

SUMMARY
A novel adenosine receptor, the A3 receptor, has recently been cloned. We have systematically investigated the hitherto largely unexplored structure-activity relationships (SARs) for binding at A3 receptors, using 125I-N6-2-(4-aminophenyl)ethyladenosine as a radioligand and membranes from Chinese hamster ovary cells stably transfected with the rat A3-cDNA. As is the case for A1 and A2a, receptors, substitutions at the N6 and 5′ positions of adenosine, the prototypic agonist ligand, may yield fairly potent compounds. However, the highest affinity and A3 selectivity is found for N6,5′-disubstituted compounds, in contrast to A1 and A2a receptors. Thus, N6-benzyladenosine-5′-N-ethylcarboxamide is highly potent (Ki, 6.8 nM) and moderately selective (13- and 14-fold versus A1 and A2a). The N6 region of the A3 receptor also appears to tolerate hydrophilic substitutions, in sharp contrast to the other subtypes. Potencies of N6,5′-disubstituted compounds in inhibition of adenylate cyclase via A3 receptors parallel their high affinity in the binding assay. None of the typical xanthine or nonxanthine (A1/A2) antagonists tested show any appreciable affinity for rat A3 receptors. 1,3-Dialkylxanthines did not antagonize the A3 agonist-induced inhibition of adenylate cyclase. A His residue in helix 6 that is absent in A3 receptors but present in A1/A2 receptors may be causal in this respect. In a molecular model for the rat A3 receptor, this mutation, together with an increased bulkiness of residues surrounding the ligand, make antagonist binding unfavorable when compared with a previously developed A1 receptor model. Second, this A3 receptor model predicted similarities with A1 and A2 receptors in the binding requirements for the ribose moiety and that xanthine-7-ribosides would bind to rat A3 receptors. This hypothesis was supported experimentally by the moderate affinity (Ki 6 μM) of 7-riboside of 1,3-dibutylxanthine, which appears to be a partial agonist at rat A3 receptors. The model presented here, which is consistent with the detailed SAR found in this study, may serve to suggest future chemical modification, site-directed mutagenesis, and SAR studies to further define essential characteristics of the ligand-receptor interaction and to develop even more potent and selective A3 receptor ligands.
Adenosine receptors, belonging to the superfamily of the G protein-coupled receptors, are generally divided into two major subclasses, A1 and A2, on the basis of the following: (i) the differential affinities of a number of adenosine receptor agonists and antagonists; (ii) their primary structures; (iii) the second messenger systems to which they couple. Thus, A2 receptors (which can be further subdivided into A2a and A2b) stimulate adenylate cyclase, whereas A1 receptors may couple to a variety of second messenger systems, including inhibition of adenylate cyclase, inhibition or stimulation of phosphoinositol turnover, activation of guanylate cyclase, activation of potassium influx, and inhibition of calcium influx (1, 2). A recent addition to the adenosine receptor family has been the A3 receptor, which was cloned from rat brain (3) and rat testis (4) and which was first recognized as an adenosine receptor on the basis of its primary structure. In the putative transmembrane domains, it shows 58% identity with the canine A1 receptor and 57% with the canine A2a receptor. Like the A1 receptor, it is negatively coupled to adenylate cyclase (3).
The physiological role of the A3 receptor is mostly unexplored. Its distribution is fairly limited, and it is found primarily in the central nervous system (3), testes (4), heart (3), and the immune system, where it appears to be involved in the modulation of release from mast cells or other cells of mediators of the immediate hypersensitivity reaction (5). Activation of A3 receptors also appears to cause xanthine-insensitive hypotensive response in pithed rats (44). In terms of therapeutic potential, a principal deficiency of A1- and A2a-selective agents has been their propensity for side effects, due to the ubiquitous nature of these receptors. However, the limited distribution of A3 receptors raises hopes that A3-selective compounds may be more useful as potential therapeutic agents.
Few ligands for this novel receptor have been reported.1 Some nonselective N6-substituted adenosine derivatives have been described as agonists for the A3 receptor, including APNEA (N6-2-(4-aminophenyl)ethyladenosine), which has been used successfully as a radioligand in its iodinated form (3). Curiously, xanthines (classical A1 and A2 antagonists) do not appear to bind to this receptor (3). Because the SAR at A3 receptors is practically unexplored, we have systematically investigated a wide range of purine and nonpurine agents for affinity in binding to arrive at leads for achieving selectivity. We have integrated these pharmacological findings with insights derived from molecular modeling of A1 receptors to present a binding site model unique for A3 receptors.
Materials and Methods
Chemicals
F-12 (Ham’s) medium, fetal bovine serum, and penicillin/streptomycin were from Life Technologies, Inc. (Gaithersburg, MD) [125I] APNEA was prepared as described previously (6). Adenosine deaminase (ADA) was from Boehringer Mannheim (Indianapolis, IN). Composition of lysis buffer was as follows: 10 mM Tris, 5 mM EDTA, pH 7.4, at 5 °. 50/10/1 buffer was as follows: 50 mM Tris, 10 mM MgCl2, 1 mM EDTA, pH 8.26, at 5 °. Displaces were from RBI (Natick, MA) except xanthine, inosine, 8-chlorotheophylline (Aldrich, Milwaukee, WI); 1,9-dimethylxanthine, 3,9-dimethylxanthine (Fluka, Ronkonkoma, NY); 3,7-dimethylxanthine, 8-bromoadenosine, adenosine-N-oxide, α-D-adenosine, 2′-deoxyadenosine, 3′-deoxyadenosine, 5′-deoxyadenosine, 5′-deoxy-5′-methylthioadenosine, 5′-deoxy-5′-isobutylthioadenosine, S-adenosylmethionine, guanosine, 6-thioguanosine, 6-thiopurine riboside (Sigma); AMP (Boehringer Mannheim); xanthosine, uridine, thymidine, cytidine (Janssen/Spectrum, Gardens, CA). The following compounds were gifts, which are gratefully acknowledged: 1,3-dibutylxanthine, 1,3-dihexylxanthine, 1,3-dibenzylxanthine, 8-cyclohexycaffeine, 7-benzyltheophylline, N6-dimethyladenosine, 3-deazaadenosine, 7-deazaadenosine, β-L-adenosine, 2′-0-methyladenosine, adenine-β-D-arabinofuranoside, xylofuranosyladenosine, β-D-psicofuranosyladenine, 5′-deoxy-5′-aminoadenosine, 5′-carboxamidoadenosine, 2-thio-3-propylxanthine, 1-propyl-8-cyclopentylxanthine (Dr. J. Daly, NIH, Bethesda, MD); N6-cyclohexylNECA, 9-ethyl-N6-cyclopentyladenine, N6-dimethylNECA, N6-benzyl-N6-methyladenosine (Dr. R. Olsson, University of South Florida, Tampa, FL); CP 66713 (Dr. R. Sarges, Pfizer, Groton, CT); CGS 15943, (Dr. J. Francis, Ciba-Geigy, Summit, NJ). The syntheses of the following compounds have been described previously: theophylline-7-riboside, 1,3-dipropylxanthine-7-riboside, 1,3-dibutylxanthine-7 riboside (7); imidazo[4,5-c]quinolin-4-amine (8); N6-2-sulfoethyladenosine, N6-4-sulfophenyladenosine, N6-3-(4-sulfophenyl)-propyladenosine, N6-4-(4-sulfophenyl)butyladenosine (9). All other materials were from standard local sources and of the highest grade commercially available.
Synthesis
N6-Benzyladenosine-N1-oxide
N6-Benzyladenosine (25 mg, 70 μmol) and m-chloroperbenzoic acid (38 mg, 220 μmol) were dissolved in 1 ml of acetic acid. The solution was stirred at room temperature for 2 days. The solvent was evaporated under a stream of nitrogen, and the residue was dissolved in a minimum of methanol and chromatographed on a silica plate (250 μ) using acetonitrile:water, 4:1 (v/v). The UV-absorbing band at RF = 0.53 was extracted with methanol to provide 7.3 mg (28% yield). Mass spectrum and 1H-NMR spectrum were consistent with the assigned structure.
Adenosine-5′-N-ethyluronamide-N1-oxide
Adenosine-5′-N-ethyluronamide-N1-oxide was synthesized by a method similar to N6-benzyladenosine-N1-oxide. Following recrystallization from hot methanol/ether, the pure product was obtained in 28% yield. Mass spectrum (electron impact, peaks at 324 (m), 308) and 1H-NMR spectrum were consistent with the assigned structure.
N6-Benzyladenosine-5′-N-ethyluronamide
To a solution of NECA (50 mg, 0.162 mmol) in DMF (1 ml) was added benzyl bromide (56 ml, 0.47 mmol), and the solution was stirred for 2 days at 40 ° while protected from moisture. DMF was removed in vacuo giving a syrup that crystallized when acetone and ether were added. The solvent was removed by decantation, and the solid was dried and dissolved in methanol (2 ml). K2CO3 (10 mg) was added and warmed under reflux overnight. The reaction mixture was cooled, filtered, and evaporated. The product was purified by preparative TLC (CHCl3:MeOH, 13:2) in 42% yield. Melting point, 170–173 °. 1H-NMR (in Me2SO-d6) was as follows: δ 1.06 (t, J = 7 Hz, 3H, CH3), 3.20 (m, 2H, CH2), 4.13 (t, J = 4Hz, 1H, H-3′), 4.30 (s, 1H, H-4′), 4.62 (m, 1H, H-2′), 4.71 (broad s, 2H, N6-CH2Ph), 5.53 (d, J = 7 Hz, 1H, OH-2′), 5.73 (d, J = 4 Hz, 1H, OH-3′), 5.96 (d, J = 8 Hz, 1H, H-1′), 7.30 (m, 5H, Phenyl), 8.25 (s, 1H, H-2), 8.42 (s, 1H, H-8), 8.55 (broad s, 1H, N6H-CH2Ph), 8.86 (t, J = 5 Hz, 1H, NH-Et). Mass Spectrum (CI-NH3): m/z 399 (MH+, base).
Inosine-5′-N-ethyluronamide (NECI)
2′,3′-O-Isopropylideneinosine-5′-uronic acid (20 mg, 62 μmol) (10), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (25 mg, 130 μmol) and N-hydroxysuccinimide (13 mg, 112 μmol) were dissolved in a minimum volume of dimethylformamide. Ethylamine (70% in water, 7 μl) was added and after 1 hr of stirring, was cooled to 0 °, and was precipitated with water to give 14 mg (65% yield). The product (10 mg, 29 μmol) was heated in 1 N HCl for 2 hr at 60 °. After cooling and neutralizing with NaHCO3, it was purified twice using reverse phase SepPak cartridges with water as eluant. Lyophilization of the fraction afforded 6.95 mg (78% yield) of an amorphous solid Melting point, 168°C (d). 1H-NMR (in Me2SO-d6) was as follows: δ 1.03 (t, J = 7 Hz, 3H, CH3), 3.17 (m, 2H, CH2), 4.15 (broad s, 1H, H-3′), 4.30 (s, 1H, H-4′), 4.54 (m, 1H, H-2′), 5.61 (broad s, 1H, OH), 5.68 (broad s, 1H, OH), 5.96 (d, J = 7 Hz, 1H, H-1′), 8.08 (s, 1H, H-2), 8.39 (s, 1H, H-8). Mass spectrum (CI-NH3): m/z 310 (MH+, base).
Cell culture and membrane preparation
CHO cells stably expressing the rat A3 receptor (3) were grown in F-12 medium containing 10% fetal bovine serum and penicillin/streptomycin (100 units/ml and 100 μg/ml, respectively) at 37 ° in a 5% CO2 atmosphere. When cells had reached confluency, they were washed twice with 10 ml of ice-cold lysis buffer. After addition of 5 ml of lysis buffer, cells were mechanically scraped and homogenized in an ice-cold Dounce homogenizer (20 strokes by hand). The suspension was centrifuged at 43,000 × g for 10 min. The pellet was resuspended in the minimum volume of ice-cold 50 mM Tris/10 mM GCl2/1 mM EDTA (pH 8.26 at 5°) buffer required for the binding assay and homogenized in a Dounce homogenizer. Typically, six to eight 175-cm2 flasks were used for a 48-tube assay. ADA was added to a final concentration of 3 units/ml, and the suspension was incubated at 37 ° for 15 min; the membrane suspension was subsequently kept on ice until use. When large batches (~100 flasks) were processed homogenization was performed with a Polytron (Brinkman, Luzern, Switzerland), and further work-up was as described above. The preparation was stored at −70 ° and retained its [125I] APNEA binding properties for at least 1 month.
Radioligand binding assay
Binding of [125I]APNEA to CHO cells stably transfected with the rat A3 receptor clone was performed essentially as described (6). Assays were performed in 50/10/1 buffer in glass tubes and contained 100 μl of the membrane suspension, 50 μl of [125I] APNEA (final concentration 0.5 nM), and 50 μl of inhibitor. Inhibitors were routinely dissolved in Me2SO and were then diluted with buffer; final Me2SO concentrations never exceeded 1%. This concentration did not influence [128I]APNEA binding. Incubations were carried out in duplicate for 1 hr at 37 ° and were terminated by rapid filtration over Whatman GF/B filters, using a Brandell cell harvester (Brandell, Gaithersburg, MD). Tubes were washed three times with 3 ml of buffer. Radioactivity was determined in a Beckman γ 5500B γ-counter. Nonspecific binding was determined in the presence of 40 μM R-PIA. Ki values were calculated according to Cheng and Prusoff (11), assuming a Kd for [l25I]APNEA of 17 nM (3).
The level of nonspecific binding with [125I]APNEA in transfected CHO cells was 20–30%. There was some variability in the Hill coefficients (range from 0.8 to 1.2). Untransfected CHO cells displayed a low level of binding displacable by 100 μM R-PIA (at 5 nM [125I]APNEA, only 5–10% of the level of specific A3 binding in transfected cells), but this binding did not have pharmacological characteristics of adenosine receptors.
Binding of [3H]PIA to A1 receptors from rat brain membranes and of [3H]CGS 21680 to A2a receptors from rat striatal membranes was performed as described previously (9).
Adenylate cyclase assay
Adenylate cyclase was assayed in membranes from CHO cells stably expressing the rat A3 receptor, prepared as above, using a modification of previously reported methods (18). Maximal inhibition of adenylate cyclase activity corresponded to ~40% of total activity under conditions of stimulation (typically by 6–8-fold) in the presence of 1 μM forskolin.
Membranes were resuspended in 75 mM Tris, 200 mM NaCl, 1.25 mM MgCl2, pH 8.12, at 4°C (TNM buffer) to give a final concentration of 0.1 mg/ml, and 2 units/ml adenosine deaminase was added. For the cyclase assay, the TMN buffer was supplemented with 140 μM dATP, 5 μM GTP, 30 units/ml creatine kinase, 5 mM creatine phosphate, 2.2 mM dithiothreitol, 100 μM papaverine, and 1.5 μCi of [α-32P] ATP. Then 40 μl of the membrane suspension, 40 μl of the supplemented buffer, and 20 μl of a solution of forskolin and the test compound (initially dissolved in Me2SO then diluted in TMN buffer) were combined and incubated at 30 ° for 15 min, followed by termination by addition of a stop solution containing 20,000 cpm/ml [3H]cyclic AMP. The final concentration of Me2SO did not exceed 1%, which had no effect on adenylate cyclase. Labeled cyclic AMP was isolated by chromatography on Dowex 50 and alumina columns, and 32P was measured using scintillation counting.
Molecular modeling
Structures were built using Quanta (version 3.3; Polygen, Waltham, MA), and molecular mechanics calculations were performed with CHARMm (version 21.2; Harvard College, Boston, MA), running on a Silicon Graphics Indigo XZ 4000 workstation. An A3 receptor model was built with the model of the A1 receptor we recently proposed as a starting point (12). The latter model is based on the well defined structure of the seven-transmembrane domain protein bacteriorhodopsin and assumes that the spatial orientation of the α-helical domains of G protein-coupled receptors is similar to that of bacteriorhodopsin. An initial structure for the A3 receptor was generated by copying the backbone coordinates (and side chain coordinates where applicable) of the relevant residues in the A1 model to their A3 counterparts. Initial bad contacts within the individual helices were relieved by energy minimization using 200 steps of steepest descents, followed by 200 steps of an adopted basis Newton Raphson. Subsequently, the model was minimized using 500 steps of steepest descents, followed by an adopted basis Newton Raphson minimization until the rms energy gradient was less than 0.01 kcal/mol Å, while keeping the backbone positions fixed. As a last step, the model was energy-minimized under the same conditions with backbone constraints of 5 kcal/mol. Adenosine and other ligands were docked into the presumed binding site starting with the orientation that CPA assumes in the A1 model (12), followed by a two-step energy minimization as described above. Calculations were performed using an extended atom approach and without explicit water, however, a distance-dependent dielectric constant was used to account for the screening effect of solvent.
Results and Discussion
SAR for adenosine derivatives
Binding data for a variety of adenosine derivatives, as well as a number of nucleosides having bases other than adenine are given in Table 1. Representative binding curves for three purine-substituted adenosine derivatives are shown in Fig. 1. The affinity of adenosine itself cannot be accurately determined in this binding assay, due to the presence of adenosine deaminase, which is required to degrade endogenously generated adenosine. Hence, it is not possible to directly compare the affinities of adenosine with the derivatives tested here. The affinity of adenosine has previously been estimated at 30 μM (3), but this value should be taken as only a rough approximation.
TABLE 1. Affinities of selected compounds at A1 A2a and A3 receptors, indicated as either Ki, (nM) or percent displacement at a concentration of 100 μM, unless otherwise indicated.
Values expressed as means ± S.E. were all measured in this study (three to five experiments). Ki, values at A1 and A2a receptors provided without S.E. are taken from the literature as indicated.
| Compound | A1 affinitya | A2a affinityb | A3 affinityc | Reference |
|---|---|---|---|---|
| Purine and 5′-modified adenosines | ||||
| 1. ADACd | 0.85 nM | 210 nM | 281 ± 51 nM | (22) |
| 2. R-PIA | 1.2 nM | 124 nM | 158 ± 52 nM | (23) |
| 3. S-PIA | 49.3 nM | 1,820 nM | 920 ± 311 nM | (23) |
| 4. CPA | 0.59 nM | 462 nM | 240 ± 36 nM | (23) |
| 5. CHA | 1.3 nM | 514 nM | 167 ± 26 nM | (23) |
| 6. N6-Phenyladenosine | 4.62 nM | 663 nM | 802 ± 279 nM | (23) |
| 7. N6-Benzyladenosine | 120 nM | 285 nM | 120 ± 20 nM | (23) |
| 8. N6-Phenethyladenosine | 12.7 nM | 161 nM | 240 ± 58 nM | (24) |
| 9. N6-Dimethyladenosine | 10,000 nM | 28,900 ± 8,500 nM | 32,500 ± 5,100 nM | (10) |
| 10. DPMA | 142 nM | 4.4 nM | 3,570 ± 1,700 nM | (25) |
| 11. N6-(2-Sulfoethyl)adenosine | 41% | 0% | 32,400 ± 7,600 nM | (9) |
| 12. N6-(p-Sulfophenyl)adenosine | 74 nM | 8,900 nM | 526 ± 142 nM | (9) |
| 13. N6-3-(p-Sulfophenyl)propyladenosine | 610 nM | 3,840 nM | 844 ± 67 nM | (9) |
| 14. N6-4-(p-Sulfophenyl)butyladenosine | 432 nM | 11,300 nM | 808 ± 116 nM | (9) |
| 15. 1-Deaza-2-chloro-N6-CPA | 1.6 nM | 13,200 nM | 770 ± 234 nM | (26) |
| 16. 2-Chloroadenosine | 9.3 nM | 63 nM | 1,890 ± 900 nM | (23) |
| 17. 2-Chloro-N6-CPA | 0.6 nM | 950 nM | 237 ± 71 nM | (27) |
| 18. 2-(Phenylamino)adenosine | 560 nM | 119 nM | 4,390 ± 1,170 nM | (23) |
| 19. CGS 21680 | 2,600 nM | 15 nM | 584 ± 32 nM | (28) |
| 20. NECA | 6.3 nM | 10.3 nM | 113 ± 34 nM | (23) |
| 21. N6-CyclohexylNECA | 0.43 nM | 170 nM | 16.0 ± 5.4 nM | (29) |
| 22. N6-BenzylNECA | 87.3 ± 13.9 nM | 95.3 ± 24.6 nM | 6.8 ± 2.5 nM | |
| 23. N6-DimethylNECA | 9,600 nM | 13,500 ± 3,600 nM | 2,260 ± 490 nM | (10) |
| 24. N6-Benzyl-N6-methyladenosine | 7,600 ± 1,900 nM | 40,100 ± 6,200 nM | 78.4 ± 4.6% | |
| 25. 8-Bromoadenosine | 41.5 ± 3.2% | 22,700 ± 5,100 nM | 31.3 ± 6.0% | |
| 26. 3-Deazaadenosine | 21,500 nM | 59,800 ± 4,600 nM | 61,700 ± 34,500 nM | (30) |
| 27. 7-Deazaadenosine (tubercidine) | >100,000 nM | 48.3 ± 0.4% | 38.9 ± 17.7% | (30) |
| 28. Adenosine-N1-oxide | 246 ± 31 nM | 328 ± 60 nM | 3,090 ± 1,910 nM | |
| 29. NECA-N1-oxide | 154 ± 20 nM | 101 ± 19 nM | 468 ± 58 nM | |
| 30. N6-Benzyladenosine-N1-oxide | 864 ± 88 nM | 8,530 ± 1,250 nM | 7,250 ± 1,680 nM | |
| ribose-modified adenosines | ||||
| 31. β-L-Adenosine | 29,000 ± 4,700 nM | 25.4 ± 1.1% | 9.5 ± 4.2% | |
| 32. α-D-Adenosine | 350,000 nM | 128,000 ± 25,000 nM | 14.2 ± 6.5% | (31) |
| 33. 2′-Deoxyadenosine | 30.9 ± 8.0% | 38.9 ± 2.9% | 28.3 ± 2.3% | |
| 34. 2′-O-Methyladenosine | 29.4 ± 7.5% | 49.0 ± 5.0% | 42.9 ± 9.4% | |
| 35. 3′-Deoxyadenosine (cordycepin) | 5.8 ± 2.8% | 26.3 ± 3.4% | 32.7 ± 2.0% | |
| 36. 5′-Deoxyadenosine | 269 ± 135 nM | 596 ± 54 nM | 2,830 ± 460 nM | |
| 37. 5′-Deoxy-5′-aminoadenosine | 42,700 ± 6,000 nM | 38,500 ± 3,800 nM | 20.6 ± 2.2% | |
| 38. 5′-Deoxy-5′-methylthioadenosine | 281 nM | 1,100 nM | 1,420 ± 530 nM | (23) |
| 39. 5′-Deoxy-5′-isobutylthioadenosine | 1,140 ± 130 nM | 6,890 ± 1,750 nM | 3,630 ± 360 nM | |
| 40. S-Adenosylmethionine | 675 ± 87 nM | 2,780 ± 250 nM | 2,470 ± 450 nM | |
| 41. AMP | –* | 57.5 ± 4.0% | 17.2 ± 6.3% | |
| 42. Adenine-β-D-arabinofuranoside | 20.2 ± 8.4% | 26.0 ± 8.4% | 23.7 ± 3.8% | |
| 43. β-D-Psicofuranosyladenine | 36.1 ± 4.9% | 51.5 ± 7.4% | 21.1 ± 0.9% | |
| non-adenosine nucleosides | ||||
| 44. Xanthosine | 9.1 ± 2.4% | 8.5 ± 2.5% | 23.4 ± 8.8% | |
| 45. Uridine | 14.3 ± 6.9% | 2.8 ± 5.2% | 18.9 ± 2.8% | |
| 46. Thymidine | 23.4 ± 2.5% | 1.7 ± 3.4% | 21.3 ± 4.9% | |
| 47. Cytidine | 18.0 ± 1.2% | 16.0 ± 1.5% | 24.5 ± 10.2% | |
| 48. Inosine | 16,700 ± 2,900 nM | 50,000 ± 12,700 nM | 45,100 ± 38,800 nM | |
| 49. Guanosine | 27,800 ± 9,600 nM | 85,100 ± 15,700 nM | 98,500 ± 28,700 nM | |
| 50. (4-Nitrobenzyl)-6-thioguanosine | 15,000 ± 3,500 nM | 48,500 ± 11,300 nM | 40,700 ± 26,300 nM | |
| 51. 6-Thioguanosine | 44.2 ± 2.3% | 27.7 ± 5.8% | 44.8 ± 18.1% | |
| 52. 6-Thiopurine riboside | 61.2 ± 3.9% | 33.6 ± 3.3% | 41.9 ± 5.0% | |
| 53. NECI | 43.7 ± 10.3% | 30.6 ± 2.3% | 5,000 ± 1,150 nM | |
| non-xanthine adenosine antagonists | ||||
| 54. CP 66713 | 270 nM | 21 nM | 29.7 ± 7.8% | (32) |
| 55. CGS 15943 | 21 nM | 3.3 nM | 38.0 ± 14.5% | (33) |
| 56. IQA | 1,600 nM | 1,400 nM | 32.6 ± 10.8% | (8) |
| 57. 9-Ethyl-N6-cyclopentyladenine | 440 nM | 17,000 nM | 30.4 ± 9.1% | (34) |
| 58. EHNA | 455 ± 10 nM | 59.6 ± 2.8% | 57.5 ± 14.3% | |
| 59. Amiloride | 11,000 nM | 17,000 nM | 22.0 ± 3.5% | (35) |
| 60. Xanthine | 298,000 nM | 16.2 ± 2.6% | 14.0 ± 7.9% | (36) |
| 61. 1-MethylX | 11,400 nM | 36,200 nM | 11.1 ± 1.6% | (23) |
| 62. 3-MethylX | 35,000 nM | 38.0 ± 0.9% | 18.1 ± 6.7% | (37) |
| 63. 7-MethylX | 52.3 ± 7.9% | 37.7 ± 4.9% | 16.4 ± 9.6% | |
| 64. 9-MethylX | 26.6 ± 3.2% | 16.1 ± 1.9% | 22.8 ± 9.5% | |
| 65. 1,3-DimethylX (theophylline) | 8,500 nM | 25,000 nM | 23.1 ± 9.5% | (23) |
| 66. 1,7-DimethylX (paraxanthine) | 30,000 nM | 19,400 ± 3500 nM | 15.5 ± 12.1% | (38) |
| 67. 1,9-DimethylX | 29.4 ± 1.6% | 6.0 ± 6.3% | 17.0 ± 7.9% | |
| 68. 3,7-DimethylX (theobromine) | 83,400 nM | 187,000 nM | 19.9 ± 7.1% | (23) |
| 69. 3,9-DimethylX | 19.7 ± 7.9% | 4.2 ± 5.9% | 19.0 ± 6.8% | |
| 70. 1-Methyl-3-ButylX | 7,000 nM | 16,000 nM | 30.1 ± 12.4% | (23) |
| 71. 1,3-DibutylX | 500 nM | 2,930 ± 700 nM | 143,000 ± 29,000 nM | (39) |
| 72. 1,3-DihexylX | 1,260 ± 90 nM | 14.3 ± 3.0% (10 μM) | 9.2 ± 6.5% (10 μM) | |
| 73. 1,3-DibenzylX | 2,000 nM | 3.61 ± 0.94% (10 μM) | 20.3 ± 8.5% (10 μM) | (39) |
| 74. 1,3,7-TrimethylX (caffeine) | 29,000 nM | 48,000 nM | 30.1 ± 12.4% | (23) |
| 75. 1,3,9-TrimethylX (isoC) | >1,000,000 nM | 14.4 ± 5.7% | 13.2 ± 12.4% | (40) |
| 76. 2-Thio-3-propylX | 26,100 ± 1,500 nM | 32,500 ± 4,800 nM | 27.7 ± 11.3% | |
| 7-substituted alkylxanthines | ||||
| 77. 7-BenzylT | 6,000 nM | 46,000 | 29.7 ± 0.2% | (41) |
| 78. 7-β-HydroxyethylT | 105,000 nM | 17,400 ± 900 | 21.1 ± 13.3% | (39) |
| 79. T-7-Riboside | 27,000 ± 3,200 nM | n.t. | 89,400 ± 13,400 nM | |
| 80. 1,3-DipropylX-7-riboside | 15,900 ± 1,800 nM | 32.0 ± 1.1% | 81,200 ± 10,700 nM | |
| 81. 1,3-DibutylX-7-riboside | 4,190 ± 1030 nM | 19,500 ± 4,200 nM | 6,030 ± 2,320 nM | |
| 8-substituted alkylxanthines | ||||
| 82. 8-PhenylT | 86 nM | 850 nM | 12.0 ± 6.0% | (23) |
| 83. 8-CyclopentylT | 11 nM | 1,400 nM | 38.7 ± 2.5% | (23) |
| 84. 8-Cyclopentyl-1-propylX | 226 ± 37 nM | 48,700 ± 5,000 nM | 22.6 ± 7.7% | |
| 85. 8-Cyctopentyl-1,3-dipropylX | 0.46 nM | 340 nM | 18.7 ± 2.9% (10 μM) | (42) |
| 86. 8-CyclohexylC | 28,000 nM | 10,400 ± 2,600 nM | 35.2 ± 1.8% | (37) |
| 87. 8-ChloroT | 30.2 ± 6.7% | 24.7 ± 3.9% | 16.8 ± 9.5% | |
| 88. XAC | 1.2 nM | 63 nM | 7.1 ± 0.9% | (43,22) |
| 89. 8-(3-Chlorostryryl)C (CSC) | 28,200 nM | 54 nM | 4.2 ± 5.1% (10 μM) | (46) |
| 90. 8-Sulfophenyl-1,3-dipropylX | 140 nM | 790 nM | 21.9 ± 6.2% | (47) |
Displacement of [3H]PIA (or [3H]CHA) binding from rat brain membranes.
Displacement of [3H]CGS 21680 (or [3H]NECA in the presence of 50 nM CPA) from rat striatal membranes.
Displacement of [125I]APNEA binding from membranes of CHO cells stably transfected with the rat A3-cDNA.
The abbreviations used are: ADAC, N6-[4-[[[4-[[[(2-aminoethyl)amino]carbonyl]methyl]anilino]carbonyl]methyl]phenyl]adenosine; DPMA, N6-[2-(3,5-dimethyoxyphenyl)-2-(2-methylphenyl)ethyl]adenosine; IQA, imidazo[4,5-c]quinolin-4-amine; EHNA, erythro-9-(2-hydroxy-3-nonyl)adenine; X, xanthine; T, theophylline; C, caffeine; n.t., not tested.
AMP displayed an extremely high slope factor (3.62 ± 0.39 μM) in A1 displacement. The apparent Ki, was 47.5 ± 6.5 μM.
Fig. 1.
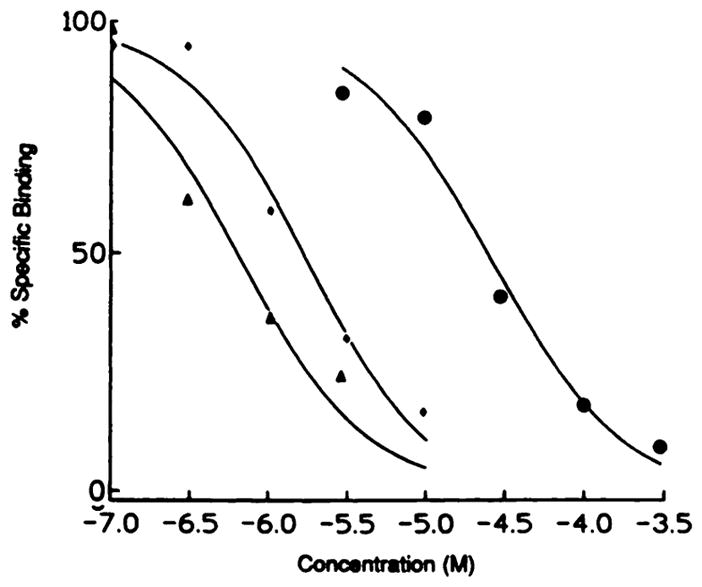
Displacement by three purine-modified adenosine analogues of specific binding of [125I]APNEA (0.5 nM) from membranes from CHO cells transfected with rat A3 receptors. The assay was carried out as described under Materials and Methods. Competitors were as follows (Hill coefficients in parentheses): ◆,2-chloroadenosine (1.10); △, 1-deaza-2-chloro-N6-cyclotopentyladenosine (1.07); and ●, inosine (1.03). The curves are representative of single experiments in which each point is determined in triplicate.
As is the case for A1 receptors, the most potent compounds at the A3 receptor are N6-substituted and/or 5′-N-ethylcarboxamide-substituted adenosine derivatives. There are, however, profound differences between the N6 topology of A1 and A3 receptors. In general, the affinities of N6-substituted adenosines are much higher at A1 than at A3 receptors, with a few notable exceptions. It appears that the selectivity ratio is proportional to A1 affinity; the higher the affinity at A1, the higher the selectivity for A1 versus A3. This indicates that at the A1 receptor, N6 substituents can interact with a receptor region that is not present in the A3 receptor. The stereoselectivity characteristic of the N6 region of A1 and A2 receptors is maintained at A3, albeit that R-PIA is only 6-fold more potent than S-PIA.
In a series of N6-aryl(alkyl)-substituted compounds, N6-benzyladenosine is more potent (Ki, 120 nM) than N6-phenyladenosine (Ki, 802 nM) or N6-phenethyladenosine (Ki, 240 nM). This is surprising given the poor affinity of the former compound at A1 (Ki, 120 nM) and at A2a (Ki, 285 nM) receptors, whereas the phenyl and phenethyl derivatives have affinities in the lower nanomolar range at A1 receptors (Table 1). Thus, N6-benzyladenosine is essentially nonselective.
Another significant difference with A1 and A2a receptors is that introduction of a p-sulfo group in N6-phenyladenosine slightly enhances affinity (N6-(p-sulfophenyl)adenosine, Ki, 526 nM), in sharp contrast to A1 and A2a receptors, where a sulfo group drastically reduces affinity (Table 1). Two other N6-sulfo derivatives, N6-3-(4-sulfophenyl)propyladenosine and N6-4-(4-sulfophenyl)butyladenosine have affinities in the same range (Table 1), but the 2-sulfoethyl derivative, which has a shorter N6 substituent, is considerably less potent (Ki, 32.4 μM). Because the polar sulfo group is apparently better tolerated at A3 than at A1 and A2a receptors, sulfo substitution shifts affinity in the direction of A3 selectivity.
The A1-selective N6-cycloalkyl derivatives, CHA and CPA, are also among the more potent compounds at A3 (Ki, 167 and 240 nM, respectively), as is the N6-functionalized congener N6-[4-[[[4-[ [ [(2-aminoethyl)amino]carbonyl]methyl]anilino]carbonyl]methyl]phenyl]adenosine (Ki, 281 nM). The A2-selective N6-substituted compound, N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)ethyl]adenosine, has moderate potency at A3 (Ki, 3.57 μM).
The affinity of N6-dimethyladenosine is quite low (Ki, 32.5 μM), similar to the poor affinity of this compound at A1 and A2a receptors. The affinity of N6-benzyl-N6-methyladenosine and N6-dimethylNECA is also considerably lower than the parent compounds N6-benzyladenosine and NECA but not as drastically reduced as at A1 and A2 receptors. Thus, although disubstitution at N6 reduces affinity, it enhances selectivity for A3 versus A1 and A2a receptors (e.g., N6-dimethylNECA is 4-fold selective versus A1 and 6-fold versus A2a).
Similar to other adenosine receptor subtypes, NECA (5′-N-ethylcarboxamide adenosine) is relatively potent (Ki, 113 nM). However, unlike A1 and A2a receptors (10), the effects of N6 and C5′ substitutions appear to reinforce each other. Thus, the 5′-N-ethylcarboxamide of CHA is considerably more potent than either CHA (Ki, 167 nM, 10-fold) or NECA (Ki, 113 nM, 7-fold), and, with a Ki of 16 nM, it is a highly potent compound at A3 receptors. Likewise, N6-dimethylNECA is 14-fold more potent than N6-dimethyladenosine. These findings prompted us to synthesize the NECA analogue of N6-benzyladenosine. Because N6-benzyladenosine is more or less equipotent at all three subtypes, and the 5′-N-ethyl substituent apparently boosts affinity at A3 receptors more than at A1 and A2a, it was expected that N6-benzylNECA would be a potent and somewhat A3-selective agonist. Indeed, N6-benzylNECA has the highest A3 affinity of all compounds tested in this study, with a Ki value of 6.8 nM, 18-fold more potent than the parent compound N6-benzyladenosine. This is also the most A3-selective compound found in the present study (13-fold versus A1 and 14-fold versus A2a). This compound may prove useful in the pharmacological characterization of A3 receptors, e.g., as a radioligand and as a lead for the further development of more selective A3 agonists.
Certain C2 modifications may result in A2a-selective agonists (1). At A3 receptors, 2-chloroadenosine and 2-phenylaminoadenosine are of intermediate potency (Ki, 1.9 and 4.4 μM, respectively). An N6-substituted derivative (2-chloro-N6-CPA, Ki, 237 nM) and one bearing a 5′-N-ethylcarboxamide group (CGS 21680, Ki, 584 nM) are more potent C2-substituted derivatives. Thus, 2 substitution is tolerated to a degree at the A3 receptor.
With regard to modifications of the ribose moiety, both the L-enantiomer and the α-anomer of adenosine are virtually inactive (IC50 ≫ 100 μM), similar to other adenosine receptors (13). Psicofuranosyladenine, which contains an extra CH2OH group at Cl′, is also very weak. 2′-Deoxy-, 2′-O-methyl, and 3′-deoxyadenosine all have low affinity (IC50 > 100 μM), and inversion of the stereochemistry of the 2′-OH group (adenine-β-D-arabinofuranoside) similarly results in a low affinity compound. Thus, the presence of the 2′-OH in the S-configuration and the 3′-OH appears to be essential for high affinity. This has also been shown for other adenosine receptor subtypes (reviewed in Ref. 1). The 5′ position is more amenable to modifications than the 2′ or the 3′ position. The 5′-deoxy derivative of adenosine is moderately potent (Ki, 2.83 μM), and, as stated, the 5′-N-ethylcarboxamide derivative (NECA) is one of the more potent compounds tested (Ki, 113 nM). Some 5′-deoxyadenosine derivatives, including those with methylthio-, isobutylthio-, and methionine substituents have affinities in the lower micromolar range, whereas 5′-deoxy-5′-aminoadenosine and AMP (bearing a 5′-phosphate group) are virtually inactive IC50 > 100 μM. This parallels the affinities of these compounds at A1 and A2a receptors (Table 1). In all, ribose SAR for the A3 receptor is comparable with A1 and A2a receptors, suggesting that the ribose domain may be quite similar in all three receptor subtypes.
Some adenosine derivatives not commonly used in adenosine receptor studies were tested. 6-Thiopurine riboside and 8-bromoadenosine both have low affinity (IC50 > 100 μM) at A3, similar to A1 and A2a receptors. A bulky 8 substituent forces the ribose moiety in a predominantly syn conformation, which is a likely explanation for the inactivity of 8-bromoadenosine. The same has been shown for A1 receptors (7). 7-Deazaadenosine has a IC50 ≫ 100 μM, which indicates the importance of N7. 3-Deazaadenosine is slightly more potent, with a Ki of 62 μM. l-Deazaadenosine was not available for testing, but 1-deaza-2-chloro-CPA, (Ki, 770 nM) is only 3-fold less potent than 2-chloro-CPA (Ki, 237 nM). This suggests that the presence of N1 is not crucial, and it is consistent with the profile at other adenosine receptor subtypes, where N1-deazaadenosine > N3-deazaadenosine > N7-deazaadenosine (reviewed in Ref. 1). The N1-oxides of adenosine, NECA, and N6-benzyladenosine are moderately potent compounds (Ki, 3.09, 0.47, and 7.25 μM, respectively) but less potent than at A1 and A2a receptors (Table 1).
Of the unsubstituted nonadenine nucleosides tested, only inosine (Ki, 45 μM) and guanosine (Ki, 99 μM) show some affinity for the A3 receptor. The 5′-N-ethylcarboxamide derivative of inosine (NECI) is more potent (Ki, 5 μM), which is consistent with the affinity-enhancing effect of the 5′-carboxamido substituent of NECA. NECI is also selective for A3 receptors, having an IC50 larger than 100 μM at A1 and A2a receptors. An adenosine transport inhibitor, (4-nitrobenzyl)-6-thioguanosine (Ki, 41 μM), is slightly more potent than the parent compound, guanosine.
SAR for nonadenosine derivatives
According to Zhou et al. (3), xanthines do not appear to displace [125I]APNEA binding to A3 receptors. We first tested a variety of nonxanthines known to act as antagonists at A1 and/or A2a receptors, including CGS 15943, CP 66713, 1H-imidazo[4,5-c]quinolinamine, 9-ethylcyclopentyladenine, and amiloride (Table 1). None of these appeared to be particularly potent, with Ki values in the range of 100 μM or larger. The adenosine deaminase inhibitor, erythro-9-(2-hydroxy-3-nonyl)adenine, was somewhat more potent with an IC50 slightly better than 100 μM (57.5% displacement at 100 μM), erythro-9-(2-Hydroxy-3-nonyl)adenine was of moderate affinity at A1 receptors.
We therefore turned to a more detailed study of xanthine SAR (Table 1) than in the original paper (3). Xanthine was found to be a very weak displacer of [125I]APNEA binding (14% at 100 μM). Substitutions at the 1 and the 3 positions enhance affinity. Compared with theophylline (1,3-dimethylxanthine, 23.1% at 100 μM), 1,3-dibutylxanthine is more potent (Ki, 143 μM). 1,3-Dihexylxanthine (9.2% at 10 μM) and 1,3-dibenzylxanthine (20.3% at 10 μM) also seem more potent than theophylline, but their limited solubility precludes direct comparison. This profile is similar, but not identical, to other adenosine receptor subtypes, where propyl and butyl are optimal, and benzyl is slightly less potent (1). Still, even the most potent of these 1,3-substituted xanthines is quite weak at rat A3 receptors.
Due to the limited solubility of even the most potent xanthine, it was not feasible to compare precisely the degree of maximal displacement of the xanthines and the adenosine derivatives. Unfortunately, the more water-soluble xanthines, such as XAC (positively charged at physiological pH) and 8-(p-sulfophenyl)-1,3-dipropylxanthine (negatively charged at physiological pH) did not bind appreciably to rat A3 receptors.
Unlike A1 and A2a receptors, 8 substituents do not appear to contribute much to affinity and none of the 8-substituted xanthine derivatives tested is particularly potent (Table 1). This is surprising, because at A1 and A2a receptors, affinities of N6-substituted adenosines and xanthines similarly substituted at the 8 position closely parallel each other, suggesting that N6 and C8 substituents interact with the same receptor domain (1, 14). Clearly, this is a subject for further investigation. Substitutions at the 7 position appear to be tolerated, in contrast to A1 receptors, where 7 substituents tend to diminish affinity (7); e.g., both caffeine and 7-benzyltheophylline are slightly more potent at A3 receptors than the 7-unsubstituted parent compound, theophylline. The A2a-selective antagonist 8-(3-chlorostyryl)caffeine (47) did not inhibit binding at rat A3 receptors.
Molecular modeling and prediction of affinity of xanthine-7-ribosides
Like other G protein-coupled receptors, the amino acid sequence of A3 receptors contains seven hydrophobic stretches of approximately 25 residues that are believed to traverse the cell membrane as α-helices (3). No detailed three-dimensional structures (X-ray or NMR) for any of the G protein-coupled receptors are known, but it is now well accepted that the structure of bacteriorhodopsin, which has been solved by cryo-electron microscopy (15), is a suitable starting point for the modeling of G protein-coupled receptors (16). We have recently described a model for the A1 adenosine receptor (12), and, here, we present a similar model for the A3 receptor. Details of the building of the model are given under Materials and Methods.
A close-up of the proposed binding site of the A3 receptor with N6-benzyladenosine as the ligand is shown in Fig. 2. This model is based on pharmacological observations and analogies with the A1 receptor and is consistent with the SAR described above. N6-Benzyladenosine was chosen as a typical agonist for the A3 receptor, because it is relatively potent (Table 1). In the A1 model (12), CPA was chosen as a typical high affinity A1- selective agent for the purpose of docking to the binding site. There, CPA is coordinated by two histidine residues in helices VI and VII, whose involvement is in full agreement with both chemical modification studies (17) and site-directed mutagenesis (18). It should be mentioned here that another quite different A1 receptor model has been developed in which no specific interaction with histidine residues was proposed (48).
Fig. 2.

The proposed adenosine binding site of the A3 receptor, with N6-benzyladenosine as the ligand. The ligand is shown as a ball-and-stick representation (thick bonds) and the receptor is shown in liquorice bond style (thinner bonds). The 2′- and 3′-OH may form a hydrogen bond with His274, and 5′-OH can form a hydrogen bond with Ser277.
There are a number of similarities as well as some substantial differences between our models for the A1 and A3 receptors. As in the A1 model, the agonist ligand is present in the anti conformation (χ, the torsion angle of the glycosidic bond is 76°), consistent with earlier modeling and NMR studies (19). The model proposes several points of interaction between the receptor and the ribose moiety. Hydrogen bonds could be formed between the 2′- and 3′-OH groups and His274 in helix VII and between the 5′-OH group and Ser277 in helix VII. There are equivalent interactions with His278 and Ser281 in the A1 receptor model. Thus, the ribose binding domain seems to be quite similar for A3 and A1 receptors, in good agreement with the agonist SAR described here; the same appears to be true for A2a receptors (45). In the present model, the side chain of Phe184 (helix V) is located near the glycosidic bond of receptor-bound N6-benzyladenosine. The same Phe residue and Thr96 (helix III) appear to be in proximity to the C5′ region.
A major structural difference is that the A3 receptor does not contain the histidine residue in helix VI that is common to all A1 and A2 receptors cloned so far (20) and that has been shown to have an effect on both agonist and antagonist binding to A1 receptors (18). In particular, the His of helix VI has been shown to be important for antagonist affinity, a finding that suggests a linkage between the absence of this His residue and the lack of high affinity binding of antagonists such as XAC at rat A3 receptors. In the model of agonists binding to A1 receptors (12), this residue forms a hydrogen bond with N6-H. In the A3 receptor, a serine residue (Ser249) is found in the analogous position, and it could be argued that serine could serve a similar function as a hydrogen bond acceptor. However, due to a different orientation of the purine moiety in the A3 binding site, this serine seems to be too far from N6-H (~7 Å) to be able to form this bond. The reason for the different orientation is as follows. The majority of the amino acid residues that are different between A1 and A3 receptors occur in the immediate vicinity (within 5 Å) of the agonist ligand, and many of the A3 residues are considerably more bulky (e.g., Phe95) than their A1 counterparts. This results in a ligand binding environment (for both the purine and ribose domains) that is much more constricted than is the case for the A1 receptor (12). In Fig. 3 the binding orientations of CPA to the A1 receptor and of N6-benzyladenosine to the A3 receptor are shown to illustrate these differences. They provide a tentative explanation for the apparent low affinity of xanthines and nonxanthine A1/A2 antagonists. In addition, the ribose moiety that likely serves to anchor the native ligand adenosine to the receptor is absent in these compounds.
Fig. 3.

Comparison of CPA as bound in the A1 model (left) and N6-benzyladenosine in the A3 model (right). Color coding is as follows: yellow, ligand; blue, H.I; orange, H.II; green, H.III; red, H.IV; purple, H.V; white, H.VI; pink, H.VII. The A1 model is identical to that proposed in Ref. 12. See Results for a discussion of specific residues proposed to be in proximity to the bound N6-benzyladenosine.
As in the A1 model, a hydrophobic pocket directed toward the extracellular space, capable of accommodating large N6 substituents, is present. Fig. 2 shows the binding environment of the benzyl substituent of N6-benzyladenosine. Indeed, a number of N6-substituted adenosine derivatives, including the long chain functionalized congener N6-[4-[[[4-[[[(2-aminoethyl)amino]carbonyl]methyl]anilino]carbonyl]methyl]-phenyl]adenosine, have considerable affinity for the A3 receptor (Table 1). Amino acid residues in proximity to the N6-benzyl ring according this model are the side chains of Phe181 (hydrophobic) and Asp177 (anionic) and the backbone atoms of Tyr178 (all in helix V). Due to the rather constricted agonist binding domain, the N6 region is much closer to the membrane surface than in the A1 model. It might be hypothesized that the paucity of hydrophobic residues for an N6 substituent to interact with, because of the proximity of the N6 region to the membrane surface, accounts for the rather moderate affinity of N6-substituted adenosines, in comparison with the much higher affinities of a number of similar agonists at A1 and A2a receptors. This would also agree well with a polar sulfo substituent being tolerated close to the N6 region of A3 but not A1 or A2a receptors. A second (hydrophobic) pocket is present adjacent to C2, which can accommodate fairly large C2 substituents. Again, this agrees well with the considerable potency of some C2-substituted agonists, e.g., CGS 21680 (Table 1). The backbone of Tyr256 (helix VI) is situated near the purine C2 position of N6-benzyladenosine.
It should be noted here that due to the relative scarcity of pharmacological and structural data for this novel receptor, of necessity, there are uncertainties in this model. The question about the involvement of Ser249 in binding of the purine moiety and whether the different agonist orientations in the A1 and A3 models will hold up should be addressed experimentally. Chemical modification studies, site-directed mutagenesis, and more SAR work will be needed to further define essential characteristics of the ligand-receptor interaction, which, in turn, may lead to a more refined model. At this stage, the A3 model should mainly be seen as a starting point to generate ideas for experiments, and its usefulness is illustrated by the following.
The notion from the modeling studies that the primary point of interaction between receptor and ligand appears to be the ribose moiety of agonists, combined with the observation that 7 substitution apparently is tolerated, prompted us to test some xanthine-7 ribosides that were previously synthesized (7). It was reasoned that the ribose hydroxyl groups might serve to anchor the xanthine nucleus to the receptor. Indeed, of the compounds tested, 1,3-dibutylxanthine-7 riboside (DBXR) was quite potent (Ki, 6μM), almost 25-fold more potent than the parent 1,3-dibutylxanthine, which was one of the most potent xanthines tested (Table 1). Affinity at A1 and A2a receptors is 4.19 and 19.5 μM, respectively, so this compound has only very moderate selectivity.
Fig. 4 illustrates a proposed model for DBXR binding to the A3 receptor. According to this model, the 1,3-dialkyl substituents of DBXR are located in hydrophobic regions near the exofacial surface of the A3 receptor. Specifically, the N1 -butyl chain is located near the side chain of Tyr256 of helix VI, and the N3-butyl chain is located near the side chain of Phe181 of helix VI.
Fig. 4.

Proposed mode of binding of 1,3-dibutylxanthine-7-riboside to the A3 receptor. Details are as for Fig. 1. The N1-butyl chain is located near the side chain of Tyr256 of helix VI, and the N3-butyl chain is located near the side chain of Phe181 of helix VI.
Effects on adenylate cyclase
The effects of key compounds on the inhibition of adenylate cyclase in CHO cells stably expressing the rat A3 receptor was measured (Fig. 5). Indeed, adenosine derivatives N6-cyclohexylNECA and N6-benzylNECA proved to be agonists at A3 receptors, with full efficacy, as observed with R-PIA and NECA (data not shown). The maximal inhibition of adenylate cyclase elicited by N6-cyclohexylNECA was 45.7 ± 2.1%. The IC50 determined for this compound was 1.30 ± 0.31 μM. N6-BenzylNECA was nearly as potent (IC50 1.61 μM) with a maximal inhibition of 47.6 ± 6.3%.
Fig. 5.
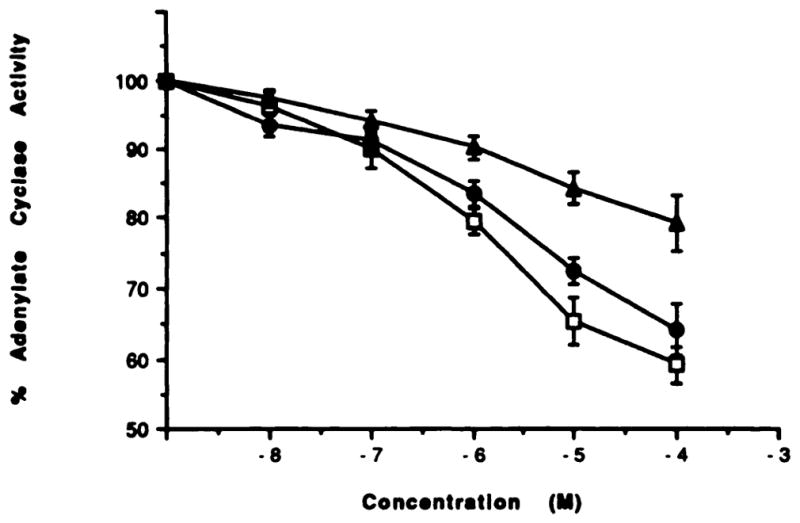
Inhibition of adenylate cyclase in membranes from CHO cells transfected with rat A3 receptors. The assay was carried out as described under Materials and Methods. Each data point is shown as mean ± S.E. for four to seven determinations. Agents were as follows (number of separate experiments in parentheses): triangles, 1,3-dibutylxanthine-7-riboside (5); circles, N6-benzylNECA (7); and squares, N6-cyclohexyl-NECA (4). In these cell membranes, a KD value for binding of [125I]APNEA to A3 receptors was 5.67 ± 0.73 nM with a Bmax of 1.51 ± 0.40 pmol/mg protein.
At A1 receptors, xanthine-7 ribosides have been shown to act as antagonists or partial agonists (7, 21). At rat A3 receptors, 1,3-dibutylxanthine-7 riboside did inhibit adenylate cyclase, but the dose-response curve was more shallow than for the adenosine derivatives (only 20.9 ± 4.0% inhibition at 100 μM); thus, it appears to be a partial agonist. The inosine derivative NECI was so weak in inhibiting adenylate cyclase that a full-dose response was not able to be measured; at a concentration of 10−4 M, it inhibited adenylate cyclase by 9.4 ± 3.8% (n = 7). Although the majority of the compounds were not assayed in this functional assay, the rank order of potency paralleled the order of potency in displacing the specific binding of radioligand at A3 receptors.
1,3-Dibutylxanthine at 100 μM neither antagonized the action of an adenosine agonist (N6-cyclohexylNECA) acting at A3 receptors nor itself inhibited adenylate cyclase in the transfected CHO cells. Theophylline was also unable to antagonize the inhibition of adenylate cyclase elicited by N6-benzylNECA. N6-BenzylNECA alone had an IC50 value of 1.35 ± 0.65 μM, with maximal inhibition of 28.8 ± 0.9% (100 μM). In the presence of 1 mM theophylline, the IC50 value was 0.91 ± 0.1 μM, with a maximal inhibition of 34.2 ± 1.5%. Theophylline had no effect on the basal level of adenylate cyclase or on how much forskolin was able to stimulate.
Conclusions
N6-BenzylNECA was identified as the first highly potent and moderately A3-selective agonist. Combined with the other SAR differences found between A3 and A1/A2 affinity, such as polar substituents being tolerated in the N6 region, it should provide a good lead toward the development of even more potent and selective A3 agonists.
Mutation experiments have shown that the His residue in the sixth transmembrane helix of both A1 and A2 receptors is involved in the high affinity binding of antagonists such as XAC (18). The complete inactivity of xanthines at rat A3 receptors, which lack that His, is consistent with this model. This A3 receptor molecular model has also been in part validated with the moderate affinity of 1,3-dibutylxanthine-7 riboside, which appears to act as a partial agonist. This study did not identify any A3 antagonists among a wide range of the known A1/A2 receptor antagonists.
The predictions of the computer-generated model for the binding site must be tested through further efforts in ligand synthesis and modification of the receptor structure through site-directed mutagenesis of A3 receptors. More elaborate SAR studies to further define optimal substituents for interaction with the N6 and C5′ regions of the A3 adenosine receptor are currently underway.
Acknowledgments
We thank Dr. Arthur Jacobson and the Scientific Resources Computer Center at the National Institutes of Health for use of computer facilities and Drs. John Daly and Bilha Fischer for useful suggestions.
ABBREVIATIONS
- APNEA
N6-2-(4-aminophenyl)ethyladenosine
- CGS 15943
9-chloro-2-(2-furyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine
- CGS 21680
2-[4-(2-carboxyethyl)phenyl]ethylamino-5′-N-ethlcarboxamidoadenosine
- CHA
N6-cyclohexyladenosine
- CPA
N6-cyclopentyladenosine
- DBXR
1,3-dibutylxanthine-7 riboside
- NECA
5′-N-ethylcarboxamidoadenosine
- NECI
5′-N-ethylcarboxamidoinosine
- R/S-PIA
N6-[(R/S)-1-methyl-2-phenylethyl]adenosine
- SAR
structure-activity relationships
- XAC
8-[-4-[[[[(2-aminoethyl)amino]carbonyl]methyl]oxy]phenyl]-1,3-dipropylxanthine
- Me2SO
dimethylsulfoxide
- CHO
Chinese hamster ovary
Footnotes
During preparation of this paper, the cloning of the sheep A3 receptor was reported (Linden et al., Mol. Pharmacol., 1993, 44:524-532). At this receptor, certain xanthine derivatives do bind and act as antagonists, albeit in most cases with diminished affinity relative to A1 receptors.
References
- 1.van Galen PJM, Stiles GL, Michaels G, Jacobson KA. Adenosine A1 and A2 receptors: structure-function relationships. Med Res Rev. 1992;12:423–471. doi: 10.1002/med.2610120502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson KA, van Galen PJM, Williams M. Perspective. Adenosine receptors: pharmacology, structure-activity relationships and therapeutic potential. J Med Chem. 1992;35:407–422. doi: 10.1021/jm00081a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou QY, Li CY, Olah ME, Johnson RA, Stiles GL, Civelli O. Molecular cloning and characterization of an adenosine receptor: the A3 adenosine receptor. Proc Natl Acad Sci U S A. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyerhof W, Müller-Brechlin R, Richter D. Molecular cloning of a novel putative G protein-coupled receptor expressed during rat spermiogenesis. FEBS Lett. 1991;284:155–160. doi: 10.1016/0014-5793(91)80674-r. [DOI] [PubMed] [Google Scholar]
- 5.Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3 adenosine receptor is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J Biol Chem. 1993;268:16887–16890. [PubMed] [Google Scholar]
- 6.Stiles GL, Daly DT, Olsson RA. The A1 adenosine receptor. Identification of the binding subunit by photoaffinity cross-linking. J Biol Chem. 1985;260:10806–10811. [PubMed] [Google Scholar]
- 7.van Galen PJM, Ijzerman AP, Soudijn W. Xanthine-7 ribosides as adenosine receptor antagonists. Nucleos Nucleot. 1990;9:275–291. [Google Scholar]
- 8.van Galen PJM, Nissen P, van Wijngaarden I, Ijzerman AP, Soudijn W. 1H-imidazo(4,5-c]qninolin-4-amines: novel nonxanthine adenosine antagonists. J Med Chem. 1991;34:1202–1206. doi: 10.1021/jm00107a046. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson KA, Nikodijevic O, Ji XD, Berkich DA, Eveleth D, Dean RL, Hiramatsu K, Kassell NF, van Galen PJM, Lee KS, Bartus RT, Daly JW, Lanoue KF, Maillard M. Synthesis and biological activity of N6-(p-sulfophenyl)alkyl and N6-sulfoalkyl derivatives of adenosine. Water-soluble and peripherally selective adenosine agonists. J Med Chem. 1992;35:4143–4149. doi: 10.1021/jm00100a020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsson RA, Kusachi S, Thompson RD, Ukena D, Padgett W, Daly JW. N6-substituted N-alkyladenosine-5′-uronamides: bifunctional ligands having recognition groups for A1 and A2 adenosine receptors. J Med Chem. 1986;29:1683–1689. doi: 10.1021/jm00159a020. [DOI] [PubMed] [Google Scholar]
- 11.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 12.Ijzerman AP, van Galen PJM, Jacobson KA. Molecular modeling of adenosine receptors. I. The ligand binding site on the A1 receptor. Drug Design Dev. 1992;9:49–68. [PMC free article] [PubMed] [Google Scholar]
- 13.Brims RF. Adenosine receptor activation in human fibroblasts: nucleoside agonists and antagonists. Can J Physiol Pharmacol. 1980;58:673–691. doi: 10.1139/y80-110. [DOI] [PubMed] [Google Scholar]
- 14.van der Wenden EM, Ijzerman AP, Soudijn W. A steric and electrostatic comparison of 3 models for the agonist/antagonist binding site on the adenosine A1 receptor. J Med Chem. 1992;35:629–635. doi: 10.1021/jm00082a003. [DOI] [PubMed] [Google Scholar]
- 15.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckman E, Downing KH. Model for the structure of bacteriorhodopsin based on high resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 16.Hibert MF, Trumpp-Kallmeyer S, Hoflack J, Bruinvels A. This is not a G protein-coupled receptor. Trends Pharmacol Sci. 1993;14:7–12. doi: 10.1016/0165-6147(93)90106-t. [DOI] [PubMed] [Google Scholar]
- 17.Klotz KN, Lohse MJ, Schwabe U. Chemical modification of A1 adenosine receptors in rat brain membranes. Evidence for histidine in different domains of the ligand binding site. J Biol Chem. 1988;263:17522–17526. [PubMed] [Google Scholar]
- 18.Olah ME, Ren HZ, Ostrowski J, Jacobson KA, Stiles GL. Cloning, expression, and characterization of the unique bovine-A1 adenosine receptor. Studies on the ligand binding site by site-directed mutagenesis. J Biol Chem. 1992;267:10764–10770. [PMC free article] [PubMed] [Google Scholar]
- 19.van Galen PJM, Ijzerman AP, Soudijn W. Xanthine-7 ribosides as adenosine receptor antagonists. Further evidence for adenosine’s anti mode of binding. Nucleos Nucleot. 1991;10:1191–1193. [Google Scholar]
- 20.Linden J, Tucker AL, Robeva AS, Graber SG, Munshi R. Properties of recombinant adenosine receptors. Drug Dev Res. 1993;28:232–236. [Google Scholar]
- 21.Borea PA, Varani K, Gardenghi A, Bertolasi V, van Galen PJM, Ijzerman AP. Theophylline-7 riboside: a partial agonist for adenosine A1, receptors. Int J Purine Pyrimidine Res. 1992;3:65. [Google Scholar]
- 22.Daly JW, Jacobson KA. Molecular probes for adenosine receptors. In: Ribeiro JA, editor. Adenosine Receptors in the Nervous System. Taylor & Francis; London: 1989. pp. 41–52. [Google Scholar]
- 23.Bruns RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- 24.Bridges AJ, Moos WH, Szotek DL, Trivedi BK, Bristol JA, Heffher TG, Bruns RF, Downs DA. N6-(2,2-diphenylethyl)adenosine, a novel adenosine receptor agonist with antipsychotic-like activity. J Med Chem. 1987;30:1709–1711. doi: 10.1021/jm00393a003. [DOI] [PubMed] [Google Scholar]
- 25.Trivedi BK. Structure-activity relationships for adenosine agonists. In: Jacobson KA, Daly JW, Manganiello V, editors. Purines in Cellular Signalling. Targets for New Drugs. Springer; New York: 1990. pp. 136–145. [Google Scholar]
- 26.Cristalli G, Franchetti P, Grifantini M, Vittori S, Klotz KN, Lohse MJ. Adenosine receptor agonists: synthesis and biological evaluation of 1-deaza analogues of adenosine derivatives. J Med Chem. 1988;31:1179–1183. doi: 10.1021/jm00401a018. [DOI] [PubMed] [Google Scholar]
- 27.Thompson RD, Secunda S, Daly JW, Olsson RA. Activity of N6-2-chloroadenosines at A1 adenosine and A2 adenosine Receptors. J Med Chem. 1991;34:3388–3390. doi: 10.1021/jm00116a007. [DOI] [PubMed] [Google Scholar]
- 28.Francis JE, Webb RL, Ghai GR, Hutchison AJ, Moskal MA, Dejesus R, Yokoyama R, Rovinski SL, Contardo N, Dotson R, Barclay B, Stone GA, Jarvis MF. Highly selective adenosine- A2 receptor agonists in a series of N-alkylated 2-aminoadenosines. J Med Chem. 1991;34:2570–2579. doi: 10.1021/jm00112a035. [DOI] [PubMed] [Google Scholar]
- 29.Daly JW, Padgett WL. Agonist activity of 2- and 5′-substituted adenosine analogs and their N6-cycloalkyl derivatives at A1-adenosine and A2-adenosine receptors coupled to adenylate cyclase. Biochem Pharmacol. 1992;43:1089–1093. doi: 10.1016/0006-2952(92)90616-q. [DOI] [PubMed] [Google Scholar]
- 30.Cristalli G, Grifantini M, Vittori S. Adenosine and 2-chloroadenosine deaza analogues as adenosine receptor antagonists. Nucleos Nucleot. 1985;4:625–639. [Google Scholar]
- 31.Lohse MJ, Klotz KN, Diekmann E, Friedrich K, Schwabe U. 2′,3′-Dideoxy-N6-cyclohexyladenosine: an adenosine derivative with antagonist properties at adenosine receptors. Eur J Pharmacol. 1988;156:157–160. doi: 10.1016/0014-2999(88)90158-6. [DOI] [PubMed] [Google Scholar]
- 32.Sarges R, Howard HR, Browne RG, Lebel LA, Seymour PA, Koe BK. 4-Amino[l,2,4]triazolo[4,3-a]quinoxalines. A novel class of potent adenosine receptor antagonists and potential rapid-onset antidepressants. J Med Chem. 1990;33:2240–2254. doi: 10.1021/jm00170a031. [DOI] [PubMed] [Google Scholar]
- 33.Francis JE, Cash WD, Psychoyos S, Ghai G, Wenk P, Friedmann RC, Atkins C, Warren V, Furness P, Hyun JL, Stone GA, Desai M, Williams M. Structure-activity profile of a series of novel triazoloquinazoline adenosine antagonists. J Med Chem. 1988;31:1014–1020. doi: 10.1021/jm00400a022. [DOI] [PubMed] [Google Scholar]
- 34.Thompson RD, Secunda S, Daly JW, Olsson RA. N6,9-Disubstituted adenines. Potent, selective antagonists at the A1-adenosine receptor. J Med Chem. 1991;34:2877–2882. doi: 10.1021/jm00113a029. [DOI] [PubMed] [Google Scholar]
- 35.Garritsen A, Ijzerman AP, Tulp MT, Cragoe EJ, Soudijn W. Receptor binding profile of amiloride provides no evidence for a link between receptor and the Na+/H+ exchanger but indicates a common structure on receptor proteins. J Recept Res. 1991;11:891–907. doi: 10.3109/10799899109064686. [DOI] [PubMed] [Google Scholar]
- 36.Schwabe U, Ukena D, Lohse MJ. Xanthine derivatives as antagonists at A1 and A2 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1985;330:212–221. doi: 10.1007/BF00572436. [DOI] [PubMed] [Google Scholar]
- 37.Shamim MT, Ukena D, Padgett WL, Daly JW. Effects of 8-phenyl and 8-cycloalkyl substituents on the activity of mono-, di-, and trisubstituted alkylxanthines with substitution at the 1, 3, and 7 positions. J Med Chem. 1989;32:1231–1237. doi: 10.1021/jm00126a014. [DOI] [PubMed] [Google Scholar]
- 38.Daly JW, Butts-Lamb P, Padgett W. Subclasses of adenosine receptors in the central nervous system: interaction with caffeine and related methylxanthines. Cell Mol Neurobiol. 1983;3:69–80. doi: 10.1007/BF00734999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daly JW, Padgett WL, Shamim MT. Analogues of caffeine and theophylline: effect of structural alterations on affinity at adenosine receptors. J Med Chem. 1986;29:1305–1308. doi: 10.1021/jm00157a035. [DOI] [PubMed] [Google Scholar]
- 40.Schneller SW, Ibay AC, Christ WJ, Bruns RF. Linear and proximal benzo-separated alkylated xanthines as adenosine-receptor antagonists. J Med Chem. 1989;32:2247–2254. doi: 10.1021/jm00130a004. [DOI] [PubMed] [Google Scholar]
- 41.Daly JW, Hide I, Bridson PK. Imidazodiazepinediones: a new class of adenosine receptor antagonists. J Med Chem. 1990;33:2818–2821. doi: 10.1021/jm00172a022. [DOI] [PubMed] [Google Scholar]
- 42.Linden J. Structure and function of A1 adenosine receptors. FASEB J. 1991;5:2668–2676. doi: 10.1096/fasebj.5.12.1916091. [DOI] [PubMed] [Google Scholar]
- 43.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of 1,3-dialkylxanthines: preparation of analogues with high affinity for adenosine receptors. J Med Chem. 1985;28:1334–1340. doi: 10.1021/jm00147a038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fozard JR, Carruthere AM. Adenosine A3 receptors mediate hypotension in the angiotensin II-supported circulation of the pithed rat. Br J Pharmacol. 1993;109:3–5. doi: 10.1111/j.1476-5381.1993.tb13522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ijzerman AP, van der Wenden EM, van Galen PJM, Jacobson KA. Molecular modeling of adenosine receptors. II. The ligand binding site on the A2 receptor. Eur J Pharmacol. doi: 10.1016/0922-4106(94)90124-4. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacobson KA, Shi D, Gallo-Rodriguez C, Manning M, Müller C, Daly JW, Neumeyer JL, Kiriasis L, Pfleiderer L. Effect of trifluoromethyl and other substituents on activity of xanthines at adenosine receptors. J Med Chem. 1993;36:2639–2644. doi: 10.1021/jm00070a007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jacobson KA, Gallo-Rodriguez C, Melman N, Fischer B, Maillard M, van Bergen A, van Galen PJM, Karton Y. Structure-activity relationships of 8-styrylxanthines as A2-selective adenosine antagonists. J Med Chem. 1993;36:1333–1342. doi: 10.1021/jm00062a005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dudley MW, Peet NP, Demeter DA, Weintraub HJR, Ijzerman AP, Nordvall G, van Galen PJM, Jacobson KA. Adenosine A1 receptor and ligand molecular modeling. Drug Dev Res. 1993;28:237–243. doi: 10.1002/ddr.430280309. [DOI] [PMC free article] [PubMed] [Google Scholar]