

Abstract
Synthetic cavitands and protein cavities have been widely studied as models for ligand recognition. Here we investigate the Met102 → His substitution in the artificial L99A cavity in T4 lysozyme as a Kemp eliminase. The resulting enzyme had kcat/KM = 0.43 M-1 s-1 and a (kcat/KM)/kuncat = 107 at pH 5.0. The crystal structure of this enzyme was determined at 1.30 Å, as were the structures of four complexes of substrate and product analogs. The absence of ordered waters or hydrogen bonding interactions, and the presence of a common catalytic base (His102) in an otherwise hydrophobic, buried cavity, facilitated detailed analysis of the reaction mechanism and its optimization. Subsequent substitutions increased eliminase activity by an additional four-fold. As activity-enhancing substitutions were engineered into the cavity, protein stability decreased, consistent with the stability-function trade-off hypothesis. This and related model cavities may provide templates for studying protein design principles in radically simplified environments.
Keywords: enzyme design, model cavity
Model host-guest systems for protein–ligand recognition (1) and catalysis have been intensely studied since Cram and Lehn introduced the first synthetic cavitands (2). More recently, cavity sites in proteins themselves have been explored as model systems (3–6). These protein cavities are characterized by burial from bulk water (simplifying the treatment of dielectric interfaces) and the dominance of a single type of ligand–protein interaction. Such dominating interactions can be nonpolar complementarity, as in the engineered core cavity in T4 lysozyme L99A (3), or ionic bonding, as in the cavity in cytochrome C peroxidase (CCP) W191G, among others (4, 6). In their simplicity they mimic many of the synthetic cavitands. In contrast to the cavitands, model proteins have the advantages of ready solubility in aqueous media, easy introduction of functionality by site-directed mutation, and of being readily over-expressed, crystallized, and assayed. These features have made them attractive for exploring the physical bases of ligand–protein binding, often in cycles of computation and experiment (7–10).
The recent successes in de novo enzyme design inspired us to wonder whether these model cavity sites might be illuminating for understanding substrate recognition and catalysis. Whereas recent efforts to design Kemp eliminases have been tour de force successes, and semi-quantitative calculations have illuminated the origins of the catalytic effects (11, 12), the newly designed enzymes often retain the complexities of the native, or native-like, protein structures from which they were designed or evolved (13–21). As with natural enzyme sites, the designed active sites bring to bear a mix of charged, polar, and apolar residues in a complex dielectric and steric environment, typically open to bulk solvent. Like native enzymes, the designed active sites can undergo substantial conformational change along the reaction coordinate (18). Such complexity undoubtedly reflects the optimization for catalysis by which these sites have been selected, but it can interfere with subsequent mechanistic analysis.
We therefore decided to explore catalysis of an elementary reaction in a simple protein cavity site. The small substrate of the Kemp elimination (Fig. 1) (9, 22), whose reactivity is enhanced by a polar, aprotic environment (23), made it well-suited to such a cavity. Among the simplest of these cavities is the fully apolar site in T4 lysozyme (Fig. 2A), created by the core substitution Leu99 → Ala. Exposure of the substrate to even high concentrations of the L99A cavity mutant had little or no effect on the Kemp elimination reaction rate. We therefore introduced a histidine into the cavity to act as a catalytic base. The resulting stability insult to the folded form of the enzyme demanded the introduction of stability-restoring substitutions in other parts of the protein to regain the stably folded form. This led to a cavity site bearing a buried histidine. The construct bound multiple substrate and product analogs along the reaction coordinate, and was well suited to crystallographic structure determination. These structures, in turn, enabled an initial dissection of the bases of recognition and catalysis, and subsequent residue substitutions to optimize them. Here, too, cycles of catalytic and stability optimization were required. The result was a construct that lent itself not only to catalysis, but to structural analysis of mechanism, biophysical trade-offs between stability and function, and future optimization for efficiency for this and other reactions.
Fig. 1.
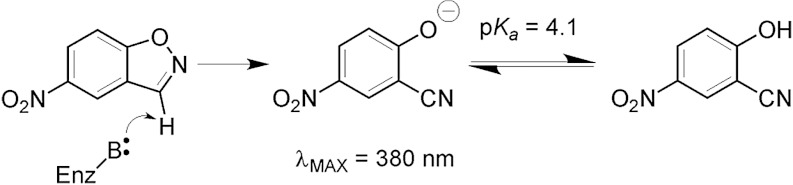
The Kemp elimination.
Fig. 2.

Structures of the model cavity sites. For panels B–F, the structures of the L99A/M102H† cavity site are shown as a blue surface with bound ligands (carbons white, nitrogens blue, oxygens red, sulfur yellow). Fo - Fc density (calculated after refinement but before the introduction of the ligand) at 3σ is shown as black mesh. (A) The molecular surface of the L99A cavity (PDB ID 181L) (inner) embedded within the overall surface of the protein (outer). The surfaces of ionic residues are shown in red, polar residues in yellow and nonpolar residues in green. Several cavity lining residues are rendered as sticks (oxygens red, nitrogens dark blue, carbons pale blue). The polar atoms of Tyr88 and Ser117 are oriented away from the cavity, which is almost entirely apolar in nature. (B) L99A/M102H† complexed with 2-mercaptoethanol at 1.30 Å. The hydrogen bond observed in all the structures between Sδ of Met106 and Nε2 of His102 is shown. (C) L99A/M102H† complexed with nitrobenzene at 1.64 Å. (D) L99A/M102H† complexed with 4-nitrophenol at 1.54 Å. (E) L99A/M102H† complexed with 2-cyanophenol at 1.49 Å. (F) L99A/M102H† complexed with benzisoxazole at 1.64 Å, modeled in the catalytically competent pose showing the 3.3 Å distance between Nδ1 of His102 and the acidic carbon of the Kemp substrate. Figures rendered with PyMol.
Results
The L99A cavity in T4 lysozyme is about 150 Å3 in volume and wholly buried from solvent (3); it accommodates ligands such as benzene and indole with only modest structural adjustments (3). Simple modeling suggested that the Met102 → His substitution into this cavity would only perturb it slightly. Molecular docking suggested that the benzisoxazole Kemp substrate, the ring-opened product, and their analogs could be accommodated by this cavity in such a way that His102 could act as a catalytic base. Based on published experience (6) with Met102 → Glu, however, we worried that the L99A/M102H† mutant protein would be grossly unstable, owing to the insult incurred by burying a highly polar residue in an otherwise apolar cavity. This concern was born out by subsequent results.
To overcome this problem, we looked to restabilize the enzyme by substitutions at positions distant from the cavity. Here we were helped by lysozyme’s status as a heavily studied model system for protein structure and stability, due to the efforts of Matthews and coworkers (24). Looking for substitutions that would increase the stability of the protein without grossly perturbing the cavity site, we introduced a series of substitutions, often in or around the native lysozyme active site, which ordinarily recognizes the muramyl peptide. These included Thr21 → Cys, Ser38 → Asp, Glu108 → Val, Ser117 → Val, Thr142 → Cys, and Asn144 → Asp. Taken together, these substitutions stabilize the native protein by over 6.5 kcal/mol (SI Appendix, Table S1) (24). Because they largely occur around, and occlude, the original active site, they result in a lysozyme largely inactive against the native muramyl peptide substrate. Engineered into this background, the L99A/M102H† construct (SI Appendix, Table S4) could be over-expressed, purified to homogeneity, crystallized to high resolution, and was stable enough to assay.
At this point, we began to examine the Kemp eliminase activity of what were by now several cavity constructs (Fig. 3). The L99A construct had minimal activity (kcat/KM = 0.017 M-1 s-1, Table 1), barely above spontaneous substrate decomposition at pH 5.0. Intriguingly, the L99A/M102Q construct did have modest activity (kcat/KM = 0.26 M-1 s-1, Table 1). The introduction of the Met102 → Gln substitution introduces a single polar atom, that of the Gln side chain carbonyl oxygen, into the otherwise apolar cavity (4); it is this increased polarity of the cavity that likely explains the activity of this destabilized mutant. However, subsequent addition of the same stabilizing substitutions that enabled the expression of L99A/M102H† to give L99A/M102Q† largely eliminated measurable activity (kcat/KM = 0.007 M-1 s-1, Table 1), probably due to stiffening of the structure and reduced access to the cavity site. These distal substitutions stabilize the protein without much affecting its structure (RMSD = 0.6 Å between L99A and L99A/M102H†, SI Appendix, Fig. S6). Finally, the Gln102 → His substitution, in the stabilized background to give L99A/M102H†, restored activity to a kcat/KM = 0.43 M-1 s-1 (Table 1, SI Appendix, Fig. S5). Though the enzyme will ultimately be inhibited by the product of the reaction (4-nitrophenol, a product analog, has a Kd of 2 μM), the enzyme catalyzed multiple substrate turnovers (SI Appendix, Fig. S7)
Fig. 3.

The relationship between Kemp eliminase activity and protein stability of the lysozyme cavity mutants. The number of substitutions required to go from WT T4 lysozyme to the specific cavity mutant is represented against the change in stability (ΔΔG, kcal/mol) between that mutant and WT. The residues present in the N-terminal hexahistidine tag are not counted towards the mutation number. The Kemp eliminase activity [kcat/KM (M-1 s-1)] of each construct is represented by the size of the circles (see Table 1 and SI Appendix, Table S1). Black arrows represent mutations that were intended to increase stability, red arrows indicate mutations that were intended to increase activity, and dashed black and red lines indicate mutations of both kinds were included in that step.
Table 1.
kcat/KM values for the cavity mutants
| Cavity |
kcat/KM(M-1 s-1) |
| L99A | 1.7 × 10-2 ± 2.1 × 10-3 |
| L99A/M102Q | 2.6 × 10-1 ± 8.5 × 10-2 |
| L99A/M102Q† | 6.7 × 10-3 ± 5.9 × 10-3 |
| L99A/M102H† | 4.3 × 10-1 ± 1.6 × 10-1 |
| L99A/M102E† | 1.4 × 10-2 ± 1.9 × 10-3 |
| A99L/M102H† | 4.9 × 10-2 ± 2.1 × 10-3 |
| L99A/M102H†/M106A | 7.7 × 10-1 ± 2.3 × 10-1 |
| L99A/M102H†/M106D | 9.2 × 10-1 ± 3.0 × 10-1 |
| L99A/M102H†/L118Q | 1.4 × 100 ± 2.9 × 10-1 |
| L99A/M102H‡ | 1.8 × 100 ± 5.7 × 10-2 |
Errors represent standard deviations (see also SI Appendix, Table S1). L99A/M102E† described previously (6).
The pH-rate profile of L99A/M102H† is flat from pH 7.7 down to pH 5.0, where titration of the product chromophore begins (Fig. 1 and SI Appendix, Fig. S4), suggesting that the catalytic His102 is neutral over this range, as might be expected for a buried histidine (25). Because the spontaneous base-catalyzed reaction is slower at lower pH-values, the enzymatic rate acceleration over the spontaneous rate in imidazole-containing buffer, 7 × 107, is greater at pH 5 (at pH 7.3, the background rate is 50- to 200-fold greater, depending on the buffer used, so the rate acceleration is correspondingly less; SI Appendix, Table S3 and Fig. S3). Consistent with the view that catalysis occurs in the cavity, a revertant substitution of Ala99 → Leu (A99L/M102H†), which largely eliminates the cavity, loses most of its activity (Table 1). Meanwhile, its stability is increased by 5.6 kcal/mol (ΔTm = 13.3 °C, SI Appendix, Table S1), and is in fact higher than that of the WT protein, owing to the many stabilizing substitutions added to the native lysozyme active site.
Lysozyme is a model system for crystallography and biophysics, and we were able to determine the structure of L99A/M102H† to 1.30 Å (Fig. 2B and SI Appendix, Table S2). Notwithstanding the addition of the N-terminal his-tag and the six stabilizing substitutions, its structure closely resembles that of the original L99A, certainly in the region of the cavity. The major differences are near the stabilizing Cys21 and Cys142 (Fig. 4), where a disulfide bond has been engineered (24). As in L99A, the approximately 150 Å3 cavity of L99A/M102H† remained isolated from bulk solvent (26). The Nδ1 atom of His102 points into the cavity, oriented by a hydrogen bond between the presumably protonated Nε2 of His102 and Sδ of Met106 (3.16 Å apart, Fig. 2B). In this structure a crystallizing additive, 2-mercaptoethanol, was bound in the cavity, and one of its hydroxyls hydrogen bonds to His102 Nδ1 at a distance of 2.8 Å.
Fig. 4.

Sacrificing an ancestral active site to engineer a new one. (A) The active site of T4 lysozyme (Left) provides an enzymatic function at the cost of reducing the stability of the protein structure (Right) The axes on the charts are purely qualitative to illustrate the stability-function trade-off. (B) Stability-restoring substitutions (such as E11F, D20N,T21C, and T142C) were engineered into the muramyl peptide site of lysozyme, eliminating the enzyme's activity as a hydrolase. (C) The restored stability of the lysozyme protein can now be traded for a new function, in this case Kemp eliminase activity with the introduction of the M102H mutation.
The X-ray structures of L99A/M102H† in complex with substrate and product analogs, including benzisoxazole, nitrobenzene, 2-cyanophenol, and 4-nitrophenol, illuminated the mechanism of this rudimentary enzyme (21). Nitrobenzene and 4-nitrophenol both place their nitro groups away from His102 (Fig. 2 C and D). Conversely, the phenolic hydroxyl of the ligands 4-nitrophenol and 2-cyanophenol both hydrogen bond to the Nδ1 of His102 (2.66 and 2.68 Å, respectively, Fig. 2 D and E). These poses do not represent the product immediately after catalysis, but likely a collapse away from this geometry, as may be necessary for multiple turnovers, as observed among some natural enzymes (27). The complexes nevertheless emphasize the role of the Nδ2 of His102 as a proton acceptor. Correspondingly, the Nε2 of His102 always appears to hydrogen bond with the Sδ of Met106, suggesting that the Nδ1, which points into the cavity, is unprotonated and is available as a base. Consistent with such a role, in the co-complex of benzisoxazole bound to the L99A/M102H† cavity, the heterocyclic ring is proximal to His102 (Fig. 2F). Whereas the electron density of the ligand itself cannot resolve whether the ligand carbon or the oxygen is closest to His102, we have modeled it in the catalytically competent pose where the hydrogen on the ring carbon is available to be abstracted by the His102 Nδ1, which is 3.3 Å away. Even here, the angle for proton abstraction is imperfect, but a modest reorientation would allow for in-line approach in the activated state. Molecular docking suggests that the full nitro-bearing substrate may be accommodated in this pose, though the docking van der Waals energies do reflect some strain, implying that there may be modest conformational change with this subststrate (SI Appendix, Fig. S8 and Table S5).
These structures suggested opportunities for optimizing the simple cavity for catalysis. Reasoning that the elimination reaction would be improved by substitutions that stabilized the histidine catalytic base in an orientation competent for catalysis, we sought hydrogen bonding opportunities that would better fix Nδ1 in such a position. This led to the substitutions Met106 → Ala and Met106 → Asp in the L99A/M102H† background, where the introduced solvent, in the case of the alanine, and carboxylate, in the case of the aspartate, might be better able to orient the Nε2 hydrogen bond donor of the His102. Because the Kemp elimination is accelerated by polar, aprotic solvents, we also sought to introduce more polarity into the cavity itself by making the further substitutions Val → 103Asn, Leu118 → Gln, Leu121 → Gln, which were expected to contribute via the carbonyl groups they introduce to site, increasing polarity, or in the case of Gln118, by forming a stabilizing hydrogen bond with the developing charge on the transition state oxygen.
As observed on the introduction of the original Leu99 → Ala cavity (-5.2 kcal/mol, ΔTm = 14.4 °C), and of the L99A/M102H† (-3.9 kcal/mol, SI Appendix, Table S1), all of these substitutions reduced the stability of the protein (Fig. 3 and SI Appendix, Table S1). Indeed, Val103 → Asn and Leu121 → Gln were destabilizing enough to result in insoluble protein. However, those substitutions that were not fatal all increased Kemp eliminase activity. The space-making Met106 → Ala mutation, which allows the catalytic His102 to hydrogen bond with bulk solvent, increased the activity by 1.8-fold, at a cost of a slight 0.1 kcal/mol loss in stability. More effective was positioning an efficient hydrogen bond acceptor near His102 by the Met106 → Asp mutation, which increased activity 2.1-fold at a cost of 0.5 kcal/mol of protein stability. The largest activity increase came from the addition of cavity polarity by the Leu118 → Gln substitution, which increased the activity by 3.3-fold at a cost of 0.7 kcal/mol stability.
To enable further enzyme optimization, we introduced a second round of stabilizing substitutions. We combined the new activating substitutions with the known stabilizing substitutions Glu11 → Phe and Asp20 → Asn (24); both replace key catalytic groups in the native lysozyme resulting in an enzyme where the native lysozyme activity as a muramidase has been completely extinguished. Conversely, these substitutions increase protein stability by 1.7 and 1.4 kcal/mol, respectively, consistent with the stability-function trade-off hypothesis (28–31). In addition to the six stabilizing substitutions in L99A/M102H†, this construct also contained the substitutions Glu11 → Phe, Asp20 → Asn, Met106 → Asp, Leu118 → Gln, which will be referred to as L99A/M102H‡. This construct could be expressed and purified, and had a Tm of 57.3 °C (versus 52.8 °C for L99A); its activity as a Kemp eliminase was increased to a kcat/KM of 1.8 M-1 s-1, a four-fold increase over that of L99A/M102H† and more than 100-fold more active than the original L99A cavity (Fig. 3 and Table 1).
Discussion
Two observations from this study merit emphasis. First, a simple, fully enclosed cavity site was developed that enabled straightforward optimization of simple catalysis, that of the Kemp elimination, and detailed analysis of mechanism. These cavities, though in many ways more primitive than earlier sites developed for this activity, are among those least dependent on the repurposing of a pre-existing catalytic site. Whereas the activities of these designed enzymes were far below those of natural enzymes, their proficiency was substantial and similar to other designed Kemp eliminases, at least at pH 5.0 where the reaction is dominated by enzyme catalysis (SI Appendix, Table S3). The tractability of this site made determination of its structure possible, while its simplicity facilitated analysis of the factors determining substrate recognition and catalysis. We suspect that these and related cavities (3, 6) may be further optimized and will lend themselves to the development and design of still other activities. Second, as activity increased in the cavity enzymes, their stability fell. This was as anticipated by the stability-function trade-off hypothesis (28–30, 32), which is well-exemplified by the strain-inducing substitutions introduced here to gain activity.
A longstanding justification for enzyme design is that success demonstrates understanding of mechanism (11, 17, 33, 34). As impressive as recent design efforts have been, the resulting enzymes have retained much of the complexity of the templates on which they were built, and have resisted full analysis of how structure encodes function (35). By comparison, in these cavities, the origins of catalysis seem simpler and the specific groups and geometries that contribute to it are readily grasped. The role of His102 as a base is born out by the X-ray structures of the complexes, where the residue interacts with the electrophilic center of a close analog of the substrate, or with the corresponding groups on the product (Fig. 2). The role of Met106 as an organizing group for His102 is also clear from the five X-ray structures in which the two residues hydrogen bond (e.g., Fig. 2 B and F). It is also supported by increases to activity from substitutions that increase the preference of His102 to adopt this catalytically competent orientation, such as Met106 → Asp, presumably by direct interaction, or Met106 → Ala, by solvent interaction. Similarly, the Kemp reaction is strongly promoted in polar, aprotic media, and so the introduction of a glutamine into the site, via the Leu118 → Gln substitution, increased activity 3.3-fold; this Gln would be positioned to hydrogen bond with the oxygen of the Kemp isoxazole, stabilizing the negative charge that develops during the reaction.
If the specifics of substrate recognition seem clear in this rudimentary cavity, so too is the general constraint of protein stability on enzyme activity optimization. The stability-function hypothesis (28–30) suggests that the preorganization necessary for substrate recognition and catalysis comes at a cost to the intrinsic stability of the protein. Maximizing activity demands preorganized sites into which a substrate fits, where charged and polar groups are at once sequestered from bulk solvent and positioned to favorably interact with a substrate (36, 37), and often unfavorably with one another, and where hydrophobic patches are exposed to ligand binding. Such preorganization for recognition and catalysis comes at the cost of protein stability, and engineering a new site, or optimizing an existing one by design or by in vitro (38, 39) or natural evolution (40, 32) will often destabilize the overall protein. The cavity enzymes illuminate this trade-off with unusual clarity. The very creation of the cavity site in the L99A mutant disrupts packing in the heart of the hydrophobic core of the protein, reducing protein stability by 5.2 kcal/mol (24). Introducing a histidine into this buried, apolar site further destabilizes the protein, here by 3.9 kcal/mol (SI Appendix, Table S1). Every case where a substitution increased activity it also reduced stability, often requiring restabilization of the protein at distal sites. These distal, restabilizing substitutions in turn illustrate the other side of the stability-function trade-off, as most of these were engineered into the natural, muramylpeptide catalytic site of lysozyme. Thus, the Thr21 → Cys/Thr142 → Cys disulfide, whose formation increases stability by 2.8 kcal/mol (24), joins the N-terminal and C-terminal lobes of lysozyme, preventing domain movement that is linked to progression along the reaction coordinate, deactivating the enzyme. Similarly, Ser117 → Val disrupts recognition of the stem-peptide of the cell-wall substrate of native lysozyme, reducing activity 10-fold (29). Because the substituted valine better packs with the rest of the enzyme, and is more hydrophobic, stability is increased by 2 kcal/mol (29). In a final round of stabilizing substitutions, Glu11 → Phe and Asp20 → Asn substituted the key catalytic residues of the native lysozyme function, resulting in a protein that has lost cell-wall hydrolytic activity entirely but is stabilized by 1.7 and 1.4 kcal/mol, respectively (24). In short, engineering an entirely new active site into lysozyme (L99A/M102H‡) came at a cost of 10.3 kcal/mol in stability, a price reimbursed by stabilizing substitutions that largely eliminated the native catalytic site (Fig. 4). Such a strategy of eliminating an original catalytic site to create a wholly new one may be broadly useful, or even necessary, in the field.
Certain cautions should not escape the reader’s attention. The activity of the best of these cavity enzymes remains modest: 2.5 orders of magnitude worse than the best computationally designed enzymes, and 4.5 orders of magnitude worse than those designs that were further optimized by in vitro evolution, certainly at neutral pH (SI Appendix, Table S3). We suspect that deployment of true design algorithms, rather than the chemical intuition used here, may better optimize this and related sites, likely with less stability insult to the protein than we ourselves incurred. Also, whereas the stability-function trade-off is well-illustrated in this cavity, we do not claim that it is a fully necessary constraint; rather, it is a common restraint on enzyme activity and evolution. Finally, whereas we suspect that this and related cavities can be used to support still other activities, the cavity’s steric constraints, even allowing some opening to solvent, will eventually limit the size of the substrates that it can accommodate and how evolvable the site may ever be.
These caveats should not obscure the key observations from this paper: into a simple model cavity rudimentary catalytic activity may be engineered, and in such a site the bases of activity, and its constraints, are laid bare. Undoubtedly, the radical simplicity of such a cavity will limit its optimization and the sort of activities that it can support. This same simplicity and restriction, however, enables one to dissect the contributions to catalysis in a way that few sites allow and can illuminate general principles without the complications that typically entangle more involved sites. As proteinaceous sites, these cavities reflect the environments and functional repertoires of natural enzymes—they are soluble in aqueous buffer, unlike many synthetic host-guest systems, and they use the same amino acids and access the same substitutions. Like synthetic host-guest systems, they eliminate many of the complicating features of natural or even designed sites, allowing one to isolate key contributions to recognition and activity. They may be pragmatic templates for detailed hypothesis testing in the field.
Materials and Methods
The cloning, expression, and purification of the model enzymes and their kinetic, thermodynamic, and structural analysis, are described in the SI Appendix. Briefly, T4 lysozyme L99A was cloned into pET-28 and mutations were introduced by overlap extension. The protein was expressed in E. coli and purified by Ni-NTA chromatography. Measurement of Kemp eliminase activity was performed as described (22) using a constant ionic strength buffer that contained imidazole to estimate its effect on the spontaneous reaction (41) (SI Appendix, Fig. S3). The thermodynamic stability of the proteins was monitored using CD spectra (6). Crystals were grown by hanging drop, reflections were measured at ALS beamline 8.3.1 and the structure determined by standard methods (SI Appendix, Table S2) and have been deposited as PDB IDs 4E97, 4EKP, 4EKQ, 4EKR, and 4EKS.
Supplementary Material
ACKNOWLEDGMENTS.
Supported by NIH grant GM63815. We thank Drs. C. Laggner, O. Eidam, M. Korczynska, and J. Irwin for reading this manuscript, and O. Eidam for assistance with crystallography.
Footnotes
References
- 1.Chang CE, Gilson MK. Free energy, entropy, and induced fit in host-guest recognition: Calculations with the second-generation mining minima algorithm. J Am Chem Soc. 2004;126:13156–13164. doi: 10.1021/ja047115d. [DOI] [PubMed] [Google Scholar]
- 2.Cram DJ, Cram JM. Cyclophane chemistry—Bent and battered benzene rings. Acc Chem Res. 1971;4:204–213. [Google Scholar]
- 3.Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355:371–373. doi: 10.1038/355371a0. [DOI] [PubMed] [Google Scholar]
- 4.Wei BQQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 5.Cellitti J, et al. Exploring subdomain cooperativity in T4 lysozyme I: Structural and energetic studies of a circular permutant and protein fragment. Protein Sci. 2007;16:842–851. doi: 10.1110/ps.062628607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu LJ, Baase WA, Michael MM, Matthews BW. Use of stabilizing mutations to engineer a charged group within a ligand-binding hydrophobic cavity in T4 lysozyme. Biochemistry. 2009;48:8842–8851. doi: 10.1021/bi900685j. [DOI] [PubMed] [Google Scholar]
- 7.Alexandrova AN, Rothlisberger D, Baker D, Jorgensen WL. Catalytic mechanism and performance of computationally designed enzymes for Kemp elimination. J Am Chem Soc. 2008;130:15907–15915. doi: 10.1021/ja804040s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvizo O, Allen BD, Mayo SL. Computational protein design promises to revolutionize protein engineering. Biotechniques. 2007;42:31–39. doi: 10.2144/000112336. [DOI] [PubMed] [Google Scholar]
- 9.Acevedo O, Jorgensen WL. Advances in quantum and molecular mechanical (QM/MM) simulations for organic and enzymatic reactions. Acc Chem Res. 2010;43:142–151. doi: 10.1021/ar900171c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brustad EM, Arnold FH. Optimizing non-natural protein function with directed evolution. Curr Opin Chem Biol. 2011;15:201–210. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frushicheva MP, Cao J, Chu ZT, Warshel A. Exploring challenges in rational enzyme design by simulating the catalysis in artificial kemp eliminase. Proc Natl Acad Sci USA. 2010;107:16869–16874. doi: 10.1073/pnas.1010381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frushicheva MP, Cao J, Warshel A. Challenges and advances in validating enzyme design proposals: The case of Kemp eliminase catalysis. Biochemistry. 2011;50:3849–3858. doi: 10.1021/bi200063a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baker D. An exciting but challenging road ahead for computational enzyme design. Protein Sci. 2010;19:1817–1819. doi: 10.1002/pro.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khersonsky O, et al. Evolutionary optimization of computationally designed enzymes: Kemp eliminases of the KE07 series. J Mol Biol. 2010;396:1025–1042. doi: 10.1016/j.jmb.2009.12.031. [DOI] [PubMed] [Google Scholar]
- 15.Lassila JK, Baker D, Herschlag D. Origins of catalysis by computationally designed retroaldolase enzymes. Proc Natl Acad Sci USA. 2010;107:4937–4942. doi: 10.1073/pnas.0913638107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorn SN, Daniels RG, Auditor MT, Hilvert D. Large rate accelerations in antibody catalysis by strategic use of haptenic charge. Nature. 1995;373:228–230. doi: 10.1038/373228a0. [DOI] [PubMed] [Google Scholar]
- 17.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–194. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 18.Korendovych IV, et al. Design of a switchable eliminase. Proc Natl Acad Sci USA. 2011;108:6823–6827. doi: 10.1073/pnas.1018191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debler EW, et al. Structural origins of efficient proton abstraction from carbon by a catalytic antibody. Proc Natl Acad Sci USA. 2005;102:4984–4989. doi: 10.1073/pnas.0409207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Privett HK, et al. Iterative approach to computational enzyme design. Proc Natl Acad Sci USA. 2012;109:3790–3795. doi: 10.1073/pnas.1118082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khersonsky O, et al. Optimization of the in-silico-designed Kemp eliminase KE70 by computational design and directed evolution. J Mol Biol. 2011;407:391–412. doi: 10.1016/j.jmb.2011.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casey ML, Kemp DS, Paul KG, Cox DD. Physical organic-chemistry of benzisoxazoles. 1. Mechanism of base-catalyzed decomposition of benzisoxazoles. J Org Chem. 1973;38:2294–2301. [Google Scholar]
- 23.Hollfelder F, Kirby AJ, Tawfik DS. Off-the-shelf proteins that rival tailor-made antibodies as catalysts. Nature. 1996;383:60–63. doi: 10.1038/383060a0. [DOI] [PubMed] [Google Scholar]
- 24.Baase WA, Liu LJ, Tronrud DE, Matthews BW. Lessons from the lysozyme of phage T4. Protein Sci. 2010;19:631–641. doi: 10.1002/pro.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno BE. Large shifts in pKa values of lysine residues buried inside a protein. Proc Natl Acad Sci USA. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dundas J, et al. CASTp: Computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beadle BM, Trehan I, Focia PJ, Shoichet BK. Structural milestones in the reaction pathway of an amide hydrolase: Substrate, acyl, and product complexes of cephalothin with AmpC beta-lactamase. Structure. 2002;10:413–424. doi: 10.1016/s0969-2126(02)00725-6. [DOI] [PubMed] [Google Scholar]
- 28.Beadle BM, Shoichet BK. Structural bases of stability-function tradeoffs in enzymes. J Mol Biol. 2002;321:285–296. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 29.Shoichet BK, Baase WA, Kuroki R, Matthews BW. A relationship between protein stability and protein function. Proc Natl Acad Sci USA. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber C, Buckle AM, Fersht AR. Stability and function—2 constraints in the evolution of barstar and other proteins. Structure. 1994;2:945–951. doi: 10.1016/s0969-2126(94)00096-4. [DOI] [PubMed] [Google Scholar]
- 31.Tokuriki N, Stricher F, Serrano L, Tawfik DS. How protein stability and new functions trade off. PLoS Comput Biol. 2008;4:e1000002. doi: 10.1371/journal.pcbi.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang XJ, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 33.Alexandrova AN, Jorgensen WL. Origin of the activity drop with the E50Dvariant of catalytic antibody 34E4 for Kemp elimination. J Phys Chem B. 2009;113:497–504. doi: 10.1021/jp8076084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zanghellini A, et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 2006;15:2785–2794. doi: 10.1110/ps.062353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khersonsky O, et al. Bridging the gaps in design methodologies by evolutionary optimization of the stability and proficiency of designed Kemp eliminase KE59. Proc Natl Acad Sci USA. 2012;109:10358–10363. doi: 10.1073/pnas.1121063109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Warshel A, Sussman F, Hwang JK. Evaluation of catalytic free-energies in genetically modified proteins. J Mol Biol. 1988;201:139–159. doi: 10.1016/0022-2836(88)90445-7. [DOI] [PubMed] [Google Scholar]
- 37.Warshel A. Energetics of enzyme catalysis. Proc Natl Acad Sci USA. 1978;75:5250–5254. doi: 10.1073/pnas.75.11.5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr Opin Struc Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 39.Levin KB, et al. Following evolutionary paths to protein–protein interactions with high affinity and selectivity. Nat Struct Mol Biol. 2009;16:1049–1067. doi: 10.1038/nsmb.1670. [DOI] [PubMed] [Google Scholar]
- 40.Thomas VL, McReynolds AC, Shoichet BK. Structural bases for stability-function tradeoffs in antibiotic resistance. J Mol Biol. 2010;396:47–59. doi: 10.1016/j.jmb.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellis KJ, Morrison JF. Buffers of constant ionic-strength for studying pH-dependent processes. Method Enzymol. 1982;87:405–426. doi: 10.1016/s0076-6879(82)87025-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.