

Abstract
As part of our search for selective and CNS-active thyrotropin-releasing hormone (TRH) analogues, we synthesized a set of 44 new analogues in which His and pGlu residues were modified or replaced. The analogues were evaluated as agonists at TRH-R1 and TRH-R2 in cells in vitro, and in vivo in mice for analeptic and anticonvulsant activities. Several analogues bound to TRH-R1 and TRH-R2 with good to moderate affinities, and are full agonists at both receptor subtypes. Specifically, analogue 21 a (R = CH3) exhibited binding affinities (Ki values) of 0.17 μM for TRH-R1 and 0.016 μM for TRH-R2; it is 10-fold less potent than TRH in binding to TRH-R1 and equipotent with TRH in binding to TRH-R2. Compound 21 a, the most selective agonist, activated TRH-R2 with a potency (EC50 value) of 0.0021 μM, but activated TRH-R1 at EC50 = 0.05 μM, and exhibited 24-fold selectivity for TRH-R2 over TRH-R1. The newly synthesized TRH analogues were also evaluated in vivo to assess their potencies in antagonism of barbiturate-induced sleeping time, and several analogues displayed potent analeptic activity. Specifically, analogues 21 a,b and 22 a,b decreased sleeping time by nearly 50 % more than TRH. These analogues also displayed potent anticonvulsant activity and provided significant protection against PTZ-induced seizures, but failed to provide any protection in MES-induced seizures at 10 μmol kg−1. The results of this study provide evidence that TRH analogues that show selectivity for TRH-R2 over TRH-R1 possess potent CNS activity.
Keywords: CNS, peptides, pharmacology, receptors, thyrotropin-releasing hormone
Introduction
Thyrotropin-releasing hormone (TRH, L-pGlu-L-His-L-ProNH2, 1) is generated from a large precursor protein that contains multiple repeats of the TRH progenitor tetrapeptide Gln-His-Pro-Gly, and is the first hypothalamic hypophysiotropic neuropeptide to have had its sequence chemically elucidated.[1–5] TRH plays a central role in the endocrine system and regulates several neurobiological activities.[6] It exhibits antidepressant, analeptic, and neuroprotective effects and is considered a promising lead for the discovery of analogues in the treatment of epilepsy, motor neuron diseases, spinal cord trauma, and Alzheimer’s disease.[7–9] The rhodopsin/β-adrenergic receptor subfamily of G-protein-coupled TRH receptors (TRH-R) in brain are ubiquitously distributed and found at highest densities in limbic structures, especially the amygdala and hypothalamus and in lower densities in brain stem and cerebellum.[10, 11] At present, two TRH receptor subtypes (TRH-R1 and TRH-R2) have been found in several species and exhibit an overall identity of ~50 %.[12–19] Studies conducted earlier demonstrated that TRH-R1 is highly expressed in the anterior pituitary, the neuroendocrine brain regions, the autonomic nervous system, and the visceral brain stem regions. TRH-R2 is expressed in the cerebellar cortex, mesencephalon, thalamic and subthalamic regions, and the brain stem.[17, 19] It has therefore been hypothesized that TRH-R2 could mediate some central nervous system (CNS) effects of TRH, whereas TRH-R1 mediates its thyroid-stimulating hormone (TSH)-releasing effects.
All three amino acid residues of TRH play a role in eliciting its physiological response(s), with pGlu and His residues accounting for almost all of TRH binding energy and affinity. In our previous studies, several modified amino acids or carboxylic acid derived scaffolds were incorporated independently or simultaneously into the sequence of TRH at pGlu and His to synthesize a library of analogues.[20–27] The results of these studies showed that TRH analogues in which His is modified at the N1(τ) or C2 positions of the imidazole ring with various bulky hydrophobic groups exhibit selectivity for TRH-R2. The TRH analogues in which pGlu is replaced with (1R)- or (1S)-3-oxo-1-cyclopentanecarboxylic acid (Ocp), and His is substituted at the C2 position, bind with high affinity (Ki) to both TRH-R1 and TRH-R2, whereas TRH analogues in which pGlu is replaced with L-pyro-2-aminoadipic acid (pAad), and His is substituted at the N1(τ) or C2 positions, exhibit good potency and selective activation of TRH-R2 over TRH-R1. These studies indicate that independent or simultaneous change of pGlu and His residues in TRH alters its receptor binding affinity and signal transducing potency.

In continuation, herein we report the synthesis of four new series of TRH analogues that involve independent or simultaneous modifications at the pGlu and His residues of the parent peptide in order to gain additional insight into the structure–activity relationships and biological activities (Figure 1). These new series allowed a comparison of carboxylic acids at the N-terminal residue and the effects of substitution at the His residue on the binding affinity and potency of the modified peptidomimetics. The His residue was replaced with either N1(τ) substituted or 1,2-disubstituted-L-His, whereas the pGlu residue was replaced with (1R)-3-Ocp or L-pAad. In addition to their receptor binding studies, the synthesized TRH analogues were also evaluated in vivo in mice for CNS activity on the antagonism of pentobarbital-induced sleep duration as a primary screen. The most promising analogues were then evaluated in the selected models of epilepsy to assess their promise as future drugs in the treatment of epilepsy.
Figure 1.
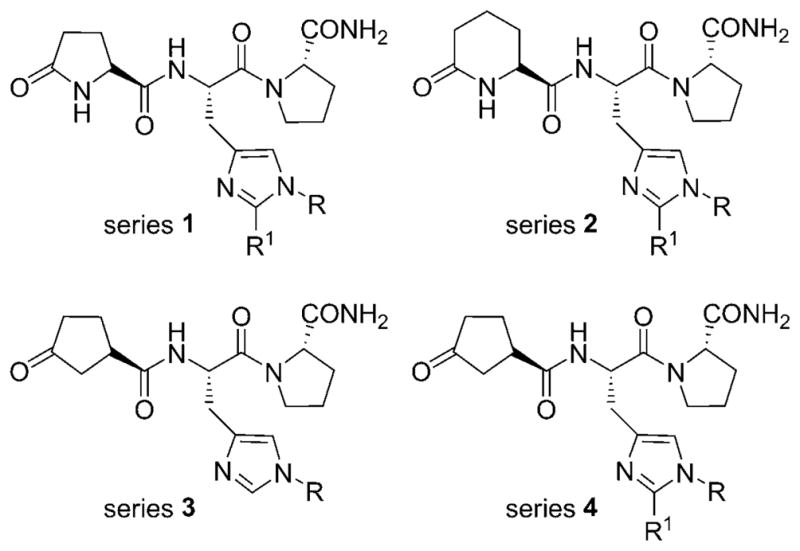
General structures of the four series of synthesized TRH analogues.
Results and Discussion
Chemistry
Boc-1,2-dialkyl-L-His-OH compounds 6 a–m were prepared from commercially available L-His-OMe using a multistep synthetic procedure as discussed previously.[28–30] L-pAadOPfp (15) was synthesized from commercially available L-Lys(Z)-OH in four steps as described earlier.[31] Boc-1-alkyl-L-His-OH compounds 17 a–e were synthesized from Boc-L-His-OH in one step as reported.[32] (1R)-3-Ocp-OH (20) was synthesized from 4-vinylcyclohexene in four steps and chemically resolved using a well-established procedure.[33–35]
A solid-phase peptide synthesis protocol was used for the synthesis of targeted tripeptides 10 a–g (Scheme 1). Briefly, a 4-methylbenzhydrylamine-functionalized resin (MBHA·HCl, 2) was neutralized with N,N-diisopropylethylamine (DIPEA) and subsequently coupled with the preformed O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU)-activated ester of Boc-L-Pro-OH (3) in N,N-dimethylformamide (DMF) to produce a protected amino acid linked resin. The resin-bound Boc-protected amino acid was deprotected with trifluoroacetic acid (TFA), followed by neutralization with DIPEA, to provide free amino acid linked resin 5. This was then subjected to further coupling and deprotection cycles with Boc-L-His(1-methyl-2-alkyl)-OH 6 a–g and Z-L-pGlu-OH 8, respectively, to yield peptide resin 9. The intermediate coupling steps were monitored by Kaiser’s test. Cleavage of the peptide from the resin support was carried out by using a solution of trifluoromethanesulfonic acid (TFMSA) in TFA in the presence of thioanisole and 1,2-ethanedithiol (EDT) as scavengers to yield peptide amides 10 a–g.
Scheme 1.
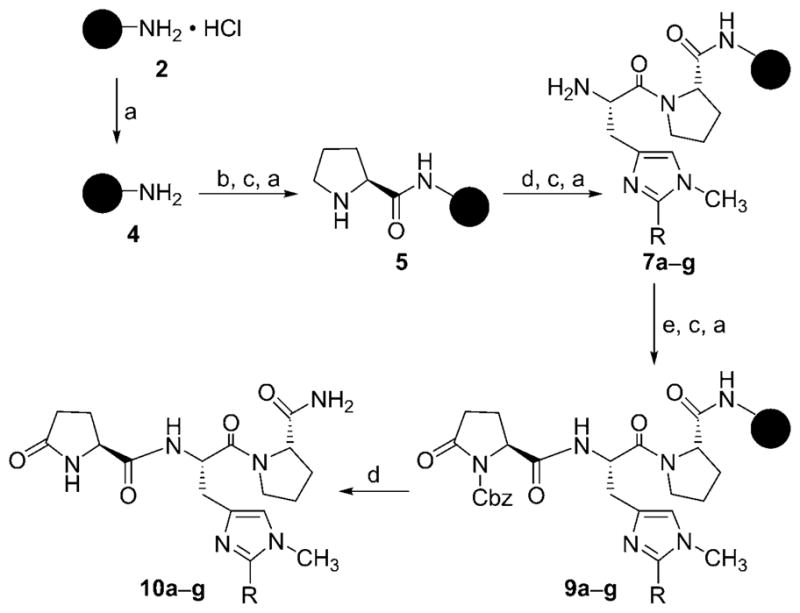
Reagents and conditions: a) 10 % DIPEA, CH2Cl2, RT, 5 min; b) Boc-L-Pro-OH (3), TBTU, DMF, 10 % DIPEA in CH2Cl2, RT, 2 h; c) 20 % TFA in CH2Cl2, RT, 20 min; d) Boc-L-His(1-methyl-2-alkyl)-OH (6 a–g), TBTU, DMF, 10 % DIPEA in CH2Cl2, RT, 2 h; e) Z-L-pGlu-OH (8), TBTU, 10 % DIPEA in CH2Cl2, RT, 2 h; f) TFMSA, EDT, thioanisole, TFA, 4 °C–RT, 2 h.
All subsequent peptides were synthesized by solution-phase synthesis to decrease consumption of the large excess of modified amino acids required for solid-phase synthesis. Reaction of L-Pro-OH 11 with Boc-L-His(1,2-dialkyl)-OH compounds 6 a–m in the presence of 1,3-diisopropylcarbodiimide (DIC) and N-hydroxy-5-norbornene-endo-2,3-dicarboximide (HONB) in DMF yielded dipeptides 12 a–m (Scheme 2). Deprotection of the Boc group with TFA produced dipeptide salts 13 a–m. The free dipeptides were obtained in situ by reaction of 13 a–m with a solution of ammonia. The intermediate dipeptides were not isolated; their immediate coupling with L-pGlu-OTcp (14) or L-pAad-OPfp (15) in DMF produced the desired TRH analogues (10 h–n and 16 a–m, respectively series 1 and 2).
Scheme 2.

Reagents and conditions: a) DIC, HONB, DMF, 4 °C, 36 h; b) 40 % TFA, CH2Cl2, 0 °C, 30 min; c) 7 N NH3 in CH3OH, 0 °C, 10 min; d) L-pGlu-OTcp (14) or L-pAad-OPfp (15), DMF, 4 °C, 36 h.
We encountered low yields in using a similar strategy to carry out the synthesis of TRH analogues 21 a–e and 22 a–l (series 3 and 4). In these cases, coupling of dipeptides 12 or 19 with (1R)-3-Ocp-OH (20) using DIC and HONB provided a complex mixture containing tripeptides in poor yields. We observed that the use of 1-hydroxybenzotriazole (HOBt) as an auxiliary nucleophile in place of HONB provided better results. Thus, reaction of dipeptides 12 or 19 with 20 using DIC/HOBt produced tripeptides 21 a–e and 22 a–l in satisfactory yields (Scheme 3 and Scheme 4).
Scheme 3.

Reagents and conditions: a) DIC, HONB, DMF, 4 °C, 36 h; b) 40 % TFA, CH2Cl2, 0 °C, 30 min; c) 7 N NH3 in CH3OH, 0 °C, 10 min; d) (1R)-3-Ocp-OH (20), DIC, HOBt, DMF, 4 °C, 36 h.
Scheme 4.

Reagents and conditions: a) 40 % TFA, CH2Cl2, 0 °C, 30 min; b) 7 N NH3 in CH3OH, 0°C, 10 min; c) (1R)-3-Ocp-OH (20), DIC, HOBt, DMF, 4 °C, 36 h.
Pharmacology at TRH-R1 and TRH-R2
Synthesized TRH analogues were examined for their affinities for TRH-R1 and TRH-R2 and their abilities to serve as agonists of the receptors.[36] Affinities, reported as Ki values (μM), were determined by measuring the concentration of the analogue required to compete with [3H][Nτ(1)-Me-His]TRH at 2 nM for receptor binding. [Nτ(1)-Me-His]TRH is known to bind TRH-R1 and TRH-R2 with affinities higher than TRH. The agonist behavior of the analogues was tested in HEK 293EM cells stably expressing TRH-R1 or TRH-R2 by incubating the cells with various doses of the analogues as described.[21, 37] The extent of agonist behavior was then determined by measuring signaling through a reporter gene, and the data are reported as EC50 values (μM) in Table 1.
Table 1.
Binding affinity (Ki) and signaling (activation) potencies (EC50) produced by TRH analogues (series 1–4) for TRH-R1 and TRH-R2.

| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compd | R | R1 | R2 | TRH-R1 | Ki [μM][a] TRH-R2 | Fold Selectivity | TRH-R1 | EC50 [μM] TRH-R2[b] | Fold Selectivity |
| Series 1: | |||||||||
| 10 a | L-pGlu | CH3 | C3H7 | 0.32 (0.24–0.35) | 0.17 (0.11–0.23) | 1.9 | 0.29 (0.18–0.47) | 0.027 (0.015–0.050) | 11 |
| 10 b | L-pGlu | CH3 | CH(CH3)2 | >50 | >50 | – | >50 | >50 | – |
| 10 c | L-pGlu | CH3 | C(CH3)3 | >50 | >50 | – | >50 | >50 | – |
| 10 d | L-pGlu | CH3 | c-C3H5 | 11.0 (9.00–13.2) | >50 | 4.5 | >50 | >50 | – |
| 10 e | L-pGlu | CH3 | c-C5H9 | >50 | >50 | – | >50 | >50 | – |
| 10 f | L-pGlu | CH3 | c-C6H11 | 0.34 (0.28–0.38) | 0.14 (0.10–0.22) | 2.4 | 0.34 (0.18–0.51) | 0.48 (0.31–0.68) | 1.4 |
| 10 g | L-pGlu | CH3 | adamantan-1-yl | >50 | >50 | – | >50 | >50 | – |
| 10 h | L-pGlu | CH2C6H5 | C3H7 | – | – | – | – | – | – |
| 10 i | L-pGlu | CH2C6H5 | CH(CH3)2 | >50 | >50 | – | >50 | >50 | – |
| 10 j | L-pGlu | CH2C6H5 | C(CH3)3 | >50 | >50 | – | >50 | >50 | – |
| 10 k | L-pGlu | CH2C6H5 | c-C3H5 | – | – | – | – | – | – |
| 10 l | L-pGlu | CH2C6H5 | c-C5H9 | – | – | – | – | – | – |
| 10 m | L-pGlu | CH2C6H5 | c-C6H11 | >50 | >50 | – | >50 | >50 | – |
| 10 n | L-pGlu | CH2C6H5 | adamantan-1-yl | >50 | >50 | – | >50 | >50 | – |
| Series 2: | |||||||||
| 16 a | L-pAad | CH2C6H5 | C3H7 | >50 | >50 | – | >50 | >50 | – |
| 16 b | L-pAad | CH2C6H5 | CH(CH3)2 | – | – | – | – | – | – |
| 16 c | L-pAad | CH2C6H5 | C(CH3)3 | – | – | – | – | – | – |
| 16 d | L-pAad | CH2C6H5 | c-C3H5 | >50 | >50 | – | >50 | >50 | – |
| 16 e | L-pAad | CH2C6H5 | c-C5H9 | – | – | – | – | – | – |
| 16 f | L-pAad | CH2C6H5 | c-C6H11 | >50 | >50 | – | >50 | >50 | – |
| 16 g | L-pAad | CH2C6H5 | adamantan-1-yl | >50 | >50 | – | >50 | >50 | – |
| 16 h | L-pAad | CH3 | C3H7 | – | – | – | – | – | – |
| 16 i | L-pAad | CH3 | CH(CH3)2 | – | – | – | – | – | – |
| 16 j | L-pAad | CH3 | C(CH3)3 | >50 | >50 | – | >50 | >50 | – |
| 16 k | L-pAad | CH3 | c-C5H9 | – | – | – | – | – | – |
| 16 l | L-pAad | CH3 | c-C6H11 | >50 | >50 | – | >50 | >50 | – |
| 16 m | L-pAad | CH3 | adamantan-1-yl | – | – | – | – | – | – |
| Series 3: | |||||||||
| 21 a | (R)-Ocp | CH3 | H | 0.17 (0.11–0.26) | 0.016 (0.007–0.027) | 10.6 | 0.05 (0.03–0.09) | 0.0021 (0.0009–0.0035) | 24 |
| 21 b | (R)-Ocp | C2H5 | H | 0.10 (0.07–0.14) | 0.32 (0.28–0.37) | 3.2 | 0.14 (0.06–0.20) | 0.030 (0.012–0.051) | 4.7 |
| 21 c | (R)-Ocp | C3H7 | H | 8.1 (6.1–13.2) | 12.4 (6.8–16.4) | 1.5 | 4.6 (3.7–9.1) | 1.2 (0.54–2.22) | 4 |
| 21 d | (R)-Ocp | CH(CH3)2 | H | >50 | >50 | – | >50 | >50 | – |
| 21 e | (R)-Ocp | CH2C6H5 | H | 10.2 (8.1–12.2) | 14.4 (10.8–20.6) | 1.4 | 3.5 (2.5–7.8) | 1.4 (0.44–2.2) | 2.5 |
| Series 4: | |||||||||
| 22 a | (R)-Ocp | CH2C6H5 | C3H7 | 0.08 (0.03–0.14) | 0.35 (0.27–0.45) | 4.4 | 0.10 (0.04–0.18) | 0.015 (0.011–0.047) | 6.6 |
| 22 b | (R)-Ocp | CH2C6H5 | CH(CH3)2 | 9.6 (5.1–12.2) | 10.2 (6.4–15.4) | 1 | 3.6 (3.2–8.1) | 1.1 (0.32–1.30) | 3.2 |
| 22 c | (R)-Ocp | CH2C6H5 | C(CH3)3 | – | – | – | – | – | – |
| 22 d | (R)-Ocp | CH2C6H5 | c-C3H5 | – | – | – | – | – | – |
| 22 e | (R)-Ocp | CH2C6H5 | c-C5H9 | – | – | – | – | – | – |
| 22 f | (R)-Ocp | CH2C6H5 | c-C6H11 | 7.8 (6.4–12.7) | 9.3 (6.5–12.8) | 1.2 | 2.7 (1.5–6.2) | 0.97 (0.52–1.34) | 2.7 |
| 22 g | (R)-Ocp | CH2C6H5 | adamantan-1-yl | – | – | – | – | – | – |
| 22 h | (R)-Ocp | CH3 | C3H7 | >50 | >50 | – | >50 | >50 | – |
| 22 i | (R)-Ocp | CH3 | CH(CH3)2 | >50 | >50 | – | – | >50 | – |
| 22 j | (R)-Ocp | CH3 | C(CH3)3 | – | – | – | – | – | – |
| 22 k | (R)-Ocp | CH3 | c-C6H11 | – | – | – | – | – | – |
| 22 l | (R)-Ocp | CH3 | adamantan-1-yl | >50 | >50 | – | >50 | >50 | – |
| TRH | 0.02 (0.01–0.03) | 0.02 (0.01–0.03) | – | 0.01 (0.008–0.015) | 0.003 (0.002–0.007) | – | |||
| [Nτ(1)-Me-His]-TRH | 0.003 (0.002–0.005) | 0.003 (0.002–0.005) | – | 0.0021 (0.002–0.005) | 0.0002 (0.0002–0.0005) | – | |||
For binding, cells were incubated with 1 nM [3H]Nτ(1)-Me-His-TRH in the absence or presence of various doses of unlabeled TRH analogues for 1 h at 37 °C.
For signaling, cells transfected with TRH-R1 or TRH-R2 and a CREB–luciferase reporter were incubated with various doses of TRH analogues for 6 h at 37 °C, and luciferase activity was measured. Experiments were performed with intact HEK 293 cells. All data represent the mean ±SD of duplicate determinations in three experiments; −: not determined.
TRH analogues of series 1 generally exhibited weak binding affinities for TRH-R1 and TRH-R2 (Table 1). Analogue 10 a (R = C3H7) displayed modest affinities for TRH-R1 and TRH-R2 (Ki = 0.32 and 0.17 μM, respectively), resulting in a ~16–17-fold decrease in binding affinity relative to TRH. However, 10 a displayed promising agonist potency at TRH-R2 (EC50 = 0.027 μM) over TRH-R1 (EC50 = 0.29 μM) and has 11-fold selectivity for TRH-R2. Introduction of a bulkier and more lipophilic substituent at the C2 position of the His residue (compounds 10 b–e) yielded ligands with poor binding affinities and selectivities. Interestingly, analogue 10 f, with a cyclohexyl group at the C2 position of His, exhibited moderate binding affinity (TRH-R1: Ki = 0.34 μM; TRH-R2: Ki = 0.14 μM) similar to that of 10 a, but is not a selective agonist (TRH-R1: EC50 = 0.34 μM; TRH-R2: EC50 = 0.48 μM). To probe the steric requirements, we replaced the 1-methyl group in analogues 10 a–f with a 1-benzyl group. The resulting TRH analogues 10 h–n were found to be inactive at both receptor subtypes. Replacement of pGlu with pAad and attachment of a 1,2-dialkyl group on the His residue also resulted in inactive analogues, 16 a–m. These results provide an initial insight into the minimum and maximum size requirement of the groups placed on the His residue in combination with pGlu and its replacement by pAad in TRH peptides.
Simultaneous replacement of the His residue with 1-alkyl-and 1,2-dialkyl-His residues, and pGlu with its bioisosteric replacement (1R)-3-Ocp, provided ligands with the best results (series 3 and 4). Placement of a methyl group at the Nτ(1) position of His provided analogue 21 a [R =(1R)-3-Ocp, R1 =CH3, R2 =H], which exhibits high binding affinities (Ki = 0.17 and Ki = 0.016 μM at TRH-R1 and TRH-R2, respectively). Moreover, analogue 21 a displays potent agonist activity at TRH-R2 with high potency (EC50 = 0.0021 μM), which is ~ 1.4-fold higher than that of TRH, and exhibits 24-fold selectivity for TRH-R2. Replacement of a methyl group with an ethyl group as in analogue 21 b [R =(1R)-3-Ocp, R1 =CH3, R2 =C2H5] resulted in higher binding affinity for TRH-R1 (Ki = 0.10 μM). Analogue 21 b exhibited agonist potency (EC50 = 0.030 μM) lower than that of 21 a and was moderately selective for TRH-R2 with 4.7-fold selectivity. The binding affinities and activation potencies were further decreased with bulkier substituents placed at the Nτ(1) position of the His residue (analogues 21 c and 21 e). These analogues also exhibited modest selectivity of 2.5–4-fold for TRH-R2. These results are consistent with our previous findings that the ring NH group of pGlu is not necessary for the binding of the peptide to TRH receptors, and that modulation of the His residue at the N1 or C2 positions imparts selectivity toward TRH-R2.[20–24]
Finally, TRH analogues 22 a–g (series 4) in which the His residue is replaced with 1-benzyl-2-alkyl-His and pGlu is replaced with (1R)-3-Ocp exhibited moderate binding affinities and activation potencies. For example, analogue 22 a [R =(1R)-3-Ocp, R1 =CH2C6H5, R2 =CH2CH2CH3] exhibited Ki values of 0.08 and 0.35 μM to TRH-R1 and TRH-R2, respectively. Similarly, analogues 22 b [R =(1R)-3-Ocp, R1 =C6H5CH2, R2 =CH(CH3)2] and 22 f [R =(1R)-3-Ocp, R1 =C6H5CH2, R2 =c-C6H11] exhibited modest binding affinities for TRH-R1 and TRH-R2. Analogue 22 a also exhibited modest activation potency (EC50 = 0.015 μM) and has 6.6-fold specificity for TRH-R2. Analogues 22 b and 22 f were also found to have 3.2-and 2.7-fold respective selectivity in activation potencies (EC50) for TRH-R2. All TRH analogues 22 h–l, in which His is replaced with 1-methyl-2-alkyl-His, and pGlu is replaced with (1R)-3-Ocp, were found to be inactive at both TRH receptors (Table 1).
Antagonism of pentobarbital-induced sleeping time
One of the best documented CNS effects of TRH is its analeptic action manifested by the decrease in barbiturate narcosis. TRH is an effective analeptic agent, which reduces pentobarbital-induced sleeping time by 50 % or more following peripheral administration of high doses or central injection of lower doses in rats, rabbits, and monkeys.[38, 39] The synthesized TRH analogues were evaluated in vivo by using the antagonism of a pentobarbital-induced sleeping time model as described.[40] TRH analogues were injected intravenously through the tail vein at a dose of 10 μmol kg−1 (equivalent to 3.7 mg kg−1 TRH). Ten minutes after administration of the TRH analogue, each animal received 50 mg kg−1 sodium pentobarbital intraperitoneally. The sleeping time was recorded as the time elapsed from the onset of loss of righting reflex until it returned (Table 2).
Table 2.
Results of pentobarbital-induced sleeping time experiment in mice for TRH analogues (series 1–4).[a]

| |||||
|---|---|---|---|---|---|
| Compd | R | R1 | R2 | Sleep Time [min] | Decrease in Sleep Time [%] |
| Series 1: | |||||
| 10 a | L-pGlu | CH3 | C3H7 | 41.7 ± 6.1[b] | 52.7 |
| 10 b | L-pGlu | CH3 | CH(CH3)2 | 66.2 ± 9.8 | 25.1 |
| 10 c | L-pGlu | CH3 | C(CH3)3 | 59.6 ± 5.5 | 32.5 |
| 10 d | L-pGlu | CH3 | c-C3H5 | 51.4 ± 5.1[b] | 41.8 |
| 10 e | L-pGlu | CH3 | c-C5H9 | 48.2 ± 10.1[b] | 45.4 |
| 10 f | L-pGlu | CH3 | c-C6H11 | 43 ± 8.1[b] | 51.3 |
| 10 g | L-pGlu | CH3 | adamantan-1-yl | 66.9 ± 5.3 | 24.2 |
| 10 h | L-pGlu | CH2C6H5 | C3H7 | 105.1 ± 5.76 | −18.8 |
| 10 i | L-pGlu | CH2C6H5 | CH(CH3)2 | 157.2 ± 17[b,c] | −77.8 |
| 10 j | L-pGlu | CH2C6H5 | C(CH3)3 | 141.9 ± 23.9[b,c] | −60.5 |
| 10 k | L-pGlu | CH2C6H5 | c-C3H5 | 147.1 ± 12.7[b,c] | −66.4 |
| 10 l | L-pGlu | CH2C6H5 | c-C5H9 | 163.3 ± 18.7[b,c] | −84.7 |
| 10 m | L-pGlu | CH2C6H5 | c-C6H11 | 136.9 ± 26.7[b,c] | −54.9 |
| 10 n | L-pGlu | CH2C6H5 | adamantan-1-yl | 223.6 ± 22.3[b,c] | −152.9 |
| Series 2: | |||||
| 16 a | L-pAad | CH2C6H5 | C3H7 | 69.3 ± 7.3 | 22.8 |
| 16 b | L-pAad | CH2C6H5 | CH(CH3)2 | ND | – |
| 16 c | L-pAad | CH2C6H5 | C(CH3)3 | ND | – |
| 16 d | L-pAad | CH2C6H5 | c-C3H5 | 250.5 ± 23.1 | −178.7 |
| 16 e | L-pAad | CH2C6H5 | c-C5H9 | 95.5 ± 4.7 | −6.2 |
| 16 f | L-pAad | CH2C6H5 | c-C6H11 | 81.0 ± 3.1 | 9.8 |
| 16 g | L-pAad | CH2C6H5 | adamantan-1-yl | 87.5 ± 5.0 | 2.6 |
| 16 h | L-pAad | CH3 | C3H7 | 98.1 ± 9.4 | −9.1 |
| 16 i | L-pAad | CH3 | CH(CH3)2 | 124.3 ± 5.8 | −38.2 |
| 16 j | L-pAad | CH3 | C(CH3)3 | 93.1 ± 4.5 | −3.6 |
| 16 k | L-pAad | CH3 | c-C5H9 | 108.1 ± 14.4 | −20.2 |
| 16 l | L-pAad | CH3 | c-C6H11 | 103.2 ± 9.1 | −14.8 |
| 16 m | L-pAad | CH3 | adamantan-1-yl | 76.0 ± 10.9 | 15.4 |
| Series 3: | |||||
| 21 a | (R)-Ocp | CH3 | H | 28.2 ± 1.7[b,c] | 68.6 |
| 21 b | (R)-Ocp | C2H5 | H | 27.8 ± 4.8[b,c] | 69 |
| 21 c | (R)-Ocp | C3H7 | H | 34.5 ± 1.4[b,c] | 61.6 |
| 21 d | (R)-Ocp | CH(CH3)2 | H | 123.2 ± 5.9 | −36.8 |
| 21 e | (R)-Ocp | CH2C6H5 | H | 35.8 ± 1.9[b,c] | 60.1 |
| Series 4: | |||||
| 22 a | (R)-Ocp | CH2C6H5 | C3H7 | 26.6 ± 1.1[b,c] | 70.3 |
| 22 b | (R)-Ocp | CH2C6H5 | CH(CH3)2 | 32.7 ± 6.1[b,c] | 63.6 |
| 22 c | (R)-Ocp | CH2C6H5 | C(CH3)3 | 91.6 ± 3.8 | −1.9 |
| 22 d | (R)-Ocp | CH2C6H5 | c-C3H5 | 104.9 ± 5.5 | −16.7 |
| 22 e | (R)-Ocp | CH2C6H5 | c-C5H9 | ND | – |
| 22 f | (R)-Ocp | CH2C6H5 | c-C6H11 | 31.7 ± 2.3[b,c] | 64.7 |
| 22 g | (R)-Ocp | CH2C6H5 | adamantan-1-yl | ND | – |
| 22 h | (R)-Ocp | CH3 | C3H7 | 80.2 ± 2.4 | 10.7 |
| 22 i | (R)-Ocp | CH3 | CH(CH3)2 | 39.8 ± 5.5[b,c] | 55.6 |
| 22 j | (R)-Ocp | CH3 | C(CH3)3 | 89.8 ± 3.0 | −0.01 |
| 22 k | (R)-Ocp | CH3 | c-C6H11 | 106.2 ± 3.0 | −18.1 |
| 22 l | (R)-Ocp | CH3 | adamantan-1-yl | 32.6 ± 5.5[b,c] | 63.6 |
| control | 89.8 ± 6.8 | 0.00 | |||
| TRH | 52.6 ± 4[b] | 41.4 | |||
Pentobarbital was injected (50 mg kg−1 i.p.) 10 min after i.v. injection of the TRH/TRH analogues. Sleep time was recorded as the time elapsed from the onset of loss to regaining of the righting reflex. Six to eight Swiss albino mice (body weight 20–30 g) were used in each group. Values are expressed as the mean ± SEM; ND: not determined.
p < 0.05 relative to control using one-way ANOVA followed by post hoc Tukey test.
p < 0.05 relative to TRH using one-way ANOVA followed by post hoc Tukey test.
Analogues 10 a, 10 b, 10 d, 10 e, and 10 f (series 1) showed a significant decrease in sleeping time as compared with the control group; however, their effects were not significantly different from those of the TRH-treated group. All other analogues of this series were ineffective in decreasing sleep time. Similarly, none of the analogues from series 2 showed positive results in decreasing sleep time. Interestingly, some of the analogues from series 1 and 2 were found to increase the sleeping time significantly over control and TRH. In particular, analogues 10 h, 10 i, 10 j, 10 k, 10 l, 10 m, and 10 n of series 1, and 16 d of series 2 increased the sleeping time significantly relative to the control group. The observed potentiation of pentobarbital-induced sleep activity may be caused by alterations in receptor interactions due to the specific properties of the substitution on the His residue of the TRH analogues. This altered interaction may potentiate the action of pentobarbital, possibly by γ-aminobutyric acid (GABA)-mediated postsynaptic inhibition through an allosteric modification of GABA receptors, or by enhancement of K+ conductance or inhibition of Ca2+ conductance in neurons.[41–48] Most of the synthesized analogues of series 3 and 4 showed positive results when evaluated in vivo for antagonism of pentobarbital-induced sleeping time in mice. The sleeping time after administration of analogues 21 a, 21 b, 21 c, 22 a, 22 b, 22 f, and 22 l were 28.2 ± 1.7, 27.8 ± 4.8, 34.4 ± 1.4, 26.6 ± 1.1, 32.6 ± 6.1, 31.7 ± 2.3, and 32.6 ± 5.5 min, respectively. The decrease in sleeping time caused by these analogues was significantly different from control (89.1 ± 4.6 min) as well as from the TRH-treated group (52.6 ± 3.5 min). Analogues 21 a and 21 b of series 3 and 22 a of series 4 were found to be the most potent analogues, effecting decreases in sleep time of 68.3, 68.8, and 70.1 % relative to control. The analeptic effects of these TRH analogues may be due to the enhancement of arousal or excitatory activity of TRH. TRH is well known for this activity, which may be mediated through neuromodulation of various neurotransmitters such as acetylcholine, as it directly affects the septohippocampal cholinergic pathway.[1, 2] In addition to cholinergic activity, blockade of leaking K+ current has also been proposed for this particular activity.[49]
As evident from the receptor binding studies and preliminary in vivo screening using the pentobarbital-based sleep time assay, TRH peptides that show good analeptic activity bind with moderate affinities to TRH-R1 and TRH-R2, but exhibit selective activation at TRH-R2, supporting our hypothesis that TRH-R2-selective analogues exhibit potent CNS effects. The outcome of these results show that several TRH analogues (21 a, 21 b, 21 c, 22 a, 22 b, 22 f, and 22 l) are more potent than TRH in their ability to decrease pentobarbital-induced sleeping time in mice, which in turn prompted us to undertake further evaluation of these compounds in various models of epilepsy. The analogues were selected on the basis of their observed biological activity and availability.
Anticonvulsant activity
Previous pharmacological studies have demonstrated that TRH and its analogues inhibit pentylenetetrazole (PTZ)- or glutamate-induced seizures in rat.[17, 50] Therefore, selected compounds 21 a–c and 22 a,b were evaluated for anticonvulsant activity against PTZ-induced seizures in mice according to a method described earlier.[51] The compounds were dissolved in normal saline and administered at doses of 10 μmol kg−1 i.v. After 10 min, animals were treated with PTZ (65 mg kg−1 i.p.) and observed for 30 min after PTZ challenge. The results of these experiments are listed in Table 3. Compounds 21 a,b and 22 a,b provided significant protection against clonic seizures induced by PTZ (between 224 ± 5 and 149 ± 8 s, compared with TRH: 79 ± 7 s), whereas analogue 21 c is inactive and did not show any protection of PTZ-induced seizures. The selected TRH analogues were also evaluated for maximal electric shock (MES)-induced epilepsy using a protocol described earlier.[52] Treatment with TRH analogues at doses of 10 μmol kg−1 i.v. was done 10 min prior to the administration of MES at 45 mA for 0.2 s. The duration of tonic hind limb extension was recorded after MES, and the results are listed in Table 3. None of the tested analogues showed protection in the MES model. The effectiveness of TRH analogues in PTZ- but not in MES-induced seizure indicates their potential in clinical absence seizures. TRH is known to provide protection against seizures induced by kainic acid and glutamate, supporting the observed potency of TRH analogues in PTZ-induced seizures.[50, 53] TRH was also shown to increase GABA release, and this may contribute to its antiepileptic action.[54] However, additional studies are required to explore the exact mechanism of action of these analogues.
Table 3.
Effect of TRH and analogues on pentylenetetrazole (PTZ) and maximal electric shock (MES)-induced seizures.[a]
| PTZ: Clonic Convulsions | MES: Hind Limb Extension | ||
|---|---|---|---|
| Compd | Onset [s] | Frequency [min−1] | Duration [s] |
| saline | 67 ± 5 | 1.3 ± 0.16 | 13.6 ± 1.10 |
| TRH | 79 ± 7 | 1.3 ± 0.49 | 11.2 ± 0.50 |
| 21 a | 153 ± 7*, ** | 1.3 ± 0.33 | 10.0 ± 0.60 |
| 21 b | 149 ± 8*, ** | 1.3 ± 0.21 | 11.3 ± 1.4 |
| 21 c | 68 ± 3 | 1.3 ± 0.21 | 12.3 ± 0.90 |
| 22 a | 179 ± 8*, ** | 1.60 ± 0.06 | 12.0 ± 0.40 |
| 22 b | 224 ± 53*, ** | 1.20 ± 0.20 | 11.0 ± 0.70 |
Mice (6–9 per group) were treated with saline, TRH, or compounds (21 a–c and 22 a,b) at 10 μmol kg−1 i.v. 10 min prior to administration of PTZ at 65 mg kg−1 i.p. or MES seizure, and were observed for 1 h. Values are expressed as the mean ± SEM; statistical analysis was done with one-way ANOVA followed by Tukey test:
p <0.05 versus saline and
p <0.05 versus TRH-treated group.
Conclusions
In the present study, we probed the structural requirements of His and pGlu residues in receptor binding and biological activity by synthesizing new TRH analogues in which His residue was replaced with 1-alkyl-L-His/1,2-dialkyl-L-His; while pGlu residue was either retained or replaced with (1R)-3-Ocp/L-pAad. Analogues in which pGlu residue was replaced with (1R)-3-Ocp and His was substituted at the N-1/C-2 positions displayed the best pharmacological profile. Compounds 21 a, 21 b, and 22 a were all found to be full agonists with good to high functional selectivity for TRH-R2. At the same time, analogue 21 a exhibited highest binding affinity among the tested analogues. Analeptic activity evaluation using pentobarbital-induced sleeping time model revealed that some of the tested analogues were more potent compared than TRH. Analogues 21 a, 21 b, 21 c, 22 a, 22 b and 22 f produced 61 to 70 % reduction in sleeping time compared to control. The best analeptic profile was displayed by analogues 21 a, 21 b, and 22 a by reducing the sleeping time to almost 50 % more than TRH. To our surprise, some analogues like 10 l, 10 n and 16 d increased the sleeping time significantly. The results of this study indicate TRH-R2 as a plausible receptor subtype mediating the analeptic effects of TRH and its analogues. Analogues 21 a, 21 b, 21 c, 22 a, and 22 b were also investigated against PTZ- and MES-induced seizures in mice, which reflect the generalized and partial seizures that commonly occur in humans. Compounds 21 a, 21 b, 22 a and 22 b provide significant protection against PTZ-induced clonic seizures. However, all analogues failed to show protection against seizures induced by MES, suggesting their beneficial effects in the generalized form of seizures. To what extent the systemically active TRH analogues may have improved the BBB permeability remains unanswered. As clear from the results of receptor binding and in vivo evaluation, TRH peptides (21 a, 21 b, 22 a and 22 b) showing good analeptic/anticonvulsant activity bind with moderate affinities to TRH-R1 and TRH-R2, but exhibited selectivities for TRH-R2, which is possibly responsible for the CNS effects of TRH. In conclusion, these analogues are potent and functionally selective ligands and displayed potent analeptic and anticonvulsant activity when tested in vivo in mice. For the first time, we have shown that TRH-R2 selective analogues displayed potent CNS profile (anti-epileptic activity), thereby making them promising candidates for further development for the prophylactic management of epilepsy.
Experimental Section
Chemicals
Amino acids used in this study were purchased from Novabiochem (Germany) or ChemImpex International (Wood Dale, IL, USA). All other chemicals were purchased from Aldrich Chemical Ltd. (Milwaukee, WI, USA). Solvents used for peptide synthesis purification were acquired from commercial sources, were of analytical or HPLC grade, and were used without further purification unless otherwise stated.
Materials and methods
Peptides were routinely checked for their purity on pre-coated silica gel G254 TLC plates (Merck), and the spots were visualized by UV light and then by exposure to I2 vapors. Column chromatography was carried out on Merck silica gel (230–400 mesh) or neutral alumina. IR spectra (ν̃max in cm−1) were recorded on a Nicolet FTIR Impact 410 instrument either as neat or KBr pellets. 1H and 13C NMR spectra were recorded on a 300 MHz Bruker FTNMR (Avance DPX 300) spectrometer using tetramethylsilane (TMS) as internal standard, and chemical shift values (δ) are reported in ppm. MS data were recorded on an HRMS (Finnigan Mat LCQ) spectrometer (APCI/ESI). Elemental analyses were recorded on an Elementar Vario EL spectrometer. The results of elemental analyses were within ±0.4 % of theoretical values. Optical rotations were recorded on a PerkinElmer 241MC polarimeter. All final peptides were checked for their homogeneity on a Shimadzu SPD-M20A HPLC system using a Supelcosil™ LC-8 column, 5 μm (25 cm × 4.6 mm i.d.). The peptides were analyzed by using an isocratic solvent system of CH3CN/H2O/TFA (ratios as indicated) at a flow rate of 1 mL min−1.
General method for the synthesis of L-pGlu-L-His(1-methyl-2-alkyl)-L-ProNH2 (10 a–g)
4-Methylbenzhydrylamine (MBHA·HCl) resin (2, 300 mg, 0.31 mmol) was swelled in CH2Cl2 (5 mL) for 5 min, and then neutralized with DIPEA (10 % solution in CH2Cl2) for 5 min followed by washing with DMF (2 0 10 mL) and CH2Cl2 (10 mL). The reaction of the first amino acid Boc-L-Pro-OH (3) was done in the presence of the coupling reagent TBTU and activator DIPEA (10 % solution in CH2Cl2) for 2 h. The Boc group was cleaved with TFA (20 % solution in CH2Cl2) for 20 min. The amino acid linked resin was again neutralized with DIPEA as described above to afford 5. Reaction of 5 with pre-formed TBTU esters of Boc-L-His(1-methyl-2-alkyl)-OH 6 a–g for 2 h in DMF followed by removal of Boc group with TFA (20 % solution in CH2Cl2) and finally neutralization with DIPEA (10 % in CH2Cl2) afforded dipeptides 7 a–g. Upon coupling with Z-L-pGlu-OH (8) as described above, the dipeptides 7 a–g produced desired peptide resins 9 a–g. All coupling reactions were monitored quantitatively by Kaiser’s test for completion. The dry peptide-linked resin 9 a–g was taken in a two-necked round-bottom flask equipped with a drying tube and a rubber septum. A cleavage cocktail of EDT (1 mL), thioanisole (2 mL), TFA (10 mL), and TFMSA (1.0 mL) were added to the reaction vessel through the septum, and reaction mixture was stirred for 10 min at 4°C followed by stirring at ambient temperature for another 2 h. The crude peptide was separated from the solid support by filtration, and the resin was washed with TFA (3 × 4 mL). The solvent was removed under reduced pressure, and the residue was neutralized with a saturated solution of ammonium bicarbonate. The nonpolar impurities were removed by extracting the aqueous layer with Et2O (3 × 10 mL). The aqueous layer was evaporated under reduced pressure to afford crude peptide, which upon purification using column chromatography over neutral alumina using CHCl3/CH3OH (4:1) as eluent, produced 10 a–g.
L-pGlu-L-His(1-methyl-2-propyl)-L-ProNH2 (10 a)
mp: 77–80 °C (dec.); 1H NMR (CD3OD): δ= 7.16 (s, 1 H), 4.90 (m, 1 H), 4.46 (m, 1 H), 4.21 (m, 1 H), 3.79 (m, 1 H), 3.73 (s, 3 H), 3.54 (m, 1 H), 2.95 (m, 2 H), 2.87 (t, 2 H, J =7.8 Hz), 2.43–1.92 (m, 8 H), 1.75 (m, 2 H), 1.01 (t, 3 H, J =7.8 Hz); 13C NMR (CD3OD): δ = 14.51, 21.72, 24.33, 25.16, 29.33, 29.38, 30.60, 31.83, 32.85, 46.81, 51.13, 55.27, 60.20, 118.47, 135.22, 147.45, 169.46, 171.39, 177.40, 177.59; MS (APCI): m/z 419 [M+1]+; Anal. calcd for C20H30N6O4 (418.5): C 57.40, H 7.23, N 20.08, found: C 57.82, H 6.77, N 20.37; Rf = 0.57 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 4.60 min, purity: 92.2 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pGlu-L-His(1-methyl-2-isopropyl)-L-ProNH2 (10 b)
mp: 92–94 °C (dec.); 1H NMR (CD3OD): δ= 7.22 (s, 1 H), 4.97 (m, 1 H), 4.46 (m, 1 H), 4.23 (m, 1 H), 3.75 (m, 1 H), 3.79 (s, 3 H), 3.59 (m, 1 H), 3.38 (m, 1 H), 3.13 (m, 2 H), 2.43–1.91 (m, 8 H), 1.40 (m, 6 H); 13C NMR (CD3OD): δ=22.36, 24.33, 25.16, 29.18, 29.23, 33.11, 46.81, 51.20, 55.27, 60.20, 118.16, 136.32, 152.94, 169.66, 171.20, 177.50, 177.70; MS (APCI): m/z 419 [M+1]+; Anal. calcd for C20H30N6O4 (418.5): C 57.40, H 7.23, N 20.08, found: 57.53, H 7.42, N 20.44; Rf = 0.56 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 4.47 min, purity: 95.2 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pGlu-L-His(1-methyl-2-tert-butyl)-L-ProNH2 (10 c)
mp: 85–87 °C (dec.); 1H NMR (CD3OD): δ= 6.79 (s, 1 H), 4.87 (m, 1 H), 4.39 (m, 1 H), 4.18 (m, 1 H), 3.77 (m, 1 H), 3.74 (s, 3 H), 3.56 (m, 1 H), 2.93 (m, 2 H), 2.45–1.96 (m, 8 H), 1.40 (m, 9 H); 13C NMR (CD3OD): δ=24.40, 25.16, 28.35, 29.23, 29.38, 31.68, 32.56, 35.14, 46.81, 51.28, 55.27, 60.20, 117.97, 135.44, 154.42, 169.67, 170.65, 176.80, 177.21; MS (APCI): m/z 433 [M+1]+; Anal. calcd for C21H32N6O4 (432.2): C 58.32, H 7.46, N, 19.43, found: C 58.77, H 7.29, N 19.76; Rf =0.60 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR =4.64 min, purity: 93.8 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pGlu-L-His(1-methyl-2-cyclopropyl)-L-ProNH2 (10 d)
mp: 64–66°C (dec.); 1H NMR (CD3OD): δ= 6.81 (s, 1 H), 4.76 (m, 1 H), 4.38 (m, 1 H), 4.17 (m, 1 H), 3.77 (m, 1 H), 3.65 (s, 3 H), 3.49 (m, 1 H), 2.88 (m, 2 H), 2.44–1.87 (m, 8 H), 2.14 (m, 1 H), 0.92 (m, 4 H); 13C NMR (CD3OD): δ=7.81, 8.51, 24.33, 25.16, 29.33, 29.38, 31.83, 33.27, 46.81, 51.19, 55.27, 60.20, 117.33, 136.66, 151.70, 169.66, 171.39, 177.40, 177.59; MS (APCI): m/z 417 [M+1]+; Anal. calcd for C20H28N6O4 (416.5): C 57.68, H 6.78, N 20.18, found: C 57.79, H 6.93, N 20.29; Rf = 0.62 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 4.23 min, purity: 97.5 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pGlu-L-His(1-methyl-2-cyclopentyl)-L-ProNH2 (10 e)
mp: 100–101 °C (dec.); 1H NMR (CD3OD): δ=6.82 (s, 1 H), 4.80 (m, 1 H), 4.38 (m, 1 H), 4.18 (m, 1 H), 3.78 (m, 1 H), 3.58 (s, 3 H), 3.32 (m, 1 H), 3.16 (m, 1 H), 2.93 (m, 2 H), 2.47–1.26 (m, 16 H); 13C NMR (CD3OD): δ= 24.33, 24.76, 25.16, 29.23, 29.38, 31.68, 33.04, 33.42, 46.81, 51.25, 55.27, 60.20, 118.08, 134.20, 150.20, 168.50, 170.65, 176.90, 178.20; MS (APCI): m/z 445 [M+1]+; Anal. calcd for C22H32N6O4 (444.5): C 59.44, H 7.26, N 18.91, found: C 59.81, H 7.62, N 18.73; Rf = 0.64 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 5.80 min, purity: 99.1 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pGlu-L-His(1-methyl-2-cyclohexyl)-L-ProNH2 (10 f)
mp: 110–112 °C (dec.); 1H NMR (CD3OD): δ= 6.81 (s, 1 H), 4.82 (m, 1 H), 4.39 (m, 1 H), 4.17 (m, 1 H), 3.79 (m, 1 H), 3.59 (s, 3 H), 3.49 (m, 1 H), 2.93 (m, 2 H), 2.77 (m, 1 H), 2.41–1.26 (m, 18 H); 13C NMR (CD3OD): δ= 23.98, 25.10, 26.15, 26.85, 29.21, 32.05, 33.09, 34.07, 46.46, 51.10, 54.80, 60.60, 118.00 134.33 149.49 169.66, 171.39, 177.40, 178.10; MS (APCI): m/z 459 [M+1]+; Anal. calcd for C23H34N6O4 (458.6): C 60.24, H 7.47, N 18.33, found: C 60.29, H 7.78, N 17.98; Rf = 0.68 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 7.10 min, purity: 97.1 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pGlu-L-His[1-methyl-2-(adamantan-1-yl)]-L-ProNH2 (10 g)
mp: 121–123 °C (dec.); 1H NMR (CD3OD): δ =6.72 (s, 1 H), 4.64 (m, 1 H), 4.39 (m, 1 H), 4.17 (m, 1 H), 3.82 (m, 1 H), 3.77 (s, 3 H), 3.53 (m, 1 H), 2.91 (m, 2 H), 2.46–2.24 (m, 4 H), 2.21–1.81 (m, 19 H); 13C NMR (CD3OD): δ= 25.10, 25.1, 29.3, 29.6, 31.80, 35.06, 36.01, 39.55, 40.39, 46.81, 51.34, 55.30, 60.10, 117.94, 135.84, 149.60, 169.67, 171.45, 177.28, 177.60; MS (APCI): m/z 511 [M+1]+; Anal. calcd for C27H38N6O4 (510.6): C 63.51, H 7.50, N 16.46, found: C 63.89, H 7.22, N 16.91; Rf =0.72 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 21.22 min, purity: 94.2 % [CH3CN/H2O/TFA (20:80:0.1 %)].
General method for the synthesis of l-pGlu-l-His(1-benzyl-2-alkyl)-L-ProNH2 (10 h–n)
The dipeptide salt (13 a–g, 1 mmol) was neutralized by addition of a solution of 7 N NH3 in CH3OH (10 mL) for 10 min at 0 °C. The solvent was evaporated under reduced pressure to afford free dipeptides. A solution of free peptides (0.75 mmol) in DMF (4 mL) was cooled to 4 °C, and L-pGlu-OTcp (14, 0.81 mmol) was added, and the reaction mixture was stirred at 4 °C for 36 h. The solvent was removed under reduced pressure, and the resulting residue was purified by column chromatography over neutral alumina using CHCl3/CH3OH (4:1) as eluent to provide tripeptides (10h–n).
L-pGlu-L-His(1-benzyl-2-propyl)-L-ProNH2 (10 h)
Yield: 18 %; mp: 102–104 °C (dec.); 1H NMR (CD3OD): δ=7.33 (m, 3 H), 7.18 (m, 2 H), 6.95 (s, 1 H), 5.14 (s, 2 H), 4.80 (m, 1 H), 4.41 (m, 1 H), 4.18 (m, 1 H), 3.78 (m, 1 H), 3.42 (m, 1 H), 2.98 (m, 2 H), 2.62 (t, 2 H, J =7.5 Hz), 2.42–1.94 (m, 8 H), 1.59 (m, 2 H), 0.91 (t, 3 H, J =7.2 Hz); MS (APCI): m/z 495 [M+1]+; Anal. calcd for C26H34N6O4 (494.6): C 63.14, H 6.93, N 16.99, found: C 62.95, H 7.08, N 17.14; Rf =0.47 [CH3OH/25 % NH4OH/CHCl3 (18:2:80)]; HPLC: tR =4.05 min, purity: 94.2 % [CH3CN/H2O/TFA (50:50:0.1 %)].
L-pGlu-L-His(1-benzyl-2-isopropyl)-L-ProNH2 (10 i)
Yield: 19 %; mp: 95–97 °C (dec.); 1H NMR (CDCl3): δ= 7.36 (m, 3 H), 7.04 (m, 2 H), 6.68 (s, 1 H), 5.02 (s, 2 H), 4.81 (m, 1 H), 4.47 (m, 1 H), 4.15 (m, 1 H), 3.70 (m, 1 H), 3.27 (m, 1 H), 2.99 (m, 3 H), 2.45–1.88 (m, 8 H), 1.18 (m, 6 H); MS (APCI): m/z 495 [M+1]+; Anal. calcd for C26H34N6O4 (494.6): C 63.14, H 6.93, N 16.99, found: C 63.38, H 7.24, N 16.82; Rf = 0.45 [CH3OH/25 % NH4OH/CHCl3 (18:2:80)]; HPLC: tR = 4.04 min, purity: 96.8 % [CH3CN/H2O/TFA (50:50:0.1 %)].
L-pGlu-L-His(1-benzyl-2-tert-butyl)-L-ProNH2 (10 j)
Yield: 12 %; mp: 104–106 °C (dec.); 1H NMR (CD3OD): δ= 7.30 (m, 3 H), 7.07 (m, 2 H), 6.79 (s, 1 H), 5.35 (m, 2 H), 4.81 (m, 1 H), 4.39 (m, 1 H), 4.15 (m, 1 H), 3.78 (m, 1 H), 3.56 (m, 1 H), 2.94 (m, 2 H), 2.40–1.95 (m, 8 H), 1.36 (s, 9 H); MS (APCI): m/z 509 [M+1]+; Anal. calcd for C27H36N6O4 (508.3): C 63.76, H 7.13, N 16.52, found: C 64.12, H 6.89, N 16.59; Rf = 0.45 [CH3OH/25 % NH4OH/CHCl3 (14:2:84)]; HPLC: tR =4.07 min, purity: 95.1 % [CH3CN/H2O/TFA (50:50:0.1 %)].
L-pGlu-L-His(1-benzyl-2-cyclopropyl)-L-ProNH2 (10 k)
Yield: 13 %; mp: 84–86 °C (dec.); 1H NMR (CD3OD): δ =7.29 (m, 5 H), 6.96 (s, 1 H), 5.24 (m, 2 H), 4.78 (m, 1 H), 4.39 (m, 1 H), 4.16 (m, 1 H), 3.76 (m, 1 H), 3.43 (m, 1 H), 2.93 (m, 2 H), 2.40–1.93 (m, 8 H), 1.83 (m, 1 H), 0.90 (m, 4 H); MS (APCI): m/z 493 [M+1]+; Anal. calcd for C26H32N6O4 (492.6): C 63.40, H 6.55, N 17.06, found: C 63.44, H 6.69, N 16.94; Rf = 0.55 [CH3OH/25 % NH4OH/CHCl3 (14:2:84)]; HPLC: tR = 4.02 min, purity: 95.0 % [CH3CN/H2O/TFA (50:50:0.1 %)].
L-pGlu-L-His(1-benzyl-2-cyclopentyl)-L-ProNH2 (10 l)
Yield: 27 %; mp: 110–112 °C (dec.); 1H NMR (CD3OD): δ= 7.30 (m, 3 H), 7.15 (m, 2 H), 6.92 (s, 1 H), 5.15 (s, 2 H), 4.80 (m, 1 H), 4.39 (m, 1 H), 4.16 (m, 1 H), 3.77 (m, 1 H), 3.47 (m, 1 H), 2.96 (m, 3 H), 2.30–1.28 (m, 16); MS (APCI): m/z 521 [M+1]+; Anal. calcd for C28H36N6O4 (520.6): C 64.60, H 6.97, N 16.14, found: C 64.89, H 7.11, N 15.98; Rf = 0.40 [CH3OH/25 % NH4OH/CHCl3 (14:2:84)]; HPLC: tR = 4.58 min, purity: 98.5 % [CH3CN/H2O/TFA (50:50:0.1 %)].
L-pGlu-L-His(1-benzyl-2-cyclohexyl)-L-ProNH2 (10 m)
Yield: 23 %; mp: 120–122 °C (dec.); 1H NMR (CDCl3): δ7.34 (m, 3 H), 7.06 (m, 2 H), 6.67 (s, 1 H), 5.15 (s, 2 H), 4.89 (m, 1 H), 4.46 (m, 1 H), 4.14 (m, 1 H), 3.69 (m, 1 H), 3.26 (m, 1 H), 2.97 (m, 2 H), 2.56 (m, 1 H), 2.47–1.22 (m, 18 H); MS (APCI): m/z 535 [M+1]+; Anal. calcd for C29H38N6O4 (534.7): C 65.15, H 7.16, N 15.72, found: C 64.65, H 6.98, N 16.20; Rf = 0.40 [CH3OH/25 % NH4OH/CHCl3 (14:2:84)]; HPLC: tR = 4.77 min, purity: 92.8 % [CH3CN/H2O/TFA (50:50:0.1 %)].
L-pGlu-L-His[1-benzyl-2-(adamantan-1-yl)]-L-ProNH2 (10 n)
Yield: 13 %; mp: 116–118 °C (dec.); 1H NMR (CD3OD): δ =7.29 (m, 3 H), 7.04 (m, 2 H), 6.77 (s, 1 H), 5.41 (m, 2 H), 4.80 (m, 1 H), 4.39 (m, 1 H), 4.15 (m, 1 H), 3.78 (m, 1 H), 3.57 (m, 1 H), 2.95 (m, 2 H), 2.44–1.28 (m, 23 H); MS (APCI): m/z 587 [M+1]+; Anal. calcd for C33H42N6O4 (586.6): C 67.55, H 7.22, N 14.32, found: C 67.88, H 7.66, N 14.57; Rf = 0.60 [CH3OH/25 % NH4OH/CHCl3 (14:2:84)]; HPLC: tR =5.92 min, purity: 92.8 % [CH3CN/H2O/TFA (50:50:0.1 %)].
General method for the synthesis of L-pAad-l-His(1,2-di-alkyl)-L-ProNH2 (16 a–m)
The dipeptide salt (13 a–m, 1 mmol) was neutralized by addition of a solution of 7 N NH3 in CH3OH (10 mL) for 10 min at 0 °C. A solution of free peptide in DMF (4 mL) was cooled to 4°C, and pAad-OPfp (15, 0.81 mmol) was added, and the reaction mixture was stirred at 4 °C for 36 h. The solvent was removed under reduced pressure, and the resulting residue was purified by column chromatography over neutral alumina using CH3OH/CHCl3 (2:8) as eluent to afford tripeptides 16 a–m.
L-pAad-L-His(1-benzyl-2-propyl)-L-ProNH2 (16 a)
Yield: 26 %; mp: 74–76 °C (dec.); 1H NMR (CD3OD): δ= 7.31 (m, 3 H), 7.16 (m, 2 H), 6.93 (s, 1 H), 5.12 (s, 2 H), 4.79 (m, 1 H), 4.40 (m, 1 H), 4.00 (m, 1 H), 3.81 (m, 1 H), 3.44 (m, 1 H), 2.95 (m, 2 H), 2.59 (t, 2 H, J =7.5 Hz), 2.29–1.66 (m, 10 H), 1.58 (m, 2 H, CH2), 0.89 (t, 3 H, J =7.2 Hz); MS (APCI): m/z 509 [M+1]+; Anal. calcd for C27H36N6O4 (508.6): C 63.76, H 7.13, N 16.52, found: C 64.07, H 7.31, N 16.19; Rf =0.48 [CH3OH/25 % NH4OH/CHCl3 (16:2:82)]; HPLC: tR = 4.71 min, purity: 99.8 % [CH3CN/H2O/TFA (40:60:0.1 %)].
L-pAad-L-His(1-benzyl-2-isopropyl)-L-ProNH2 (16 b)
Yield: 13 %; mp: 68–70 °C (dec.); 1H NMR (CD3OD): δ= 7.31 (m, 3 H), 7.14 (m, 2 H), 6.92 (s, 1 H), 5.15 (s, 2 H), 4.82 (m, 1 H), 4.38 (m, 1 H), 4.01 (m, 1 H), 3.79 (m, 1 H), 3.45 (m, 1 H), 3.04 (m, 1 H), 2.97 (m, 2 H), 2.29–1.68 (m, 10 H), 1.18 (m, 6 H); MS (APCI): m/z 509 [M+1]+; Anal. calcd for C27H36N6O4 (508.6): C 63.76, H 7.13, N 16.52, found: C 64.07, H 7.31, N 16.19; Rf = 0.46 [CH3OH/25 % NH4OH/CHCl3 (16:2:82)]; HPLC: tR = 4.63 min, purity: 95.9 % [CH3CN/H2O/TFA (40:60:0.1 %)].
L-pAad-L-His(1-benzyl-2-tert-butyl)-L-ProNH2 (16 c)
Yield: 19 %; mp: 77–80 °C (dec.); 1H NMR (CD3OD): δ= 7.29 (m, 3 H), 7.07 (m, 2 H), 6.81 (s, 1 H), 5.36 (s, 2 H), 4.81 (m, 1 H), 4.39 (m, 1 H), 4.00 (m, 1 H), 3.81 (m, 1 H), 3.58 (m, 1 H), 2.96 (m, 2 H), 2.30–1.70 (m, 10 H), 1.37 (s, 9 H); MS (APCI): m/z 523 [M+1]+; Anal. calcd for C28H38N6O4 (522.6): C 64.35, H 7.33, N 16.08, found: C 64.46, H 7.55, N 15.79; Rf = 0.42 [CH3OH/25 % NH4OH/CHCl3 (16:2:82)]; HPLC: tR =4.82 min, purity: 98.9 % [CH3CN/H2O/TFA (40:60:0.1 %)].
L-pAad-L-His(1-benzyl-2-cyclopropyl)-L-ProNH2 (16 d)
Yield: 24 %; mp: 72–74 °C (dec.); 1H NMR (CD3OD): δ= 7.27 (m, 3 H), 7.11 (m, 2 H), 6.94 (s, 1 H), 5.22 (s, 2 H), 4.77 (m, 1 H), 4.37 (m, 1 H), 3.99 (m, 1 H), 3.75 (m, 1 H), 3.43 (m, 1 H), 2.92 (m, 2 H), 2.28–1.67 (m, 11 H), 0.88 (m, 4 H); MS (APCI): m/z 507 [M+1]+; Anal. calcd for C27H34N6O4 (506.6): C 64.01, H 6.76, N 16.59, found: C 63.96, H 6.64, N 16.51; Rf =0.54 [CH3OH/25 % NH4OH/CHCl3 (16:2:82)]; HPLC: tR = 4.51 min, purity: 96.6 % [CH3CN/H2O/TFA (40:60:0.1 %)].
L-pAad-L-His(1-benzyl-2-cyclopentyl)-L-ProNH2 (16 e)
Yield: 28 %; mp: 80–83 °C (dec.); 1H NMR (CD3OD): δ= 7.31 (m, 3 H), 7.14 (m, 2 H), 6.93 (s, 1 H), 4.88 (s, 2 H), 4.81 (m, 1 H), 4.38 (m, 1 H), 4.01 (m, 1 H), 3.75 (m, 1 H), 3.43 (m, 1 H), 3.05 (m, 1 H), 2.96 (m, 2 H), 2.28–1.60 (m, 18 H); MS (APCI): m/z 535 [M+1]+; Anal. calcd for C29H38N6O4 (534.7): C 65.15, H 7.16, N 15.72, found: C 65.34, H 6.94, N 16.07; Rf =0.58 [CH3OH/25 % NH4OH/CHCl3 (16:2:82)]; HPLC: tR = 5.39 min, purity: 97.8 % [CH3CN/H2O/TFA (40:60:0.1 %)].
L-pAad-L-His(1-benzyl-2-cyclohexyl)-L-ProNH2 (16 f)
Yield: 23 %; mp: 88–90 °C (dec.); 1H NMR (CD3OD): δ= 7.31 (m, 3 H), 7.16 (m, 2 H), 6.92 (s, 1 H), 5.14 (s, 2 H), 4.81 (m, 1 H), 4.38 (m, 1 H), 4.01 (m, 1 H), 3.78 (m, 1 H), 3.45 (m, 1 H), 2.96 (m, 2 H), 2.82 (m, 1 H, CH), 2.29–1.28 (m, 20 H); MS (APCI): m/z 549 [M+1]+; Anal. calcd for C30H40N6O4 (548.7): C 65.67, H 7.35, N 15.32, found: C 65.98, H 7.23, N 14.97; Rf =0.62 [CH3OH/25 % NH4OH/CHCl3 (16:2:82)]; HPLC: tR = 6.32 min, purity: 93.1 % [CH3CN/H2O/TFA (40:60:0.1 %)].
L-pAad-L-His[1-benzyl-2-(adamantan-1-yl)]-L-ProNH2 (16 g)
Yield: 17 %; mp: 96–98 °C (dec.); 1H NMR (CD3OD): δ= 7.29 (m, 3 H), 7.02 (m, 2 H), 6.76 (s, 1 H), 5.37 (m, 2 H), 4.71 (m, 1 H), 4.38 (m, 1 H), 3.99 (m, 1 H), 3.80 (m, 1 H), 3.58 (m, 1 H), 2.94 (m, 2 H), 2.27 (m, 4 H), 2.14–1.22 (m, 21 H); MS (APCI): m/z 601 [M+1]+; Anal. calcd for C34H44N6O4 (600.3): C 67.98, H 7.38, N 13.99, found: C 68.25, H 7.18, N 13.50; Rf = 0.52 [CH3OH/25 % NH4OH/CHCl3 (8:2:90)]; HPLC: tR = 9.49 min, purity: 94.4 % [CH3CN/H2O/TFA (40:60:0.1 %)].
L-pAad-L-His(1-methyl-2-propyl)-L-ProNH2 (16 h)
Yield: 32 %; mp: 64–66 °C (dec.); 1H NMR (CDCl3): δ=8.19 (bs, 1 H), 6.63 (s, 1 H), 4.77 (m, 1 H), 4.50 (m, 1 H), 4.02 (m, 1 H), 3.67 (m, 1 H), 3.52 (s, 3 H), 3.21 (m, 1 H), 2.97 (m, 2 H), 2.53 (t, 2 H, J =7.5 Hz), 2.42–1.72 (m, 10 H), 1.58 (m, 2 H, CH2), 0.90 (t, 3 H, J =7.2 Hz); MS (APCI): m/z 433 [M+1]+; Anal. calcd for C21H32N6O4 (432.5): C 58.32, H 7.46, N 19.43, found: C 58.64, H 7.57, N 19.11; Rf = 0.55 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR =4.93 min, purity: 94.9 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pAad-L-His(1-methyl-2-isopropyl)-L-ProNH2 (16 i)
Yield: 38 %; mp: 70–71 °C (dec.); 1H NMR (CD3OD): δ=7.16 (s, 1 H), 4.96 (m, 1 H), 4.45 (m, 1 H), 4.05 (m, 1 H), 3.81 (m, 1 H), 3.77 (s, 3 H), 3.56 (m, 1 H), 3.37 (m, 1 H), 3.11 (m, 2 H), 2.28 (m, 4 H), 2.09–1.72 (m, 6 H), 1.38 (m, 6 H); MS (APCI): m/z 433 [M+1]+; Anal. calcd for C21H32N6O4 (432.5): C 58.32, H 7.46, N 19.43, found: C 58.44, H 7.59, N 19.58; Rf = 0.55 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 4.85 min, purity: 95.5 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pAad-L-His(1-methyl-2-tert-butyl)-L-Pro-NH2 (16 j)
Yield: 38 %; mp: 74–75 °C (dec.); 1H NMR (CD3OD): δ=7.20 (s, 1 H), 5.00 (m, 1 H), 4.43 (m, 1 H), 4.05 (m, 1 H), 3.93 (s, 3 H), 3.77 (m, 1 H), 3.59 (m, 1 H), 3.09 (m, 2 H), 2.45–1.72 (m, 10 H), 1.52 (s, 9 H); MS (APCI): m/z 447 [M+1]+; Anal. calcd for C22H34N6O4 (446.5): C 59.17, H 7.67, N 18.82, found: C 58.99, H 7.53, N 19.11; Rf = 0.57 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR =5.42 min, purity: 99.2 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pAad-L-His(1-methyl-2-cyclopentyl)-L-ProNH2 (16 k)
Yield: 27 %; mp: 77–79 °C (dec.); 1H NMR (CD3OD): δ=6.83 (s, 1 H), 4.82 (m, 1 H), 4.38 (m, 1 H), 4.02 (m, 1 H), 3.81 (m, 1 H), 3.59 (s, 3 H), 3.55 (m, 1 H), 3.17 (m, 1 H), 2.93 (m, 2 H), 2.27–1.28 (m, 18 H); MS (APCI): m/z 459 [M+1]+; Anal. calcd for C23H34N6O4 (458.7): C 60.24, H 7.47, N 18.33, found: C 60.22, H 7.42, N 18.31; Rf = 0.60 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR =6.98 min, purity: 96.5 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pAad-L-His(1-methyl-2-cyclohexyl)-L-ProNH2 (16 l)
Yield: 22 %; mp: 82–84 °C (dec.); 1H NMR (CD3OD): δ=6.77 (s, 1 H), 4.80 (m, 1 H), 4.36 (m, 1 H), 4.02 (m, 1 H), 3.81 (m, 1 H), 3.57 (s, 3 H), 3.53 (m, 1 H), 2.92 (m, 2 H), 2.73 (m, 1 H), 2.30–1.26 (m, 20 H); MS (APCI): m/z 473 [M+1]+; Anal. calcd for C24H36N6O4 (472.7): C 61.00, H 7.68, N 17.78, found: C 61.15, H 7.98, N 17.47; Rf = 0.62 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR =8.95 min, purity: 97.8 % [CH3CN/H2O/TFA (20:80:0.1 %)].
L-pAad-L-His[1-methyl-2-(adamantan-1-yl)]-L-ProNH2 (16 m)
Yield: 15 %; mp: 88–90 °C (dec.); 1H NMR (CDCl3): δ= 6.61 (s, 1 H), 4.83 (m, 1 H), 4.46 (m, 1 H), 4.00 (m, 1 H), 3.73 (s, 3 H), 3.66 (m, 1 H), 3.31 (m, 1 H), 2.91 (m, 2 H), 2.35 (m, 4 H), 2.18–1.25 (m, 21 H); MS (APCI): m/z 525 [M+1]+; Anal. calcd for C28H40N6O4 (524.6): C 64.10, H 7.68, N 16.02, found: C 63.92, H 7.96, N 15.73; Rf =0.59 [CH3OH/25 % NH4OH/CHCl3 (20:2:78)]; HPLC: tR = 24.78 min, purity: 94.7 % [CH3CN/H2O/TFA (20:80:0.1 %)].
Synthesis of (1R)-3-Ocp-l-His(1-alkyl)-L-ProNH2 (21 a–e)
The dipeptide salt (19a–e, 1 mmol) was neutralized with a solution of 7 N NH3 in CH3OH (10 mL) for 10 at 0 °C min. HOBt (1.1 mmol) was added to a solution of free dipeptide in anhydrous DMF (10 mL). The reaction mixture was cooled to −10 °C, and DIC (1.1 mmol) was added in one portion. The reaction mixture was stirred for an additional 5 min at −10 °C. (1R)-3-Ocp-OH (20, 1 mmol) was then added to the reaction mixture, and stirring was continued for an additional 36 h at 4°C. The solvent was evaporated under reduced pressure, and the crude product was purified by column chromatography on neutral alumina using CH3OH/CHCl3 (10:90) as eluent to afford tripeptides 21a–e.
(1 R)-3-Ocp-L-His(1-methyl)-L-ProNH2 (21 a)
Yield: 30 %; mp: 101–103 °C (dec.); 1H NMR (CD3OD): δ= 7.50 (s, 1 H), 6.94 (s, 1 H), 4.79 (m, 1 H), 4.39 (m, 1 H), 3.72 (m, 1 H), 3.67 (s, 3 H), 3.43 (m, 1 H), 3.05 (m, 1 H), 2.96 (m, 2 H), 2.35–1.91 (m, 10 H); 13C NMR (CD3OD): δ= 24.57, 25.84, 29.38, 31.97, 33.02, 37.57, 40.16, 42.36, 46.81, 50.51, 60.20, 117.33, 137.31, 139.77, 169.40, 174.84, 177.59, 216.11; MS (APCI): m/z 376 [M+1]+; Anal. calcd for C18H25N5O4 (375.4): C 57.59, H 6.71, N 18.65, found: C 57.97, H 6.45, N 18.51; Rf = 0.44 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR = 3.98 min, purity: 99.5 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His(1-ethyl)-L-ProNH2 (21 b)
Yield: 49 %; mp: 96–98°C (dec.); 1H NMR (CD3OD): δ= 7.58 (s, 1 H), 7.03 (s, 1 H), 4.80 (m, 1 H), 4.39 (m, 1 H), 4.00 (q, 2 H, J =7.2 Hz), 3.75 (m, 1 H), 3.38 (m, 1 H), 3.06 (m, 1 H), 2.96 (m, 2 H), 2.34–1.90 (m, 10 H), 1.40 (t, 3 H, J = 7.2 Hz); 13C NMR (CD3OD): δ=15.72, 24.10, 26.20, 29.70, 31.67, 38.10, 39.95, 41.34, 42.70, 46.60, 50.99, 60.45, 115.35, 135.84, 138.74, 169.27, 174.0, 177.10, 215.87; MS (APCI): m/z 390 [M+1]+; Anal. calcd for C19H27N5O4 (389.5): C 58.60, H 6.99, N 17.98, found: C 58.91, H 7.07, N 17.61; Rf = 0.48 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR = 4.30 min, purity: 97.4 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His(1-propyl)-L-ProNH2 (21 c)
Yield: 31 %; mp: 90–92°C (dec.); 1H NMR (CD3OD): δ= 7.56 (s, 1 H), 7.01 (s, 1 H), 4.78 (m, 1 H), 4.40 (m, 1 H), 3.92 (t, 2 H, J =6.6 Hz), 3.78 (m, 1 H), 3.40 (m, 1 H), 3.06 (m, 1 H), 2.96 (m, 2 H), 2.35–1.95 (m, 10 H), 1.79 (m, 2 H), 0.89 (t, 3 H, J =6.9 Hz); 13C NMR (CD3OD): δ= 11.05, 23.64, 24.08, 25.94, 29.08, 32.10, 37.12, 40.44, 42.97, 46.50, 48.24, 51.01, 60.20, 116.19, 136.01, 139.60, 168.86, 174.53, 177.07, 216.50; MS (APCI): m/z 404 [M+1]+; Anal. calcd for C20H29N5O4 (403.5): C 59.54, H 7.24, N 17.36, found: C 59.83, H 7.49, N 17.39; Rf =0.55 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR =4.95 min, purity: 99.6 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His(1-isopropyl)-L-ProNH2 (21 d)
Yield: 20 %; mp: 88–90 °C (dec.); 1H NMR (CD3OD): δ= 7.64 (s, 1 H), 7.11 (s, 1 H), 4.80 (m, 1 H), 4.40 (m, 1 H), 3.77 (m, 1 H), 3.36 (m, 1 H), 3.04 (m, 1 H), 2.96 (m, 3 H), 2.35–1.95 (m, 10 H), 1.46 (d, 6 H, J =6.0 Hz); 13C NMR (CD3OD): δ= 24.33, 25.84, 26.46, 29.38, 33.09, 37.57, 40.16, 42.36, 46.81, 47.95, 51.08, 60.20, 112.02, 133.40, 137.63, 169.49, 174.84, 177.84, 177.51, 216.25; MS (APCI): m/z 404 [M+1]+; Anal. calcd for C20H29N5O4 (403.5): C 59.54, H 7.24, N 17.36, found: C 59.68, H 6.96, N 17.74; Rf = 0.53 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR = 4.79 min, purity: 97.6 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His(1-benzyl)-L-ProNH2 (21 e)
Yield: 18 %; mp: 96–98°C (dec.); 1H NMR (CDCl3): 7.71 (s, 1 H), 7.35 (m, 5 H), 7.05 (s, 1 H), 5.19 (m, 2 H), 4.80 (m, 1 H), 4.44 (m, 1 H), 3.79 (m, 1 H), 3.35 (m, 1 H), 3.07 (m, 1 H), 2.96 (m, 2 H), 2.35–1.71 (m, 10 H); 13C NMR (CD3OD): δ=23.87, 24.84, 29.38, 32.00, 37.30, 39.95, 42.05, 42.81, 50.10, 50.94, 60.59, 117.19, 127.13, 128.20, 128.99, 136.18, 137.63, 139.05, 169.27, 174.57, 177.59, 216.05; MS (APCI): m/z 452 [M+1]+; Anal. calcd for C24H29N5O4 (451.5): C 63.84, H 6.47, N 15.51, found: C 64.06, H 6.20, N 15.65; Rf = 0.59 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR = 8.43 min, purity: 97.0 % [CH3CN/H2O/TFA (20:80:0.1 %)].
General method for the synthesis of (1R)-3-Ocp-L-His(1,2-di-alkyl)-L-ProNH2 (22 a–l)
Protected dipeptide (12, 0.73 mmol) was treated with a solution of 40 % TFA in CH2Cl2 (15 mL) at 4 °C for 30 min. The salt was neutralized with a solution of 7 N NH3 in CH3OH (10 mL) for 10 min at 0 °C. HOBt (1.1 mmol) was added to a solution of free dipeptides in anhydrous DMF (10 mL). The reaction mixture was cooled to −10 °C, and DIC (1.1 mmol) was added in one portion. The reaction mixture was stirred for an additional 5 min at −10 °C. (1R)-3-Ocp-OH (20, 1 mmol) was then added to the reaction mixture, and stirring was continued for an additional 36 h at 4°C. The solvent was removed, and the crude product was purified by column chromatography on neutral alumina using CH3OH/CHCl3 (10:90) to afford tripeptides 22a–l.
(1R)-3-Ocp-L-His(1-benzyl-2-propyl)-L-ProNH2 (22 a)
Yield: 27 %; mp: 110–112 °C (dec.); 1H NMR (CD3OD): δ= 7.31 (m, 3 H), 7.17 (m, 2 H), 6.93 (s, 1 H), 5.13 (s, 2 H), 4.81 (m, 1 H), 4.39 (m, 1 H), 3.78 (m, 1 H), 3.46 (m, 1 H), 3.03 (m, 1 H), 2.95 (m, 2 H), 2.61 (t, 2 H, J = 7.8 Hz), 2.34–1.93 (m, 10 H), 1.57 (m, 2 H), 0.89 (t, 3 H, J =7.2 Hz); 13C NMR (CD3OD): δ= 14.44, 21.69, 24.33, 25.84, 29.38, 29.88, 32.20, 37.57, 40.16, 42.36, 46.81, 48.60, 51.06, 60.20, 117.97, 127.15, 128.82, 128.20, 135.48, 135.65, 148.95, 169.28, 174.64, 177.59, 216.11; MS (APCI): m/z 494 [M+1]+; Anal. calcd for C27H35N5O4 (493.6): C 65.70, H 7.15, N 14.19, found: 65.94, H 7.29, N 14.28; Rf = 0.52 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR = 5.14 min, purity: 99.0 % [CH3CN/H2O/TFA (40:60:0.1 %)].
(1R)-3-Ocp-L-His(1-benzyl-2-isopropyl)-L-ProNH2 (22 b)
Yield: 24 %; mp: 120–122 °C (dec.); 1H NMR (CD3OD): δ=7.30 (m, 3 H), 7.16 (m, 2 H), 6.90 (s, 1 H), 5.15 (s, 2 H), 4.83 (m, 1 H), 4.38 (m, 1 H), 3.81 (m, 1 H), 3.51 (m, 1 H), 3.05 (m, 2 H), 2.95 (m, 2 H), 2.34–1.93 (m, 10 H), 1.17 (d, 6 H, 2 0 J =6.9 Hz); 13C NMR (CD3OD): δ= 22.20, 24.17, 25.80, 28.50, 29.31, 33.19, 37.54, 40.10, 42.48, 46.98, 49.35, 51.13, 60.60, 116.20, 127.06, 128.43, 128.50, 134.14, 135.05, 153.87, 169.50, 174.37, 177.55, 215.13; MS (APCI): m/z 494 [M+1]+; Anal. calcd for C27H35N5O4 (493.6): C 65.70, H 7.15, N 14.19, found: 65.79, H 7.38, N 14.23; Rf = 0.50 [CH3OH/25 % NH4OH/CHCl3 (10:2:88)]; HPLC: tR = 4.95 min, purity: 99.4 % [CH3CN/H2O/TFA (40:60:0.1 %)].
(1R)-3-Ocp-L-His(1-benzyl-2-tert-butyl)-L-ProNH2 (22 c)
Yield: 10 %; mp: 126–128 °C (dec.); 1H NMR (CD3OD): δ=7.30 (m, 3 H), 7.08 (m, 2 H), 6.90 (s, 1 H), 5.36 (m, 2 H), 4.81 (m, 1 H), 4.39 (m, 1 H), 3.79 (m, 1 H), 3.56 (m, 1 H), 3.03 (m, 1 H), 2.94 (m, 2 H), 2.35–1.95 (m, 10 H), 1.38 (s, 9 H); 13C NMR (CD3OD): δ= 24.15, 25.89, 28.28, 29.34, 32.05, 32.86, 37.57, 40.10, 43.36, 46.70, 50.34, 51.20, 60.16, 115.90, 126.96, 128.13, 128.76, 135.00, 135.83, 154.98, 169.49, 174.52, 177.51, 216.15; MS (APCI): m/z 508 [M+1]+; Anal. calcd for C28H37N5O4 (507.6): C 66.25, H 7.35, N 13.80, found: 66.36, H 7.04, N 14.17; Rf = 0.56 [CH3OH/25 % NH4OH/CHCl3 (10:2:88)]; HPLC: tR = 5.26 min, purity: 95.5 % [CH3CN/H2O/TFA (40:60:0.1 %)].
(1R)-3-Ocp-L-His(1-benzyl-2-cyclopropyl)-L-ProNH2 (22 d)
Yield: 29 %; mp: 116–118 °C (dec.); 1H NMR (CD3OD): δ =7.32 (m, 3 H), 7.21 (m, 2 H), 6.83 (s, 1 H), 5.23 (s, 2 H), 4.77 (m, 1 H), 4.36 (m, 1 H), 3.76 (m, 1 H), 3.44 (m, 1 H), 3.00 (m, 1 H), 2.92 (m, 2 H), 2.40–1.92 (m, 10 H), 1.84 (m, 1 H), 0.88 (m, 4 H); 13C NMR (CD3OD): δ=8.52, 8.87, 24.13, 25.80, 29.30, 32.10, 37.26, 40.19, 42.40, 46.81, 49.57, 51.11, 60.17, 115.79, 127.34, 128.20, 128.82, 134.79, 136.59, 152.02, 169.54, 174.64, 177.77, 216.30; MS (APCI): m/z 492 [M+1]+; Anal. calcd for C27H33N5O4 (491.6): C 65.97, H 6.77, N 14.25, found: C 66.38, H 7.16, N 13.85; Rf = 0.53 [CH3OH/25 % NH4OH/CHCl3 (10:2:88)]; HPLC: tR = 4.72 min, purity: 96.0 % [CH3CN/H2O/TFA (40:60:0.1 %)].
(1R)-3-Ocp-L-His(1-benzyl-2-cyclopentyl)-L-ProNH2 (22 e)
Yield: 23 %; mp: 122–124 °C (dec.); 1H NMR (CD3OD): δ =7.31 (m, 3 H), 7.15 (m, 2 H), 6.91 (s, 1 H), 5.14 (s, 2 H), 4.71 (m, 1 H), 4.36 (m, 1 H), 3.81 (m, 1 H), 3.50 (m, 1 H), 3.06 (m, 2 H), 2.96 (m, 2 H), 2.34–1.60 (m, 18 H); 13C NMR (CD3OD): δ= 24.78, 25.84, 29.38, 31.82, 37.50, 39.15, 40.16, 42.36, 46.71, 48.88, 51.17, 60.32, 116.44, 127.13, 128.43, 128.65, 134.41, 135.17, 150.33, 169.50, 174.64, 177.58, 214.13; MS (APCI): m/z 520 [M+1]+; Anal. calcd for C29H37N5O4 (519.6): C 67.03, H 7.18, N 13.48, found: C 67.50, H 7.47, N 13.03; Rf =0.57 [CH3OH/25 % NH4OH/CHCl3 (10:2:88)]; HPLC: tR = 5.98 min, purity: 93.0 % [CH3CN/H2O/TFA (40:60:0.1 %)].
(1R)-3-Ocp-L-His(1-benzyl-2-cyclohexyl)-L-ProNH2 (22 f)
Yield: 24 %; mp: 130–132 °C (dec.); 1H NMR (CD3OD): δ =7.31 (m, 3 H), 7.16 (m, 2 H), 6.90 (s, 1 H), 5.14 (s, 2 H), 4.85 (m, 1 H), 4.37 (m, 1 H), 3.80 (m, 1 H), 3.47 (m, 1 H), 3.02 (m, 1 H), 2.94 (m, 2 H), 2.70 (m, 1 H), 2.34–1.28 (m, 20 H); 13C NMR (CD3OD): δ =24.30, 25.76, 26.13, 27.03, 29.13, 32.01, 32.23, 35.14, 37.57, 39.96, 42.12, 46.81, 49.08, 51.04, 60.03, 116.36, 127.13, 128.20, 128.82, 134.57, 135.29, 150.08, 169.34, 174.64, 177.59, 216.12; MS (APCI): m/z 534 [M+1]+; Anal. calcd for C30H39N5O4 (533.7): C 67.52, H 7.37, N 13.12, found: C 67.64, H 7.50, N 13.38; Rf = 0.58 [CH3OH/25 % NH4OH/CHCl3 (10:2:88)]; HPLC: tR = 7.27 min, purity: 99.3 % [CH3CN/H2O/TFA (40:60:0.1 %)].
(1R)-3-Ocp-L-His[1-benzyl-2-(adamantan-1-yl)]-L-ProNH2 (22 g)
Yield: 18 %; mp: 136–138 °C (dec.); 1H NMR (CD3OD): δ= 7.38 (m, 3 H), 7.15 (m, 2 H), 7.04 (s, 1 H), 5.56 (s, 2 H), 4.97 (m, 1 H), 4.42 (m, 1 H), 3.72 (m, 1 H), 3.59 (m, 1 H), 3.06 (m, 2 H), 3.00 (m, 1 H), 2.33–1.28 (m, 25 H); 13C NMR (CD3OD): δ=24.34, 25.84, 29.31, 29.38, 32.16, 35.99, 37.44, 40.16, 40.96, 42.36, 46.65, 50.35, 51.26, 59.12, 115.87, 127.05, 128.76, 135.04, 136.55, 151.17, 169.49, 174.12, 177.14, 215.80; MS (APCI): m/z 586 [M+1]+; Anal. calcd for C34H43N5O4 (585.7): C 69.72, H 7.40, N 11.96, found: C 69.38, H 7.69, N 12.29; Rf = 0.62 [CH3OH/25 % NH4OH/CHCl3 (10:2:88)]; HPLC: tR = 11.07 min, purity: 98.3 % [CH3CN/H2O/TFA (40:60:0.1 %)].
(1R)-3-Ocp-L-His(1-methyl-2-propyl)-L-ProNH2 (22 h)
Yield: 28 %; mp: 100–103 °C (dec.); 1H NMR (CD3OD): δ=6.81 (s, 1 H), 4.77 (m, 1 H), 4.39 (m, 1 H), 3.78 (m, 1 H), 3.57 (s, 3 H), 3.45 (m, 1 H), 3.08 (m, 1 H), 2.96 (m, 2 H), 2.64 (t, 2 H, J =7.5 Hz), 2.35–1.94 (m, 10 H), 1.67 (m, 2 H), 0.96 (t, 3 H, J =7.2 Hz); MS (APCI): m/z 418 [M+1]+; Anal. calcd for C21H31N5O4 (417.5): C 60.41, H 7.48, N 16.77, found: C 60.52, H 7.61, N 17.10; Rf = 0.43 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR = 5.46 min, purity: 98.4 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His(1-methyl-2-isopropyl)-L-ProNH2 (22 i)
Yield: 31 %; mp: 96–98 °C (dec.); 1H NMR (CD3OD): δ = 6.80 (s, 1 H), 4.80 (m, 1 H), 4.39 (m, 1 H), 3.79 (m, 1 H), 3.59 (s, 3 H), 3.50 (m, 1 H), 3.11 (m, 2 H), 2.93 (m, 2 H), 2.36–1.93 (m, 10 H), 1.28 (m, 6 H); MS (APCI): m/z 418 [M+1]+; Anal. calcd for C21H31N5O4 (417.5): C 60.41, H 7.48, N 16.77, found: C 60.25, H 7.37, N 16.61; Rf =0.42 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR =5.17 min, purity: 98.3 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His(1-methyl-2-tert-butyl)-L-ProNH2 (22 j)
Yield: 35 %; mp: 104–106 °C (dec.); 1H NMR (CD3OD): δ= 6.78 (s, 1 H), 4.79 (m, 1 H), 4.37 (m, 1 H), 3.78 (m, 1 H), 3.73 (s, 3 H), 3.53 (m, 1 H), 3.07 (m, 1 H), 2.89 (m, 2 H), 2.36–1.89 (m, 10 H), 1.40 (s, 9 H, 3 × CH3); MS (APCI): m/z 432 [M+1]+; Anal. calcd for C22H33N5O4 (431.5): C 61.23, H 7.71, N 16.23; C 60.99, H 8.09, N 15.97; Rf = 0.46 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR =5.88 min, purity: 99.7 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His(1-methyl-2-cyclohexyl)-L-ProNH2 (22 k)
Yield: 19 %; mp: 110–112 °C (dec.); 1H NMR (CD3OD): δ =6.78 (s, 1 H), 4.80 (m, 1 H), 4.38 (m, 1 H), 3.79 (m, 1 H), 3.57 (s, 3 H), 3.47 (m, 1 H), 3.08 (m, 1 H), 2.90 (m, 2 H), 2.74 (m, 1 H), 2.35–1.28 (m, 20 H); MS (APCI): m/z 458 [M+1]+; Anal. calcd for C24H35N5O4 (457.6): C 63.00, H 7.71, N 15.31, found: C 63.08, H 7.78, N 15.36; Rf =0.54 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR =10.45 min, purity: 96.6 % [CH3CN/H2O/TFA (20:80:0.1 %)].
(1R)-3-Ocp-L-His[1-methyl-2-(adamantan-1-yl)]-L-ProNH2 (22 l)
Yield: 25 %; mp: 118–120 °C (dec.); 1H NMR (CD3OD): δ=6.75 (s, 1 H), 4.79 (m, 1 H), 4.37 (m, 1 H), 3.79 (m, 1 H), 3.77 (s, 3 H), 3.54 (m, 1 H), 3.08 (m, 1 H), 2.89 (m, 2 H), 2.34 (m, 2 H), 2.29–1.81 (m, 23 H); MS (APCI): m/z 510 [M+1]+; Anal. calcd for C28H39N5O4 (509.6): C 65.99, H 7.71, N 13.74, found: C 66.28, H 8.05, N 13.35; Rf = 0.59 [CH3OH/25 % NH4OH/CHCl3 (12:2:86)]; HPLC: tR =26.14 min, purity: 95.1 % [CH3CN/H2O/TFA (20:80:0.1 %)].
Receptor binding assays
Receptor binding studies were carried out according to a procedure described previously.[36] Briefly, HEK 293EM cells stably expressing either TRH-R1 or TRH-R2 were grown in Dulbecco’s modified Eagle’s medium containing 10 % fetal bovine serum (FBS) and 200 mg mL−1 hygromycin. For equilibrium binding experiments, cells were seeded into 24-well plates (1.5 × 105 cells per well). After 48 h, cells were incubated at 37 °C for 1 h with [3H][N(1)-Me-His]TRH (MeTRH, 2 nM) in Hanks’ balanced salt solution, pH 7.4, and various doses of TRH analogues. Apparent inhibitory constants (Ki) were derived from curves fitted by nonlinear regression analysis and drawn with the Prism version 3 software package (GraphPad Software, Inc.) using the formula , in which IC50 is the concentration of unlabeled analogue that half-competes and Kd is the equilibrium dissociation constant for [3H][N(1)-Me-His]TRH.
Luciferase activity assays
On the day prior to transfection, cells stably expressing either TRH-R1 or TRH-R2 were seeded into 24-well plates (1.5 × 105 cells per well). After 16 h, the medium was aspirated, and the cells (~50 % confluent) were co-transfected with plasmid DNA encoding CREB and the CREB-activated luciferase gene (PathDetect CREB trans-Reporting System™, Stratagene) using the calcium phosphate method. On the second day, 6 h before the assay, medium containing 10 % FBS was changed to medium containing 1 % FBS, and various concentrations of TRH and TRH analogues were added to the medium. Luciferase activity was measured 24 h after transfection. Cells were washed with phosphate-buffered saline and lysed with 0.2 mL lysis buffer (25 mM Gly-Gly, pH 7.8, 15 mM MgSO4, 4 mM EGTA, 1 mM dithiothreitol, and 1 % Triton X-100). Cell lysates (0.025 mL) were combined automatically with 0.125 mL reaction buffer (25 mM Gly-Gly, pH 7.8, 15 mM MgSO4, 4 mM EGTA, 1 mM dithiothreitol, 15 mM KH2PO4, and 2 mM ATP) and 0.025 mL luciferin (0.4 mM) in reaction buffer, and the luminescence was measured for 10 s in a TR717 Microplate luminometer (Tropix, Bedford, MA, USA). The luciferase activity levels detected by this assay reflect the activation of signaling by TRH analogues.
Analeptic activity
Male Swiss albino mice (20–30 g) were procured from the Central Animal Facility (CAF), NIPER, S.A.S. Nagar. Animals were housed in a room at 22 ± 2°C with 12 h light/dark cycles and were allowed free access to pellet food and water. All procedures used in this study were approved by the Institutional Animal Ethics Committee (IAEC), NIPER (experimental protocol approval numbers IAEC/08/43 and IAEC/09/01) and were conducted in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.
The analeptic activities of TRH analogues were studied by the antagonism of pentobarbital-induced sleep time in albino Swiss mice as described elsewhere.[40] Test compounds were dissolved in vehicle (saline). Vehicle (5 mL kg−1) or TRH/TRH analogues (10 μmol kg−1, equivalent to 3.7 mgkg−1 TRH) were injected via the tail vein. Ten minutes after treatment, each animal received sodium pentobarbital 50 mg kg−1 i.p. The sleeping time was recorded as the time elapsed from the onset of loss of righting reflex until it returned. The sleep time in treatment groups was compared with control group by one-way analysis of variance (ANOVA) followed by post hoc Tukey test at p < 0.05. Decrease in sleep time was calculated with Equation (1):
| (1) |
for which S =sleep time in compound-treated group, and C = sleeping time in vehicle-treated group.
Anticonvulsant activity
Animal seizure tests included one chemically induced seizure episode test and one for electrically induced seizures; these were conducted according to protocols described elsewhere.[51, 52] For the chemically induced convulsion test, administration of pentylenetetrazole (PTZ, 65 mgkg−1 i.p.) was used to induce clonic seizures, reflecting a generalized seizure condition in humans. TRH/TRH analogues (10 μmol kg−1 i.v.) were injected into mice (n =6–8 per group) via the tail vein, 10 min prior to PTZ administration, whereas the control group was administered saline. The animals were observed for 10 min after PTZ challenge. Onset of, and latency to, generalized clonic convulsions with falling was recorded.
The electrical test employed was the maximal electric shock (MES) seizure test. The MES-induced seizure model was used to assess the potential of drugs against partial seizures. Seizures were induced in mice by applying a current of 45 mA through a corneal electrode for 0.2 s as described elsewhere with slight modification.[52] Mice (n =6–8 per group) were treated with saline or TRH (10 μmol kg−1) or TRH analogues (10 μmol kg−1 i.v.). Ten minutes later, seizures were induced by MES, and the mice were observed for duration of tonic hind limb extension in mice.
The Supporting Information contains detailed experimental procedures and spectral data for intermediates 12a–m, 13 a–m, 18 a–e, and 19a–e.
Supplementary Material
Acknowledgments
This work is supported by the Council of Scientific and Industrial Research, New Delhi (Grant No. 01(1861)/03/EMR-II). V.M. thanks the Department of Science and Technology, New Delhi (Grant No. SR/FTP/CS-79/2005), for the award of a young scientist fellowship. C.L.M. thanks the University Grant Commission for the award of a senior research fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201000481: detailed experimental procedures and spectral data for intermediates 12 a–m, 13 a–m, 18 a–e, and 19 a–e.
References
- 1.Horita A, Carino MA, Lai H. Ann Rev Pharmacol Toxicol. 1986;26:311–332. doi: 10.1146/annurev.pa.26.040186.001523. [DOI] [PubMed] [Google Scholar]
- 2.Nillni EA, Sevarino KA. Endocr Rev. 1999;20:599–648. doi: 10.1210/edrv.20.5.0379. [DOI] [PubMed] [Google Scholar]
- 3.Burgus R, Dunn JF, Desiderio D, Guillemin R. C R Acad Sci Hebd Seances Acad Sci D. 1969;269:1870–1873. [PubMed] [Google Scholar]
- 4.Boler J, Enzmann F, Folkers K, Bowers CY, Schally AV. Biochem Biophys Res Commun. 1969;37:705–710. doi: 10.1016/0006-291x(69)90868-7. [DOI] [PubMed] [Google Scholar]
- 5.Schaner P, Todd RB, Seidah NG, Nillini EA. J Biol Chem. 1997;272:19958–19968. doi: 10.1074/jbc.272.32.19958. [DOI] [PubMed] [Google Scholar]
- 6.Prokai L. Prog Drug Res. 2002;59:133–169. doi: 10.1007/978-3-0348-8171-5_5. [DOI] [PubMed] [Google Scholar]
- 7.Griffiths EC. Clin Sci. 1987;73:449–457. doi: 10.1042/cs0730449. [DOI] [PubMed] [Google Scholar]
- 8.Kelly JA. Essays Biochem. 1995;30:133–149. [PubMed] [Google Scholar]
- 9.Yamamoto M, Sudoh K, Sasamata M. Eur J Pharmacol. 1991;192:165–167. doi: 10.1016/0014-2999(91)90084-4. [DOI] [PubMed] [Google Scholar]
- 10.Gershengorn MC, Osman R. Endocrinology. 2001;142:2–10. doi: 10.1210/endo.142.1.7919. [DOI] [PubMed] [Google Scholar]
- 11.Bidaud I, Lory P, Nicolas M, Bulant M, Ladram A. Eur J Bio Chem. 2002;269:4566–4576. doi: 10.1046/j.1432-1033.2002.03152.x. [DOI] [PubMed] [Google Scholar]
- 12.Duthie SM, Taylor PL, Anderson L, Cook J, Eidne KA. Mol Cell Endocrinol. 1993;95:R11–R15. doi: 10.1016/0303-7207(93)90043-j. [DOI] [PubMed] [Google Scholar]
- 13.Cao J, O’Donnell D, Vu H, Payza K, Pou C, Godbout C, Jakob A, Pelletier M, Lembo P, Ahmad S, Walker P. J Biol Chem. 1998;273:32281–32287. doi: 10.1074/jbc.273.48.32281. [DOI] [PubMed] [Google Scholar]
- 14.De La Pena P, Delgado LM, del Camino D, Barros F. J Biol Chem. 1992;267:25 703–25 708. [PubMed] [Google Scholar]
- 15.Itadani H, Nakamura T, Itoh J, Iwaasa H, Kanatani A, Borkowski J, Ihara M, Ohta M. Biochem Biophys Res Commun. 1998;250:68–71. doi: 10.1006/bbrc.1998.9268. [DOI] [PubMed] [Google Scholar]
- 16.Sun Y, Lu X, Gershengorn MC. J Mol Endocrinol. 2003;30:87–97. doi: 10.1677/jme.0.0300087. [DOI] [PubMed] [Google Scholar]
- 17.Monga V, Meena CL, Kaur N, Jain R. Curr Med Chem. 2008;15:2718–2733. doi: 10.2174/092986708786242912. [DOI] [PubMed] [Google Scholar]
- 18.Ogawa N, Yamawaki Y, Kuroda H, Ofuji T, Itoga E, Kito S. Brain Res. 1981;205:169–174. doi: 10.1016/0006-8993(81)90728-9. [DOI] [PubMed] [Google Scholar]
- 19.Burt DR, Taylor RL. Endocrinology. 1980;106:1416–1423. doi: 10.1210/endo-106-5-1416. [DOI] [PubMed] [Google Scholar]
- 20.Jain R, Singh J, Perlman JH, Gershengorn MC. Bioorg Med Chem. 2002;10:189–194. doi: 10.1016/s0968-0896(01)00265-6. [DOI] [PubMed] [Google Scholar]
- 21.Engel S, Neumann S, Kaur K, Monga V, Jain R, Northup J, Gershengorn MC. J Biol Chem. 2006;281:13103–13109. doi: 10.1074/jbc.M600440200. [DOI] [PubMed] [Google Scholar]
- 22.Kaur N, Monga V, Josan JS, Lu X, Gershengorn MC, Jain R. Bioorg Med Chem. 2006;14:5981–5988. doi: 10.1016/j.bmc.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 23.Kaur N, Lu X, Gershengorn MC, Jain R. J Med Chem. 2005;48:6162–6165. doi: 10.1021/jm0505462. [DOI] [PubMed] [Google Scholar]
- 24.Kaur N, Monga V, Lu X, Gershengorn MC, Jain R. Bioorg Med Chem. 2007;15:433–443. doi: 10.1016/j.bmc.2006.09.045. [DOI] [PubMed] [Google Scholar]
- 25.Rajput SK, Krishnamoorthy S, Pawar C, Kaur N, Monga V, Meena CL, Jain R, Sharma SS. Epilepsy Behav. 2009;14:48–53. doi: 10.1016/j.yebeh.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Rajput SK, Singh JN, Ingole S, Jain G, Kaur N, Monga V, Meena CL, Jain R, Sharma SS. Epilepsy Res. 2009;87:223–233. doi: 10.1016/j.eplepsyres.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 27.Monga V, Meena CL, Kaur N, Kumar S, Pawar C, Sharma SS, Jain R. J Heterocyclic Chem. 2008;45:1603–1608. [Google Scholar]
- 28.Jain R, Cohen LA. Tetrahedron. 1997;53:2365–2370. [Google Scholar]
- 29.Cohen LA, Jain R. Tetrahedron. 1996;52:5363–5370. [Google Scholar]
- 30.Jain R, Cohen LA. Tetrahedron. 1997;53:4539–4548. [Google Scholar]
- 31.Szirtes T, Kisfaludy L, Palosi E, Szporny L. J Med Chem. 1986;29:1654–1658. doi: 10.1021/jm00159a015. [DOI] [PubMed] [Google Scholar]
- 32.Kaur N, Monga V, Jain R. Tetrahedron Lett. 2004;45:6883–6885. [Google Scholar]
- 33.Stephani RA, Rowe WB, Gass JD, Meister A. Biochemistry. 1972;11:4094–4094. doi: 10.1021/bi00772a011. [DOI] [PubMed] [Google Scholar]
- 34.Toki K. Bull Chem Soc Jpn. 1958;31:333–335. [Google Scholar]
- 35.Curry K, Peet MJ, Magnuson DSK, McLennan H. J Med Chem. 1988;31:864–867. doi: 10.1021/jm00399a030. [DOI] [PubMed] [Google Scholar]
- 36.Perlman JH, Thaw CN, Laakkonen L, Bowers CY, Osman R, Gershengorn MC. J Biol Chem. 1994;269:1610–1631. [PubMed] [Google Scholar]
- 37.Lu X, Huang W, Worthington S, Drabik P, Osman R, Gershengorn MC. Mol Pharmacol. 2004;66:1192–1200. doi: 10.1124/mol.104.000349. [DOI] [PubMed] [Google Scholar]
- 38.Breeze G, Cott J, Cooper B, Prange A, Lipton M, Plotniko N. J Pharmcol Exp Ther. 1975;193:11–22. [PMC free article] [PubMed] [Google Scholar]
- 39.Horita A, Carino M. Pharmacol Biochem Behav. 1976;5:111–116. doi: 10.1016/0091-3057(76)90337-3. [DOI] [PubMed] [Google Scholar]
- 40.Wu J, Yoon SH, Wu WM, Bodor N. J Pharm Pharmacol. 2002;54:945–950. doi: 10.1211/002235702760089063. [DOI] [PubMed] [Google Scholar]
- 41.Barker JL, Ransom BR. J Physiol. 1978;280:355–372. doi: 10.1113/jphysiol.1978.sp012388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathers DA, Barker JL. Science. 1980;209:507–509. doi: 10.1126/science.6248961. [DOI] [PubMed] [Google Scholar]
- 43.Chu QP, Wang LE, Cui XY, Fu HZ, Lin ZB, Lin SQ, Zhang YH. Pharmacol Biochem Behav. 2007;86:693–698. doi: 10.1016/j.pbb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 44.Nicoll RA, Madison DV. Science. 1982;217:1055–1057. doi: 10.1126/science.7112112. [DOI] [PubMed] [Google Scholar]
- 45.O’Beirne M, Gurevich N, Carlen P. Can J Physiol Pharmacol. 1987;65:36–41. doi: 10.1139/y87-007. [DOI] [PubMed] [Google Scholar]
- 46.Sirois JE, Pancrazio JJ, Bayliss DA. J Physiol. 1998;512:851–862. doi: 10.1111/j.1469-7793.1998.851bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.French-Mullen JMH, Barker JL, Rogawski MA. J Neurosci. 1993;13:3211–3221. doi: 10.1523/JNEUROSCI.13-08-03211.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Werz MA, Macdonald RL. Mol Pharmacol. 1985;28:269–277. [PubMed] [Google Scholar]
- 49.Colson AO, Gershengorn MC. Mini-Rev Med Chem. 2006;6:221–226. doi: 10.2174/138955706775476019. [DOI] [PubMed] [Google Scholar]
- 50.Ogawa N, Hirose Y, Mori A, Kajita S, Sato M. Regul Pept. 1985;12:249–256. doi: 10.1016/0167-0115(85)90066-7. [DOI] [PubMed] [Google Scholar]
- 51.Malhotra J, Gupta YK. Br J Pharmacol. 1997;120:282–288. doi: 10.1038/sj.bjp.0700869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Manocha A, Mediratta PK, Sharma KK. Pharmacol Biochem Behav. 2003;76:111–117. doi: 10.1016/s0091-3057(03)00218-1. [DOI] [PubMed] [Google Scholar]
- 53.Jaworska-Feil L, Kajta M, Budziszewska B, Leskiewicz M, Lason W. Epilepsy Res. 2001;43:67–73. doi: 10.1016/s0920-1211(00)00178-9. [DOI] [PubMed] [Google Scholar]
- 54.Deng PY, Porter JE, Shin HS, Lei S. J Physiol. 2006;577:497–511. doi: 10.1113/jphysiol.2006.118141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.