

Abstract
Water oxidation by cyanobacteria, algae, and plants is pivotal in oxygenic photosynthesis, the process that powers life on Earth, and is the paradigm for engineering solar fuel–production systems. Each complete reaction cycle of photosynthetic water oxidation requires the removal of four electrons and four protons from the catalytic site, a manganese–calcium complex and its protein environment in photosystem II. In time-resolved photothermal beam deflection experiments, we monitored apparent volume changes of the photosystem II protein associated with charge creation by light-induced electron transfer (contraction) and charge-compensating proton relocation (expansion). Two previously invisible proton removal steps were detected, thereby filling two gaps in the basic reaction-cycle model of photosynthetic water oxidation. In the S2 → S3 transition of the classical S-state cycle, an intermediate is formed by deprotonation clearly before electron transfer to the oxidant (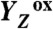 ). The rate-determining elementary step (τ, approximately 30 µs at 20 °C) in the long-distance proton relocation toward the protein–water interface is characterized by a high activation energy (Ea = 0.46 ± 0.05 eV) and strong H/D kinetic isotope effect (approximately 6). The characteristics of a proton transfer step during the S0 → S1 transition are similar (τ, approximately 100 µs; Ea = 0.34 ± 0.08 eV; kinetic isotope effect, approximately 3); however, the proton removal from the Mn complex proceeds after electron transfer to
). The rate-determining elementary step (τ, approximately 30 µs at 20 °C) in the long-distance proton relocation toward the protein–water interface is characterized by a high activation energy (Ea = 0.46 ± 0.05 eV) and strong H/D kinetic isotope effect (approximately 6). The characteristics of a proton transfer step during the S0 → S1 transition are similar (τ, approximately 100 µs; Ea = 0.34 ± 0.08 eV; kinetic isotope effect, approximately 3); however, the proton removal from the Mn complex proceeds after electron transfer to 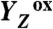 . By discovery of the transient formation of two further intermediate states in the reaction cycle of photosynthetic water oxidation, a temporal sequence of strictly alternating removal of electrons and protons from the catalytic site is established.
. By discovery of the transient formation of two further intermediate states in the reaction cycle of photosynthetic water oxidation, a temporal sequence of strictly alternating removal of electrons and protons from the catalytic site is established.
Keywords: oxygen evolution, manganese complex, photothermal spectroscopy, proton-coupled electron transfer, water splitting
In oxygenic photosynthesis, plants, algae, and cyanobacteria oxidize water at the manganese–calcium (Mn4Ca) complex of photosystem II (PSII) (1–3). This process has shaped the atmosphere by massive O2 formation (from water) and the biosphere by facilitating the large-scale production of primary biomass and energy-rich carbohydrates (4), but is still insufficiently understood. Improved insight into photosynthetic water oxidation could promote the development of biomimetic systems for direct production of solar fuels (3, 5–9).
In PSII, the absorption of a light quantum results in oxidation of a specific tyrosine (10), YZ [redox-active tyrosine residue (Tyr161) in the D1 subunit of PSII], which functions as the oxidant in the redox chemistry of water oxidation (9, 11, 12):
| [1] |
The actual catalyst facilitating the reaction described by Eq. 1 is the Mn4Ca complex bound to the proteins of PSII (13–15) (Fig. 1A). Four electrons are removed sequentially from the Mn complex [that is, the Mn4Ca(μ–O)n core and its ligand environment] by electron transfer to  , resulting in accumulation of four oxidizing equivalents before the onset of O─O bond formation and O2 liberation, as described by Kok’s classical S-state cycle (16, 17) (Fig. 1B, inner circle of S-states). Four protons are removed by deprotonation of the Mn complex and relocation toward the aqueous phase of the thylakoid lumen (18–20).
, resulting in accumulation of four oxidizing equivalents before the onset of O─O bond formation and O2 liberation, as described by Kok’s classical S-state cycle (16, 17) (Fig. 1B, inner circle of S-states). Four protons are removed by deprotonation of the Mn complex and relocation toward the aqueous phase of the thylakoid lumen (18–20).
Fig. 1.

Photosystem II (A) and reaction cycle of water oxidation (B). In A, crucial redox cofactors and dimensions of the PSII complex are shown (15). Red arrows connect redox cofactors of the ET chain, including the primary electron donor (P680), the primary pheophytin acceptor (Phe), the primary (QA) and secondary (QB) quinone acceptors, and, at the electron donor side, a redox-active tyrosine (YZ) and the Mn complex. Water molecules resolved in the crystallographic model (Protein Data Bank entry 3ARC; ref. 15) are shown as red dots; the indicated distances illustrate relevant dimensions. In B, the classical Kok model (16) (inner circle, including states S4 and S4′; ref. 22) is extended to describe both oxidation of the Mn complex by ET to the YZ radical and proton removal from the Mn complex or its ligand environment by long-distance proton transfer. Coupling of the ET step to local proton shifts is not covered by the shown framework model. The subscripts indicate the number of oxidation equivalents accumulated at the Mn complex; the superscripts indicate the charge relative to the dark-stable S1-state (+, positive; n, neutral). The proton release steps in the S0 → S1 and S2 → S3 transitions have not been tracked in time-resolved experiments before, but now these steps are detected in the PBD experiments; the indicated time constants result from the present study.
The location of the Mn complex at the interface between the membrane-intrinsic part of PSII and the extrinsic lumenal proteins (13–15) (Fig. 1A) implies long-distance proton relocation toward the aqueous phase (approximately 30 Å), occurring within tens or hundreds of microseconds along chains of water molecules and ionic residues (21). The interrelation between electron transfer and protonation dynamics (that is, the relocation of protons on various time and length scales) is functionally crucial (1, 22–25). Our study aims to identify the basic sequence of electron transfer (ET) and long-distance proton relocation in the water oxidation cycle.
The electron transfer from the Mn complex to 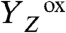 (22, 26–29) and the proton release (i.e., the appearance of protons in the aqueous phase) (18–20), have been investigated extensively. It was found (inter alia) that the observable proton release often does not reflect the removal of a proton from the Mn complex because electrostatically induced deprotonation of residues at the lumenal periphery of the protein masks the protein-intrinsic proton removal (20, 30). Nonetheless, it was possible to determine the “intrinsic proton release pattern” (i.e., the number of protons removed in each of the classical S-state transitions from the Mn complex), which is: 1 H+ in S0 → S1, 0 H+ in S1 → S2, 1 H+ in S2 → S3, and 2 H+ in S3 → S0 (18, 19, 31). The appearance of protons in the aqueous bulk phase rapidly after
(22, 26–29) and the proton release (i.e., the appearance of protons in the aqueous phase) (18–20), have been investigated extensively. It was found (inter alia) that the observable proton release often does not reflect the removal of a proton from the Mn complex because electrostatically induced deprotonation of residues at the lumenal periphery of the protein masks the protein-intrinsic proton removal (20, 30). Nonetheless, it was possible to determine the “intrinsic proton release pattern” (i.e., the number of protons removed in each of the classical S-state transitions from the Mn complex), which is: 1 H+ in S0 → S1, 0 H+ in S1 → S2, 1 H+ in S2 → S3, and 2 H+ in S3 → S0 (18, 19, 31). The appearance of protons in the aqueous bulk phase rapidly after 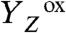 formation and prior to its reduction by ET from the Mn complex has been established firmly (19, 20, 30), but likely does not reflect the deprotonation of a chemical group (e.g., a substrate water molecule) at the Mn complex (18, 19, 31). This implies that the time-resolved detection of proton release into the aqueous phase cannot be employed to decide whether the proton removal from the Mn complex precedes the ET to
formation and prior to its reduction by ET from the Mn complex has been established firmly (19, 20, 30), but likely does not reflect the deprotonation of a chemical group (e.g., a substrate water molecule) at the Mn complex (18, 19, 31). This implies that the time-resolved detection of proton release into the aqueous phase cannot be employed to decide whether the proton removal from the Mn complex precedes the ET to 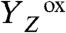 .
.
The temporal sequence of electron and proton removal steps after rapid  formation therefore has remained largely obscure, with one notable exception: Today there is strong experimental support that, after formation of the S3
formation therefore has remained largely obscure, with one notable exception: Today there is strong experimental support that, after formation of the S3
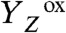 state in the S3 → S0 transition, a proton is removed from the Mn complex before onset of the electron transfer to
state in the S3 → S0 transition, a proton is removed from the Mn complex before onset of the electron transfer to 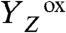 (22, 26, 32). This finding has led to an extension of the S-state cycle involving formation of a distinct S4 state by deprotonation (inner circle of Fig. 1B) before S4 formation by electron transfer (22, 17). Later, this reaction cycle was extended further to include each of the four protons (24, 33) (outer circle of Fig. 1B). However, the proposed sequence of events has remained hypothetical, in particular because the proton removal from the Mn complex in the
(22, 26, 32). This finding has led to an extension of the S-state cycle involving formation of a distinct S4 state by deprotonation (inner circle of Fig. 1B) before S4 formation by electron transfer (22, 17). Later, this reaction cycle was extended further to include each of the four protons (24, 33) (outer circle of Fig. 1B). However, the proposed sequence of events has remained hypothetical, in particular because the proton removal from the Mn complex in the  and
and  transitions could not be tracked in time-resolved experiments.
transitions could not be tracked in time-resolved experiments.
To detect proton removal from the Mn complex, we employ a photothermal beam deflection (PBD) experiment exploiting the high sensitivity of the PBD signal to density changes (34–38), which in the following are discussed in terms of apparent volume changes of the protein. Expansion and contraction of the PSII complex were monitored with microsecond resolution at a precision of about 2 Å3 per PSII. For comparison, the average volume of one water molecule in aqueous solution is approximately 30 Å3. In photoacoustic or photothermal measurements, it is usually found that a volume contraction results from the (light-induced) deposition of charges at the electron donor and/or acceptor, as has been shown for synthetic molecules (39, 40) and photosystems (41, 42). Also in PSII, the light-induced formation of the primary quinone acceptor (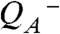 ) and of
) and of 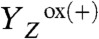 is associated with a volume contraction (37, 43). The decharging of the donor side by removal of a proton results in an expansion that reverts the
is associated with a volume contraction (37, 43). The decharging of the donor side by removal of a proton results in an expansion that reverts the 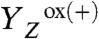 contribution to the preceding contraction. This enables monitoring of proton removal from the Mn complex by measuring the concomitant volume expansion in the PBD experiment, thereby revealing the temporal sequence of electron and proton removal steps in the classical S0 → S1 and S2 → S3 transitions.
contribution to the preceding contraction. This enables monitoring of proton removal from the Mn complex by measuring the concomitant volume expansion in the PBD experiment, thereby revealing the temporal sequence of electron and proton removal steps in the classical S0 → S1 and S2 → S3 transitions.
Results
For insight into the individual S-state transitions, PBD measurements were combined with the following laser-flash protocol (Fig. S1): Dark-adapted PSII membrane particles were excited by a sequence of n saturating ns-laser flashes (532 nm, 10 mJ cm-2). Each flash populated predominantly a specific stable/semistable S-state of Kok’s classical reaction cycle (16, 17), namely S1 (n = 0, dark-adapted PSII), S2 (n = 1), S3 (n = 2), S0 (n = 3), and again S1 (n = 4). Subsequently, a single subsaturating ns-flash (0.1 mJ cm-2) was applied and the PBD signal induced thereby was recorded. The subsaturating flash initiated predominantly the following transitions: S1 → S2 (flash 1), S2 → S3 (flash 2), S3 → S0 (flash 3), and S0 → S1 (flash 4). The measured PBD signals were corrected for imperfect advancement in the S-state cycle (S-state mixing) using previously established procedures (26, 44) (Fig. S2). We note that all central conclusions of this work are independent of the details of the correction procedure. Corrected PBD transients for each of the four transitions between semistable S-states are shown in Fig. 2 and discussed below.
Fig. 2.

Flash-induced PBD signals and volume changes: S1 → S2 (A, A′), S2 → S3 (B, B′), 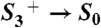 (C, C′), and S0 → S1 (D, D′). Thin lines, experimental data; thick lines, simulations using a step-shaped function for the rapid jump caused by
(C, C′), and S0 → S1 (D, D′). Thin lines, experimental data; thick lines, simulations using a step-shaped function for the rapid jump caused by 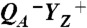 formation and single-exponential functions for the slower signal contributions. (Right) Schematic illustration of volume changes deduced from the analysis of the temperature dependence of the PBD signals (time constants for about 20 °C; see Fig. 3).
formation and single-exponential functions for the slower signal contributions. (Right) Schematic illustration of volume changes deduced from the analysis of the temperature dependence of the PBD signals (time constants for about 20 °C; see Fig. 3).
The instantaneous rise observed after each flash (at 25 °C) is attributable to the rapid light-induced processes that result in reduction of QA and oxidation of the tyrosine donor (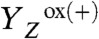 formation) (12). At 12 °C, the magnitude of the instantaneous rise was smaller than at 25 °C, and at 1 °C a decay was observed (Fig. 2B). This behavior results from the temperature-dependent thermal (ΔQ) and temperature-independent nonthermal (ΔV) contributions to the PBD signal (34–36) associated with the
formation) (12). At 12 °C, the magnitude of the instantaneous rise was smaller than at 25 °C, and at 1 °C a decay was observed (Fig. 2B). This behavior results from the temperature-dependent thermal (ΔQ) and temperature-independent nonthermal (ΔV) contributions to the PBD signal (34–36) associated with the 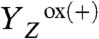
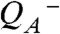 formation. Evaluation of the temperature dependence of the rapid phase in comparison to a calorimetric standard (Figs. S3–S5) yielded an apparent volume contraction (ΔV) by about -12 Å3, in reasonable agreement with previous estimates (37, 43). For a possible contribution to the PBD signals associated with interquinone electron transfer, see Figs. S6 and S7 and Table S1. [We note that no ΔV values presented herein were corrected for the effective quantum yield, Φeff, of the light-induced transition, which could be as low as 50% (SI Text). This means that the volume changes per PSII complex may be twice as large as the values documented herein.]
formation. Evaluation of the temperature dependence of the rapid phase in comparison to a calorimetric standard (Figs. S3–S5) yielded an apparent volume contraction (ΔV) by about -12 Å3, in reasonable agreement with previous estimates (37, 43). For a possible contribution to the PBD signals associated with interquinone electron transfer, see Figs. S6 and S7 and Table S1. [We note that no ΔV values presented herein were corrected for the effective quantum yield, Φeff, of the light-induced transition, which could be as low as 50% (SI Text). This means that the volume changes per PSII complex may be twice as large as the values documented herein.]
In the S1 → S2 transition (flash 1), the instantaneous 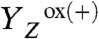
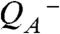 signal (< 10 μs) was followed by an exponential decay with a time constant of about 100 μs at 20 °C (Figs. 2 and 3, and Table 1). The amplitude of the decay phase depended only weakly on the temperature, suggesting that it mostly originates from a volume change (ΔV) of the PSII sample; its negative amplitude (PBD-signal decrease) indicates a contraction. The weak temperature dependence of the signal magnitude suggests a small contribution to the signal from a positive ΔQ (heat release) (Table 1). The decay became only slightly slower at lower temperatures, and the Arrhenius plot of the rate constants (Fig. 3B) revealed a small activation energy (Table 2). From PBD transients measured in D2O (Fig. 4), a minor H/D kinetic isotope effect (KIE) of 1.3 was determined. (The KIE is the ratio of time constants determined in D2O or H2O; KIE = τD/τH = kH/kD.) Time constant, activation energy, and KIE agree well with figures previously determined for the ET from the Mn complex to
signal (< 10 μs) was followed by an exponential decay with a time constant of about 100 μs at 20 °C (Figs. 2 and 3, and Table 1). The amplitude of the decay phase depended only weakly on the temperature, suggesting that it mostly originates from a volume change (ΔV) of the PSII sample; its negative amplitude (PBD-signal decrease) indicates a contraction. The weak temperature dependence of the signal magnitude suggests a small contribution to the signal from a positive ΔQ (heat release) (Table 1). The decay became only slightly slower at lower temperatures, and the Arrhenius plot of the rate constants (Fig. 3B) revealed a small activation energy (Table 2). From PBD transients measured in D2O (Fig. 4), a minor H/D kinetic isotope effect (KIE) of 1.3 was determined. (The KIE is the ratio of time constants determined in D2O or H2O; KIE = τD/τH = kH/kD.) Time constant, activation energy, and KIE agree well with figures previously determined for the ET from the Mn complex to 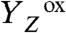 in the S1 → S2 transition (22, 26, 27) (Fig. 1B).
in the S1 → S2 transition (22, 26, 27) (Fig. 1B).
Fig. 3.

Temperature dependence of the PBD signals of the four resolved transitions. (A) Temperature dependence of the amplitudes as obtained by an exponential simulation (symbols, experimental data; lines obtained by a fit). The dotted lines show the 1σ error ranges of the fit curves. The bars represent the nonthermal part of the PBD signal (volume change ΔV) that corresponds to the PBD amplitude at -14 °C (T0 = -14 ± 1 °C; SI Text). (B) Arrhenius plots of the rate constants (k = τ-1, left y axis; time constants, τ, on right y axis). The symbols indicate the experimentally determined values; the lines are fit curves used for determination of the respective activation energy shown in Table 2.
Table 1.
Time constants (τ at 20 °C), volume changes (ΔV), and heat release (ΔQ) of four transitions resolved in PBD measurements
| Flash no. | Transition | τ (μs) | ΔV*, † (A3) | ΔQ † (meV) |
| 1 | 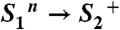 |
98 ± 3 | -6.1 ± 0.4 | 160 ± 50 |
| 2 | 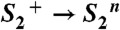 |
29 ± 2 | +4.4 ± 1.1 | 190 ± 120 |
| 3 | S3 → S0 | 1,960 ± 90 | +15 ± 2.5 | 60 ± 250 |
| 4 | 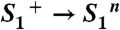 |
94 ± 8 | +3.5 ± 1.3 | 40 ± 140 |
The given parameters were determined from data shown in Fig. 3; error ranges correspond to uncertainty of the fit result at the 1σ level.
*The value of ΔV denotes apparent volume changes calculated using SI Text, Eq. S1. Negative or positive signs correspond to contractions or expansions, respectively.
†The figures given for ΔV and ΔQ were calculated for a single PSII complex after absorption of one light quantum (without correction for nonunity quantum yield).
Table 2.
Kinetic parameters of ET (e-) and rate-determining proton transfer (PT, H+) during the reaction cycle of PSII water oxidation at approximately 20 °C

Fig. 4.

Comparison of PBD signals on the four S-transitions measured for PSII membranes in H2O (black) and D2O (red) at 20 °C. Thin lines, experimental data; thick lines, simulations with single-exponential functions plus offset, except for the S0 → S1 transition, for which a double-exponential function plus offset was used (Fig. S7 and Table S2). The respective rate constant ratio is indicated (KIE = kH2O/kD2O). The PBD amplitudes differ because of different thermoelastic properties of H2O and D2O.
The large contraction associated with the ET from the Mn complex to 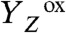 is specifically observed in the S1 → S2 transition. For S2 → S3, such a contraction paralleling the ET step was not detectable (Fig. 2B and Fig. S7). In the S0 → S1 transition, a small contraction might be coupled to the ET step (Fig. S4). We note that the contraction associated with the ET in the S1 → S2 transition, which likely is reversed in the S3 → S0 transition (see below), could reflect an interesting new mode of coupling the ET step to nuclear rearrangements, possibly related to changes in the protein backbone conformation suggested by FTIR data (45).
is specifically observed in the S1 → S2 transition. For S2 → S3, such a contraction paralleling the ET step was not detectable (Fig. 2B and Fig. S7). In the S0 → S1 transition, a small contraction might be coupled to the ET step (Fig. S4). We note that the contraction associated with the ET in the S1 → S2 transition, which likely is reversed in the S3 → S0 transition (see below), could reflect an interesting new mode of coupling the ET step to nuclear rearrangements, possibly related to changes in the protein backbone conformation suggested by FTIR data (45).
In the S2 → S3 transition (flash 2), the instantaneous 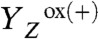
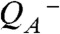 rise was followed by an exponentially rising phase with a time constant of about 30 μs (at 20 °C; Figs. 2 and 3). This rise was roughly 10 times faster than the ET from the Mn complex to
rise was followed by an exponentially rising phase with a time constant of about 30 μs (at 20 °C; Figs. 2 and 3). This rise was roughly 10 times faster than the ET from the Mn complex to 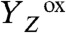 (30 μs versus 300 μs; Table 2) and is thus assignable to a process that precedes the ET step. Its almost temperature-independent signal amplitude (Fig. 3A) indicates that the rising phase originates mostly from a volume expansion (Table 1). A striking feature of the rapid rise was its large KIE of close to 6 (Fig. 4 and Table 2), facilitating the assignment to a process involving proton movements. We emphasize that at all temperatures, and also in D2O, the apparent volume expansion clearly preceded the
(30 μs versus 300 μs; Table 2) and is thus assignable to a process that precedes the ET step. Its almost temperature-independent signal amplitude (Fig. 3A) indicates that the rising phase originates mostly from a volume expansion (Table 1). A striking feature of the rapid rise was its large KIE of close to 6 (Fig. 4 and Table 2), facilitating the assignment to a process involving proton movements. We emphasize that at all temperatures, and also in D2O, the apparent volume expansion clearly preceded the 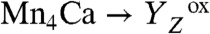 ET step (Table 2). Thus, we conclude that in the classical S2 → S3 transition, a proton relocation precedes the ET from the Mn complex to
ET step (Table 2). Thus, we conclude that in the classical S2 → S3 transition, a proton relocation precedes the ET from the Mn complex to  .
.
In the transition S3 → S0 + O2 (flash 3), a prominent millisecond rise (τ around 2 ms at 20 °C) that resulted mostly from a volume expansion by about 15 Å3 was visible (Figs. 2 and 3A, and Table 1). The moderate activation energy and small KIE of the millisecond phase (Figs. 3 and 4, and Table 2) were similar to the respective values for the ET step ( ) and the concomitant dioxygen formation (
) and the concomitant dioxygen formation ( ) (26, 27, 46, 47).
) (26, 27, 46, 47).
Using time-resolved X-ray spectroscopy and near-UV measurements to monitor the oxidation state of the Mn complex, it has been found that an apparent lag phase of an approximately 200-μs duration precedes the millisecond rise of O2 formation (22, 26, 27, 32). A similar lag phase was not discernable in the PBD transients. Instead, a rising phase with a similar τ value to the previously observed lag phase could be detected by simulation of summed PBD transients (Fig. S5). However, this phase was not sufficiently well-resolved for analysis of its temperature dependence and quantitative determination of ΔQ and ΔV. Conservatively, we conclude that the PBD data are compatible with a volume expansion associated with proton release in the  transition. Thus, we propose that the overall extent of the expansion in the S3 → S0 transition is explainable by three additive contributions—namely, proton removal from the Mn complex prior to the ET, reversal of the contractions associated with previous manganese oxidation (in the S1 → S2 transition), and removal of a second proton from the Mn complex after the ET (more quantitative considerations appear in SI Text).
transition. Thus, we propose that the overall extent of the expansion in the S3 → S0 transition is explainable by three additive contributions—namely, proton removal from the Mn complex prior to the ET, reversal of the contractions associated with previous manganese oxidation (in the S1 → S2 transition), and removal of a second proton from the Mn complex after the ET (more quantitative considerations appear in SI Text).
In the S0 → S1 transition (flash 4), the initial signal increase was followed by an exponentially rising phase with a time constant of approximately 100 μs (at 20 °C). The amplitude of this signal rise was almost temperature-independent, indicating a volume expansion (Fig. 3A), similar to the signal rise in the  transition. Also, further parameters of this phase in the S0 → S1 transition were similar—namely, its large activation energy, its large KIE (approximately 3), and the ΔV magnitude (Tables 1 and 2, and Table S2). The 100-μs rise in the PBD signal was slower than the ET from the Mn complex to
transition. Also, further parameters of this phase in the S0 → S1 transition were similar—namely, its large activation energy, its large KIE (approximately 3), and the ΔV magnitude (Tables 1 and 2, and Table S2). The 100-μs rise in the PBD signal was slower than the ET from the Mn complex to 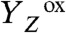 (approximately 40 μs in refs. 22 and 29) (Table 2), implying that the underlying process occurred after the ET step—that is, after the
(approximately 40 μs in refs. 22 and 29) (Table 2), implying that the underlying process occurred after the ET step—that is, after the  transition (Fig. 1B).
transition (Fig. 1B).
We note that vastly different values have been reported for the ET rate constant of the S0 → S1 transition, ranging from about 40 μs to 300 μs (at approximately 20 °C) (27–29, 32, 48). For the same PSII samples used herein, we have previously determined a value of approximately 40 μs (22), suggesting that the ET step in the S0 → S1 transition is faster than the proton removal step already at room temperature, and even more so at lower temperatures and in D2O (27, 28). In conclusion, the volume expansion observed in the classical S0 → S1 transition results from a proton relocation (the  transition in Fig. 1B) that takes place after manganese oxidation by ET to
transition in Fig. 1B) that takes place after manganese oxidation by ET to 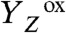 .
.
Discussion
In the course of the classical S2 → S3 transition, a volume expansion precedes the ET step and can be assigned to the removal of a proton from the Mn complex in the  transition of the extended S-state cycle shown in Fig. 1B. This is a central finding of our investigation because it directly supports the proton-first ET in the S2 → S3 transition and establishes a close analogy to the first steps (
transition of the extended S-state cycle shown in Fig. 1B. This is a central finding of our investigation because it directly supports the proton-first ET in the S2 → S3 transition and establishes a close analogy to the first steps (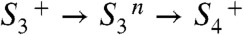 ) in the S3 → S0 + O2 transition (22). This finding implies that a previously undetected intermediate state (
) in the S3 → S0 + O2 transition (22). This finding implies that a previously undetected intermediate state (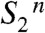 ) is transiently formed by deprotonation. Why this proton removal step is associated with a volume expansion is explained straightforwardly: The removal of a positive charge (H+) from the donor side of PSII reverts the preceding volume contraction caused by charging of the donor side by
) is transiently formed by deprotonation. Why this proton removal step is associated with a volume expansion is explained straightforwardly: The removal of a positive charge (H+) from the donor side of PSII reverts the preceding volume contraction caused by charging of the donor side by 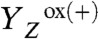 formation. Detection of this proton removal step (t1/2 of only approximately 20 μs at pH 6.2) by analysis of electrochromic absorption changes of the PSII pigments may be feasible, but has not been achieved yet (28, 29, 32), presumably because of technical limitations.
formation. Detection of this proton removal step (t1/2 of only approximately 20 μs at pH 6.2) by analysis of electrochromic absorption changes of the PSII pigments may be feasible, but has not been achieved yet (28, 29, 32), presumably because of technical limitations.
In the course of the classical S0 → S1 transition, a volume expansion occurs after the ET step and is assigned to proton removal from the Mn complex in the  transition. In analogy to the
transition. In analogy to the  transition, this expansion is explained by removal of a positive charge (H+) from the donor side of PSII, thereby reversing the contraction caused by
transition, this expansion is explained by removal of a positive charge (H+) from the donor side of PSII, thereby reversing the contraction caused by 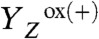 formation. As opposed to the
formation. As opposed to the  transition, the deprotonation in the course of the classical S0 → S1 transition proceeds after oxidation of the Mn complex (after
transition, the deprotonation in the course of the classical S0 → S1 transition proceeds after oxidation of the Mn complex (after  formation). This result directly supports the extended S-state cycle scheme (Fig. 1B) by revealing transient formation of the
formation). This result directly supports the extended S-state cycle scheme (Fig. 1B) by revealing transient formation of the 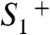 intermediate.
intermediate.
The kinetic parameters determined for proton removal from the Mn complex relate to the slowest step in the sequence of all elementary proton transfer steps of the long-distance relocation of a proton from the Mn complex toward the aqueous phase. Presently, the site and physicochemical nature of the rate-determining steps are unknown. We determined similarly large activation energies and KIE values for proton removal from the 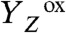
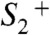 and
and 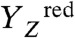
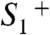 intermediates, pointing to the formation of a transition state of relatively high energy and proton tunneling across a sizeable energetic barrier; a more elaborate analysis of the thermodynamic and kinetic parameters may provide deeper insight (49, 50). Interestingly, activation energy and KIE for proton removal from the
intermediates, pointing to the formation of a transition state of relatively high energy and proton tunneling across a sizeable energetic barrier; a more elaborate analysis of the thermodynamic and kinetic parameters may provide deeper insight (49, 50). Interestingly, activation energy and KIE for proton removal from the 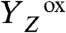
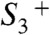 intermediate are smaller, suggesting that site and/or nature of the rate-determining proton transfer may differ. Identification of pathways and modes of proton transfer within PSII could be approached experimentally (e.g., by time-resolved infrared spectroscopy) (51, 52).
intermediate are smaller, suggesting that site and/or nature of the rate-determining proton transfer may differ. Identification of pathways and modes of proton transfer within PSII could be approached experimentally (e.g., by time-resolved infrared spectroscopy) (51, 52).
In extension of the classical S-state cycle model, we have suggested a basic reaction-cycle model (24, 33) that involves nine intermediate states and describes the temporal sequence of light-induced YZ oxidation, electron transfer to 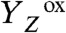 , proton removal from the Mn complex, and O2 formation (Fig. 1B). However, with respect to the proton removal steps, the supporting evidence remained largely circumstantial. Only the
, proton removal from the Mn complex, and O2 formation (Fig. 1B). However, with respect to the proton removal steps, the supporting evidence remained largely circumstantial. Only the  transition (or S3 → S4; ref. 22) had become detectable in time-resolved experiments (22, 26, 27, 32, 53, 54). Now, we have followed the charge-compensating proton removal in the classical S0 → S1 and S2 → S3 transitions and have determined the kinetic parameters of these essential reaction steps (see Table 2).
transition (or S3 → S4; ref. 22) had become detectable in time-resolved experiments (22, 26, 27, 32, 53, 54). Now, we have followed the charge-compensating proton removal in the classical S0 → S1 and S2 → S3 transitions and have determined the kinetic parameters of these essential reaction steps (see Table 2).
On these grounds and based on earlier investigations (reviewed in refs. 2 and 24), we propose the basic reactions sequence for the transitions from 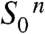 to the
to the 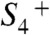 state outlined in the following (Figs. 1B and 5): (i) Starting in the most reduced semistable S-state,
state outlined in the following (Figs. 1B and 5): (i) Starting in the most reduced semistable S-state, 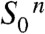 , absorption of a photon by PSII induces rapid YZ oxidation (< 1 μs) followed by ET from the Mn complex to
, absorption of a photon by PSII induces rapid YZ oxidation (< 1 μs) followed by ET from the Mn complex to  (40 μs in Fig. 1B), resulting in
(40 μs in Fig. 1B), resulting in 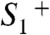 formation. Mn oxidation in the
formation. Mn oxidation in the  transition lowers the pK of a Mn ligand, possibly a bridging hydroxide (44, 55, 56), to a value around 3.3 (57). The proton is removed from the Mn complex and relocated toward the lumen only after
transition lowers the pK of a Mn ligand, possibly a bridging hydroxide (44, 55, 56), to a value around 3.3 (57). The proton is removed from the Mn complex and relocated toward the lumen only after 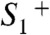 formation in the
formation in the  transition. (ii) The next absorbed photon induces
transition. (ii) The next absorbed photon induces  oxidation followed by a MnIII → MnIV oxidation in the
oxidation followed by a MnIII → MnIV oxidation in the  transition. The oxidation of the Mn complex lowers the pK values of ligand groups, but not to an extent sufficient for deprotonation. (iii) Oxidation of the Mn complex without any charge-compensating chemical change raises its redox potential to a level that prohibits a second oxidation by
transition. The oxidation of the Mn complex lowers the pK values of ligand groups, but not to an extent sufficient for deprotonation. (iii) Oxidation of the Mn complex without any charge-compensating chemical change raises its redox potential to a level that prohibits a second oxidation by 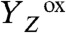 (1, 2, 7). In the
(1, 2, 7). In the 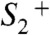 state, this redox-potential problem initially prevents oxidation of the Mn complex by
state, this redox-potential problem initially prevents oxidation of the Mn complex by 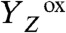 . However,
. However, 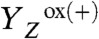 drives the removal of a proton from the Mn complex, resulting in formation of the
drives the removal of a proton from the Mn complex, resulting in formation of the 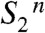 state within about 30 μs. We propose that the proton is removed from the cluster of water molecules indicated in Fig. 5. Thereby created is a proton vacancy that is effectively delocalized within the water cluster (on the μs time scale), but likely resides mostly on the water molecule close to
state within about 30 μs. We propose that the proton is removed from the cluster of water molecules indicated in Fig. 5. Thereby created is a proton vacancy that is effectively delocalized within the water cluster (on the μs time scale), but likely resides mostly on the water molecule close to 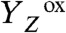 , which is coordinated to the Ca ion of the Mn complex. In the subsequent ET step (
, which is coordinated to the Ca ion of the Mn complex. In the subsequent ET step ( ), MnIII→IV oxidation of Mn1 (58–60) is directly coupled to proton transfer to the previously deprotonated water cluster. Such a concerted electron–proton transfer is in line with a comparably large H/D isotope effect for this ET step, and is well-suited to solve the redox-potential problem mentioned above. In the proton removal step preceding the ET, deprotonation of D1–Asp61 can be excluded because the S2 → S3 transition is not severely affected in the Asp–Asn mutant (61, 62). A central role of CP43–Arg357 (63) is unlikely because this residue is close to neither YZ nor Mn1. Thus, we consider deprotonation of the water cluster interconnecting YZ and the Mn complex to be the most plausible option, which is supported by the importance of the pK value of the water molecules coordinated to the Ca ion (64). (iv) In the
), MnIII→IV oxidation of Mn1 (58–60) is directly coupled to proton transfer to the previously deprotonated water cluster. Such a concerted electron–proton transfer is in line with a comparably large H/D isotope effect for this ET step, and is well-suited to solve the redox-potential problem mentioned above. In the proton removal step preceding the ET, deprotonation of D1–Asp61 can be excluded because the S2 → S3 transition is not severely affected in the Asp–Asn mutant (61, 62). A central role of CP43–Arg357 (63) is unlikely because this residue is close to neither YZ nor Mn1. Thus, we consider deprotonation of the water cluster interconnecting YZ and the Mn complex to be the most plausible option, which is supported by the importance of the pK value of the water molecules coordinated to the Ca ion (64). (iv) In the 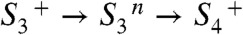 transitions, the basic temporal sequence of proton removal and electron transfer is similar to the one described above for the
transitions, the basic temporal sequence of proton removal and electron transfer is similar to the one described above for the 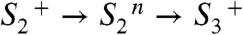 transitions. However, the key players are others. As opposed to the S2 → S3 transition, the proton removal step likely involves D1–Asp61 (62). Therefore, and in line with the clear differences in H/D isotope effect (KIE) and activation energy (Table 2), we propose that proton removal in the
transitions. However, the key players are others. As opposed to the S2 → S3 transition, the proton removal step likely involves D1–Asp61 (62). Therefore, and in line with the clear differences in H/D isotope effect (KIE) and activation energy (Table 2), we propose that proton removal in the  transition proceeds along a path that includes D1–Asp61 (14, 21), whereas proton removal in the
transition proceeds along a path that includes D1–Asp61 (14, 21), whereas proton removal in the  transition proceeds along another path starting close to D1–Tyr161/His190 (15, 65). In comparison to the ET of the
transition proceeds along another path starting close to D1–Tyr161/His190 (15, 65). In comparison to the ET of the  transition, the
transition, the  transition may be coupled to more extensive chemical changes suitable to initiate O─O bond formation. Gaining insight into the presently merely hypothetical
transition may be coupled to more extensive chemical changes suitable to initiate O─O bond formation. Gaining insight into the presently merely hypothetical  and
and 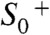 intermediates represents a central challenge in future research on photosynthetic water oxidation.
intermediates represents a central challenge in future research on photosynthetic water oxidation.
Fig. 5.

Sequence of events in the classical S2 → S3 transition of photosynthetic water oxidation. The Mn4CaO5 cluster, the redox-active tyrosine (Tyr161), and the key groups of the surrounding hydrogen-bonded network (15) are shown. All indicated amino acid residues are from the D1 subunit of PSII, with exception of CP43–Arg357. (Water molecules, HxO, are indicated as red spheres; putative H-bonds as broken lines that connect H-bond donor and acceptor. Of all the protons, only the phenolic proton is shown as a grey sphere.) The grey mesh outlines a water cluster that includes 4 HxO in the first coordination sphere of manganese (Mn4), as well as the calcium (Ca) and three second-sphere water molecules. Within less than 100 ns after absorption of a photon and oxidation of the primary chlorophyll donor of PSII (P680), Tyr161 (YZ) is oxidized by P680+ (“1st”). 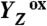 formation results in a rearrangement of the shown H-bonded network (completed within less than 1 µs), likely involving a shift of the phenolic proton to His190 and lowering of pK values for deprotonation of the water molecules in the outlined cluster (grey mesh). A proton is removed from the Mn complex/YZ environment within about 30 µs, as evidenced by the PBD results presented herein, and a proton vacancy is supposedly created within the outlined water cluster (“2nd”). In the ET to
formation results in a rearrangement of the shown H-bonded network (completed within less than 1 µs), likely involving a shift of the phenolic proton to His190 and lowering of pK values for deprotonation of the water molecules in the outlined cluster (grey mesh). A proton is removed from the Mn complex/YZ environment within about 30 µs, as evidenced by the PBD results presented herein, and a proton vacancy is supposedly created within the outlined water cluster (“2nd”). In the ET to 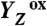 (about 300 µs), Mn oxidation is directly coupled to a proton transfer step involving the previously created proton vacancy of the water cluster (concerted electron–proton transfer) (“3rd”).
(about 300 µs), Mn oxidation is directly coupled to a proton transfer step involving the previously created proton vacancy of the water cluster (concerted electron–proton transfer) (“3rd”).
In conclusion, the basic sequence of events in the reaction cycle of water oxidation has now been established for the six transitions leading from 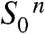 to
to 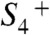 . This sequence is characterized by the strictly alternating removal of electrons and protons from the Mn complex. Three intermediate states of the catalytic metal center (
. This sequence is characterized by the strictly alternating removal of electrons and protons from the Mn complex. Three intermediate states of the catalytic metal center (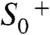 ,
, 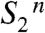 ,
, 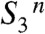 ) and its immediate ligand environment are transiently formed. On these grounds, strategies can be developed for characterization of the new reaction intermediates at the atomistic level. The extended S-state cycle model (Fig. 1B) may serve as a framework for the future design of experimental and theoretical investigations on photosynthetic water oxidation. It may also be worthwhile to scrutinize synthetic systems for catalysis of water oxidation within a conceptual framework that involves redox-potential leveling and local accumulation of four oxidizing equivalents by alternating electron and proton removal from the catalytic site.
) and its immediate ligand environment are transiently formed. On these grounds, strategies can be developed for characterization of the new reaction intermediates at the atomistic level. The extended S-state cycle model (Fig. 1B) may serve as a framework for the future design of experimental and theoretical investigations on photosynthetic water oxidation. It may also be worthwhile to scrutinize synthetic systems for catalysis of water oxidation within a conceptual framework that involves redox-potential leveling and local accumulation of four oxidizing equivalents by alternating electron and proton removal from the catalytic site.
Materials and Methods
PSII membrane particles prepared from spinach were resuspended in a buffered solution (80 μg chlorophyll per mL; pH/pD of 6.2), and 20 μM of 2,6-dichloro-p-benzoquinone (DCBQ) were added as artificial electron acceptor. The PBD experiments were carried out and analyzed as described previously (37, 38). In the PBD experiments presented herein, 100 mL of PSII suspension were kept in a dark reservoir on ice (gently stirred) and pumped first through a thermostated laboratory-built heat exchanger and then into the sample compartment of the thermostated flow-through cuvette (3-mm optical path). Then, the respective laser-flash protocol was applied (0–4 saturating ns-laser flashes plus a single subsaturating flash; 532 nm; see Fig. S1). The PBD signal induced by the nonsaturating laser flash was recorded. In the next pump cycle, the flow-through cuvette was filled again with a fresh sample of dark-adapted PSII. Extensive signal averaging was applied; about 4,000 measurements were needed for obtaining the data in Fig. 2 (SI Text).
Supplementary Material
ACKNOWLEDGMENTS.
We thank M. Fünning for preparation of the PSII membrane particles. We acknowledge support from the Berlin Cluster of Excellence on Unifying Concepts in Catalysis (UniCat), the European Union (7th Framework Program; SOLAR-H2 consortium, Grant 212508), the German “Bundesministerium für Bildung und Forschung” (BMBF; H2 Design Cell consortium, Grant 03SF0355D), and the Volkswagen Foundation (Grant I/77-575). M.H. thanks the Deutsche Forschungsgemeinschaft for a Heisenberg fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206266109/-/DCSupplemental.
References
- 1.McEvoy JP, Brudvig GW. Water-splitting chemistry of photosystem II. Chem Rev. 2006;106:4455–4483. doi: 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- 2.Dau H, Haumann M. The manganese complex of photosystem II in its reaction cycle: Basic framework and possible realization at the atomic level. Coord Chem Rev. 2008;252:273–295. [Google Scholar]
- 3.Barber J. Photosynthetic energy conversion: Natural and artificial. Chem Soc Rev. 2009;38:185–196. doi: 10.1039/b802262n. [DOI] [PubMed] [Google Scholar]
- 4.Blankenship RE. Molecular Mechanisms of Photosynthesis. Oxford: Blackwell Science; 2002. [Google Scholar]
- 5.Rutherford AW, Moore TA. Mimicking photosynthesis, but just the best bits. Nature. 2008;453:449. doi: 10.1038/453449b. [DOI] [PubMed] [Google Scholar]
- 6.Magnuson A, et al. Biomimetic and microbial approaches to solar fuel generation. Acc Chem Res. 2009;42:1899–1909. doi: 10.1021/ar900127h. [DOI] [PubMed] [Google Scholar]
- 7.Dau H, et al. The mechanism of water oxidation: From electrolysis via homogeneous to biological catalysis. ChemCatChem. 2010;2:724–761. [Google Scholar]
- 8.Lubitz W, Reijerse EJ, Messinger J. Solar water-splitting into H2 and O2: Design principles of photosystem II and hydrogenases. Energy Environ Sci. 2008;1:15–31. [Google Scholar]
- 9.Dau H, Zaharieva I. Principles, efficiency, and blueprint character of solar-energy conversion in photosynthetic water oxidation. Acc Chem Res. 2009;42:1861–1870. doi: 10.1021/ar900225y. [DOI] [PubMed] [Google Scholar]
- 10.Barry BA, Babcock GT. Tyrosine radicals are involved in the photosynthetic oxygen-evolving system. Proc Natl Acad Sci USA. 1987;84:7099–7103. doi: 10.1073/pnas.84.20.7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Styring S, Sjöholm J, Mamedov F. Two tyrosines that changed the world: Interfacing the oxidizing power of photochemistry to water splitting in photosystem II. Biochim Biophys Acta. 2012;1817:76–87. doi: 10.1016/j.bbabio.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Rappaport F, Diner BA. Primary photochemistry and energetics leading to the oxidation of the (Mn)4Ca cluster and to the evolution of molecular oxygen in photosystem II. Coord Chem Rev. 2008;252:259–272. [Google Scholar]
- 13.Zouni A, et al. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution. Nature. 2001;409:739–743. doi: 10.1038/35055589. [DOI] [PubMed] [Google Scholar]
- 14.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 15.Umena Y, Kawakami K, Shen J-R, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 16.Kok B, Forbush B, McGloin M. Cooperation of charges in photosynthetic O2 evolution, I. A linear four-step mechanism. Photochem Photobiol. 1970;11:457–475. doi: 10.1111/j.1751-1097.1970.tb06017.x. [DOI] [PubMed] [Google Scholar]
- 17.Dau H, Haumann M. Time-resolved X-ray spectroscopy leads to an extension of the classical S-state cycle model of photosynthetic oxygen evolution. Photosynth Res. 2007;92:327–343. doi: 10.1007/s11120-007-9141-9. [DOI] [PubMed] [Google Scholar]
- 18.Lavergne J, Junge W. Proton release during the redox cycle of the water oxidase. Photosynth Res. 1993;38:279–296. doi: 10.1007/BF00046752. [DOI] [PubMed] [Google Scholar]
- 19.Junge W, Haumann M, Ahlbrink R, Mulkidjanian A, Clausen J. Electrostatics and proton transfer in photosynthetic water oxidation. Philos Trans R Soc Lond B Biol Sci. 2002;357:1407–1418. doi: 10.1098/rstb.2002.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rappaport F, Lavergne J. Proton release during successive oxidation steps of the photosynthetic water oxidation process: Stoichiometries and pH dependence. Biochemistry. 1991;30:10004–10012. doi: 10.1021/bi00105a027. [DOI] [PubMed] [Google Scholar]
- 21.Ho FM. Uncovering channels in photosystem II by computer modelling: Current progress, future prospects, and lessons from analogous systems. Photosynth Res. 2008;98:503–522. doi: 10.1007/s11120-008-9358-2. [DOI] [PubMed] [Google Scholar]
- 22.Haumann M, et al. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science. 2005;310:1019–1021. doi: 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- 23.Hoganson CW, Babcock GT. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 1997;277:1953–1956. doi: 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
- 24.Dau H, Haumann M. Eight steps preceding O─O bond formation in oxygenic photosynthesis: A basic reaction cycle of the photosystem II manganese complex. Biochim Biophys Acta. 2007;1767:472–483. doi: 10.1016/j.bbabio.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 25.Meyer TJ, Huynh MHV, Thorp HH. The possible role of proton-coupled electron transfer (PCET) in water oxidation by photosystem II. Angew Chem Int Ed. 2007;46:5284–5304. doi: 10.1002/anie.200600917. [DOI] [PubMed] [Google Scholar]
- 26.Gerencsér L, Dau H. Water oxidation by photosystem II: H2O–D2O exchange and the influence of pH support formation of an intermediate by removal of a proton before dioxygen creation. Biochemistry. 2010;49:10098–10106. doi: 10.1021/bi101198n. [DOI] [PubMed] [Google Scholar]
- 27.Haumann M, Bögershausen O, Cherepanov D, Ahlbrink R, Junge W. Photosynthetic oxygen evolution: H/D isotope effects and the coupling between electron and proton transfer during the redox reactions at the oxidizing side of photosystem II. Photosynth Res. 1997;51:193–208. [Google Scholar]
- 28.Renger G, Hanssum B. Studies on the reaction coordinates of the water oxidase in PS II membrane fragments from spinach. FEBS Lett. 1992;299:28–32. doi: 10.1016/0014-5793(92)80092-u. [DOI] [PubMed] [Google Scholar]
- 29.Dekker JP, Plijter JJ, Ouwehand L, van Gorkom HJ. Kinetics of manganese redox transitions in the oxygen evolving apparatus of photosynthesis. Biochim Biophys Acta. 1984;767:176–179. [Google Scholar]
- 30.Haumann M, Junge W. Extent and rate of proton release by photosynthetic water oxidation in thylakoids: Electrostatic relaxation versus chemical production. Biochemistry. 1994;33:864–872. doi: 10.1021/bi00170a003. [DOI] [PubMed] [Google Scholar]
- 31.Schlodder E, Witt HT. Stoichiometry of proton release from the catalytic center in photosynthetic water oxidation. Reexamination by a glass electrode study at ph 5.5–7. J Biol Chem. 1999;274:30387–30392. doi: 10.1074/jbc.274.43.30387. [DOI] [PubMed] [Google Scholar]
- 32.Rappaport F, Blanchard-Desce M, Lavergne J. Kinetics of electron transfer and electrochromic change during the redox transition of the photosynthetic oxygen-evolving complex. Biochim Biophys Acta. 1994;1184:178–192. [Google Scholar]
- 33.Dau H, Haumann M. Reaction cycle of photosynthetic water oxidation in plants and cyanobacteria (response letter) Science. 2006;312:1471–1472. [Google Scholar]
- 34.Braslavsky SE, Heibel GE. Time-resolved photothermal and photoacoustic methods applied to photoinduced processes in solution. Chem Rev. 1992;92:1381–1410. [Google Scholar]
- 35.Falvey DE. Photothermal beam deflection calorimetry in solution photochemistry: Recent progress and future prospects. Photochem Photobiol. 1997;65:4–9. doi: 10.1111/j.1751-1097.1997.tb01870.x. [DOI] [PubMed] [Google Scholar]
- 36.Gensch T, Viappiani C. Time-resolved photothermal methods: Accessing time-resolved thermodynamics of photoinduced processes in chemistry and biology. Photochem Photobiol Sci. 2003;2:699–721. doi: 10.1039/b303177b. [DOI] [PubMed] [Google Scholar]
- 37.Krivanek R, Dau H, Haumann M. Enthalpy changes during photosynthetic water oxidation tracked by time-resolved calorimetry using a photothermal beam deflection technique. Biophys J. 2008;94:1890–1903. doi: 10.1529/biophysj.107.117085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klauss A, Krivanek R, Dau H, Haumann M. Energetics and kinetics of photosynthetic water oxidation studied by photothermal beam deflection (PBD) experiments. Photosynth Res. 2009;102:499–509. doi: 10.1007/s11120-009-9417-3. [DOI] [PubMed] [Google Scholar]
- 39.Feitelson J, Mauzerall D. Enthalpy and electrostriction in the electron-transfer reaction between triplet zinc uroporphyrin and ferricyanide. J Phys Chem B. 2002;106:9674–9678. [Google Scholar]
- 40.Rizzi AC, et al. Entropic changes control the charge separation process in triads mimicking photosynthetic charge separation. J Phys Chem A. 2008;112:4215–4223. doi: 10.1021/jp712008b. [DOI] [PubMed] [Google Scholar]
- 41.Hou HJM, Mauzerall D. Listening to PS II: Enthalpy, entropy, and volume changes. J Photochem Photobiol B. 2011;104:357–365. doi: 10.1016/j.jphotobiol.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Mauzerall D, Hou JM, Boichenko VA. Volume changes and electrostriction in the primary photoreactions of various photosynthetic systems: Estimation of dielectric coefficient in bacterial reaction centers and of the observed volume changes with the Drude–Nernst equation. Photosynth Res. 2002;74:173–180. doi: 10.1023/A:1020903525973. [DOI] [PubMed] [Google Scholar]
- 43.Hou JM, Boichenko VA, Diner BA, Mauzerall D. Thermodynamics of electron transfer in oxygenic photosynthetic reaction centers: Volume change, enthalpy, and entropy of electron-transfer reactions in manganese-depleted photosystem II core complexes. Biochemistry. 2001;40:7117–7125. doi: 10.1021/bi010373s. [DOI] [PubMed] [Google Scholar]
- 44.Haumann M, et al. Structural and oxidation state changes of the photosystem II manganese complex in four transitions of the water oxidation cycle (S0 → S1, S1 → S2, S2 → S3, and S3,4 → S0) characterized by X-ray absorption spectroscopy at 20 K and room temperature. Biochemistry. 2005;44:1894–1908. doi: 10.1021/bi048697e. [DOI] [PubMed] [Google Scholar]
- 45.Noguchi T. Fourier transform infrared analysis of the photosynthetic oxygen-evolving center. Coord Chem Rev. 2008;252:336–346. [Google Scholar]
- 46.Buchta J, Grabolle M, Dau H. Photosynthetic dioxygen formation studied by time-resolved delayed fluorescence measurements: Method, rationale, and results on the activation energy of dioxygen formation. Biochim Biophys Acta. 2007;1767:565–574. doi: 10.1016/j.bbabio.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Karge M, Irrgang KD, Renger G. Analysis of the reaction coordinate of photosynthetic water oxidation by kinetic measurements of 355 nm absorption changes at different temperatures in photosystem II preparations suspended in either H2O or D2O. Biochemistry. 1997;36:8904–8913. doi: 10.1021/bi962342g. [DOI] [PubMed] [Google Scholar]
- 48.Razeghifard MR, Klughammer C, Pace RJ. Electron paramagnetic resonance kinetic studies of the S states in spinach thylakoids. Biochemistry. 1997;36:86–92. doi: 10.1021/bi9614287. [DOI] [PubMed] [Google Scholar]
- 49.Hatcher E, Soudackov A, Hammes-Schiffer S. Comparison of dynamical aspects of nonadiabatic electron, proton, and proton-coupled electron transfer reactions. Chem Phys. 2005;319:93–100. doi: 10.1063/1.1814635. [DOI] [PubMed] [Google Scholar]
- 50.Krishtalik LI. The mechanism of the proton transfer: An outline. Biochim Biophys Acta. 2000;1458:6–27. doi: 10.1016/s0005-2728(00)00057-8. [DOI] [PubMed] [Google Scholar]
- 51.Garczarek F, Gerwert K. Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature. 2006;439:109–112. doi: 10.1038/nature04231. [DOI] [PubMed] [Google Scholar]
- 52.Barry BA, et al. Time-resolved vibrational spectroscopy detects protein-based intermediates in the photosynthetic oxygen-evolving cycle. Proc Natl Acad Sci USA. 2006;103:7288–7291. doi: 10.1073/pnas.0600216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rappaport F, Ishida N, Sugiura M, Boussac A. Ca2+ determines the entropy changes associated with the formation of transition states during water oxidation by photosystem II. Energy Environ Sci. 2011;4:2520–2524. [Google Scholar]
- 54.Razeghifard MR, Pace RJ. EPR kinetic studies of oxygen release in thylakoids and PSII membranes: An intermediate in the S3 to S0 transition. Biochemistry. 1999;38:1252–1257. doi: 10.1021/bi9811765. [DOI] [PubMed] [Google Scholar]
- 55.Dau H, Iuzzolino L, Dittmer J. The tetra-manganese complex of photosystem II during its redox cycle: X-ray absorption results and mechanistic implications. Biochim Biophys Acta. 2001;1503:24–39. doi: 10.1016/s0005-2728(00)00230-9. [DOI] [PubMed] [Google Scholar]
- 56.Robblee JH, et al. The Mn cluster in the S0 state of the oxygen-evolving complex of photosystem II studied by EXAFS spectroscopy: Are there three di-μ-oxo-bridged Mn2 moieties in the tetranuclear Mn complex? J Am Chem Soc. 2002;124:7459–7471. doi: 10.1021/ja011621a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaharieva I, Wichmann JM, Dau H. Thermodynamic limitations of photosynthetic water oxidation at high proton concentrations. J Biol Chem. 2011;286:18222–18228. doi: 10.1074/jbc.M111.237941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teutloff C, et al. Electronic structure of the tyrosine D radical and the water-splitting complex from pulsed ENDOR spectroscopy on photosystem II single crystals. Phys Chem Chem Phys. 2009;11:6715–6726. doi: 10.1039/b908093g. [DOI] [PubMed] [Google Scholar]
- 59.Siegbahn PE. A structure-consistent mechanism for dioxygen formation in photosystem II. Chem Eur J. 2008;14:8290–8302. doi: 10.1002/chem.200800445. [DOI] [PubMed] [Google Scholar]
- 60.Ames W, et al. Theoretical evaluation of structural models of the S2 state in the oxygen evolving complex of photosystem II: Protonation states and magnetic interactions. J Am Chem Soc. 2011;133:19743–19757. doi: 10.1021/ja2041805. [DOI] [PubMed] [Google Scholar]
- 61.Hundelt M, Hays AM, Debus RJ, Junge W. Oxygenic photosystem II: The mutation D1–D61N in Synechocystis sp. PCC 6803 retards S-state transitions without affecting electron transfer from YZ to P680+ Biochemistry. 1998;37:14450–14456. doi: 10.1021/bi981164j. [DOI] [PubMed] [Google Scholar]
- 62.Dilbeck PL, et al. The mutation D1–D61N in Synechocystis sp. PCC 6803 allows the observation of pH-sensitive intermediates in the formation and release of O2 from photosystem II. Biochemistry. 2012;51:1079–1091. doi: 10.1021/bi201659f. [DOI] [PubMed] [Google Scholar]
- 63.McEvoy JP, Brudvig GW. Structure-based mechanism of photosynthetic water oxidation. Phys Chem Chem Phys. 2004;6:4754–4763. [Google Scholar]
- 64.Vrettos JS, Stone DA, Brudvig GW. Quantifying the ion selectivity of the Ca2+ site in photosystem II: Evidence for direct involvement of Ca2+ in O2 formation. Biochemistry. 2001;40:7937–7945. doi: 10.1021/bi010679z. [DOI] [PubMed] [Google Scholar]
- 65.Bondar AN, Dau H. Extended protein/water H-bond networks in photosynthetic water oxidation. Biochim Biophys Acta. 2012;1817:1177–1190. doi: 10.1016/j.bbabio.2012.03.031. [DOI] [PubMed] [Google Scholar]
- 66.Zaharieva I, Grabolle M, Chernev P, Dau H. Water oxidation in photosystem II: Energetics and kinetics of intermediates formation in S2 → S3 and S3 → S0 transitions monitored by delayed chlorophyll fluorescence; Proceedings of the Fifteenth International Conference on Photosynthesis; Beijing, China: 2013. in press. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.