

Abstract
The myeloproliferative neoplasms (MPNs) are a particularly useful model for studying mutation accumulation in neoplastic and the mechanisms of the molecular cells, understanding underlying defects our current This review summarizes acquisition. present their in patients with an MPN, and the effects of mutations targeting Janus kinase 2 (JAK2)-mediated intracellular signaling on DNA damage, and on the elimination of mutation-bearing cells by programmed cell death. Moreover, we discuss findings that suggest that the acquisition of disease-initiating mutations in hematopoietic stem cells of some MPN patients may be the consequence of an inherent genomic instability that was not previously appreciated.
Keywords: JAK2, mutation, genomic instability, clonal evolution
You jest about what you suppose to be a triviality, in asking whether the hen came first from an egg or the egg from a hen, but the point should be regarded as one of importance, one worthy of discussion, and careful discussion at that.
- Ambrosius Theodosius Macrobius (AD395-423), philosopher
Disease progression and metastatic growth in human malignancies have long been thought to involve the Darwinian selection of mutant sub-clones, such that cells with the best evolutionary fitness will over time come to dominate the neoplastic population (1, 2). This selection process is facilitated by an increase in genomic instability that results in the accumulation of additional mutations. Some of these, the so-called “driver mutations”, are directly responsible for any competitive advantage, whereas “passenger mutations” contribute nothing to the oncogenic potential of affected cells. Next-generation sequencing studies are just beginning to reveal the identity of the genetic changes that separate the predominant cells within a primary tumor from their metastatic progeny (3-6). In contrast, little is known about the genomic changes driving the earliest stages of tumor evolution. Does this process involve a linear stepwise acquisition of mutations, as suggested by Nowell (1)? Does the malignant cell-of-origin instead give rise to a diverse set of sub-clones that continue to evolve in parallel, a scenario apparent in cases of acute lymphoblastic leukemia (ALL) (7, 8)? Or, in some instances, is a single catastrophic event that generates dozens of mutations sufficient to produce a malignancy (9)?
A particularly useful model for examining the pattern of mutation accumulation in neoplastic cells, and the mechanisms underlying their acquisition, are the myeloproliferative neoplasms (MPNs). These are a group of related hematologic disorders that includes polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (MF) and chronic myeloid leukemia (CML), and several more rare entities, such as systemic mastocytosis and chronic neutrophilic leukemia (10, 11). The MPNs are characterized by preferential expansion of one or more morphologically normal myeloid cell types. Several aspects of MPN biology make them particularly useful in the investigation of the molecular mechanisms underlying clonal evolution. In particular, with appropriate disease management, the life expectancy of patients with ET or PV is near normal (12), permitting repeat biological sampling over many years. Sample taking can also be achieved with ease; a small aliquot of peripheral blood is sufficient to provide cells for most analyses. Finally, the ability to culture circulating hematopoietic progenitors in vitro facilitates the genetic analysis of mutations at the single-cell level, since all cells within a resulting colony are derived from the initial progenitor cell that was seeded, and can be readily retrieved from the methylcellulose in which they were propagated for subsequent analysis. Many meaningful insights into MPN biology have been obtained using this strategy (13-17).
Comprehensive reviews relating to the genomic instability associated with CML have been published (18-20), but the genomic instability associated with the Philadelphia chromosome (Ph)-negative MPNs, ET, PV and MF, has not been discussed in detail. Here, we summarize the current understanding of the molecular defects underlying these disorders, and the effects of mutations targeting Janus kinase 2 (JAK2)-mediated intracellular signaling on DNA damage and repair and on the elimination of mutation-bearing cells by apoptosis. Moreover, we discuss recent findings that suggest that acquisition of the disease-initiating mutation in MPN patients may itself result from an inherent genomic instability that had previously not been appreciated.
The molecular genetics underlying development of a human MPN
The MPNs have long been considered to arise from mutations acquired within a hematopoietic stem cell (HSC). This concept was initially based upon studies of the isoform expression patterns of polypeptides encoded by the X chromosome-encoded glucose-6-phosphate dehydrogenase locus (G6PD) in informative female patients (21, 22), which demonstrated that cells of the malignant clone all had the same X-inactivation pattern, and were likely to arise from a common HSC. Recent analyses, following the identification of most of the molecular lesions associated with the MPNs, confirm that these mutations can be detected in primitive hematopoietic stem or progenitor cells obtained from patients (23-26).
Little was known about the molecular pathogenesis of the Ph-negative MPNs until seven years ago, when the presence of a one-nucleotide change within JAK2 exon 14 was identified in the majority of patients (13, 27-29). As a consequence, the valine residue located at position 617 of JAK2 is substituted with a phenylalanine (“V617F”). This change is neither a single nucleotide polymorphism (SNP) nor an inherited mutation, since it can be detected in patient granulocytes but not matched T-lymphocytes, which are not part of the malignant clone in the chronic phase of these disorders. The JAK2V617F mutant allele is present in 50-60% of ET and MF patients and in 95% of PV patients, but occurs only rarely in other myeloid disorders (30-32). Theoretical models of JAK2 structure predict that the V617F substitution induces a conformational shift that alleviates repressive interactions between its JH1 and JH2 (or kinase and pseudokinase) domains (33). In support of this hypothesis, the JAK2V617F mutation is present in cells of the erythropoietin (EPO)-independent erythroid colonies that are a hallmark feature of these disorders (13, 14). JAK2V617F expression also confers factor independence to cytokine-dependent cell-lines (27-29), and results in constitutive JAK2 phosphorylation and consequent activation of the STAT, AKT and ERK signaling pathways. JAK2V617F expression in retroviral transduction/transplantation models and in transgenic mice recapitulates most aspects of human ET and/or PV (27, 34, 35), strongly arguing that it alone is sufficient to produce an MPN phenotype.
Ph-negative MPN patients lacking the JAK2V617F mutation may nonetheless bear mutations that result in deregulated JAK2 signaling. The vast majority of JAK2V617F-negative PV patients have alternative JAK2 mutations (36-39), which consist of a wide variety of deletions, insertions and/or amino acid substitutions that target residues located adjacent to the JH2 domain (40). These “exon 12” mutations constitutively activate JAK2-mediated signaling in vitro and in vivo (36). About 10% of JAK2V617F-negative ET and MF patients instead carry activating mutations in MPL, the receptor for thrombopoietin (TPO), which utilizes JAK2 for intracellular signaling (41-43). These predominantly target residue 515, which is part of a juxta-membrane amphipathic motif that acts to maintain MPL in a constitutively inactive configuration (44); others mutations target residue 505, located in the trans-membrane domain, resulting in TPO-independent receptor dimerization (45). Occasional JAK2V617F-negative MPN patients have mutations in SH2B3 (also called LNK; (46)), which negatively regulates JAK2 signaling by recruiting phosphatases to the activated JAK2. Loss-of-function LNK mutations therefore augment and sustain JAK2 signaling. A third of ET and MF patients do not have mutations in JAK2, MPL or SH2B3; the molecular pathogenesis of these cases remains unclear. The observation in pre-clinical studies of ATP-competitive JAK2 inhibitors that these agents inhibit the in vitro viability of cells from JAK2V617F-positive and JAK2V617F-negative patients suggests that the unidentified mutations associated with ET and MF likely occur in genes involved in the regulation of JAK/STAT signaling (47, 48). Candidate gene sequencing has thus far excluded JAK1, JAK3, TYK2, STAT5A or STAT5B as frequent targets for mutation (31).
Other acquired mutations associated with an MPN: a link to epigenetic regulation
In the last three years, inactivating mutations in the TET2, ASXL1, DNMT3A or EZH2 genes have been detected in a subset of patients with an MPN (49-52). Strikingly, these mutations may be detected in patients with other myeloid malignancies, including myelodysplasia (MDS), chronic myelomonocytic leukemia (CMML) or de novo acute myeloid leukemia (AML) (53-56). TET2, ASXL1, DNMT3A and EZH2 are all involved in the epigenetic regulation of gene transcription, although the role of these proteins in MPN disease initiation or progression has not been fully elucidated. Conditional knockout mouse strains have nonetheless provided intriguing insights into the consequences of absent DNMT3A or TET2 expression (57-59). In particular, Dnmt3a-null, Tet2-null and Tet2-haploinsufficient HSCs have enhanced self-renewal activity in vivo, suggesting that inactivating TET2 or DNMT3A mutations acquired by patients provide affected cells of the malignant clone with a competitive advantage over those without the mutation. In contrast, Asxl1-deficient murine HSCs are unable to outcompete their wild-type equivalents in competitive reconstitution assays (60); additional studies will be required to elucidate the role of this protein in normal and malignant hematopoiesis.
MPN disease progression in vivo: a pattern of mutation accumulation over time
The first evidence that abnormalities in DNA repair plays an important role in the progression of the Ph chromosome-negative MPNs was provided by the description of acquired uniparental disomy of chromosome 9p in a relatively high proportion (~30%) of PV patients (61). With the discovery of the JAK2V617F mutation several years later, it soon became apparent that most JAK2V617F-positive PV patients were homozygous for this mutation: when the genotype of individual hematopoietic colonies was determined, all those patients with PV were found to have a subset of cells with biallelic JAK2V617F mutations (14). As this homozygosity could be detected in erythroid and granulocytic cells from the same patient, it is likely that acquisition of the second JAK2V617F mutation occurred in a JAK2V617F-positive early myeloid progenitor, if not in an HSC. In contrast, only wildtype and JAK2V617F-heterozygous cells were detected in patients with ET, although JAK2V617F-homozygous cells were noted in two such patients that had subsequently experienced polycythemic transformation (14). Collectively, these data support the proposal that JAK2V617F-positive ET and PV form a disease continuum (62), with duplication of the mutant JAK2 allele driving disease transformation from thrombocythemia to polycythemia. Murine models of JAK2V617F-positive MPNs, in which the copy number (35) or the expression level (63) of the JAK2V617F transgene was shown to dictate the phenotype observed, provide experimental support for this “dosage hypothesis”.
On the basis of these and other studies, a simple model for disease initiation and progression in the Ph-negative MPNs might be proposed (Figure 1). Acquisition of a single JAK2V617F mutation in an HSC leads to the development of a phenotype consistent with a diagnosis of ET. Duplication of the mutant JAK2 allele results in a polycythemic phenotype in the vast majority of cases. Rare JAK2V617F-positive PV patients may have monoallelic JAK2 mutations, where the compound mutant allele (for example, the V615LV617F allele described in a later section) is a stronger gain-of-function mutation than JAK2V617F alone. Heterozygous mutations within JAK2 exon 12 are also sufficient to generate a polycythemic phenotype, whereas patients with an MPL mutation present with ET. Whilst homozygous JAK2 exon 12 or MPL mutations occur in rare instances (42, 64), there is no indication that mutation homozygosity profoundly changes the disease phenotype. Acquisition of additional mutations would initiate transformation from ET or PV to MF (Figure 1). Inactivating mutations that target ASXL1, TET2, EZH2 or DNMT3A are unlikely to underlie this process, as they can be readily detected in MPN patients that have not developed myelofibrosis (49-51). Several groups are currently using whole genome or exome sequence analysis to identify the mutations associated with myelofibrotic transformation.
Figure 1. Mutation accumulation in the MPNs.
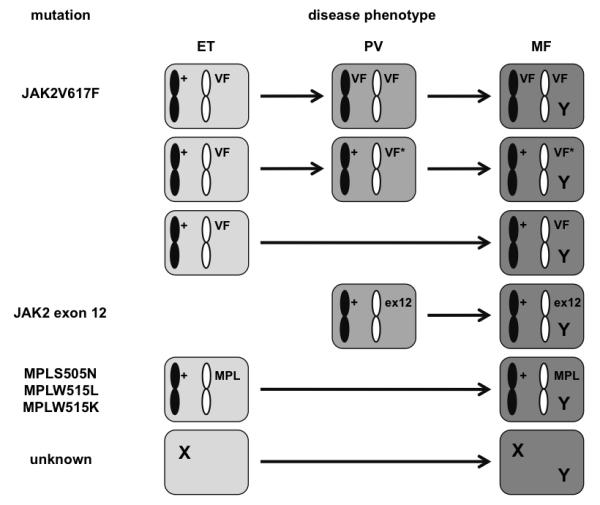
A simple model for disease initiation and progression to myelofibrosis in the Ph-negative MPNs can be proposed. Patients diagnosed with essential thrombocythemia (ET) may have either a single acquired JAK2V617F (VF) allele, an MPLS505N, MPLW515L or MPLW515K mutation (MPL), or one or more mutation(s) affecting unknown targets (collectively referred to here as an X). Duplication of the JAK2V617F allele or the acquisition of additional mutations within the affected JAK2 allele (VF*) would instead produce a polycythemic phenotype, as would the acquisition of a mono-allelic JAK2 exon 12 (ex12) mutation. The acquisition over time of one or more “driver” mutations (Y), the identities of which are currently uncertain, would initiate the transformation to myelofibrosis.
JAK2V617F expression is associated with increased DNA damage in vitro and in vivo
The pattern of mutation accumulation over time observed in MPN patients raises the possibility that these individuals may have defective DNA damage repair; several in vitro and in vivo studies support this hypothesis. JAK2V617F expression was found to appreciably enhance the frequency of spontaneous homologous recombination (HR) events in Ba/F3 pro-B cells, with a four-fold increase in incidence compared to that in cells engineered to express wildtype JAK2 (65). This was accompanied by an increased level of nuclear RAD51, a component of the DNA repair foci that form after the induction of double-strand breaks (66, 67). Additional indicators of genomic instability, including increased sister chromatid exchange, increased spontaneous mutation at the HPRT locus, changes in DNA ploidy, centromere abnormalities and increased DNA repair by single-strand annealing, are apparent in JAK2V617F-expressing Ba/F3 cells (65, 68). In contrast, JAK2V617F had little effect on non-homologous end joining (NHEJ) when assayed in cell-free extracts or intact cells (65). Changes in RAD51 distribution and HR frequency similar to that observed in vitro have also been noted in hematopoietic cells obtained from patients with JAK2V617F-positive PV or MF (65).
In a JAK2V617F-heterozygous conditional knock-in mouse, in which animals develop an ET-like disease soon after JAK2V617F expression is initiated, the HSC compartment appears compromised: six months after the induction of mutant JAK2 expression, mice had a two-fold reduction in the absolute number of lineage-negative, Sca1-positive, c-Kit-positive (LSK) cells compared to controls (69). Competitive reconstitution assays also indicated that these LSK cells were impaired in their long-term repopulating ability. To explain this phenomenon, investigators evaluated the level of DSBs in the total bone marrow population, as well as in the purified LSK fraction, from control and JAK2V617F knock-in animals. The intensity of γ-H2AX staining was significantly elevated in both cell populations obtained from the latter group of mice. Despite an accumulation of DNA damage, the proportion of JAK2V617F-positive LSK cells undergoing apoptosis was lower. However, in another JAK2V617F knock-in mouse model (70), quantitative or qualitative differences between the LSK compartments of JAK2V617F-heterozygous and wildtype littermates were not noted. This discrepancy might simply be attributable to subtle strain differences between the two models, but may also reflect the age of the animals analyzed in the later study (which was not specified). Interestingly, Li and colleagues detected differences in LSK frequency six months after JAK2V617F expression was induced, but these were not apparent five months earlier, suggesting any alteration in stem cell function might be subtle and only become apparent over time. Additional studies will be required to fully resolve this issue.
The molecular basis for the increase in HR events observed in JAK2V617F-expressing cells remains uncertain. A likely possibility is the STAT activation-dependent generation of reactive oxygen species (ROS), which can lead to DSB formation. Endogenous ROS levels are increased in many myeloid malignancies (71), including CML, although levels have not yet been evaluated in cells expressing JAK2V617F. Another possibility involves a non-canonical JAK signaling pathway revealed by studies of modifiers of Drosophila JAK activity (72). It has been long recognized that the JAK proteins are not located solely within the cytoplasm (73, 74), although little was known about their function(s) in the nucleus. Activated Hopscotch (Hop), the single JAK protein in flies, was recently found to disrupt gene silencing by displacing heterochromatin protein-1 (Hp1; (72)). This in turn induces the aberrant expression of otherwise silenced genes, several of which are likely to contribute to a malignant phenotype, since loss-of-function Hp1 mutants enhance the leukemic potential of the Hop mutants (72). Nuclear JAK2 is present in mammalian hematopoietic cell-lines and primary CD34+ progenitors, but is absent from mature blood cells (75). In mammalian nuclei, JAK2 directly phosphorylates histone H3 on tyrosine-41, abolishing the ability of HP1α to bind to this residue, thereby altering the chromatin structure surrounding the promoters of genes, including those of LMO2, NANOG and MYC (76-78). In addition to repressing heterochromatic gene transcription, HP1α has important roles in telomere structure and function, chromosome segregation during mitosis, the cellular response to DSBs, and the maintenance of chromatin in a state that minimizes the rates of HR and spontaneous gene conversion (79-82). Displacement of HP1α from regions of heterochromatin by activated JAK2 might therefore contribute to the effects associated with JAK2V617F expression in vitro.
JAK2V617F expression is likely to induce a “mutator phenotype” (83), with cells acquiring many additional genetic lesions, a small proportion of which may confer an in vivo advantage. Targets might include L3MBTL1, located within the region of chromosome 20q that is deleted in a small proportion of patients (84). L3MBTL1 itself plays an important role in DNA replication and genome stability: L3MBTL1-depleted cells have multiple DSBs and an increased number of repair foci that contain γH2AX, and also activate their ATM/ATR-dependent DNA damage response pathway (85). Loss of a L3MBTL1 allele might therefore enhance genomic instability in JAK2V617F-positive cells, accelerating the rate at which additional mutations occur.
JAK2V617F and BCR-ABL both inhibit apoptosis induced in response to DNA damage
A feature of cellular transformation is the prevention of DNA damage-induced apoptosis by inappropriate regulation of cell survival pathways (2). One such pathway involves modification of the anti-apoptotic factor, Bcl-xL. In normal cells, the rapid conversion of asparagine residues within Bcl-xL into isoaspartic acid occurs in response to DNA damage, disrupting its ability to sequester pro-apoptotic factors, and promoting cell death (86). This non-enzymatic conversion occurs through enhanced expression of NHE-1, a Na+/H+ ion exchange protein, thereby elevating the intra-cellular pH and facilitating Bcl-xL deamidation (87). However, intracellular NHE-1 levels do not increase in granulocytes from patients with Ph-positive CML or JAK2-mutated PV after genotoxic stress or γ-irradiation; there is no change in the levels of deamidated Bcl-xL, and programmed cell death is not apparent (88). Enforced alkalinization or NHE-1 over-expression reverses this phenomenon, restoring to normal the levels of Bcl-xL deamidation and apoptosis. Inhibition of this pathway was found to be neither a general feature of malignancy nor a property specific to all constitutively-activate tyrosine kinases: several hematopoietic cell-lines expressing mutant tyrosine kinases responded to induced DNA damage by increasing their intracellular pH and levels of deamidated Bcl-xL, whereas HEL (JAK2V617F-positive) and K562 (BCR/ABL-positive) cells did not (88). The kinase activity of JAK2 or ABL appears to be required to inhibit Bcl-xL deamidation, since treatment of granulocytes from PV patients with JAK2 inhibitors, or CML blast cells with imatinib mesylate, reversed the block on DNA damage-induced apoptosis. Investigators also found that, in the blast cells of a patient carrying an E255V BCR/ABL kinase domain mutation (which renders cells insensitive to imatinib), enforced alkalinization or NHE-1 expression readily overcame this drug resistance.
The finding that in vitro expression of BCR/ABL or JAK2V617F increases the incidence of DNA damage, and that primary cells expressing either protein subsequently fail to undergo DNA damage-induced apoptosis, together suggest that these MPN-associated mutant proteins provide a two-pronged advantage to affected cells (Figure 2): over time, subsets of daughter cells will be generated that carry additional mutations, some of which will provide an in vivo advantage over others, facilitating disease progression.
Figure 2. MPN-associated mutant kinases block normal induction of apoptosis after DNA damage.
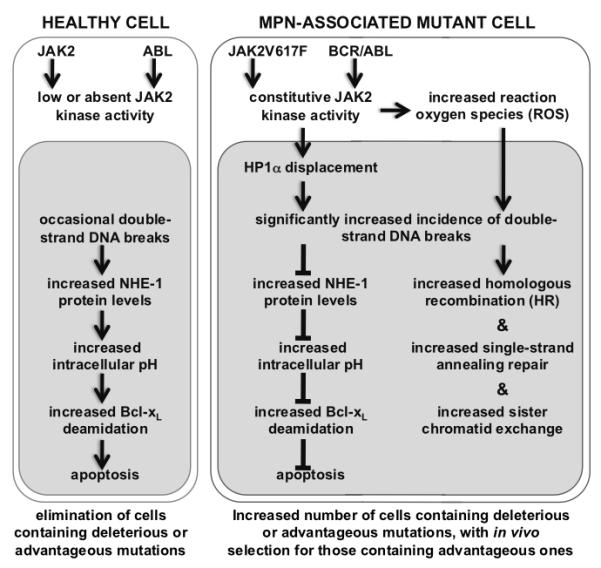
In a healthy cell (depicted on the left), the presence of DSBs induces increased NHE-1 expression, by a mechanism that remains to be elucidated. The resulting ion flux via this antiport protein increases the intracellular pH, leading to the deamidation of Bcl-xL and programmed cell death. In contrast, BCR/ABL or JAK2V617F expression in a mutated cell (depicted on the right) results in nuclear translocation of activated JAK2, phosphorylation of histone H3 on tyrosine-41, and the displacement of HP1α from heterochromatin. This, combined with the generation of reactive oxygen species (ROS) by activated JAK2, is likely to result in an increase in number of DSBs, and inhibition of apoptosis by the prevention of NHE-1 up-regulation, noted in vitro and in vivo. As a consequence of these two alterations, MPN cells would accumulate more mutations, some of which may provide an in vivo proliferative advantage.
Cases of co-existent mutations suggest HSCs in MPN patients are predisposed to mutation
It is becoming increasingly apparent from the study of JAK2V617F-positive patients that not only are cells bearing this mutation more likely to acquire additional mutations as the result of genomic instability associated with deregulated JAK2, but also that there may be inherent genomic instability in the HSCs of a proportion of the individuals that eventually acquire a JAK2V617F mutation. Several of the more important findings on this intriguing possibility are discussed in the following sections.
(1) The JAK2 locus in MPN patients is more prone to secondary mutations than might be expected
Genotyping of granulocyte DNA for the JAK2V617F mutation and a SNP present in JAK2 intron 14 revealed that this mutation had been acquired on more than one occasion in three of the 109 informative MPN cases studied (15), a frequency significantly higher than might be expected given the incidence of JAK2V617F acquisition in the general population. A similar analysis was performed using patients grouped by diagnosis (89); the JAK2V617F mutation was found to have occurred on multiple occasions in patients with ET, but not PV, indicating that there may be a disorder-specific pattern of JAK2V617F mutation. In either study, genotyping was not performed at the single-cell level (that is, using individual hematopoietic colonies), so it is unclear whether the JAK2 mutations were acquired in separate HSCs, resulting in independent JAK2V617F-heterozygous clones, or within a single HSC, producing a JAK2V617F-homozygous sub-clone, which is consistent with a diagnosis of PV. JAK2 point mutations may nevertheless be acquired on multiple occasions in patients with PV. We have identified a patient who acquired a second JAK2 mutation 5 years after presenting with JAK2V617F-positive PV (LMS, manuscript in preparation), producing a compound JAK2V615LV617F mutant allele that allowed affected cells to out-compete wildtype and JAK2V617F-positive cells over time. Colony analysis revealed a mutation pattern consistent with at least three separate genetic events: acquisition of the JAK2V617F mutation in a normal HSC, addition of a JAK2V615L mutation to the JAK2V617F allele, and the generation of JAK2V615LV617F-homozygous cells by HR.
Collectively, these studies suggest a fundamental difference in the nature of secondary JAK2 mutations that occur in JAK2V617F-positive MPN patients. One group is defined by mutations that occur in a JAK2V617F-heterozygous cell, including HR, duplication of the entire mutated chromosome 9 (with or without loss of the wildtype equivalent), or acquisition of additional base substitutions in the mutant JAK2 allele. Each event would likely be due to the mutagenic potential of JAK2V617F, result in a polycythemic phenotype, and may provide cells with an in vivo proliferative advantage. The second group is characterized by the acquisition of JAK2V617F mutations by separate HSCs, and implies the presence of a preceding mutation or natural genetic variant that predisposes an individual to acquiring a JAK2 mutation during their lifetime.
(2) Co-incident JAK2 and MPL mutations occur in distinct disease clones
Included in this second group would be those rare PV or ET JAK2V617F-positive patients that respectively carry co-incident JAK2 exon 12 or MPL mutations. Genotyping of hematopoietic colonies from these double mutation-positive patients has shown that the JAK2V617F mutation always existed in a clone distinct from that carrying a JAK2 exon 12 or an MPLW515L mutation (43, 90, 91). The mutation pattern observed could result from separate mutations within separate cells of a pre-existing mutant clone, or from separate events occurring in independent HSCs (Figure 3). Proof for the latter possibility was obtained by the expression analysis of genes that undergo random X-chromosome inactivation in individual progenitors collected from informative females (91): for example, in one patient, cells bearing the JAK2V617F mutation expressed the T allele of SNP rs11549009, located in the coding region of the X-inactivated IDS gene, whereas cells bearing an MPLW515L mutation expressed the C allele. The low likelihood of these mutational events happening in separate HSCs by chance alone suggests that an inherited allele plays a role in the subsequent acquisition of mutations.
Figure 3. Tracking the cell-of-origin in cases with co-incident MPN mutations.
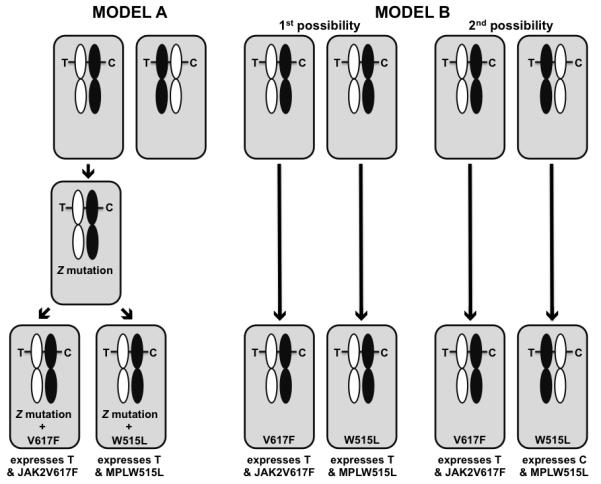
The pattern of co-existent JAK2 and MPL mutations apparent in some MPN cases could arise in several ways. In Model A, JAK2V617F-positive and MPLW515L-positive colony-forming cells would result from secondary mutations acquired separately in cells of a common founder clone. As the likelihood of co-incident mutations occurring by chance is significantly less than the observed frequency, the shared founder clone would have to carry a mutation in unknown gene(s), Z, that increases the likelihood of subsequent mutation or promotes the survival of mutant JAK2- or MPL-positive cells. In Model B, the JAK2V617F and MPLW515L mutations occur separately in independent hematopoietic stem cells. Experimental proof for the latter possibility is obtained by evaluating in informative female patients expression from loci undergoing X-inactivation. In Model A, with allele C on the imprinted X chromosome (shaded in black), all mutation-positive cells express the “T” transcript. An identical pattern would occur in half of those cases in which Model B was operative: by chance, the JAK2 and MPL mutations were acquired in unrelated stem cells that both had an active T allele (“1st possibility”). However, in the remaining 50% (“2nd possibility”), one mutation would occur in a stem cell expressing “T”, the other in a stem cell expressing “C”.
(3) JAK2V617F-positive patients may be positive for other MPN-associated mutations
There is a growing body of evidence that JAK2V617F-positive ET, PV or MF patients have an increased risk of developing BCR/ABL-positive CML. In several such cases, a Ph-negative MPN co-existent with CML became apparent only after imatinib-induced remission of the BCR/ABL-positive population; retrospective molecular analyses showed that the JAK2V617F allele was already present at the time of CML diagnosis, and was clearly not therapy-related (92-94). Two models could explain the disease history in these instances: the patient had an undiagnosed MPN that either preceded the acquisition of a t(9;22) translocation in a HSC that was part of the malignant clone, or developed CML separately as the result of transformation of a normal HSC. Unfortunately, samples that would have ruled out one of these possibilities were unavailable.
Rare examples of co-existent MF and systemic mastocytosis have also been reported (91, 95). Analysis of erythroid colonies from one patient revealed that the KITD816V mutation that is associated with systemic mastocytosis (96) and the MF-associated JAK2V617F mutation were mutually exclusive, supporting the contention that HSCs in these individuals were more prone to acquiring mutations than members of the general population.
(4) Leukemia in JAK2V617F-positive MPN patients often arises in a non-mutated HSC
Surprisingly, the mutant JAK2 allele is not present in the leukemic blast cells of about half of all JAK2V617F-positive MPN patients that develop a secondary AML (97, 98). SNP analysis of several matched MPN (pre-transformation) and AML (post-transformation) samples has shown that the apparent loss of the JAK2V617F allele during leukemic transformation is not the result of HR or gene conversion in a JAK2V617F-heterozygous cell (99). Instead, the JAK2V617F mutation and the mutation(s) driving leukemogenesis are likely to arise in separate HSCs, with the latter occurring in an HSC with a wildtype JAK2 genotype in half of cases.
(5) “Secondary” mutations seen in MPN patients can occur prior to any JAK2 mutation
Colony genotyping studies have shown that at least several of the mutations present in MPN patients that were assumed to occur secondary to the JAK2V617F mutation, including deletion of chromosome 20q (del20q) and TET2 inactivation, may in fact arise first, consistent with their being the “pre-JAK2” mutation postulated by Skoda and colleagues (28). In one study, del(20q) was found to occur in almost all hematopoietic cells from some patients, but that the mutant JAK2 allele was present in only a subset of cells (100). However, in other cases, del(20q) clearly can occur in a sub-population of cells that do not carry the JAK2V617F mutation (91) or are acquired secondary to the JAK2V617F mutation (16). Similarly, TET2 mutations do not follow a strict temporal pattern of acquisition, with the TET2 mutation arising first in almost half of the JAK2V617F-positive, TET2-mutated patients analyzed (17, 49). These data support the notion that there may underlying genomic instability in the HSCs of a subset of individuals that acquire a JAK2V617F mutation, although it cannot be ruled out that haploinsufficiency for TET2 or genes located on chromosome 20q renders HSCs more susceptible to subsequent acquisition of a JAK2 mutation.
A common JAK2 haplotype predisposes to the acquisition of mutations in JAK2 or MPL
A population-based study of almost 12000 Swedish patients with a non-familial MPN and 25000 of their first-degree relatives demonstrated that family members have a five-fold to seven-fold increased risk of developing ET, PV or MF, and a two-fold increased risk of CML (101). Genetic analyses recently provided a partial explanation for this intriguing finding: a relatively frequent JAK2 haplotype predisposes an individual to acquiring a JAK2 mutation (15, 102-104), accounting for approximately half of the risk identified in the Swedish cohort. Compared to healthy controls, the “46/1” JAK2 haplotype is significantly over-represented in individuals positive for a JAK2V617F, JAK2 exon 12 or MPL mutation. However, this JAK2 haplotype is not over-represented in patients with CML (105), suggesting that other genomic loci influence the initiation of this disorder in the first-degree relatives of MPN patients.
Two models have been proposed to explain this phenomenon: the “hypermutability” model, which suggests local mutations occur more frequently in JAK2 alleles with the 46/1 haplotype; and the “fertile ground” model, which states that the mutation rate is not dependent on haplotype, but that mutations occurring on the 46/1 haplotype provide some selective advantage over mutations occurring on other JAK2 haplotypes. Currently, the published data are such that neither model can be ruled out. Genome-wide association studies independently identified that rs10758669, a JAK2 SNP that tags the 46/1 haplotype, is being associated with Crohn’s disease (106); as this is a non-malignant disorder of the gastrointestinal tract, it would appear that the hypermutability model might not be valid, although experimental evidence has suggested that this SNP enhances susceptibility to DNA damage (107). Furthermore, the 46/1 haplotype is over-represented in ET patients with an MPL mutation (104); it is currently unclear how sequence variation on chromosome 9 increases the mutability of a locus located on another chromosome. To date, changes in JAK2 mRNA levels in granulocytes or coding sequence variants other than SNPs that lend support to the “fertile ground” hypothesis have not been linked to the 46/1 haplotype. However, healthy individuals with a 46/1 JAK2 allele do have reduced numbers of myeloid progenitors, supporting the contention that there is a functional difference between JAK2 haplotypes. One possibility that remains to be explored is that the non-mutated 46/1 allele is transcribed or translated in HSCs at higher levels than JAK2 alleles with other haplotypes. This may predispose HSCs to acquiring mutations, including those in JAK2 and MPL, since increased rates of mutation are observed in vitro following over-expression of wild-type JAK2 (65).
Concluding comments
Meaningful insights into the pattern of mutation accumulation in patients with a Ph-negative MPN have been recently provided by genotyping individual hematopoietic colonies. In many instances, these studies revealed a linear stepwise acquisition of mutations over time, involving two types of genomic target: those that result in constitutive JAK2 signaling (including JAK2, MPL and SH2B3), and those encoding epigenetic regulators (such as TET2, ASXL1, DNMT3A or EZH2). These mutations do not, however, follow a strict temporal pattern of acquisition; it may be the combination of mutations, rather than the order in which they are acquired, that is the important factor in disease progression. In a small proportion of MPN cases, the malignant cell-of-origin can instead give rise to a limited number of distinct sub-clones that continue to evolve in parallel. Clonal evolution in either patient group is likely to be assisted by the increased levels of DNA damage observed in cells carrying mutations that enhance JAK2-mediated signaling, and by the suppression of pathways that normally eliminate cells with compromised genomic integrity. Currently, it is unclear which came first: the chicken (genomic instability as a direct consequence of constitutive JAK2-mediated signaling) or the egg (an inherent predisposition to acquiring MPN-associated mutations, including JAK2V67F). Further insights into evolution in the MPNs are anticipated as next-generation sequencing methodologies identify the complete repertoire of genetic lesions underlying these disorders.
Colony genotyping studies have also revealed that acquisition of an MPN-initiating mutation in some instances may be the consequence of an inherent genomic instability within the patient’s HSCs. This intriguing possibility is supported by both epidemiological data and the identification of a common JAK2 haplotype that elevates the risk of an individual acquiring a JAK2 or MPL mutation. Any such genomic instability in hematopoietic cells of MPN patients has important implications for those treated with pharmacologic agents that inhibit JAK2 activity: although these drugs may alleviate the constitutional symptoms associated with myelofibrosis and significantly reduce the proportion of JAK2V617F-positive blood cells (108, 109), they may nonetheless fail to normalize the elevated risk of leukemic transformation seen in MPN patients.
Elucidation of the impact of constitutively active JAK2 signaling on genome instability and tumor evolution in the MPNs is likely to shed light on the mechanisms behind this process in other malignancies. JAK2 activating mutations are not only found in patients with an MPN, but also occur in a significant proportion of children with Down syndrome-associated or high-risk ALL (110), and in a number of solid tumors, including cancers of the lung and breast (3, 111). Moreover, JAK2 activation occurs in a variety of malignancies that are not known to carry JAK2 mutations; its inhibition significantly reduces constitutive STAT3 activation and suppresses the growth of breast, ovarian and prostate cancer xenografts in mice (112). Commercially available JAK2 inhibitors may therefore reduce genome instability, diminishing the metastatic potential of these tumor types, and improving patient outcome.
Acknowledgements
Research in the authors’ laboratories is funded by the Greehey Children’s Cancer Research Institute (UTHSCSA), a Congressionally-Directed Medical Research Program Bone Marrow Failure Research Program Exploration-Hypothesis Development Award (W81XWH-10-1-0314; to LMS), and a National Institutes of Aging R21 Award (5R21AG033339; to VIR).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Ding L, Ellis MJ, Li S, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109–1113. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–94. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469:356–361. doi: 10.1038/nature09650. [DOI] [PubMed] [Google Scholar]
- 8.Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469:362–367. doi: 10.1038/nature09733. [DOI] [PubMed] [Google Scholar]
- 9.Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6:372–375. [PubMed] [Google Scholar]
- 11.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri S, Stein H, Thiele J, Vardiman JW. WHO classification of tumours of haematopoietic and lymphoid tissues. IARC; Lyon: 2008. [Google Scholar]
- 12.Cervantes F, Passamonti F, Barosi G. Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukemia. 2008;22:905–914. doi: 10.1038/leu.2008.72. [DOI] [PubMed] [Google Scholar]
- 13.Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 14.Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006;108:2435–2437. doi: 10.1182/blood-2006-04-018259. [DOI] [PubMed] [Google Scholar]
- 15.Olcaydu D, Harutyunyan A, Jager R, et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet. 2009;41:450–454. doi: 10.1038/ng.341. [DOI] [PubMed] [Google Scholar]
- 16.Schaub FX, Jager R, Looser R, et al. Clonal analysis of deletions on chromosome 20q and JAK2-V617F in MPD suggests that del20q acts independently and is not one of the predisposing mutations for JAK2-V617F. Blood. 2009;113:2022–2027. doi: 10.1182/blood-2008-07-167056. [DOI] [PubMed] [Google Scholar]
- 17.Schaub FX, Looser R, Li S, et al. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood. 2010;115:2003–2007. doi: 10.1182/blood-2009-09-245381. [DOI] [PubMed] [Google Scholar]
- 18.Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7:441–453. doi: 10.1038/nrc2147. [DOI] [PubMed] [Google Scholar]
- 19.Skorski T. BCR/ABL, DNA damage and DNA repair: implications for new treatment concepts. Leuk Lymphoma. 2008;49:610–614. doi: 10.1080/03093640701859089. [DOI] [PubMed] [Google Scholar]
- 20.Burke BA, Carroll M. BCR-ABL: a multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leukemia. 2010;24:1105–1112. doi: 10.1038/leu.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fialkow P, Gartler SM, Yoshida A. Clonal origin of chronic myelocytic leukemia in man. Proc Natl Acad Sci USA. 1967;58:1468. doi: 10.1073/pnas.58.4.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamson JW, Fialkow PJ, Murphy S, Prchal JF, Steinmann L. Polycythemia vera: stem-cell and probable clonal origin of the disease. N Engl J Med. 1976;295:913–916. doi: 10.1056/NEJM197610212951702. [DOI] [PubMed] [Google Scholar]
- 23.Udomsakdi C, Eaves CJ, Swolin B, et al. Rapid decline of chronic myeloid leukemic cells in long-term culture due to a defect at the leukemic stem cell level. Proc Natl Acad Sci USA. 1992;89:6192–6196. doi: 10.1073/pnas.89.13.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sirard C, Lapidot T, Vormoor J, et al. Normal and leukemic SCID-repopulating cells (SRC) coexist in the bone marrow and peripheral blood from CML patients in chronic phase, whereas leukemic SRC are detected in blast crisis. Blood. 1996;87:1539–1548. [PubMed] [Google Scholar]
- 25.Jamieson CH, Gotlib J, Durocher JA, et al. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc Natl Acad Sci USA. 2006;103:6224–6229. doi: 10.1073/pnas.0601462103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anand S, Stedham F, Beer PA, et al. Effects of the JAK2 mutation on the hematopoietic stem and progenitor compartment in human myeloproliferative neoplasms. Blood. 2011;118:177–181. doi: 10.1182/blood-2010-12-327593. [DOI] [PubMed] [Google Scholar]
- 27.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 28.Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 29.Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;4:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 30.Levine RL, Loriaux M, Huntly BJ, et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood. 2005;106:3377–3379. doi: 10.1182/blood-2005-05-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott LM, Campbell PJ, Baxter EJ, et al. The V617F JAK2 mutation is uncommon in cancers and in myeloid malignancies other than the classic myeloproliferative disorders. Blood. 2005;106:2920–2921. doi: 10.1182/blood-2005-05-2087. [DOI] [PubMed] [Google Scholar]
- 32.Jones AV, Kreil S, Zoi K, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106:2162–2168. doi: 10.1182/blood-2005-03-1320. [DOI] [PubMed] [Google Scholar]
- 33.Kaushansky K. On the molecular origins of the chronic myeloproliferative disorders: it all makes sense. Blood. 2005;105:4187–4190. doi: 10.1182/blood-2005-03-1287. [DOI] [PubMed] [Google Scholar]
- 34.Lacout C, Pisani DF, Tulliez M, et al. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108:1652–1660. doi: 10.1182/blood-2006-02-002030. [DOI] [PubMed] [Google Scholar]
- 35.Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111:3931–3940. doi: 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 36.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356:459–468. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott LM, Beer PA, Bench AJ, Erber WN, Green AR. Prevalance of JAK2 V617F and exon 12 mutations in polycythaemia vera. Br J Haematol. 2007;139:511–512. doi: 10.1111/j.1365-2141.2007.06806.x. [DOI] [PubMed] [Google Scholar]
- 38.Pietra D, Li S, Brisci A, et al. Somatic mutations of JAK2 exon 12 in patients with JAK2 (V617F)-negative myeloproliferative disorders. Blood. 2008;111:1686–1689. doi: 10.1182/blood-2007-07-101576. [DOI] [PubMed] [Google Scholar]
- 39.Butcher CM, Hahn U, To LB, et al. Two novel JAK2 exon 12 mutations in JAK2V617F-negative polycythaemia vera patients. Leukemia. 2008;22:870–873. doi: 10.1038/sj.leu.2404971. [DOI] [PubMed] [Google Scholar]
- 40.Scott LM. The JAK2 exon 12 mutations: a comprehensive review. Am J Hematol. 2011;86:688–676. doi: 10.1002/ajh.22063. [DOI] [PubMed] [Google Scholar]
- 41.Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006;3:1140–1151. doi: 10.1371/journal.pmed.0030270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beer PA, Campbell PJ, Scott LM, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112:141–149. doi: 10.1182/blood-2008-01-131664. [DOI] [PubMed] [Google Scholar]
- 43.Pardanani A, Levine R, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472–3476. doi: 10.1182/blood-2006-04-018879. [DOI] [PubMed] [Google Scholar]
- 44.Staerk J, Lacout C, Sato T, et al. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood. 2006;107:1864–1871. doi: 10.1182/blood-2005-06-2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ding J, Komatsu H, Iida S, et al. The Asn505 mutation of the c-MPL gene, which causes familial essential thrombocythemia, induces autonomous homodimerization of the c-Mpl protein due to strong amino acid polarity. Blood. 2009;114:3325–3328. doi: 10.1182/blood-2008-04-149047. [DOI] [PubMed] [Google Scholar]
- 46.Oh ST, Simonds EF, Jones C, et al. Novel mutations in the inhibitory adaptor protein LNK drive JAK-STAT signaling in patients with myeloproliferative neoplasms. Blood. 2010;116:988–992. doi: 10.1182/blood-2010-02-270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pardanani A, Hood J, Lasho T, et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007;21:1658–1668. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]
- 48.Hexner EO, Serdikoff C, Jan M, et al. Lestaurtinib (CEP701) is a JAK2 inhibitor that suppresses JAK2/STAT5 signaling and the proliferation of primary erythroid cells from patients with myeloproliferative disorders. Blood. 2008;111:5663–5671. doi: 10.1182/blood-2007-04-083402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Delhommeau F, Dupont S, Della Valle V, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 50.Carbuccia N, Murati A, Trouplin V, et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia. 2009;23:2183–2186. doi: 10.1038/leu.2009.141. [DOI] [PubMed] [Google Scholar]
- 51.Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 52.Stegelmann F, Bullinger L, Schlenk RF, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. 2011;25:1217–1219. doi: 10.1038/leu.2011.77. [DOI] [PubMed] [Google Scholar]
- 53.Langemeijer SM, Kuiper RP, Berends M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 54.Tefferi A, Lim KH, Abdel-Wahab O, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23:1343–1345. doi: 10.1038/leu.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gelsi-Boyer V, Trouplin V, Roquain J, et al. ASXL1 mutation is associated with poor prognosis and acute transformation in chronic myelomonocytic leukaemia. Br J Haematol. 2010;151:365–375. doi: 10.1111/j.1365-2141.2010.08381.x. [DOI] [PubMed] [Google Scholar]
- 56.Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 57.Moran-Crusio K, Reavie L, Shih A, et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20:11–24. doi: 10.1016/j.ccr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Quivoron C, Couronne L, Della Valle V, et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell. 2011;20:25–38. doi: 10.1016/j.ccr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 59.Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2011;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fisher CL, Pineault N, Brookes C, et al. Loss-of-function Additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood. 2010;115:38–46. doi: 10.1182/blood-2009-07-230698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp Hematol. 2002;30:229–236. doi: 10.1016/s0301-472x(01)00789-5. [DOI] [PubMed] [Google Scholar]
- 62.Campbell PJ, Scott LM, Buck G, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005;366:1945–1953. doi: 10.1016/S0140-6736(05)67785-9. [DOI] [PubMed] [Google Scholar]
- 63.Shide K, Shimoda HK, Kumano T, et al. Development of ET, primary myelofibrosis and PV in mice expressing JAK2 V617F. Leukemia. 2008;22:87–95. doi: 10.1038/sj.leu.2405043. [DOI] [PubMed] [Google Scholar]
- 64.Percy MJ, Scott LM, Erber WN, et al. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica. 2007;92:1607–1614. doi: 10.3324/haematol.11643. [DOI] [PubMed] [Google Scholar]
- 65.Plo I, Nakadake M, Wiesmuller L, et al. JAK2 activation stimulates homologous recombination and genomic instability. Blood. 2007;112:1402–1412. doi: 10.1182/blood-2008-01-134114. [DOI] [PubMed] [Google Scholar]
- 66.Haaf T, Golub EI, Reddy G, Radding CM, Ward DC. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc Natl Acad Sci U S A. 1995;92:2298–2302. doi: 10.1073/pnas.92.6.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aten JA, Stap J, Krawczyk PM, et al. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 2004;303:92–95. doi: 10.1126/science.1088845. [DOI] [PubMed] [Google Scholar]
- 68.Fernandes MS, Reddy MM, Gonneville JR, et al. BCR-ABL promotes the frequency of mutagenic single-strand annealing DNA repair. Blood. 2009;114:1813–1819. doi: 10.1182/blood-2008-07-172148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, Spensberger D, Ahn JS, et al. JAK2 V617F impairs hematopoietic stem cell function in a conditional knock-in mouse model of JAK2 V617F-positive essential thrombocythemia. Blood. 2010;116:1528–1538. doi: 10.1182/blood-2009-12-259747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mullally A, Lane SW, Ball B, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17:584–596. doi: 10.1016/j.ccr.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270:1–9. doi: 10.1016/j.canlet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 72.Shi S, Calhoun HC, Xia F, et al. JAK signaling globally counteracts heterochromatic gene silencing. Nat Genet. 2006;38:1071–1076. doi: 10.1038/ng1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lobie PE, Ronsin B, Silvennoinen O, et al. Constitutive nuclear localization of Janus kinases 1 and 2. Endocrinology. 1996;137:4037–4045. doi: 10.1210/endo.137.9.8756581. [DOI] [PubMed] [Google Scholar]
- 74.Ram PA, Waxman DJ. Interaction of growth hormone-activated STATs with SH2-containing phosphotyrosine phosphatase SHP-1 and nuclear JAK2 tyrosine kinase. J Biol Chem. 1997;272:17694–17702. doi: 10.1074/jbc.272.28.17694. [DOI] [PubMed] [Google Scholar]
- 75.Rinaldi CR, Rinaldi P, Alagia A, et al. Preferential nuclear accumulation of JAK2V617F in CD34+ but not in granulocytic, megakaryocytic, or erythroid cells of patients with Philadelphia-negative myeloproliferative neoplasia. Blood. 2010;116:6023–6026. doi: 10.1182/blood-2010-08-302265. [DOI] [PubMed] [Google Scholar]
- 76.Rui L, Emre NC, Kruhlak MJ, et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell. 2010;18:590–605. doi: 10.1016/j.ccr.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dawson MA, Bannister AJ, Gottgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Griffiths DS, Li J, Dawson MA, et al. LIF-independent JAK signalling to chromatin in embryonic stem cells uncovered from an adult stem cell disease. Nat Cell Biol. 2010;13:13–21. doi: 10.1038/ncb2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcia-Cao M, O’Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet. 2004;36:94–99. doi: 10.1038/ng1278. [DOI] [PubMed] [Google Scholar]
- 80.Obuse C, Iwasaki O, Kiyomitsu T, et al. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol. 2004;6:1135–1141. doi: 10.1038/ncb1187. [DOI] [PubMed] [Google Scholar]
- 81.Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature. 2008;453:682–686. doi: 10.1038/nature06875. [DOI] [PubMed] [Google Scholar]
- 82.Cummings WJ, Yabuki M, Ordinario EC, et al. Chromatin structure regulates gene conversion. PLoS Biol. 2007;5:e246. doi: 10.1371/journal.pbio.0050246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Loeb LA. Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer. 2011;11:450–457. doi: 10.1038/nrc3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bench AJ, Cross NC, Huntly BJ, Nacheva EP, Green AR. Myeloproliferative disorders. Best Pract Res Clin Haematol. 2001;14:531–551. doi: 10.1053/beha.2001.0153. [DOI] [PubMed] [Google Scholar]
- 85.Gurvich N, Perna F, Farina A, et al. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc Natl Acad Sci USA. 2010;107:22552–22557. doi: 10.1073/pnas.1017092108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deverman BE, Cook BL, Manson SR, et al. Bcl-xL deamidation is a critical switch in the regulation of the response to DNA damage. Cell. 2002;111:51–62. doi: 10.1016/s0092-8674(02)00972-8. [DOI] [PubMed] [Google Scholar]
- 87.Zhao R, Oxley D, Smith TS, et al. DNA damage-induced Bcl-xL deamidation is mediated by NHE-1 antiport regulated intracellular pH. PLoS Biol. 2007;5:e1. doi: 10.1371/journal.pbio.0050001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao R, Follows GA, Beer PA, et al. Inhibition of the Bcl-xL deamidation pathway in myeloproliferative disorders. N Engl J Med. 2008;359:2778–2789. doi: 10.1056/NEJMoa0804953. [DOI] [PubMed] [Google Scholar]
- 89.Lambert JR, Everington T, Linch DC, Gale RE. In essential thrombocythemia, multiple JAK2-V617F clones are present in most mutant-positive patients: a new disease paradigm. Blood. 2009;114:3018–3023. doi: 10.1182/blood-2009-03-209916. [DOI] [PubMed] [Google Scholar]
- 90.Li S, Kralovics R, De Libero G, et al. Clonal heterogeneity in polycythemia vera patients with JAK2 exon12 and JAK2-V617F mutations. Blood. 2008;111:3863–3866. doi: 10.1182/blood-2007-09-111971. [DOI] [PubMed] [Google Scholar]
- 91.Beer PA, Jones AV, Bench AJ, et al. Clonal diversity in the myeloproliferative neoplasms: independent origins of genetically distinct clones. Br J Haematol. 2009;144:904–908. doi: 10.1111/j.1365-2141.2008.07560.x. [DOI] [PubMed] [Google Scholar]
- 92.Jallades L, Hayette S, Tigaud I, et al. Emergence of therapy-unrelated CML on a background of BCR-ABL-negative JAK2V617F-positive chronic idiopathic myelofibrosis. Leuk Res. 2008;32:1608–1610. doi: 10.1016/j.leukres.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 93.Bee PC, Gan GG, Nadarajan VS, Latiff NA, Menaka N. A man with concomitant polycythaemia vera and chronic myeloid leukemia: the dynamics of the two disorders. Int J Hematol. 2010;91:136–139. doi: 10.1007/s12185-009-0471-6. [DOI] [PubMed] [Google Scholar]
- 94.Conchon MR, Costa JL, Novaes MM, et al. Simultaneous detection of JAK2 V617F mutation and Bcr-Abl translocation in a patient with chronic myelogenous leukemia. Int J Hematol. 2008;88:243–245. doi: 10.1007/s12185-008-0131-2. [DOI] [PubMed] [Google Scholar]
- 95.Sotlar K, Bache A, Stellmacher F, et al. Systemic mastocytosis associated with chronic idiopathic myelofibrosis: a distinct subtype of systemic mastocytosis associated with a clonal hematological non-mast cell lineage disorder carrying the activating point mutations KITD816V and JAK2V617F. J Mol Diagn. 2008;10:58–66. doi: 10.2353/jmoldx.2008.070061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nagata H, Worobec AS, Oh CK, et al. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc Natl Acad Sci USA. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Campbell PJ, Baxter EJ, Beer PA, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108:3548–3555. doi: 10.1182/blood-2005-12-013748. [DOI] [PubMed] [Google Scholar]
- 98.Theocharides A, Boissinot M, Girodon F, et al. Leukemic blasts in transformed JAK2-V617F positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110:375–379. doi: 10.1182/blood-2006-12-062125. [DOI] [PubMed] [Google Scholar]
- 99.Beer PA, Delhommeau F, LeCouedic JP, et al. Two routes to leukemic transformation after a JAK2 mutation-positive myeloproliferative neoplasm. Blood. 2010;115:2891–2900. doi: 10.1182/blood-2009-08-236596. [DOI] [PubMed] [Google Scholar]
- 100.Kralovics R, Teo SS, Li S, et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108:1377–1380. doi: 10.1182/blood-2005-11-009605. [DOI] [PubMed] [Google Scholar]
- 101.Landgren O, Goldin LR, Kristinsson SY, et al. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood. 2008;112:2199–2204. doi: 10.1182/blood-2008-03-143602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jones AV, Chase A, Silver RT, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet. 2009;41:446–449. doi: 10.1038/ng.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Olcaydu D, Skoda RC, Looser R, et al. The ‘GGCC’ haplotype of JAK2 confers susceptibility to JAK2 exon 12 mutation-positive polycythemia vera. Leukemia. 2009;23:1924–1926. doi: 10.1038/leu.2009.110. [DOI] [PubMed] [Google Scholar]
- 104.Jones AV, Campbell PJ, Beer PA, et al. The JAK2 46/1 haplotype predisposes to MPL mutated myeloproliferative neoplasms. Blood. 2010;115:4517–4523. doi: 10.1182/blood-2009-08-236448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Spolverini A, Jones AV, Hochhaus A, et al. The myeloproliferative neoplasm-associated JAK2 46/1 haplotype is not overrepresented in chronic myelogenous leukemia. Ann Hematol. 2010;90:365–366. doi: 10.1007/s00277-010-1009-y. [DOI] [PubMed] [Google Scholar]
- 106.Barrett JC, Hansoul S, Nicolae DL, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ferguson LR, Han DY, Fraser AG, et al. Genetic factors in chronic inflammation: single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and Crohn’s disease in a New Zealand population. Mutat Res. 2010;690:108–115. doi: 10.1016/j.mrfmmm.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 108.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pardanani A, Gotlib JR, Jamieson C, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29:789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372:1484–1492. doi: 10.1016/S0140-6736(08)61341-0. [DOI] [PubMed] [Google Scholar]
- 111.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hedvat M, Huszar D, Herrmann A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]