, Ghazi-Noori S, Fisher EM, Holton JL, Revesz T, Rossor MN, Collinge J, Mead S, Isaacs AM The FReJA Consortium. FUS pathology defines the majority of tau-and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol. 2010;120:33–41. doi: 10.1007/s00401-010-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann M, Roeber S, Kretzchmar HA, Rademakers R, Baker M, Mackenzie IRA. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol. 2009;118:605–16. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz DG, Neumann M, Kusaka H, Yokota O, Ishihara K, Terada S, Kuroda S, Mackenzie IRA. FUS pathology in basophilic inclusion body disease. Acta Neuropathol. 2009;118:617–27. doi: 10.1007/s00401-009-0598-9. [DOI] [PubMed] [Google Scholar]
- 16.Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, Ince PG, Kamphorst W, Revesz T, Rozemuller AJM, Kumar-Singh S, Akiyama H, Baborie A, Spina S, Dickson DW, Trojanowski JQ, Mann DMA. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations: an update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aman P, Panagopoulos I, Larssen C, Fioretos T, Mencinger M, Toresson H, Hoglund M, Forster A, Rabbitts TH, Ron D, Mandahl N, Mitelman F. Expression pattern of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics. 1996;37:1–8. doi: 10.1006/geno.1996.0513. [DOI] [PubMed] [Google Scholar]
- 18.Yang S, Warraich ST, Nicholson GA, Blair IP. Fused in sarcoma/translocated in liposarcoma: a multifunctional DNA/RNA binding protein. Int J Biochem Cell Biol. 2010;42:1408–11. doi: 10.1016/j.biocel.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Andersson MK, Stahlberg A, Arvidsson Y, Olofsson A, Semb H, Stenman G, Nilsson O, Aman P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008;9:37. doi: 10.1186/1471-2121-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zinszner H, Sok J, Immanuel D, Yin Y, Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J Cell Sci. 1997;110:1741–50. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- 21.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, Than ME, Mackenzie IR, Capell A, Scmid B, Neumann M, Haass C. ALS-associated fused in sarcoma (FUS) mutations disrupttransportin-mediated nuclear import. EMBO J. 2010;29:2841–57. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brellstaff J, Lashley T, Holton JL, Lees AJ, Rossor MN, Bandopadhyay R, Revesz T. Transportin 1: a marker for FTLD-FUS. Acta Neuropathol. 2011;122:591–600. doi: 10.1007/s00401-011-0863-6. [DOI] [PubMed] [Google Scholar]
- 23.Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O, Roeber S, Kretsscmar HA, Munoz DG, Kusaka H, Yokota O, Ang L-C, Bilbao J, Rademakers R, Haass C, Mackenzie IRA. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134:2595–609. doi: 10.1093/brain/awr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cairns NJ, Grossman M, Arnold SE, Burn DJ, Jaros E, Perry RH, Duyckaerts C, Stankoff B, Pillon B, Skullerud K, Cruz-Sanchez FF, Bigio EH, Mackenzie IR, Gearing M, Juncos JL, Glass JD, Yokoo H, Nakazato Y, Mosaheb S, Thorpe JR, Uryu K, Lee VM, Trojanowski JQ. Clinical and neuropathologic variation in neuronal intermediate filament inclusion disease. Neurology. 2004;63:1376–84. doi: 10.1212/01.wnl.0000139809.16817.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malnou CE, Salem T, Brockly F, Wodrich H, Piechaczyk, Jariel-Encontre I. Heterodimerization with Ju8n family members regulates c-fos nucleocytoplasmic traffic. J Biol Chem. 2007;282:31046–59. doi: 10.1074/jbc.M702833200. [DOI] [PubMed] [Google Scholar]
- 26.Kodiha M, Banski P, Cheong H-W, Stochaj U. Dissection of the molecular mechanisms that control the nuclear accumulation of transport factors importin-α and CAS in stressed cells. Cell Mol Life Sci. 2008;65:1756–67. doi: 10.1007/s00018-008-7588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kovar H. Dr. Jekyll and Mr. Hyde: the two faces of the FUS/EWS/TAF15 protein family. Sarcoma. 2011:837474. doi: 10.1155/2011/837474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Law WJ, Cann KL, Hicks GG. TLS, EWS and TAF15: a model for transcriptional integration of gene expression. Brief Funct Genomic Proteomic. 2006;5:8–14. doi: 10.1093/bfgp/ell015. [DOI] [PubMed] [Google Scholar]
- 29.Parlich S, Zakaryan RP, Gehring H. Identification of proteins interacting with protein arginine methyltransferase 8; the Ewing sarcoma (EWS) protein binds independently of its methylation state. Proteins. 2008;72:1125–37. doi: 10.1002/prot.22004. [DOI] [PubMed] [Google Scholar]
- 30.Jobert L, Argentini M, Tora L. PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp Cell Res. 2009;315:1273–86. doi: 10.1016/j.yexcr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 31.Kwiatkowski TJ, Bosco DA, LeClerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PA, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, Kenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH., Jr Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 32.Vance C, Roegelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, Ganesalingam J, Williams KL, Tripathi V, Al-Saraj S, Al-Chalabi A, Leigh PN, Blair IP, Nicholson G, de Belleroche J, Gallo JM, Miller CC, Shaw CE. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Langenhove T, van der Zee J, Sleegers K, Engelborghs S, Vandenberghe R, Gijselinck I, Van den Broeck M, Mattheijssens M, Peeters K, De Deyn PP, Cruts M, Van Broeckhoven Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–71. doi: 10.1212/WNL.0b013e3181ccc732. [DOI] [PubMed] [Google Scholar]