

Abstract
The synthesis of halogenated analogs of 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic acid (1), known commonly as bexarotene, and their evaluation for retinoid-X-receptor (RXR)-specific agonist performance is described. Compound 1 is FDA approved to treat cutaneous T-cell lymphoma (CTCL); however, bexarotene treatment can induce hypothyroidism and elevated triglyceride levels, presumably by disrupting RXR heterodimer pathways for other nuclear receptors. The novel halogenated analogs in this study were modeled and assessed for their ability to bind to RXR and stimulate RXR homodimerization in an RXRE-mediated transcriptional assay as well as an RXR mammalian-2-hybrid assay. In an array of 8 novel compounds, 4 analogs were discovered to promote RXR-mediated transcription with comparable EC50 values as 1 and are selective RXR agonists. Our approach also uncovered a periodic trend of increased binding and homodimerization of RXR when substituting a halogen atom for a proton ortho to the carboxylic acid on 1.
Keywords: Bexarotene, Cutaneous T Cell Lymphoma, Retinoic Acid Receptor, Retinoid X Receptor, Rexinoid
Introduction
Retinoids are small molecules that interact with at least two specific nuclear receptors and regulate cellular processes such as gene transcription, proliferation and differentiation. The two major nuclear receptors that retinoids target are retinoid X receptors (RXRs) and retinoic acid receptors (RARs), and both receptors have three known subtypes: α, β and γ. [1] Retinoid receptors, in addition to other lipophilic hormone molecule receptors, are part of a larger receptor superfamily, including the vitamin D receptor (VDR) and thyroid hormone receptor (TR), all of which essentially function to promote transcription in the presence of an appropriate molecular signal. This molecular signal, usually comprised of an endogenous ligand, binds to the protein’s ligand binding pocket (LBP), inducing a conformational change that ultimately enables the protein to bind to a hormone responsive element (HRE) specific for the receptor on DNA. Many HREs are located in or proximal to the promoter regions for the genes they regulate, although a growing number of these elements can be found at large distances upstream or downstream of the gene they control. Nonetheless, HREs are typically constructed of minimal core hexad sequences consisting of half-sites interrupted by nucleotide spacers of variable length between inverted, everted or direct repeats. [2] Nuclear receptors activate transcription by binding to the HREs as heterodimers or homodimers, where each partner binds to a half-site in the element.
Although initially proposed to operate as homodimers [3], HRE high affinity binding by RAR, VDR and TR actually proceeds via an RXR heterodimer. [4] When RXR binds to its natural 9-cis retinoic acid (9-cis RA) ligand, it forms a homodimer that associates with the RXR responsive element or RXRE, but when functioning as a heterodimeric partner with other receptors, RXR can be either liganded or unliganded. For example, in the case of the RXR-VDR heterodimer, RXR is believed to be unliganded. [5] There are a few nuclear receptors, such as the liver X receptor (LXR), that form heterodimers with liganded RXR. [6] Thus, RXR often plays the role of the “master” partner, controlling the operation of several nuclear receptors through liganded and unliganded heterodimers that effect specific physiological equilibria via control of gene expression. [7]
Notably, ligand-promoted RXR homodimer transcriptional activity is suppressed for most cases where RXR is coordinated in a heterodimer with an endogenous ligand-bound receptor such as TR or VDR, and the TR and VDR partners in these RXR heterodimers are termed “nonpermissive” partners for RXR. [5] In contrast to TR and VDR, RAR forms a heterodimer with RXR when RAR binds to all-trans-retinoic acid, but the RXR partner is still capable of binding 9-cis-RA, and both retinoids act in synergy to promote RAR/RARE-mediated gene transcription. When synthetic high-affinity RXR binding ligands (rexinoids) or 9-cis-RA are available, even in the presence of the TR and VDR agonists for the TR-RXR or VDR-RXR heterodimers, the rexinoids divert RXR proteins from heterodimer formation, instead promoting RXR homodimer formation and reducing thyroid hormone and 1,25(OH)2D3 responsiveness. Altering the binding ligand structure for a given nuclear receptor (NR), especially the RXR master heteropartner ligand, produces specific NR modulators (SNuRMs) that have unique properties that exert novel influences on the NR activity. [8]
RXR selective molecule (rexinoid) SNuRMs have been recent targets for medicinal chemistry, since selective RXR activation versus RAR appears to confer chemotherapeutic effects [8] in a number of cancers without inciting concomitant negative side-effects from RAR interaction. [9] After extensive synthesis [10] of molecules modeled in part on the endogenous 9-cis RA (shown below), Ligand Pharmaceuticals, Inc., demonstrated a highly selective RXR agonist, 4-[1-(3,5,5,8,8-pentamethyltetralin-2-yl)ethenyl]benzoic acid (1) [11], commonly named bexarotene. A disilabexarotene (2) [12]Compound, modeled on 1 but substituting two silicon atoms for two carbon atoms in the aliphatic ring system, demonstrated similar RXR specific agonism.
Compound 1 has been FDA approved to treat cutaneous T-cell lymphoma (CTCL), has recently been employed off-label to treat lung cancer [13], and has been analyzed as a treatment for breast cancer [14], colon cancer [15] and other uncontrolled cell proliferation diseases, because promoted expression of RXR regulated genes appears to slow or arrest cell proliferation as well as predisposing cancerous cells to apoptosis in the presence of a chemotherapeutic agent. [14] Additionally, compound 1 and several of its analogs have been considered in non-insulin-dependent diabetes mellitus (NIDDM) mouse models. [16] Despite the ability of compound 1 to specifically activate RXR, versus RAR, the primary drawbacks to treatment with 1 include hyperlipidemia, hypothyroidism [17], and cutaneous toxicity. Many of these side-effects occur either because of antagonism of a non-permissive receptor, as in the case of TR for hypothyroidism [18], or agonism of a permissive receptor, as in the cases of LXR for hyperlipidemia [19] and RAR for cutaneous toxicity [20], at the typical dose concentration. Hence, there is ample motivation to explore novel RXR agonists that may attenuate or avoid these side effects.
There are a plethora of demonstrated RXR agonists modeled on 1. For example, cyclopropyl dienoic acid (3) [21], and several novel aza-retinoids, of which compound 4 [22] is representative, in addition to amide retinoids [23]Have all been disclosed in literature. The thiocarbamate bexarotene analog (5) [24] induces apoptosis when administered to leukemia HL-60 cells. Substituting pyridine for one of the aromatic rings has led to several analogs of 1, such as compound 6 [25], and analogs of 1 possessing unsaturation in the aliphatic ring, as in compound 7 [26], have also been reported. Studies describing the development of selective RXR agonists containing aryl-trienoic acid moieties, either alone [27], or locked by one [28] or two [29] ring systems, were disclosed by Boehm et al., for which compound 8 is representative of the latter. Acetal 9 [30] and compound 10 [7] are both selective, potent RXR agonists, with the latter serving as a model to design a potent RXR antagonist. Also, our group recently synthesized an analog of 1 bearing a fluorine atom ortho to the carboxylic acid moiety, 2-fluoro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (11) [31], that demonstrated slightly higher RXR activation in Caco-2 colon cancer cells and possessed a slightly lower Kd than 1.
Thus, our recent studies have systematically examined by modeling, synthesis, and in vitro biological evaluation eight novel analogs of 11 bearing different or additional halogen atoms, exemplified by compounds 12–19.
Herein, we report the synthesis of compounds 12–19 and evaluate their potency and activity in biologically relevant systems including mammalian 2-hybrid (M2H) assays, RAR- and RXR-response element (RARE and RXRE) transcriptional activation assays in cultured human cell lines, as well as in mutagenicity and apoptosis assays. We have assessed these novel analogs in comparison to compound 1 as well as its ketone analog (20).
Results and Discussion
Molecular Modeling
Computational docking studies were performed to help guide the selection of ligands for synthesis. Analysis of the top binders in a large library of bexarotene-like analogs showed a high preference for electron withdrawing groups ortho to the carboxylic acid of the phenyl group; nearly all the top binders in the docking studies contain a halogen or a nitro group in this position. Structural comparisons of the docked poses showed that the torsional angle between the phenyl ring and the bridge head changed by ~15 to 30° in the ortho-halogenated compounds compared to the non-halogenated compounds. The effect of this rotation was a strengthening of the interactions between Ile268 and the hydrophobic portion of the ligand, as measured by decreased distances in the poses. Halogenation did not change hydrogen bonding with Arg316, however. The degree of rotation was inversely related to the size of the halogen atom, with fluorine incurring the largest rotation and stabilization of the hydrophobic group, and iodine having a smaller rotation and stabilization. Addition of a second halogen amplified the effect. Replacement of the phenyl group by a heteroatomic ring generally decreased binding by disrupting interactions between the ring and Leu309, Ala271, and Leu326. In the docking studies ligands with methylene and cyclopropane bridge heads were predicted to bind more favorably than ligands with ketone bridge heads. Structural analyses of the docked poses showed that methylene and cyclopropane bridge heads were bound in a small hydrophobic pocket, while the ketone bridge head tended to vacate the hydrophobic pocket, disrupting interactions with the hydrophobic ring system of the ligand. This disruption led to a rotation of the ring system away from Ile268, weakening the hydrophobic interactions with this residue. The hydrophobic ring system was accommodated in a large hydrophobic pocket, with interactions between the ligand and Ile345, Ile268, and Leu436.
Given the highly favorable docking energies of the class of ortho-halogenated compounds, and the relative synthetic accessibility of this class, we decided to synthesize and test compounds 12–18, as well as compounds 19–20. Ortho-halogenated compounds 12–17 were found to be within the best 20 binders for the library, and the binding of compound 18 was also predicted to be more favorable than bexarotene. The calculated relative binding free energy for compounds 12–20 as predicted by the docking studies are shown in Fig. 1. These calculated binding free energies were merely used to score the ligands; reported values are relative to the calculated binding free energy of docked bexarotene, with negative numbers indicating better binding. An example of a low energy docking pose for compound 18 is shown in Fig. 2.
Figure 1.

Docking results for compounds 12–20; calculated binding free energies are relative to the calculated binding free energy of docked bexarotene with negative numbers indicating better binding. Triangles represent docks to 1MVC, circles to 1H9U; open symbols represent AutoDockTool charges, and closed symbols OpenBabel charges. Compounds are listed from left to right by increasing docking free energies averaged over the various protocols.
Figure 2.

Low energy docking pose for compound 18.
The Chemistry
The synthesis of compounds 12–19 started with the radical di-bromination of commercially available 3-chloro-4-methylbenzoic acid (21) and 3-bromo-4-methylbenzoic acid (22) to give dibromides 23 and 24, respectively, which were smoothly converted to aldehydes 25 and 26 upon treatment with silver nitrate in ethanol and water according to the method Kishida et al. reported. [32] We initially envisioned the synthesis of the 3,5-difluoro-formylbenzoic acid (27) [33] to follow the same route as used for aldehydes 25 and 26, beginning with the known 3,5-difluoro-4-methylbenzoic acid. [34]However, we chose to implement a three-step, one-pot Bouveault aldehyde synthesis [35] in which 3,5-difluorobenzoic acid (28) was treated with tert-butyl lithium, followed by addition of dimethylformamide and hydrolysis with hydrochloric acid to give 27 in 36% yield according to the method of Anderson and co-workers, instead. [33]Continuing with the general method of Kishida et al. [32], the carboxylic acid aldehydes 25–27 were benzyl-protected by treatment with sodium hydride and benzyl bromide to give benzyl ester aldehydes 29–31, respectively. The ester aldehydes 29–31 were oxidized with sodium chlorite to give the benzyl ester acids 32–34, respectively, whose carboxylic acid moieties were converted to methyl esters 35–37 by treatment with thionyl chloride followed by methanol. [32] The benzyl esters of 35–37 were converted to carboxylic acids 38–40 by treatment with 10% palladium on carbon and hydrogen, however, while 35 and 36 were converted smoothly to 38 and 39 at room temperature and atmospheric pressure hydrogen, benzyl ester 37 required a higher temperature of 70 °C and higher pressure of hydrogen (10–15 bar) that was safely achieved [36] to give quantitative yield of 40. The carboxylic acids 38–40 were then quantitatively converted to the acid chlorides 41–43 with thionyl chloride (Scheme 1). [32]
Scheme 1.
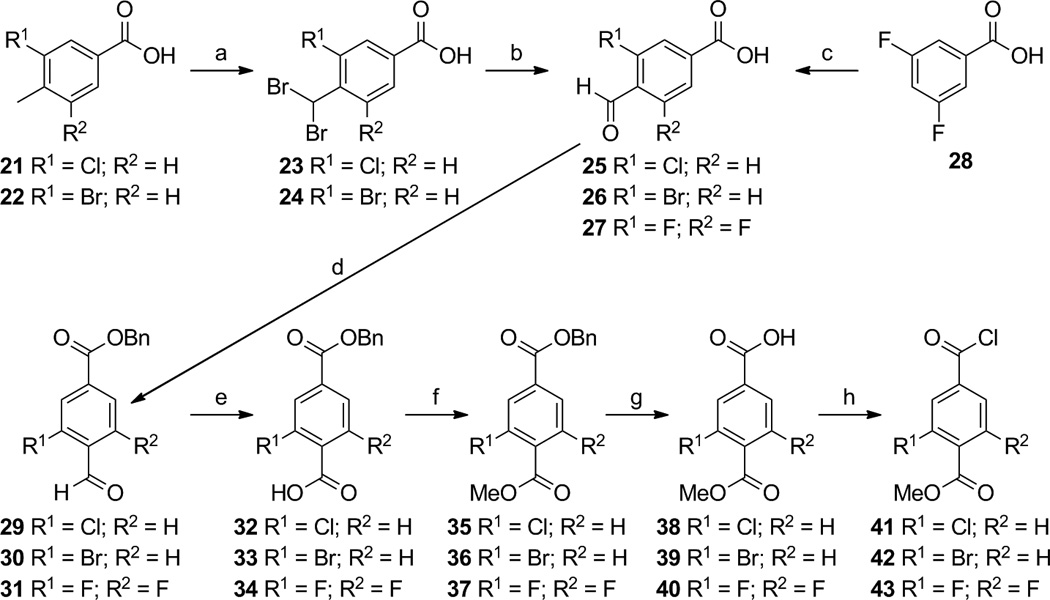
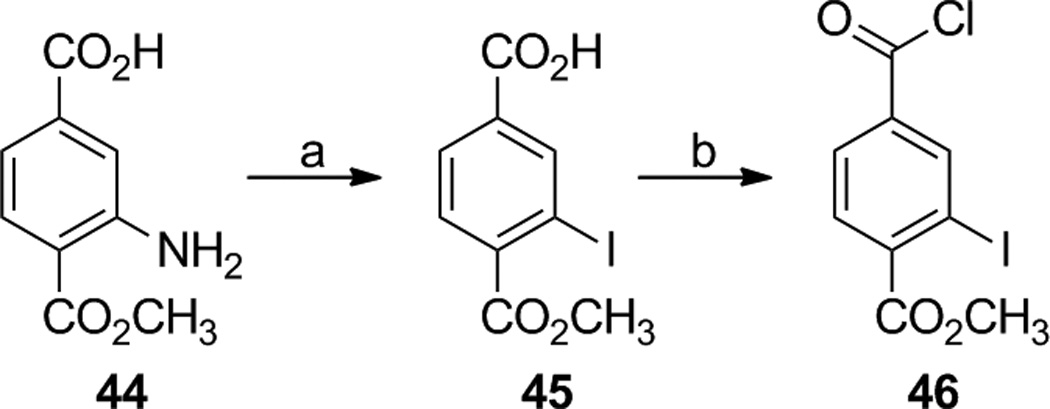
A slightly different route was taken to prepare the iodinated analogs 16 and 17. 4-(Methoxycarbonyl)-3-aminobenzoic acid (44) was treated with sodium nitrite and hydrochloric acid followed by potassium iodide to give 4-(methoxycarbonyl)-3-iodobenzoic acid (45) [37] in 77% yield, and acid 45 was converted to acid chloride 46 with thionyl chloride (Scheme 2).
Scheme 2.
Finally, 2,5-dimethyl-2,5-hexanediol was converted to 2,5-dichloro-2,5-dimethylhexane (47) [38],[11] in 73% yield by treatment with concentrated hydrochloric acid, and the dichloride 47 was reacted with toluene and catalytic aluminum chloride to give 1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalene (48) [11], [31] in 94% yield.
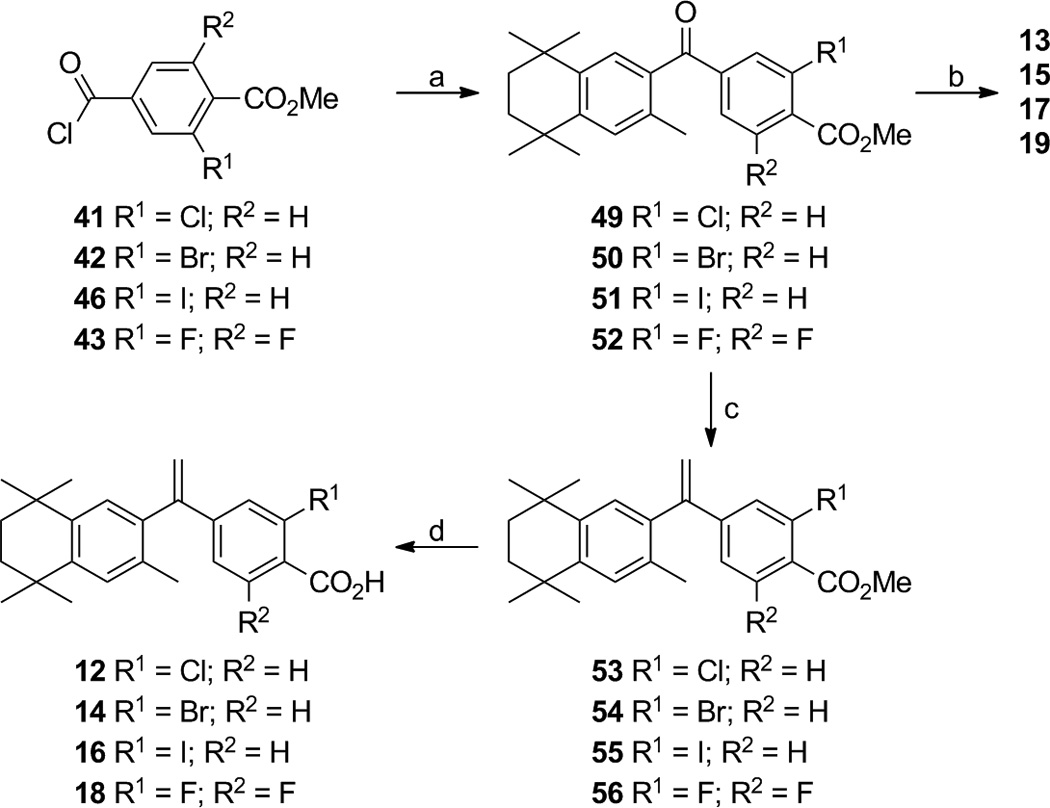
With compound 48 in hand, acid chlorides 41–43 and 46 were converted to ketones 49–52 according to the general method of Boehm and co-workers. [11] Iodo-ketone methyl-ester 51 was observed to possess a small percentage (~7%) of the chloro-ketone methyl-ester 49 by 1H-NMR analysis, and the iodine atom may be labile under reflux conditions with aluminum trichloride. Compounds 49–52 were either saponified with potassium hydroxide in methanol, followed by acidic work-up, to give analogs 13, 15, 17, and 19 respectively, or they were treated with triphenylphosphine methylide to give alkenes 53–56, respectively. The bromo-ketone 15 also possessed minor amounts of unidentifiable impurities that could not be removed by chromatography or crystallization. Alkenes 53–56 were then saponified with potassium hydroxide in methanol, followed by acidic work-up, to give analogs 12, 14, 16, and 18, respectively (Scheme 3). While the iodo-ketone acid 17 contained ~9% of the chloro-ketone acid 13 and could not be purified by column chromatography or attempted crystallization, the minor amount of chloro-alkene acid 12 could be removed from iodo-alkene 16 by crystallization from ethyl acetate.
Scheme 3.
The X-ray crystal structures of compounds 12, 14, and 16 are shown in Figure 3.
Figure 3.

The X-ray crystal structures of bexarotene analogs 12, 14, and 16 shown without hydrogen atoms, for clarity, with thermal ellipsoids at the 50 % probability level.
The X-ray crystal structures of compounds 18 and 52 are shown in Figure 4.
Figure 4.

The X-ray crystal structures of bexarotene analogs 52 and 18 shown without hydrogen atoms, for clarity, with thermal ellipsoids at the 30 % probability level. Compound 18 displayed twist-isomerism in the aliphatic ring. Hence, one twist isomer has been displayed.
Biological Assays and Rationale
A Mammalian Two-Hybrid Assay Demonstrates that Several Novel Analogs bind RXR in an Agonist Fashion
Biological evaluation of the synthetic analogs described above (compounds 12–19) was first carried out in a mammalian two-hybrid assay in human colon cancer (Caco-2) cells then repeated in a different colorectal carcinoma line (HCT-116) (Figure 5 a) and b), respectively). This assay tests the ability of the analog to bind to the recombinant human RXR receptor and induce homodimerization as measured by luciferase output.
Figure 5.


a) and b). Evaluation of potential RXR selective agonists via a mammalian two-hybrid assay in two different types of human colon cancer cells, Caco-2 (a) and HCT-116 (b), respectively. Both cell lines were transfected with pCMV-BD-hRXR binding domain vector (BD), pCMV-AD-hRXR activation domain (AD), pFR-Luc reporter gene containing BD-binding sites, and a renilla control plasmid. Cells were transfected for 7 hours utilizing a liposome-mediated transfection protocol then exposed to either the ethanol vehicle or 10−7 M compound 1 or the indicated analog. After 24 hours the cells were lysed and a luciferase assay was completed. Analog-dependent RXR binding and homodimerization, as measured by luciferase output, was compared to the parent compound 1 (value set to 1.0).
Four compounds were identified in this initial evaluation whose agonist activity ranged from 20 to 400% of the binding of compound 1 in the two separate cell lines. More specifically 12, 14, and 16 are able to bind and mediate homodimerization with about 80, 60, and 20% respectively of the parent compound’s efficiency independent of the cell line. They were also found to be significantly better than the ethanol control vehicle (P values of < 0.05 for all, using one-tailed heteroscadastic t test). Compound 18, however, was found to induce receptor binding and homodimerization better than that of the parent compound 1 to a degree considered statistically significant. Moreover, in comparison to compound 1, 18 was about 1.5 times more potent in its ability to induce RXR homodimerization than 1 in the Caco-2 cells and about 3.5 times better in HCT-116 cells (P values of < 0.05 for all, using one-tailed heteroscedastic t test). These results further support that compounds modeled after 1 can be successfully synthesized to possess RXR binding ability [31] and agonistic properties.
Novel Analogs of 1 are able to direct RXR/RXRE Homodimer Mediated Transcription
The mammalian two-hybrid assay is useful as an initial screen for RXR agonist induced homodimerization because of its accessibility, speed and sensitivity. However, because this assay employs a system that utilizes a synthetic binding domain (BD) and synthetic activating domain (AD), it is possible that within this artificial context the RXR-agonist complex may have an altered ligand or transcriptional coactivator affinity. Thus, it is important to test our collection of potential RXR agonists for the ability to mediate transcription in a more natural setting. Hence, a second screening protocol included the transfection of both Caco-2 and HCT-116 cells with human RXRα and an authentic RXRE from the naturally occurring responsive element in the rat cellular retinol binding protein II gene [39] linked to a luciferase reporter gene. The results in Figure 6 demonstrate that compounds 12, 14, 18, and 19 are able to activate RXR homodimer-mediated transcription significantly better than that of the ethanol vehicle (for compounds 12, 14, and 18 in Caco-2 cells, using a one-tailed heteroscedastic t test P values < 0.05; for these compounds in HCT-116 cells using a one-tailed heteroscedastic t test P values < 0.01; and for compound 19 in both cell lines, using a one-tailed heteroscadastic t test P value < 0.01).
Figure 6.


a) and b). Detection of potential RXR agonists via an RXRE-luciferase based system utilizing human colon cancer cells, Caco-2 (a) and HCT-116 (b), respectively. Both cell lines were transfected with hRXRα, an RXRE luciferase reporter gene, renilla control plasmid, and carrier DNA (pTZ18U). Cells were transfected for 7 hours utilizing a liposome-mediated transfection protocol then exposed to either the ethanol vehicle or 10−7 M compound 1 or the indicated analog. After 24 hours the cells were lysed and a luciferase assay was completed. Analog-dependent, RXR-mediated transcription, as measured by luciferase output, was compared to the parent compound 1 (value set to 1.0).
In the Caco-2 cells, 16 was a weak agonist, while 17 was not able to activate transcription when compared to ethanol, but both analogs did display significant activity in the HCT-116 colorectal carcinoma (using a one-tailed heteroscedastic t test P value for 16 < 0.01 and <0.001 for 17).
Importantly, compounds 12, 14, 16 and 18 displayed activity in both the mammalian two-hybrid assay (Fig. 5) and the RXRE assay (Fig. 6), thus revealing that both assays are not only valid, but generate consistent and complementary data when evaluating RXR agonists.
Determination of Ligand-Receptor Binding Affinity of the Most Active Compounds
To determine the relative binding affinity and effectiveness of our most active analogs, a mammalian two-hybrid assay was performed with ligand concentrations ranging from 10 × 10−10 M up to 0.5 × 10−5 M and EC50 values were calculated (Table 1). Notably, the difluorobexarotene analog (18) has an EC50 value of 34 +/− 6 nM in HCT116 cells versus an EC50 value of 55 +/− 6 nM for bexarotene (1). All of the other analogs possess EC50 values that mirror the results of the mammalian two-hybrid analysis in Figure 5, and the RXRE-based assays in Figure 6.
Table 1.
Determination of EC50 Values
| Compound | EC 50 value, nM (± S.D.) |
|---|---|
| Bexarotene 1 | 55 (± 6) |
| Analog 12 | 90 (± 14) |
| Analog 14 | 150 (± 18) |
| Analog 16 | 280 (± 34) |
| Analog 18 | 34 (± 6) |
| Analog 19 | 550 (± 67) |
EC50 values were determined from full dose-response curves ranging from 10−10 to 10−5 M in transfected HCT-116 cells using an RXR mammalian two-hybrid system as described in Experimental Section.
Analysis of RAR Agonist Activity by the Most Promising Compounds
Since compound 1 is known to have some “residual” retinoic acid receptor (RAR) agonist activity [31] we evaluated this group of analogs for activation of this closely related nuclear receptor. This assay utilizes the expression of human RAR and a retinoic acid responsive element (RARE)-luciferase reporter gene, and it demonstrates (Table 2) that compounds 12, 14, and 18 are comparable to 1 (P value > 0.05 for all, using a one-tailed heteroscedastic t test). Table 2 also shows that 16 and 19 possess significantly lower RAR binding affinity (P value < 0.05 for both, using a one-tailed heteroscedastic t test). All-trans retinoic acid was used as the positive control in this experiment because it is the endogenous ligand for RAR. Taken together, these results not only further support our previous findings [31] that variation of 1 with a halogen atom on the aromatic that bears the carboxylic acid may reduce the activation of RAR (analogs 16 and 19) or increase its capacity to bind and activate RXR (compound 18), but they also strongly suggest a specific periodic trend for the substitution of a proton that may relate to the analog’s degree of agonist activity.
Table 2.
Quantitation of RAR Agonist Activity
| Compound | % RAR Agonist Activity at 100 nM (± S.D.) | % RAR Agonist Activity at 1 µM (± S.D.) |
|---|---|---|
| Bexarotene 1 | 21 (4) | 19 (3) |
| Analog 12 | 13 (2) | 14 (2) |
| Analog 14 | 5 (1) | 5 (1) |
| Analog 16 | 5 (1) | 5 (1) |
| Analog 18 | 8 (1) | 9 (2) |
| Analog 19 | 4 (1) | 8 (2) |
RAR agonist activity was derived from an RAR/RARE reporter system in transfected HEK-293 cells treated with analog or all-trans retinoic acid (RA) at either 100 nM or 1 µM. The activity with analog (or Bex) divided by the activity with all-trans RA expressed as a percentage represents the RAR agonist activity.
Apoptosis in a CTCL System
As previously reported [31], bexarotene and several of its analog are capable of inducing apoptosis. We were interested in determining the pro-apoptotic ability of these new analogs in comparison to compound 1. Since compound 1 has been effectively employed in the treatment of CTCL because it induces apoptosis in the T-lymphocyte, we assayed HuT-78 lymphocytes treated with compound 1 or analogs for caspase 3 and 7 activity, a hallmark of apoptosis, and compared caspase activity to cells treated with ethanol vehicle and sodium butyrate (apoptotic positive control) (Figure 7). Similar to previously published results [31], bexarotene (1) and all of the analogs were better than vehicle control at inducing apoptosis (P values of < 0.01 using one-tailed heteroscadastic t test), after a 48 hour treatment period in our CTCL cells.
Figure 7.

Apoptosis analysis in a CTCL cell line. HuT-78 cells were treated for 48 hours with analog, ethanol vehicle, or sodium butyrate as a positive control. Cells were analyzed for apoptosis via a caspase assay (Promega Caspase-Glo 3/7 assay) according to manufacturer’s instructions. Data is an average of six independent experiments and plotted as a percentage of sodium butyrate activity (set at 100%).
Conclusions
Here we report the modeling, synthesis, and biological evaluation of several analogs of compound 1, in an extension of our earlier work. [31] We have shown that the addition of two fluorine atoms ortho to the carboxylic acid of 1 increases the ability of analog 18 to activate RXR. We have identified several new halogenated analogs of 1 that have apparent binding and biological activity similar to the parent compound, and even follow a periodic trend, with one (18) that capitalizes on the finding that adding one fluorine ortho to the carboxylic acid of 1 increases binding and activation of RXR. [31] With the fact that several RXR selective agonists are being explored to treat many conditions through biological pathways impacted by RXR, now including the demonstrated up-regulation of the apoE gene and the facilitated clearance of plaques by 1 in mouse models of Alzheimer’s Disease [40], there is motivation to develop new RXR agonists. Our results indicate that novel analogs of 1 that substitute halogen atoms on the aromatic ring bearing the carboxylic acid can likely serve as effective, and perhaps more potent, ligands for RXR, which may also have reduced RAR agonist activity (Table 2); thus, these compounds may also possess less detrimental side-effects in cutaneous T-cell lymphoma patients.
Experimental Section
Docking Studies
Docking studies using Autodock 4.2 [41] were performed using the X-ray structures of human RXRα in complex with BMS 649 [42] and RXRβ in complex with LG100268 [43], protein data bank access codes 1MVC and 1H9U, respectively. In both cases, the Arg316 and Ile268 protein residues (numbering as in 1MVC) were treated flexible. Arg316 was selected to enable hydrogen bonding with carboxylate groups on the phenyl moiety, an important interaction identified in earlier studies. [11], [31] Ile268 was selected since structural overlays of 33 different ligand-bound RXR structures in the protein data bank showed large motions of Ile268, and in all cases Ile268 made significant interactions with hydrophobic portions of the ligands. Atomic charges generated by AutoDockTools [41] were frequently overpolarized for nitro groups (leading to a net negative charge on the nitro), while charges generated by OpenBabel 2.3.0 [44] did not have this artifact. Therefore, all docks were performed with both charge models, and results for nitro-compounds were omitted for the AutoDockTools-generated charges. Docking was performed with the Lamarckian genetic algorithm using a maximum of 25,000,000 energy evaluations per docking. The number of docks was set to 250 for three torsions; this number was increased by fifty for each additional torsion, up to a maximum of 400 for six or more torsions. The docking was performed with a large library of bexarotene-like analogs. These analogs differed in substituents on the phenyl ring (including halogenated, carboxylated, hydroxylated and nitrated rings) and fused phenyl ring (including methylated, aminated, nitrated and halogenated rings), as well as changes in the identity of the bridge head between the ring systems (including methylene, ketone, and cyclopropane bridge heads, as well as the absence of a bridge head), and the identity of the aliphatic part of the fused ring system. Calculated AutoDock binding free energies were used to score the ligands.
Mammalian Two-Hybrid Assay
Caco-2 colorectal carcinoma cells were plated overnight at 70,000 cells/well in a 24 well plate and kept in minimum essential media (MEM) (Invitrogen, Carlsbad CA) enhanced with 20% fetal bovine serum (FBS) (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 100 µg/mL streptomycin, and 100 unit/mL penicillin. HCT-116 colorectal carcinoma cells were plated overnight at 70,000 cells/well in a 24 well plate and kept in DMEM enhanced with 10% FBS (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 100 µg/mL streptomycin, and 100 unit/mL penicillin. Both cell lines were co-transfected using a human RXR binding domain vector, a hRXR activation domain (AD), a luciferase reporter gene containing BD-binding sites, and a renilla control plasmid. A liposome-mediated transfection was completed according to the manufacturer’s protocol using 2 µL/well of Express-In transfection reagent (Thermo Fisher Scientific, Lafayette, CO) and allowed to incubate for 7 hours. The cells were then treated with ethanol vehicle, 1, or analogs at a final concentration of 10−7 M and incubated for 24 hours. The amount of rexinoid activity was measure by luciferase output utilizing a dual-luciferase reporter assay system according to the manufacturer’s protocol (Promega, Madison, WI) in a Sirus FB12 luminometer (Berthold Detection Systems, Zylux Corporation, Huntsville, AL). Several independent assays were conducted with triplicate samples for each treatment group.
RXRE-Mediated Transcription Assay
The RXRE assays were completed using both Caco-2 and HCT-116 cells plated at 70,000 cells/well in a 24 well plate and maintained as described above. The cells were cotransfected using 250 ng of RXRE-luciferase reporter gene (RXRE from the naturally occurring responsive element in the rat cellular retinol binding protein II gene), 20 ng of the renilla control plasmid, 50 ng of human pSG5-RXRα, and 100 ng pTZ18U carrier DNA plasmid and 2 µL/well of Express-In was again used for the liposome mediated delivery. The cells were incubated for 7 hours post-transfection and then treated with ethanol, or 10−7 M of either the parent compound or the indicated analog. After a 24-hour incubation period the amount of retinoid activity was measured using the same luciferase assay described above.
RAR/RARE-Agonist Activity Assay
An embryonic kidney cell line, HEK-293, was used for the RARE transcription assay. The cells were plated at a concentration of 70,000 cells/well and maintain as described above. The cells were allowed to incubate over night to ensure attachment to the plate surface. The transfection protocol called for 20 ng of renilla null control plasmid to monitor transfection efficiency, 30 ng of pTZ18U carrier DNA plasmid, 50 ng of the pCMX-human RARα expression vector, and 250 ng of pTK-DR5(X2)-Luc plasmid. Specifics pertaining to this RARE-containing reporter vector have been described previously as well as the sequence of the double RARE. 45 The cells were incubated for 7 hours and then treated with ethanol, all-trans retinoic acid, or analogs at final concentration ranging from 10−6 M to 10−7 M for 24 hours. After the incubation period cell were lysed and the same luciferase assay was completed that was previously described.
Apoptosis Assay
Apoptotic activity was assessed by the Caspase-Glo® 3/7 Assay (Promega, Madison, WI) according to the manufacturer’s instructions. The Caspase-Glo ® 3/7 Assay is based on the cleavage of the DEVD sequence of a luminogenic substrate by caspases 3 and 7 which results in a luminescent signal. HuT-78 (human T-cell lymphoma) cells were distributed (1×104 Cells/well) in white-walled 96-well microplates (Corning, NY) in 100 µl of medium and incubated with 500 µM sodium butyrate (NaBu), 10 µM bexarotene (Bex) or rexinoid analogs for 24 and 48h. The Caspase-Glo® 3/7 Reagent was then added to each well and incubated for an additional 1 h at room temperature. The luminescence was measured in a luminometer (Safire2, Tecan, US). NaBu, a known inducer of apoptosis in HuT-78, was used as a positive control. Each treatment group was dosed in triplicate, and at least two independent experiments were performed. Numbers were standardized to sodium butyrate (set at 100%).
Mutagenicity
We tested the compounds for mutagenicity as in Wagner et al. [31] None of the compounds are mutagenic. This assay utilized a yeast strain, D7, that is genetically engineered to change phenotype upon a genotype change. [46] We used this strain to test the mutagenicity of the compounds by solubilizing the compounds in DMSO and performing a dose/response curve with the highest concentration being 0.15% w/v comparing to DMSO control for mutagenicity, scored as a phenotype change on agar plates. [47]
Instrumentation
A 400 MHz Bruker spectrometer was used to acquire 1H NMR and 13C NMR spectra. Chemical shifts (δ) are listed in ppm against residual non-deuterated solvent peaks in a given deuterated solvent (e.g. CHCl3 in CDCl3) as an internal reference. Coupling constants (J) are reported in Hz, and the abbreviations for splitting include: s, single; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet; br, broad. All 13C NMR spectra were acquired on a Bruker instrument at 100.6 MHz. Chemical shifts (δ) are listed in ppm against deuterated solvent carbon peaks as an internal reference. High resolution mass spectra were recorded using either a JEOL GCmate(2004), a JEOL LCmate(2002) high resolution mass spectrometer or an ABI Mariner (1999) ESI-TOF mass spectrometer. HPLC traces were obtained on an Agilent 1100 LC with a Phenomenex Kinetex C18 10 cm by 2.1 mm column with 2.6u solid core particles.
General Procedures
Tetrahydrofuran, methylene chloride, diethyl ether, and benzene were dried by filtration through alumina according to the procedure described by Grubbs. [48] Removal of volatile solvents transpired under reduced pressure using a Büchi rotary evaporator and is referred to as removing solvents in vacuo. Thin layer chromatography was conducted on precoated (0.25 mm thickness) silica gel plates with 60F-254 indicator (Merck). Column chromatography was conducted using 230–400 mesh silica gel (E. Merck reagent silica gel 60). All tested compounds were analyzed for purity by combustion analysis through Columbia Analytical Services (formerly Desert Analytics in Tucson, AZ) and were found to be > 95% pure.
Benzyl-3-Chloro-4-formylbenzoate (29)
Compound 29 was synthesized according to the methods of Kishida and co-workers. [32] To a 500 mL round bottom flask charged with 3-chloro-4-methylbenzoic acid (21) (3.24 g, 19.0 mmol) was added NBS (8.00 g, 44.9 mmol), benzoylperoxide (0.23 g, 0.95 mmol), and carbon tetrachloride (37 mL). The reaction solution was heated to reflux under magnetic stirring for 36 h, cooled to room temperature, and solids were filtered and washed with carbon tetrachloride (~20 mL). The filtrate solvent was removed in vacuo and the crude 4-(dibromomethyl)-3-chlorobenzoic acid (23) was dried on high vacuum and used without further purification. To a 500 mL round bottom flask charged with crude 23 (6.23 g, 19.0 mmol) was added ethanol (48 mL), and a solution of silver nitrate (6.64 g, 39.1 mmol) in warm water (9 mL) was added dropwise while the reaction solution was stirred in an oil bath preheated to 50–55 °C. Upon addition of the silver nitrate solution, a green precipitate formed. After stirring at 50 °C for 45 min, the reaction solution was cooled to room temperature and filtered to remove the green precipitate. The filtrate solvent was concentrated in vacuo, extracted with ethyl acetate, and the combined organic extracts were washed with water and brine, dried over sodium sulfate and removed in vacuo to give crude 3-chloro-4-formylbenzoic acid (25) (3.51 g, 100%) that was used without further purification. To a 500 mL round bottom flask charged with 25 (3.51 g, 19.0 mmol) was added dry dimethylformamide (37 mL) and a 60 wt % suspension of NaH in mineral oil (0.93 g, 23 mmol) in small aliquots over 20 min. The reaction solution was stirred an additional 20 min, and benzylbromide (2.8 mL, 23 mmol) was added to the red heterogeneous solution. After stirring 5 h, the reaction solution had become homogeneous, and it was poured into 1N HCl (100 mL), extracted with ethyl acetate (100 mL, twice), and the combined organic extracts were washed with saturated NaHCO3 (75 mL) and brine, dried over sodium sulfate, and removed in vacuo to give crude 29. Crude 29 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 4:1) to give 29 (5.2 g, 99%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 10.52 (s, 1H), 8.15 (dd, J = 8.0, 1.6, 1H), 7.97–8.04 (m, 2H), 7.37–7.48 (m, 5H), 5.39 (s, 2H); IR (neat): ν = 2968, 2855, 2656, 2560, 1724, 1687, 1605, 1556, 1485 cm−1 ; GC-MS [M+] calcd for C15H11O3Cl: 274.0397, found: 274.0390.
4-((Benzyloxy)carbonyl)-2-chlorobenzoic Acid (32)
Compound 32 was prepared according to the method of Kishida and co-workers. [32] To a 100 mL round bottom flask charged with 29 (5.22 g, 19.0 mmol), sulfamic acid (1.86 g, 19.2 mmol), water (20 mL), and ACN (15 mL) was added a solution of 80% NaClO2 (1.79 g, 19.8 mmol) in water (10 mL). After stirring for 1 h, the reaction solution was poured into saturated Na2SO3 (25 mL) and 1N HCl (50 mL), and the resulting solution was extracted with ethyl acetate (50 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 32 (4.97 g, 90%) that was used without further purification. A small sample was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 1:1) to give pure 32 as a white powder, m.p. 120–122 °C: 1H NMR (400 MHz, CDCl3) δ 10.71 (br s, 1H), 8.17 (d, J = 1.6, 1H), 8.05 (d, J = 8.0, 1H), 8.02 (dd, J = 8.0, 1.6, 1H), 7.36–7.47 (m, 5H), 5.40 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 170.1, 164.4, 135.2, 134.8, 134.7, 132.4, 132.3, 132.2, 128.7, 128.6, 127.6, 67.6; IR (neat): ν = 2964, 1724, 1685, 1603, 1557, 1484, 1455 cm−1 ; LC-FAB-MS [M+ +H] calcd for C15H12O4Cl: 291.0424, found: 291.0434.
4-Benzyl 1-methyl 2-chlorobenzene-1,4-dioate (35)
Compound 35 was synthesized according to the method of Kishida and co-workers. [32] To a 100 mL round bottom flask charged with compound 32 (5.5 g, 18.9 mmol) was added SOCl2 (16.0 mL, 220 mmol) and the reaction solution was refluxed for 1h in an oil bath at 84 °C. The reaction solution was cooled to room temperature and the excess thionyl chloride was removed in vacuo to give crude 4-chlorocarbonyl-2-chlorobenzoic acid benzyl ester. The crude 4-chlorocarbonyl-2-chlorobenzoic acid in dry toluene (8 mL) was added dropwise to a solution of triethylamine (5.2 mL, 37 mmol) in methanol (53 mL, 1.3 mol) over 10 min. The reaction solution was stirred 1h and poured into 1N HCl (150 mL) and extracted with ethyl acetate (80 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 35. Crude 35 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 95:5) to give pure 35 as a white solid (5.05 g, 87%), m.p. 46–48 °C: 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 1.6, 1H), 7.98 (dd, J = 8.0, 1.6, 1H), 7.85 (d, J = 8.0, 1H), 7.36–7.46 (m, 5H), 5.38 (s, 2H), 3.95 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 165.5, 164.4, 135.3, 134.0, 133.8, 133.7, 132.0, 131.2, 128.7, 128.6, 128.4, 127.6, 67.4, 52.7; IR (neat): ν = 3033, 2958, 1735, 1716, 1560, 1498, 1482 cm−1; GC-MS [M+] calcd for C16H13O4Cl: 304.0502, found: 304.0498.
4-(Methoxycarbonyl)-3-chlorobenzoic Acid (38)
Compound 38 was synthesized according to the methods of Kishida and co-workers. [32] A 3-neck 250 mL round bottom flask charged with 35 (4.94 g, 16.2 mmol), 10% Pd/C (0.499 g), ethanol (28.0 mL), and ethyl acetate (28.0 mL) was evacuated and back-filled with hydrogen gas from a balloon three times, and the reaction solution was allowed to stir under hydrogen at room temperature overnight. The reaction solution was filtered through celite, and the solvents were removed in vacuo to give crude 38 (2.97 g, 85%) as a white crystalline solid that was used without further purification. A small sample of crude 38 was purified by recrystallization from hot ethyl acetate to give pure 38, m.p. 156–157 °C: 1H NMR (400 MHz, DMSO-d6) δ 13.64 (br s, 1H), 7.99 (d, J = 1.6, 1H), 7.95 (dd, J = 8.0, 1.6, 1H), 7.89 (d, J = 8.0, 1H), 3.89 (s, 3H); 13C NMR (100.6 MHz, DMSO-d6) δ 165.4, 165.1, 134.9, 133.7, 131.8, 131.2, 130.9, 128.0, 52.8; IR (neat): ν = 2961, 2824, 2545, 1717, 1687, 1557, 1488 cm−1; LC-APCI-MS [M+] calcd for C9H7O4Cl: 214.0033, found: 214.0027.
Methyl 2-chloro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoate (49)
Compound 49 was synthesized according to the method of Boehm and co-workers. [11] Methyl 4-(chlorocarbonyl)-2-chlorobenzoate (41) was synthesized by refluxing 4-(methoxycarbonyl)-3-chlorobenzoic acid (38) (1.35 g, 6.29 mmol) in thionyl chloride (10.0 mL, 137 mmol) in a 100 mL one-neck round bottom flask fitted with a water-cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 41 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 41 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 48 (1.38 g, 6.82 mmol) followed by a solution of crude acid chloride 41 (6.29 mmol) in DCM (15 mL). Aluminum chloride (2.0 g, 15 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and concentrated to give crude 49. Crude 49 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 92.5:7.5) to give 49 (2.22 g, 88%) as a white, crystalline solid, m.p. 97–98 °C: 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 8.0, 1H), 7.88 (d, J = 1.6, 1H), 7.70 (dd, J = 8.0, 1.6, 1H), 7.25 (s, 1H), 7.21 (s, 1H), 3.96 (s, 3H), 2.35 (s, 3H), 1.69 (s, 4H), 1.31 (s, 6H), 1.20 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ196.0, 165.7, 148.8, 142.0, 141.8, 134.8, 133.9, 133.7, 133.3, 132.3, 131.1, 129.6, 128.6, 127.8, 52.7, 34.8, 34.7, 34.3, 33.8, 31.6, 31.5, 20.0; IR (neat): ν = 2961, 2864, 1738, 1665, 1547, 1484 cm−1; GC-MS [M+] calcd for C24H27O3Cl: 398.1649, found: 398.1657.
Methyl 2-chloro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (53)
Compound 53 was synthesized according to the method of Boehm and co-workers. [11] To a 20-dram vial containing 49 (0.830 g, 2.08 mmol) and dry THF (3 mL) at room temperature with a Teflon magnetic stir-bar was slowly added a triphenylphosphonium methylide solution prepared as follows: to a 100 mL round bottom flask equipped with a Teflon magnetic stir-bar and containing dry THF (2.0 mL) was added iPr2NH (0.66 mL, 4.67 mmol) and a 2.5M solution of n-butyl lithium in hexanes (1.7 mL, 4.25 mmol), and the solution was stirred for 30 min at room temperature at which point, methyl triphenylphosphonium bromide (1.13 g, 3.19 mmol) was added and the solution was stirred an additional 20 min to provide a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (50 mL) and the aqueous solution was extracted with ethyl acetate (50 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 53 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 53 (0.283 g, 34%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.0, 1H), 7.39 (d, J = 1.6, 1H), 7.18 (dd, J = 8.0, 1.6, 1H), 7.10 (s, 1H), 7.09 (s, 1H), 5.80 (d, J = 1.2, 1H), 5.35 (d, J = 1.2, 1H), 3.92 (s, 3H), 1.95 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 165.9, 147.9, 145.7, 144.6, 142.4, 137.2, 133.9, 132.6, 131.5, 128.9, 128.2, 128.1, 128.0, 124.8, 117.7, 52.3, 35.1, 35.0, 33.9, 33.8, 31.9, 31.8, 19.9; IR (neat): ν = 2955, 2924, 2860, 1734, 1714, 1598, 1542, 1497, 1456 cm−1; GC-MS [M+] calcd for C25H29O2Cl: 396.1856, found: 396.1850.
2-Chloro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (12)
Compound 12 was synthesized following the methods of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 53 (0.353 g, 0.89 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.4 mL, 2.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (42 mL). The aqueous solution was extracted with ethyl acetate (50 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 12. Crude 12 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 12 (0.31 g, 91%) as a white crystalline solid, m.p. 200–201 °C: 1H NMR (400 MHz, CDCl3) δ 7.98 (d, J = 8.0, 1H), 7.43 (d, J = 1.6, 1H), 7.21 (dd, J = 8.0, 1.6, 1H), 7.11 (s, 1H), 7.09 (s, 1H), 5.83 (d, J = 0.8, 1H), 5.38 (d, J = 0.8, 1H), 1.96 (s, 3H), 1.71 (s, 4H), 1.31 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 170.5, 147.8, 146.8, 144.7, 142.5, 137.1, 135.0, 132.6, 132.5, 129.3, 128.2, 128.0, 126.5, 124.9, 118.2, 35.1, 34.0, 33.9, 31.9, 31.8, 19.9; IR (neat): ν = 2958, 1694, 1596, 1488 cm−1; LC-APCI-MS [M++H] calcd for C24H28O2Cl: 383.1778, found: 383.1778. Anal. Calcd for C24H27O2Cl: C 75.28; H 7.11; Cl 9.26. Found: C 75.20; H 6.93; Cl 9.60.
2-Chloro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoic acid (13)
Compound 13 was synthesized following the method of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 49 (0.353 g, 0.88 mmol) and methanol (4 mL) was added a 5M aqueous solution of potassium hydroxide (0.4 mL, 2.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The precipitate was filtered and washed with water to give crude 13 (0.335 g, 98%). Crude 13 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 13 (0.13 g, 40%) as a white crystalline solid, m.p. 174–175 °C: 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 8.0, 1H), 7.92 (d, J = 1.6, 1H), 7.74 (dd, J = 8.0, 1.6, 1H), 7.27 (s, 1H), 7.23 (s, 1H), 2.37 (s, 3H), 1.70 (s, 4H), 1.32 (s, 6H), 1.22 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 196.0, 169.9, 149.0, 142.6, 142.1, 134.9, 134.7, 133.7, 132.7, 132.1, 131.6, 129.7, 128.7, 127.9, 34.8, 34.7, 34.4, 33.9, 31.6, 31.5, 20.1; IR (neat): ν = 2955, 2926, 1704, 1667, 1543, 1458 cm−1; LC-APCI-MS [M++H] calcd for C23H26O3Cl: 385.1571, found: 385.1564. Anal. Calcd for C23H25O3Cl: C 71.77; H 6.55; Cl 9.21. Found: C 71.45; H 6.30; Cl 9.2.
Benzyl-3-bromo-4-formylbenzoate (30)
Compound 30 was synthesized according to the methods of Kishida and co-workers. [32] To a 500 mL round bottom flask charged with 3-chloro-4-methylbenzoic acid (22) (8.17 g, 38.0 mmol) was added NBS (16.10 g, 90.45 mmol), benzoylperoxide (0.45 g, 1.8 mmol), and carbon tetrachloride (74 mL). The reaction solution was heated to reflux under magnetic stirring for 36 h, cooled to room temperature, and solids were filtered and washed with carbon tetrachloride (~20 mL). The filtrate solvent was removed in vacuo and the crude 4-(dibromomethyl)-3-bromobenzoic acid (24) was dried on high vacuum and used without further purification. To a 500 mL round bottom flask charged with crude 24 (14.16 g, 38.0 mmol) was added ethanol (96 mL), and a solution of silver nitrate (13.28 g, 78.18 mmol) in warm water (18 mL) was added dropwise while the reaction solution was stirred in an oil bath preheated to 50–55 °C. Upon addition of the silver nitrate solution, a green precipitate formed. After stirring at 50 °C for 45 min, the reaction solution was cooled to room temperature and filtered to remove the green precipitate. The filtrate solvent was concentrated in vacuo, extracted with ethyl acetate, and the combined organic extracts were washed with water and brine, dried over sodium sulfate and removed in vacuo to give crude 3-bromo-4-formylbenzoic acid (26) (8.7 g, 100%) that was used without further purification. To a 500 mL round bottom flask charged with 26 (8.7 g, 38.0 mmol) was added dry dimethylformamide (74 mL) and a 60 wt % suspension of NaH in mineral oil (1.86 g, 46.5 mmol) in small aliquots over 20 min. The reaction solution was stirred an additional 20 min, and benzylbromide (5.60 mL, 46.8 mmol) was added to the red heterogeneous solution. After stirring 5 h, the reaction solution had become homogeneous, and it was poured into 1N HCl (200 mL), extracted with ethyl acetate (100 mL, twice), and the combined organic extracts were washed with saturated NaHCO3 (75 mL) and brine, dried over sodium sulfate, and removed in vacuo to give crude 30. Crude 30 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 4:1) to give 30 (12.1 g, 99%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 10.40 (d, J = 0.8, 1H), 8.33 (d, J = 1.6, 1H), 8.10–8.08 (ddd, J = 0.8, 1.6, 8.0, 1H), 7.95 (d, J = 8.0 5H), 7.37–7.47 (m, 5H), 5.39 (s, 2H); IR (neat): ν = 2962, 1723, 1683, 1601, 1556, 1479 cm−1; GC-MS [M+] calcd for C15H11O3Br: 317.9892, found: 317.9887.
4-((Benzyloxy)carbonyl)-2-bromobenzoic Acid (33)
Compound 33 was prepared according to the method of Kishida and co-workers. [32] To a 100 mL round bottom flask charged with 30 (12.1 g, 37.9 mmol), sulfamic acid (4.06 g, 41.8 mmol), water (45 mL), and ACN (32 mL) was added a solution of 80% NaClO2 (3.84 g, 42.5 mmol) in water (20 mL). After stirring for 1 h, the reaction solution was poured into saturated Na2SO3 (50 mL) and 1N HCl (100 mL), and the resulting solution was extracted with ethyl acetate (100 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 33 (10.5 g, 82%) that was used without further purification. A small sample was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 1:1) to give pure 33 as a white powder: 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 1.6, 1H), 8.08 (dd, J = 8.0, 1.6, 1H), 8.01 (d, J = 8.0, 1H), 7.34–7.47 (m, 5H), 5.39 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 170.3, 164.2, 135.6, 135.2, 134.4, 134.2, 132.1, 128.7, 128.6, 128.4, 128.2, 122.3, 67.6; IR (neat): ν = 2963, 2538, 1681, 1602, 1551, 1481 cm−1; LC-APCI-MS [M++H] calcd for C15H12O4Br: 334.9919, found: 334.9934.
4-Benzyl 1-methyl 2-bromobenzene-1,4-dioate (36)
Compound 36 was synthesized according to the method of Kishida and co-workers. [32] To a 100 mL round bottom flask charged with compound 33 (10.5 g, 31.3 mmol) was added SOCl2 (24.0 mL, 330 mmol) and the reaction solution was refluxed for 1h in an oil bath at 84 °C. The reaction solution was cooled to room temperature and the excess thionyl chloride was removed in vacuo to give crude 4-chlorocarbonyl-2-bromobenzoic acid benzyl ester. The crude 4-chlorocarbonyl-2-bromobenzoic acid in dry toluene (16 mL) was added dropwise to a solution of triethylamine (10.4 mL, 74 mmol) in methanol (106 mL, 2.6 mol) over 10 min with vigorous stirring. The reaction solution was stirred 1h and poured into 1N HCl (300 mL) and extracted with ethyl acetate (80 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 36. Crude 36 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 95:5) to give pure 36 as an oil (8.55 g, 78%): 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 1.6, 1H), 8.03 (dd, J = 8.0, 1.6, 1H), 7.80 (d, J = 8.0, 1H), 7.36–7.46 (m, 5H), 5.38 (s, 2H), 3.95 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 166.1, 164.3, 136.2, 135.3, 135.2, 133.7, 130.9, 128.7, 128.5, 128.4, 128.2, 121.4, 67.4, 52.7; IR (neat): ν = 2964, 2558, 1723, 1688, 1603, 1557, 1484 cm−1; GC-MS [M+] calcd for C16H13O4Br: 347.9997, found: 348.0013.
4-(Methoxycarbonyl)-3-bromobenzoic Acid (39)
Compound 39 was synthesized according to the methods of Kishida and co-workers. [32] A 3-neck 250 mL round bottom flask charged with 36 (8.55 g, 24.4 mmol), 10% Pd/C (1.8 g), ethanol (50.0 mL), and ethyl acetate (50.0 mL) was evacuated and back-filled with hydrogen gas from a balloon three times, and the reaction solution was allowed to stir under hydrogen at room temperature for 48 h. The reaction solution was filtered through celite, and the solvents were removed in vacuo to give crude 39 (6.15 g, 96%) as a white crystalline solid that was used without further purification. A small sample of crude 39 was purified by recrystallization from hot ethyl acetate to give pure 39, m.p. 138–140 °C: 1H NMR (400 MHz, DMSO-d6) δ 13.59 (br s, 1H), 8.15 (d, J = 1.6, 1H), 8.00 (dd, J = 8.0, 1.6, 1H), 7.85 (d, J = 8.0, 1H), 3.89 (s, 3H); 13C NMR (100.6 MHz, DMSO-d6) δ 165.8, 165.3, 136.1, 134.7, 134.0, 130.9, 128.4, 119.9, 52.8; IR (neat): ν = 2959, 2844, 2545, 1716, 1686, 1603, 1551, 1485 cm−1; LC-GC-MS [M+] calcd for C9H7O4Br: 257.9528, found: 257.9502.
Methyl 2-bromo-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoate (50)
Compound 50 was synthesized according to the method of Boehm and co-workers.[11] Methyl 4-(chlorocarbonyl)-2-bromobenzoate (42) was synthesized by refluxing 4-(methoxycarbonyl)-3-bromobenzoic acid (39) (1.64 g, 6.31 mmol) in thionyl chloride (12.0 mL, 165 mmol) in a 100 mL one-neck round bottom flask fitted with a water-cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 42 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 42 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 48 (1.38 g, 6.82 mmol) followed by a solution of crude acid chloride 42 (6.31 mmol) in DCM (15 mL). Aluminum chloride (2.20 g, 16.5 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and concentrated to give crude 50. Crude 50 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 92.5:7.5) to give 50 (2.37 g, 84%) as a white, crystalline solid, m.p. 109–111 °C: 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 1.6, 1H), 7.83 (d, J = 8.0, 1H), 7.75 (dd, J = 8.0, 1.6, 1H), 7.25 (s, 1H), 7.22 (s, 1H), 3.97 (s, 3H), 2.35 (s, 3H), 1.69 (s, 4H), 1.31 (s, 6H), 1.20 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 195.8, 166.2, 148.8, 142.0, 141.6, 135.6, 135.4, 134.8, 133.8, 130.9, 129.6, 128.7, 128.5, 121.5, 52.7, 34.8, 34.7, 34.3, 33.8, 31.6, 31.5, 20.0; IR (neat): ν = 3015, 2953, 2931, 2863, 1736, 1666, 1605, 1544, 1496, 1459 cm−1; GC-MS [M+] calcd for C24H27O3Br: 442.1144, found: 442.1135.
Methyl 2-bromo-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (54)
Compound 54 was synthesized according to the method of Boehm and co-workers. [11] To a 20-dram vial containing 50 (0.923 g, 2.08 mmol) and dry THF (3 mL) at room temperature with a Teflon magnetic stir-bar was slowly added a triphenylphosphonium methylide solution prepared as follows: to a 100 mL round bottom flask equipped with a Teflon magnetic stir-bar and containing dry THF (2.0 mL) was added iPr2NH (0.66 mL, 4.67 mmol) and a 2.5M solution of n-butyl lithium in hexanes (1.7 mL, 4.25 mmol), and the solution was stirred for 30 min at room temperature at which point, methyl triphenylphosphonium bromide (1.13 g, 3.19 mmol) was added and the solution was stirred an additional 20 min to provide a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (50 mL) and the aqueous solution was extracted with ethyl acetate (50 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 54 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 54 (0.712 g, 77%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.0, 1H), 7.62 (d, J = 1.6, 1H), 7.20 (dd, J = 8.0, 1.6, 1H), 7.10 (s, 1H), 7.08 (s, 1H), 5.80 (d, J = 1.2, 1H), 5.34 (d, J = 0.8, 1H), 3.92 (s, 3H), 1.95 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ166.3, 147.8, 145.7, 144.6, 142.4, 137.1, 132.6, 132.1, 131.3, 130.2, 128.1, 128.0, 125.4, 121.9, 117.8, 52.4, 35.1, 35.0, 33.9, 33.8, 31.9, 31.8, 20.0; IR (neat): ν = 2956, 2921, 1731, 1596, 1541, 1497, 1485 cm−1; GC-MS [M+] calcd for C25H29O2Br: 440.1351, found: 440.1379.
2-Bromo-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (14)
Compound 14 was synthesized following the methods of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 54 (0.4056 g, 0.9189 mmol) and methanol (4 mL) was added a 5M aqueous solution of potassium hydroxide (0.4 mL, 2.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (42 mL). The aqueous solution was extracted with ethyl acetate (50 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 14. Crude 14 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 14 (0.273 g, 70%) as a white crystalline solid, m.p. 199–200 °C: 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.0, 1H), 7.67 (d, J = 1.6, 1H), 7.24 (dd, J = 8.0, 1.6, 1H), 7.10 (s, 1H), 7.09 (s, 1H), 5.82 (d, J = 0.8, 1H), 5.37 (d, J = 0.8, 1H), 1.96 (s, 3H), 1.70 (s, 4H), 1.30 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 170.4, 147.7, 146.6, 144.7, 142.4, 137.0, 132.6, 132.5, 128.4, 128.2, 128.0, 125.6, 122.8, 118.3, 35.1, 35.0, 34.0, 33.9, 31.9, 31.8, 20.0; IR (neat): ν = 2959, 1697, 1594, 1484, 1452 cm−1; LC-APCI-MS [M++H] calcd for C24H28O2Br: 427.1273, found: 427.1287. Anal. Calcd for C24H27O2Br: C 67.45; H 6.37; Br 18.7. Found: C 68.16; H 6.07; Br 18.1.
2-Bromo-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoic acid (15)
Compound 15 was synthesized following the method of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 50 (0.395 g, 0.89 mmol) and methanol (4 mL) was added a 5M aqueous solution of potassium hydroxide (0.4 mL, 2.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The precipitate was filtered and washed with water to give crude 15 (0.3722 g, 97%). Crude 15 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give pure 15 (0.25 g, 65%) as a white crystalline solid, m.p. 166–167 °C: 1H NMR (400 MHz, CDCl3) δ 8.13 (d, J = 1.6, 1H), 8.05 (d, J = 8.0, 1H), 7.80 (dd, J = 8.0, 1.6, 1H), 7.27 (s, 1H), 7.23 (s, 1H), 2.38 (s, 3H), 1.70 (s, 4H), 1.32 (s, 6H), 1.21 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 195.7, 170.6, 149.0, 143.1, 142.2, 142.0, 136.4, 135.1, 133.6, 131.6, 129.7, 129.4, 129.0, 94.1, 34.8, 34.7, 34.4, 33.9, 31.6, 31.5, 20.1; IR (neat): ν = 2955, 2924, 1703, 1667, 1607, 1542, 1456 cm−1; LC-APCI-MS [M++H] calcd for C23H26O3Br: 429.1065, found: 429.1074. Anal. Calcd for C23H25O3Br: C 64.34; H 5.87; Br 18.61. Found: C 65.08; H 5.59; Br 17.1.
4-(Methoxycarbonyl)-3-iodobenzoic acid (45)
The method of Bertozzi and co-workers was followed to synthesize 45. [37] To a 100 mL round bottom flask charged with 1-mehtyl-2-aminoterephthalate (0.503 g, 2.58 mmol) was added concentrated HCl (5 mL) followed by the dropwise addition of a solution of sodium nitrite (0.185 g, 2.68 mmol) in water (1 mL), during which addition, orange gas evolved. The reaction was stirred (30 min) at room temperature, filtered through glass wool, and then a solution of potassium iodide (4.31 g, 2.6 mmol) in water (7 mL) was added, dropwise. The resulting red solution was stirred (1h) and then diluted with DCM (100 mL). The layers were separated, and the aqueous layer was washed with DCM (10 mL, twice), followed by saturated Na2SO3, water, and then brine. The aqueous layers were then back extracted with DCM (20 mL), and the combined organic layers were dried over sodium sulfate and concentrated to give crude 45. Crude 45 was dissolved in hot methanol (15 mL), and water was added (15 mL), and the resulting solution was cooled in an ice bath and the precipitate was filtered to give pure 45 (0.318 g, 40%) as a yellow powder, m.p. 160–163 °C: 1H NMR (400 MHz, CDCl3) δ 11.00 (br s, 1H), 8.68 (d, J = 1.6, 1H), 8.11 (dd, J = 8.0, 1.6, 1H), 7.83 (d, J = 8.0, 1H), 3.97 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 169.7, 166.5, 142.6, 140.1, 132.4, 130.5, 129.4, 93.3, 52.8. IR (neat): ν = 3289, 2952, 2652, 1736, 1693, 1551, 1480 cm−1.
Methyl 2-iodo-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoate (51)
Compound 51 was synthesized according to the method of Boehm and co-workers. [11] Methyl 4-(chlorocarbonyl)-2-iodobenzoate (46) was synthesized by refluxing 4-(methoxycarbonyl)-3-iodobenzoic acid (45) (1.93 g, 6.31 mmol) in thionyl chloride (10.0 mL, 137 mmol) in a 100 mL one-neck round bottom flask fitted with a water-cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 46 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 46 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 48 (1.38 g, 6.82 mmol) followed by a solution of crude acid chloride 46 (6.31 mmol) in DCM (15 mL). Aluminum chloride (2.30 g, 17.2 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to dark red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and concentrated to give crude 51. Attempted purification of crude 51 by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 92.5:7.5) gave 51 (2.91 g, 89%) that was ~ 7 mol % 49 as a white, crystalline solid, m.p. 115–116 °C: 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 1.6, 1H), 7.83 (d, J = 8.0, 1H), 7.79 (dd, J = 8.0, 1.6, 1H), 7.25 (s, 1H), 7.22 (s, 1H), 3.97 (s, 3H), 2.37 (s, 3H), 1.69 (s, 4H), 1.31 (s, 6H), 1.21 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 195.7, 166.6, 148.8, 142.5, 141.9, 141.4, 138.5, 135.0, 133.7, 130.4, 129.6, 129.3, 128.8, 93.5, 52.7, 34.8, 34.7, 34.3, 33.8, 31.6, 31.5, 20.1; IR (neat): ν = 2953, 1729, 1667, 1540, 1459 cm−1; GC-MS [M+] calcd for C24H27O3I: 490.1005, found: 490.0995.
methyl 2-iodo-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (55)
Compound 55 was synthesized according to the method of Boehm and co-workers. [11] To a 20-dram vial containing 51 (1.0123 g, 2.06 mmol) and dry THF (3 mL) at room temperature with a Teflon magnetic stir-bar was slowly added a triphenylphosphonium methylide solution prepared as follows: to a 100 mL round bottom flask equipped with a Teflon magnetic stir-bar and containing dry THF (2.0 mL) was added iPr2NH (0.66 mL, 4.67 mmol) and a 2.5M solution of n-butyl lithium in hexanes (1.7 mL, 4.25 mmol), and the solution was stirred for 30 min at room temperature at which point, methyl triphenylphosphonium bromide (1.13 g, 3.19 mmol) was added and the solution was stirred an additional 20 min to provide a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (50 mL) and the aqueous solution was extracted with ethyl acetate (50 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 55. Attempted purification of 55 by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) gave 55 (0.3502 g, 32%) that possessed ~ 9 mol% 53 as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.99 (d, J = 1.6, 1H), 7.72 (d, J = 8.0, 1H), 7.21 (dd, J = 8.0, 1.6, 1H), 7.09 (s, 1H), 7.08 (s, 1H), 5.78 (d, J = 1.2, 1H), 5.33 (d, J = 0.8, 1H), 3.92 (s, 3H), 1.95 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 166.6, 147..6, 145.6, 144.5, 142.4, 139.1, 137.2, 133.1, 132.6, 130.8, 128.1, 128.0, 126.3, 117.8, 94.4, 52.4, 35.1, 33.9, 33.8, 31.9, 31.8, 20.0; IR (neat): ν = 2957, 2926, 2862, 1726, 1664, 1590, 1541, 1497, 1456 cm−1; GC-MS [M+] calcd for C25H29O2I: 488.1213, found: 488.1222.
2-Iodo-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (16)
Compound 16 was synthesized following the methods of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 55 (0.3156 g, 0.646 mmol) and methanol (4 mL) was added a 5M aqueous solution of potassium hydroxide (0.4 mL, 2.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (42 mL). The aqueous solution was extracted with ethyl acetate (50 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 16. Crude 16 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 16 (0.105 g, 34%), and this material was crystallized from ethyl acetate to provide pure 16 (0.063, 20%) as a white crystalline solid, m.p. 199–200 °C: 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 2.0, 1H), 7.94 (d, J = 8.0, 1H), 7.25 (dd, J = 8.0, 2.0, 1H), 7.10 (s, 1H), 7.09 (s, 1H), 5.81 (d, J = 0.8, 1H), 5.37 (d, J = 1.2, 1H), 1.97 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 170.9, 147.5, 146.5, 144.6, 142.4, 139.7, 137.0, 132.5, 132.0, 131.2, 128.2, 128.0, 126.4, 118.2, 95.0, 35.0, 34.0, 33.8, 31.9, 31.8, 20.0; IR (neat): ν = 2954, 2909, 1695, 1591, 1539, 1481 cm−1; LC-APCI-MS [M++H] calcd for C24H28O2I: 475.1134, found: 475.1126. Anal. Calcd for C24H27O2I: C 60.77; H 5.74; I 26.75. Found: C 61.04; H 5.64; I 26.4.
2-Iodo-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoic acid (17)
Compound 17 was synthesized following the method of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 51 (0.437 g, 0.89 mmol) and methanol (4 mL) was added a 5M aqueous solution of potassium hydroxide (0.4 mL, 2.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The precipitate was filtered and washed with water to give crude 17 (0.4043 g, 95%). Attempted purification of crude 17 by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) gave 17 (0.34 g, 74%) that contained ~9 mol % 13 as a white crystalline solid: 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 1.6, 1H), 8.05 (d, J = 8.0, 1H), 7.84 (dd, J = 8.0, 1.6, 1H), 7.27 (s, 1H), 7.23 (s, 1H), 2.38 (s, 3H), 1.70 (s, 4H), 1.32 (s, 6H), 1.22 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 195.7, 170.6, 149.0, 143.1, 142.2, 142.0, 136.4, 135.1, 133.6, 131.6, 129.7, 129.4, 129.0, 94.1, 34.8, 34.7, 34.4, 33.9, 31.6, 31.5, 20.1; IR (neat): ν = 2958, 1703, 1661, 1542, 1458 cm−1; LC-APCI-MS [M++H] calcd for C23H26O3I: 477.0927, found: 477.0922. Anal. Calcd for C23H25O3I: C 57.99; H 5.29; I 26.64. Found: C 59.15; H 5.14; I 24.5.
3,5-Difluoro-4-formylbenzoic acid (27)
The method of Anderson and co-workers was followed to synthesize 27. [33] To a 1 L round bottom flask charged with 3,5-difluorobenzoic acid (10.00 g, 63.3 mmol) and THF (290 mL) cooled to –78 °C was added a 1.7 M solution of tert-butyl lithium in pentane (93.0 mL, 158 mmol), dropwise. The reaction was stirred at –78 °C for 30 min, and then dimethylformamide (12.4 mL, 158 mmol) was added. The reaction was allowed to stir at –78 °C for 1 h, and then stirred at 0 °C for 1 h, and then allowed to warm to room temperature and stirred for 16 h. The reaction was carefully quenched with concentrated HCl (by slow addition) until pH 1 (about 30 mL), and then concentrated in vacuo. The residue was extracted with DCM (100 mL, twice), and the organic layers were combined and concentrated in vacuo. The residue was diluted with DCM (50 mL) and then washed with saturated NaHCO3 (75 mL, twice). The aqueous extracts were acidified with concentrated HCl (18 mL), and the resulting precipitate was filtered and dried to give pure 27 (4.28 g, 36%) as a white powder, m.p. 194 – 216 °C: 1H NMR (400 MHz, DMSO-d6) δ 13.98 (br s, 1H), 10.23 (s, 1H), 7.64 (d, J = 9.2, 1H); 13C NMR (100.6 MHz, DMSO-d6) δ 184.9, 184.9, 184.8, 164.6, 164.5, 164.5, 163.3, 163.2, 160.7, 160.6, 138.4, 138.3, 138.2, 116.5, 116.4, 116.3, 113.4, 113.3, 113.2, 113.1; IR (neat): ν = 3078, 2935, 2615, 1717, 1679, 1633, 1573, 1475 cm−1; GC-MS [M+] calcd for C8H4O3F2: 186.0129, found: 186.0130.
Benzyl-3,5-difluoro-4-formylbenzoate (31)
Compound 31 was synthesized according to the methods of Kishida and co-workers. [32] To a 500 mL round bottom flask charged with 27 (8.8 g, 47.2 mmol) was added dry dimethylformamide (100 mL) and a 60 wt % suspension of NaH in mineral oil (2.93 g, 73.3 mmol) in small aliquots over 20 min. The reaction solution was stirred an additional 20 min, and benzylbromide (8.10 mL, 67.7 mmol) was added to the red heterogeneous solution. After stirring 5 h, the reaction solution had become homogeneous, and it was poured into 1N HCl (250 mL), extracted with ethyl acetate (100 mL, twice), and the combined organic extracts were washed with saturated NaHCO3 (75 mL) and brine, dried over sodium sulfate, and removed in vacuo to give crude 31. Crude 31 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 4:1) to give 31 (12.1 g, 93%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 10.32 (s, 1H), 7.66 (d, J = 11.2, 1H), 7.36–7.46 (m, 5H), 5.38 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 186.2, 186.2, 184.0, 184.0, 183.9, 164.0, 164.0, 163.1, 163.1, 163.1, 161.4, 161.3, 137.4, 137.2, 137.1, 134.8, 134.6, 128.7, 128.7, 128.7, 128.5, 128.4, 116.8, 116.7, 116.6, 113.8, 113.7, 113.7, 113.6, 113.5, 113.5, 113.0, 107.9, 107.6, 68.4, 68.0; IR (neat): ν = 3072, 2668, 2548, 1722, 1697, 1632, 1573, 1484 cm−1; GC-MS [M+] calcd for C15H10O3F2: 292.0547, found: 292.0548.
4-((Benzyloxy)carbonyl)-2,6-difluorobenzoic Acid (34)
Compound 34 was prepared according to the method of Kishida and co-workers. [32] To a 100 mL round bottom flask charged with 31 (12.1 g, 43.8 mmol), sulfamic acid (4.60 g, 47.4 mmol), water (75 mL), and ACN (38 mL) was added a solution of 80% NaClO2 (5.43 g, 60.0 mmol) in water (25 mL). After stirring for 1 h, the reaction solution was poured into saturated Na2SO3 (80 mL) and 1N HCl (150 mL), and the resulting solution was extracted with ethyl acetate (100 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 34 (11.4 g, 89%) that was used without further purification. A small sample was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 1:1) to give pure 34 as a white powder: 1H NMR (400 MHz, CDCl3) δ 9.83 (br s, 1H), 7.67 (d, J = 8.0, 1H), 7.36–7.46 (m, 5H), 7.34–7.47 (m, 5H), 5.39 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 165.6, 163.4, 163.4, 162.1, 162.0, 159.5, 159.5, 135.6, 135.5, 135.4, 134.8, 128.7, 128.4, 113.6, 113.5, 113.4, 113.3, 113.3, 113.2, 67.9; IR (neat): ν = 3071, 2896, 2668, 2548, 1723, 1694, 1632, 1572, 1484 cm−1; LC-APCI-MS [M++H] calcd for C15H10O3F2: 276.0598, found: 276.0604.
4-Benzyl 1-methyl-2,6-difluorobenzene-1,4-dioate (37)
Compound 37 was synthesized according to the method of Kishida and co-workers. [32] To a 100 mL round bottom flask charged with compound 33 (11.4 g, 39.0 mmol) was added SOCl2 (25.0 mL, 340 mmol) and the reaction solution was refluxed for 1h in an oil bath at 84 °C. The reaction solution was cooled to room temperature and the excess thionyl chloride was removed in vacuo to give crude 4-chlorocarbonyl-2,6-difluorobenzoic acid benzyl ester. The crude 4-chlorocarbonyl-2,6-difluorobenzoic acid in dry toluene (25 mL) was added dropwise to a solution of triethylamine (13.2 mL, 95 mmol) in methanol (70 mL, 1.7 mol) over 10 min with vigorous stirring. The reaction solution was stirred 1h and poured into 1N HCl (300 mL) and extracted with ethyl acetate (80 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 37. Crude 37 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 95:5) to give pure 37 as an oil (9.67 g, 81%): 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 7.6, 2H), 7.36–7.48 (m, 5H), 5.38 (s, 2H), 3.97 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 163.5, 163.4, 163.4, 161.5, 161.4, 161.2, 158.9, 158.9, 134.9, 134.6, 134.5,134.4, 128.7, 128.6, 128.4, 115.0,114.8, 114.6, 113.3, 113.3, 113.3, 113.1, 113.1, 113.0, 67.7, 53.1; IR (neat): ν = 2956, 1728, 1634, 1574, 1488 cm−1; LC-MS [M++H] calcd for C16H13O4F2: 307.0782, found: 307.0784.
4-(Methoxycarbonyl)-3,5-difluorobenzoic Acid (40)
Compound 40 was synthesized as follows. A 0.05 M solution of 37 (9.67 g, 31.6 mmol) in ethanol (630 mL) was passed through a 10% Pd/C cartridge in the ThalesNano H-cube® at 70 °C and 14 bar. The resulting solution was concentrated in vacuo to give crude 40 (6.54 g, 96%) as a white crystalline solid that was used without further purification. A small sample of crude 40 was purified by recrystallization from hot ethyl acetate to give pure 40, m.p. 148–150 °C: 1H NMR (400 MHz, CDCl3) δ 11.65 (br s, 1H), 7.67 (d, J = 7.6, 2H), 3.99 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 169.2, 161.5, 161.4, 161.1, 158.9, 158.9, 133.5, 133.4, 133.3, 115.9, 115.7, 115.5, 113.9, 113.8, 113.6, 113.6, 53.2; IR (neat): ν = 3080, 2840, 2653, 2577, 1739, 1697, 1632, 1575, 1489 cm−1; LC-MS [M++H] calcd for C9H7O4F2: 217.0312, found: 217.0300.
Methyl 2,6-difluoro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoate (52)
Compound 52 was synthesized according to the method of Boehm and co-workers. [11] Methyl 4-(chlorocarbonyl)-3,5-difluorobenzoate (43) was synthesized by refluxing 4-(methoxycarbonyl)-3,5-difluorobenzoic acid (40) (1.47 g, 6.80 mmol) in thionyl chloride (12.0 mL, 165 mmol) in a 100 mL one-neck round bottom flask fitted with a water-cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 43 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 43 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 48 (1.47 g, 7.26 mmol) followed by a solution of crude acid chloride 43 (6.80 mmol) in DCM (15 mL). Aluminum chloride (2.20 g, 16.5 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and concentrated to give crude 52. Crude 52 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 92.5:7.5) to give 52 (3.19 g, 58%) as a white, crystalline solid, m.p. 107–111 °C: 1H NMR (400 MHz, CDCl3) δ 7.37 (d, J = 8.0, 2H), 7.23 (s, 1H), 7.21 (s, 1H), 3.98 (s, 3H), 2.34 (s, 3H), 1.69 (s, 4H), 1.31 (s, 6H), 1.21 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 194.7, 161.5, 161.4, 158.9, 158.9, 149.0, 142.5, 142.5, 142.1, 136.1, 134.7, 133.3, 133.1, 129.9, 129.7, 128.3, 114.1, 113.5, 113.2, 53.1, 52.6, 34.7, 34.7, 34.4, 33.9, 33.8, 31.6, 31.5, 20.6, 20.0; IR (neat): ν = 3072, 2957, 2922, 2858, 1747, 1670, 1630, 1567, 1455 cm−1; LC-MS [M++H] calcd for C24H27O3F2: 401.1928, found: 401.1937.
Methyl 2,6-difluoro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (56)
Compound 56 was synthesized according to the method of Boehm and co-workers. [11] To a 20-dram vial containing 52 (0.814 g, 2.03 mmol) and dry THF (3 mL) at room temperature with a Teflon magnetic stir-bar was slowly added a triphenylphosphonium methylide solution prepared as follows: to a 100 mL round bottom flask equipped with a Teflon magnetic stir-bar and containing dry THF (2.0 mL) was added iPr2NH (0.66 mL, 4.67 mmol) and a 2.5M solution of n-butyl lithium in hexanes (1.7 mL, 4.25 mmol), and the solution was stirred for 30 min at room temperature at which point, methyl triphenylphosphonium bromide (1.13 g, 3.19 mmol) was added and the solution was stirred an additional 20 min to provide a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (50 mL) and the aqueous solution was extracted with ethyl acetate (50 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 56 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 56 (0.36 g, 44%) as a yellow oil: 1H NMR (400 MHz, CDCl3) δ 7.09 (s, 1H), 7.08 (s, 1H), 6.86 (dd, J = 9.2, 4, 2H), 5.80 (d, J = 3.2, 1H), 5.36 (d, J = 3.2, 1H), 3.94 (s, 3H), 1.95 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 162.0, 159.5, 159.4, 147.3, 146.6, 146.4, 144.8, 144.8, 142.5, 136.6, 136.6, 132.5, 132.1, 132.0, 131.9, 128.5, 128.4, 128.2, 127.9, 118.2, 110.1, 109.8, 109.0, 61.8, 60.3, 52.69, 35.0, 35.0, 34.0, 33.8, 31.8, 31.8, 21.0, 19.8, 14.1, 14.1; IR (neat): ν = 2958, 2924, 2862, 1728, 1629, 1557, 1497, 1456 cm−1; LC-MS [M++H] calcd for C25H29O2F2: 399.2136, found: 399.2138.
2,6-difluoro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (18)
Compound 18 was synthesized following the method of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 56 (0.3586 g, 0.8999 mmol) and methanol (4 mL) was added a 5M aqueous solution of potassium hydroxide (0.4 mL, 2.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The precipitate was filtered and washed with water to give crude 18 (0.3459 g, 100%). Crude 18 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give pure 18 (0.25 g, 72%) as a white crystalline solid, m.p. 191–192 °C: 1H NMR (400 MHz, CDCl3) δ 9.23 (br s, 1H), 7.10 (s, 1H), 7.08 (s, 1H), 6.89 (d, J = 10, 2H), 5.84 (s, 1H), 5.41(s, 1H), 1.97 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 166.6, 162.8, 162.8, 160.2, 160.2, 147.9, 147.8, 147.7, 147.2, 144.9, 142.6, 136.5, 132.5, 128.2, 128.0, 118.7, 110.4, 110.3, 110.1, 107.8, 107.6, 107.5, 35.0, 35.0, 34.0, 33.8, 31.9, 31.8, 19.8; IR (neat): ν = 3748, 2956, 2349, 1695, 1625, 1556, 1487 cm−1; LC-APCI-MS [M++H] calcd for C24H27O2F2: 385.1979, found: 385.1975. Anal. Calcd for C24H26O2F2: C, 74.98; H, 6.82; F, 9.88. Found: C, 74.74; H, 6.84; F, 9.10.
2,6-Difluoro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoic acid (19)
Compound 19 was synthesized following the method of Boehm and co-workers. [11] To a 100 mL round bottom flask charged with 52 (0.692 g, 1.73 mmol) and methanol (9 mL) was added a 5M aqueous solution of potassium hydroxide (0.8 mL, 4.0 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (42 mL). The precipitate was filtered and washed with water to give crude 19 (0.634 g, 95%). Crude 19 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 19 (0.54 g, 81%) as a white crystalline solid: 1H NMR (400 MHz, CDCl3) δ 8.54 (br s, 1H), 7.39 (d, J = 8.4, 1H), 7.25 (s, 1H), 7.22 (s, 1H), 2.34 (s, 3H), 1.69 (s, 4H), 1.31 (s, 6H), 1.22 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 194.7, 165.6, 162.0, 162.0, 159.4, 159.4, 149.2, 143.2, 142.2, 134.8, 133.2, 129.7, 128.3, 113.7, 113.4, 34.7, 34.7, 34.4, 33.9, 31.6, 31.5, 20.0; IR (neat): ν = 3498, 2956, 2923, 2522, 1674, 1632, 1571, 1487 cm−1; LC-APCI-MS [M++H] calcd for C23H25O3F2: 387.1772, found: 387.1757. Anal. Calcd for C23H24O3F2 : C, 71.49; H, 6.26; F, 9.83. Found: C, 70.53; H, 6.05; F, 7.90.
Supplementary Material
Scheme 4.

Formula 1.
Formula 2.
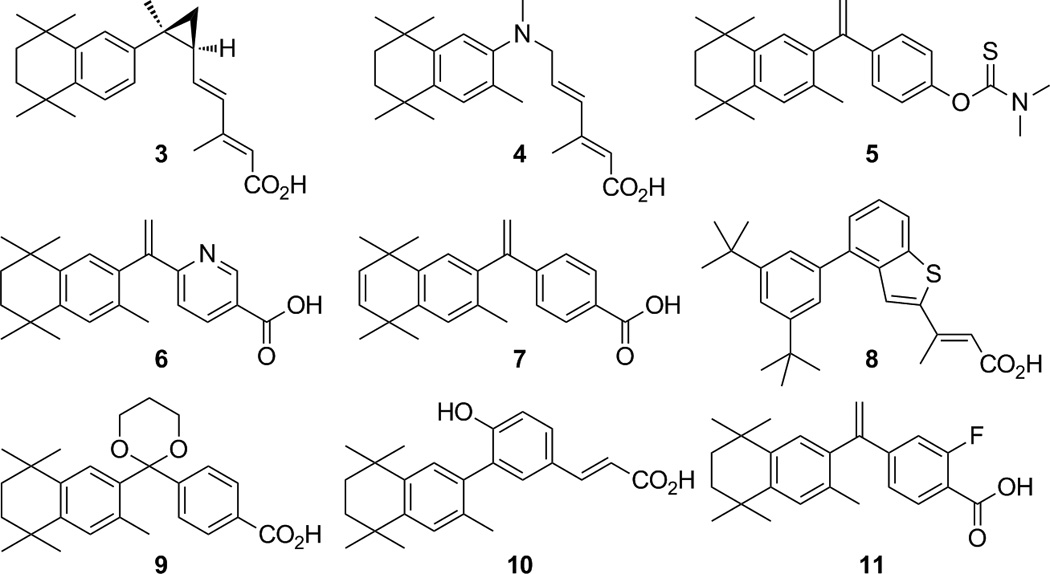
Formula 3.
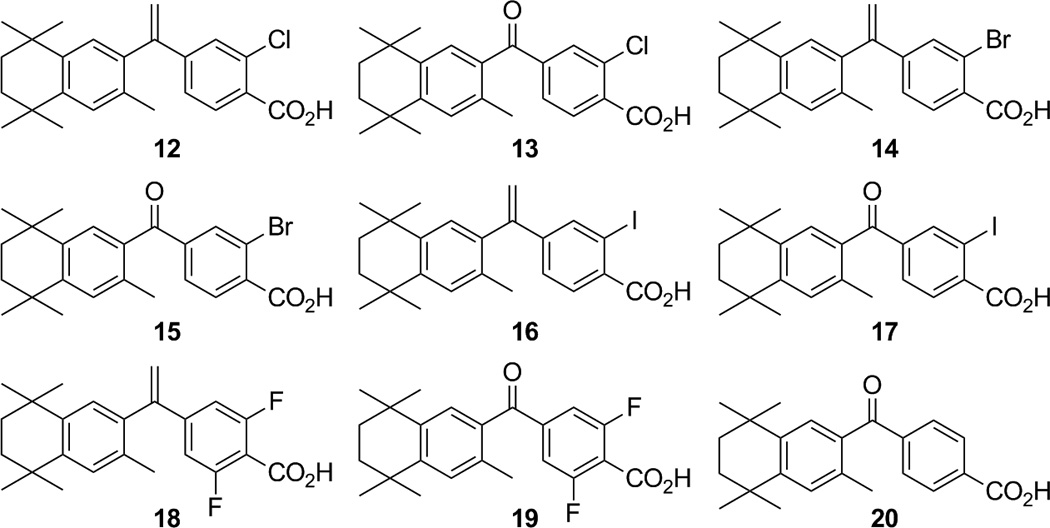
Acknowledgements
We thank the Thome Foundation and the National Cancer Institute of the National Institutes of Health for financial support of this work (1 R15 CA139364-01A2). Thanks is also given to Dr. Natalya Zolotova of the High-Resolution Mass Spectrometry Lab at ASU Tempe and Dr. John Greaves of the Mass Spectrometry Facility at UC Irvine. Julie K. Furmick wishes to thank ASU Tempe for a School of Life Sciences Undergraduate Research (SOLUR) fellowship. Drew Browder was also supported by the SOLUR Program. pCMX-hRARα and pTK-RARE(2)-Luc were generous gifts from Dr. Paul Thompson, School of Biomedical Sciences, University of Ulster, Coleraine, UK.
Abbreviations
- (h)RXR
(human) retinoid-X-receptor
- RAR
retinoic-acid-receptor
- CTCL
cutaneous T-cell lymphoma
- RXRE
retinoid-X-receptor element
- HRE
hormone response element
- LBP
ligand binding pocket
- LXR
liver X receptor
- TR
thyroid hormone receptor
- VDR
1,25-dihydroxyvitamin D receptor
- SNuRMs
specific nuclear receptor modulators
- TLC
thin layer chromatography
- FDA
Food and Drug Administration
- MEM
minimum essential media
- FBS
fetal bovine serum
- BD
binding domain
- AD
activation domain
- apoE
apolipoprotein E
Footnotes
Supporting Information Available: 1H-NMR and 13C-NMR spectra of all compounds reported in the experimentals as well as X-ray data for compounds 12, 14, 16, 18 and 52. The Xray data can be found at the Cambridge Crystallographic Data Centre under the identification numbers: 859464-859468.
References
- 1.a) Mangelsdorf DJ, Umesono K, Evans RM. The Retinoids. Orlando, FL: Academic Press; 1994. The Retinoid Receptors; pp. 319–349. [Google Scholar]; b) Leid M, Kastner P, Chambon P. Trends Biochem. Sci. 1992;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
- 2.Olefsky JM. J. Biol. Chem. 2001;276(40):36863–36864. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 3.Forman BM, Yang C-R, Au M, Casanova J, Ghysdael J, Samuels HH. Mol. Endocrinol. 1989;3:1610–1626. doi: 10.1210/mend-3-10-1610. [DOI] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Evans RM. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 5.Thompson PD, Remus LS, Hsieh J-C, Jurutka PW, Whitfield GK, Galligan MA, Encinas Dominguez C, Haussler CA, Haussler MR. J. Mol. Endocrinol. 2001;27(2):211–227. doi: 10.1677/jme.0.0270211. [DOI] [PubMed] [Google Scholar]
- 6.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, Hallen D, Johansson IC, Zachrisson K, Ogg D, Jendeberg L. EMBO J. 2003;22:4625–4633. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nahoum V, Perez E, Germain P, Rodriquez-Barrios F, Manzo F, Kammerer S, Lemaire G, Hirsch O, Royer CA, Gronemeyer H, de Lera AR, Bourguet W. PNAS. 2007;104:17323–17328. doi: 10.1073/pnas.0705356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, H. Gronemeyer H. Nature Rev. Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 9.Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu X-P, Haefner P, Dawson MI, Pfahl M. Science. 1992;258:1944–1946. doi: 10.1126/science.1335166. [DOI] [PubMed] [Google Scholar]
- 10.a) Jong L, Lehmann JM, Hobbs PD, Harlev E, Huffman JC, Pfahl M, Dawson MI. J. Med. Chem. 1993;36:2605–2613. doi: 10.1021/jm00070a003. [DOI] [PubMed] [Google Scholar]; b) Dawson MI, Jong L, Hobbs PD, Cameron JF, Chao W-R, Pfahl M, Lee M-O, Shroot B, M. Pfahl M. J. Med. Chem. 1995;38:3368–3383. doi: 10.1021/jm00017a021. [DOI] [PubMed] [Google Scholar]
- 11.Boehm MF, Zhang L, Badea BA, White SK, Mais DE, Berger E, Suto CM, Goldman ME, Heyman RA. J. Med. Chem. 1994;37:2930–2941. doi: 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]
- 12.Daiss JO, Burschka C, Mills JS, Montana JG, Showell GA, Fleming I, Gaudon C, Ivanova D, Gronemeyer H, Tacke R. Organometallics. 2005;24:3192–3199. [Google Scholar]
- 13.a) Yen W-C, Corpuz MR, Prudente RY, Cooke TA, Bissonnette RP, Negro-Vilar A, Lamph WW. Clin. Cancer Res. 2004;10:8656–8664. doi: 10.1158/1078-0432.CCR-04-0979. [DOI] [PubMed] [Google Scholar]; b) Dragnev KH, Petty WJ, Shah SJ, Lewis LD, Black CC, Memoli V, Nugent WC, Hermann T, Negro-Vilar A, Rigas JR, Dmitrovsky E. Clin. Cancer Res. 2007;13:1794–1800. doi: 10.1158/1078-0432.CCR-06-1836. [DOI] [PubMed] [Google Scholar]
- 14.Yen W-C, Prudente RY, Lamph WW. Breast Cancer Res. Treat. 2004;88:141–148. doi: 10.1007/s10549-004-1426-5. [DOI] [PubMed] [Google Scholar]
- 15.Cesario RM, Stone J, Yen W-C, Bissonnette RP, Lamph WW. Cancer Letters. 2006;240:225–233. doi: 10.1016/j.canlet.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee R, Davies PJA, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamanns LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti JR, Heyman RA. Nature. 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 17.Sherman SI, Gopal J, Haugen BR, Chiu AC, Whaley K, Nowlakha P, Duvic M. N. Engl. J. Med. 1999;340:1075–1079. doi: 10.1056/NEJM199904083401404. [DOI] [PubMed] [Google Scholar]
- 18.Li D, Li T, Wang F, Tian H, Samuels HH. Mol. Cell. Biol. 2002;22:5782–5792. doi: 10.1128/MCB.22.16.5782-5792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Murthy S, Born E, Mathur SN, Field FJ. J. Lipid Res. 2002;43:1054–1064. doi: 10.1194/jlr.m100358-jlr200. [DOI] [PubMed] [Google Scholar]; b) Field FJ, Born E, Mathur SN. J. Lipid Res. 2004;45:905–913. doi: 10.1194/jlr.M300473-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Thacher SM, Standeven AM, Athanikar J, Kopper S, Castilleja O, Escobar M, Beard RL, Chandraratna RAS. JPET. 1997;282:528–534. [PubMed] [Google Scholar]
- 21.Vuligonda V, Thacher SM, Chandraratna RAS. J. Med. Chem. 2001;44:2298–2303. doi: 10.1021/jm0100584. [DOI] [PubMed] [Google Scholar]
- 22.Farmer LJ, Marron KS, Canan Koch SS, Hwang CK, Kallel EA, Zhi L, Nadzan AM, Robertson DW, Bennani YL. Bioorg. Med. Chem. Lett. 2006;16:2352–2356. doi: 10.1016/j.bmcl.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 23.Kagechika H, Kawachi E, Hashimoto Y, Himi T, Shudo K. J. Med. Chem. 1988;31:2182–2192. doi: 10.1021/jm00119a021. [DOI] [PubMed] [Google Scholar]
- 24.Winum J-Y, Baghdiguian S, Commes T, Leydet A, Montero J-L. Bioorg. Med. Chem. Lett. 2002;12:3529–3532. doi: 10.1016/s0960-894x(02)00803-x. [DOI] [PubMed] [Google Scholar]
- 25.Boehm MF, Zhang L, Zhi L, McClurg MR, Berger E, Wagoner M, Mais DE, Suto CM, Davies PJA, Heyman RA, Nadzan AM. J. Med. Chem. 1995;38:3146–3155. doi: 10.1021/jm00016a018. [DOI] [PubMed] [Google Scholar]
- 26.Faul MM, Ratz AM, Sullivan KA, Trankle WG, Winneroski LL. J. Org. Chem. 2001;66:5772–5782. doi: 10.1021/jo0103064. [DOI] [PubMed] [Google Scholar]
- 27.Michellys PY, Ardecky RJ, Chen JH, Crombie DL, Etgen GJ, Faul MM, Faulkner AL, Grese TA, Heyman RA, Karanewsky DS, Klausing K, Leibowitz MD, Liu S, Mais DA, Mapes CM, Marschke KB, Reifel-Miller A, Ogilvie KM, Rungta D, Thompson AW, Tyhonas JS, Boehm MF. J. Med. Chem. 2003;46:2683–2696. doi: 10.1021/jm020340q. [DOI] [PubMed] [Google Scholar]
- 28.Michellys P-Y, Ardecky RJ, Chen J-H, D’Arrigo J, Grese TA, Karanewsky DS, Leibowitz MD, Liu S, Mais DA, Mapes CM, Montrose-Rafizadeh C, Ogilvie KM, Reifel-Miller A, Rungta D, Thompson AW, Tyhonas JS, Boehm MF. J. Med. Chem. 2003;46:4087–4103. doi: 10.1021/jm020401k. [DOI] [PubMed] [Google Scholar]
- 29.Michellys P-Y, Darrigo J, Grese TA, Karanewsky DS, Leibowitz MD, Mais DA, Mapes CM, Reifel-Miller A, Rungta D, Boehm MF. Bioorg. Med. Chem. Lett. 2004;14:1593–1598. doi: 10.1016/j.bmcl.2003.12.089. [DOI] [PubMed] [Google Scholar]
- 30.Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu XP, Haefner P, Dawson MI, Pfahl M. Science. 1992;258:1944–1946. doi: 10.1126/science.1335166. [DOI] [PubMed] [Google Scholar]
- 31.Wagner CE, Jurutka PW, Marshall PA, Groy TL, van der Vaart A, Ziller JW, Furmick JK, Graeber ME, Matro E, Miguel BV, Tran IT, Kwon J, Tedeschi JN, Moosavi S, Danishyar A, Philp JS, Khamees RO, Jackson JN, Grupe DK, Badshah SL, Hart JW. J. Med. Chem. 2009;52:5950–5966. doi: 10.1021/jm900496b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakaki J, Kishida M, Konishi K, Gunji H, Toyao A, Matsumoto Y, Kanazawa T, Uchiyama H, Fukaya H, Mitani H, Arai Y, Kimura M. Bioorg. Med. Chem. Lett. 2007;17:4804–4807. doi: 10.1016/j.bmcl.2007.06.080. [DOI] [PubMed] [Google Scholar]
- 33.Anderson DR, Mahoney MW, Philion DP, Rogers TE, Meyers MJ, Poda G, Hegde SG, Singh M, Reitz DB, Wu KK, Buchler IP, Xie J, Vernier WF. PCT Int. Appl. 2004 Publication Number: WO 2004058762 A1, Publication Date: 20040715. [Google Scholar]
- 34.Grohe K, Schriewer M, Zeiler H-J, Metzger KG. Number: 4762844. U.S. Patent. Issue Date: 19880809.
- 35.Bouveault L. Bull. Soc. Chim. Fr. 1904;31:1306. [Google Scholar]
- 36.The ThalesNano H-Cube® was used for this hydrogenation reaction.
- 37.Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Proc. Natl. Acad. Sci. U.S.A. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Condon FE. J. Org. Chem. 1956;21:761–763. [Google Scholar]
- 39.Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong ES, Evans RM. Cell. 1991;66:555–561. doi: 10.1016/0092-8674(81)90018-0. [DOI] [PubMed] [Google Scholar]
- 40.Cramer PE, Cirrito JR, Wesson DW, Lee CYD, Restivo JL, Goebel DA, Landreth GE. Science. 2012 Feb 9th; [Google Scholar]
- 41.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egea PF, Mitschler A, Moras D. Mol. Endocrinol. 2002;16:987–997. doi: 10.1210/mend.16.5.0823. [DOI] [PubMed] [Google Scholar]
- 43.Love JD, Gooch JT, Benko S, Li C, Nagy L, Chatterjee VKK, Evans RM, Schwabe JWR. J. Biol. Chem. 2002;277:11385–11391. doi: 10.1074/jbc.M110869200. [DOI] [PubMed] [Google Scholar]
- 44.O'Boyle NM, Banck M, James CA, Morley C, Vnadermeersch T, Hutchison GR. J. Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Forman BM, Umesono K, Chen J, Evans RM. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 46.a) Zimmerman FK. Mutat. Res. 1975;31:71–86. doi: 10.1016/0165-1161(75)90069-2. [DOI] [PubMed] [Google Scholar]; b) Zimmermann FK, Kern R, H. Rasenberger H. Mutat. Res. 1975;28:381–388. [Google Scholar]
- 47.Marshall PA. CBE-LSE. 2007;6:307–315. doi: 10.1187/cbe.06-12-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.