

Summary
Farnesyl pyrophosphate (FPP) is an intermediate in cholesterol biosynthesis, and it has also been reported to activate platelet LPA (lysophosphatidic acid) receptors. The aim of this study was to investigate the role of extracellular FPP in platelet aggregation. Human platelets were studied with light transmission aggregometry, flow cytometry and [35S]GTPγS binding assays. As shown previously, FPP could potentiate LPA-stimulated shape change. Surprisingly, FPP also acted as a selective insurmountable antagonist to ADP-induced platelet aggregation. FPP inhibited ADP-induced expression of P-selectin and the activated glycoprotein (Gp)llb/llla receptor. FPP blocked ADP-induced inhibition of cAMP accumulation and [35S]GTPγS binding in platelets. In Chinese hamster ovary cells expressing the P2Y12 receptor, FPP caused a right-ward shift of the [35S]GTPγS binding curve. In Sf9 insect cells expressing the human P2Y12 receptor, FPP showed a concentration-dependent, although incomplete inhibition of [3H]PSB-0413 binding. Docking of FPP in a P2Y12 receptor model revealed molecular similarities with ADP and a good fit into the binding pocket for ADP. In conclusion, FPP is an insurmountable antagonist of ADP-induced platelet aggregation mediated by the P2Y12 receptor. It could be an endogenous antithrombotic factor modulating the strong platelet aggregatory effects of ADP in a manner similar to the use of clopidogrel, prasugrel or ticagrelor in the treatment of ischaemic heart disease.
Keywords: ADP receptors, platelet pharmacology, platelet physiology
Introduction
Farnesyl pyrophosphate (FPP) is an intermediate in the mevalonate biosynthetic pathway, an important metabolic route controlling production of cholesterol, which is present in all eukaryotic animals (1). FPP is formed by the sequential condensation of dimethylallyl pyrophosphate (DMPP) and 3-isopentenyl pyrophosphate (IPP). Besides its function as a precursor in cholesterol synthesis, FPP also serves as a donor in post-translational isoprenylation of proteins, both directly and indirectly, by being an intermediate for geranylgeranyl pyrophosphate (GGPP) (2).
Although FPP is synthesised in the cytoplasm, it can be detected in human plasma. The steady-state plasma level was reported to be 6.6 ng/ml (17 nM), but even the mild physiological alteration caused by eating a meal has been demonstrated to increase the plasma concentration 200 fold (3). Very little is known about plasma levels of FPP during physiological or pathophysiological conditions, such as ischaemia. Since HMG-CoA reductase is the main target for statins, the major group of cholesterol-lowering medications, it is likely that statin therapy also affects the plasma FPP levels.
While the basic function of FPP seems to be structural, there have recently been several reports indicating an additional role in signalling. FPP was shown to act as a natural antagonist of the LPA2 (lysophosphatidic acid type 2) and LPA3 receptors, while it was reported to be an agonist at the structurally divergent LPA4 and LPA5 receptors (4). In an earlier report we surveyed the different G protein-coupled receptors present in platelet cDNA and found high expression levels of LPA5 and LPA4 mRNA (5). In another study Khandoga et al. (6) determined the expression level of LPA1 – LPA7, in platelets and investigated them for their involvement in aggregation. It was concluded that none of the investigated LPA receptors could be coupled with LP-mediated platelet activation. As there have been conflicting reports regarding the function of FPP on platelets, we decided to investigate this function. Somewhat surprisingly, we discovered that the main function of FPP is not as an agonist at the LPA4 and LPA5 receptors, but rather as an antagonist at the structurally related adenosine diphosphate (ADP) receptor P2Y12, which is perhaps the most important receptor in the platelet activation process. Furthermore, the molecular structure of FPP is similar to ADP in having a diphosphate side chain. Here we demonstrate that FPP attenuates platelet aggregation by antagonising the action of 2-MeSADP, both at the receptor level and on a functional level.
Methods and material
Ethics
The Ethics Committee of Lund University approved the project. All volunteers submitted written consent to participate in the study. The study conforms to the US National Institutes of Health guidelines and the Declaration of Helsinki.
Chemicals
3,7,11-Trimethyl-2,6,10-dodecatrien-1-yl pyrophosphate ammonium salt (FPP), 3,7,11-trimethyl-2,6,10-dodecatrien-1-yl monophosphate ammonium salt (FMP), all trans-3,7,11,15-tetra-methyl-2,6,10,14-hexadecatetraenyl pyrophosphate ammonium salt (GGPP), trans-3,7-dimethyl-2,6-octadienyl pyrophosphate ammonium salt (GPP), isopentenyl pyrophosphate triammonium salt (IPP), γ,γ-dimethylallyl pyrophosphate triammonium salt (DMPP), 2-methylthioadenosine-5′-diphosphate (2-MeSADP), Thrombin Receptor Activator Peptide (TRAP), epinephrine and IBMX were bought from Sigma-Aldrich (St. Louis, MO, USA). [35S]GTPγS was bought from PerkinElmer (Waltham, MA, USA). Ticagrelor (AZD6140) was a gift from AstraZeneca (R&D Mölndal, Mölndal, Sweden). Fluo4-AM was from Invitrogen Life Technologies (Carlsbad, CA, U SA). N-arachidonoylglycine was bought from Cayman Chemical (Ann Arbor, MI, USA).
Platelet preparation for light transmission aggregation (LTA)
Aggregation studies on human platelet-rich plasma (PRP) were done as previously described (7). Platelet concentration in PRP were nomalised to 300,000 cells/μl. Preparation for aggregation studies on washed platelets was performed as described (8). The final platelet pellet was suspended to give a final concentration of 300,000 cells/μl.
Flow cytometric analysis of platelet activation
Whole blood was collected into 0.129 M Na-Citrate using a Vacutainer® tube (BD, Franklin Lakes, NJ, USA). For P-selectin and GPIIb/IIIa activity, whole blood was incubated in HEPES-saline buffer (145 mM NaCl, 5 mM KCl,10 mM HEPES pH 7.4) with fluorescently labelled monoclonal antibodies against P-selectin (CD62P) and GPIIb/IIIa (PAC-1) or corresponding isotype control antibodies (BD). Whole blood was pre-incubated for 5 minutes (min) with 30 μM FPP. To induce platelet activation varying doses of ADP (between 1 μM and 20 nM) or 2MeS-ADP (between 100 nM and 0.2 nM) was added. After 15 min in the dark and at room temperature RT), 0.2 % parformaldehyde was added to fix the cells. All samples were diluted 1:20 in phosphate-buffered saline (PBS) and analysed on a Coulter EPICS XL flow cytometer (Beckman Coulter, Brea, CA, USA). The platelet population was gated based on forward and side scattered light. A threshold for non-specific fluorescence was set using the isotype control antibodies, and all events above that value were considered positive. A total of 3,000 events were collected in the platelet gate from each sample.
Calcium flux assay on platelets
Washed platelets for studies on intracellular Ca2+-release mediated through receptor stimulation was prepared as described earlier (9), with the following modifications: 5 μM Fluo4-AM was used instead of 2 μM Fluo4-AM and calcium free Hanks medium was used in the assay. Platelet count was normalised to 100,000 cells/μl. The platelets were injected, at low speed (25 μl/second [s]), into a 96-well plate where each well contained buffer solution and test substances. Fluorescence (excitation 485 nm, emission 535 nm) was recorded for 30 s in a Victor3 spectrophotometer, 1420 Multi-label Counter (PerkinElmer).
cAMP assay on platelets
PRP prepared as described earlier was incubated in the presence of 1 mM acetylic salicylic acid (ASA), 0.3 U/ml apyrase (15 min, RT). After incubation, the PRP was further incubated with 5 mM EDTA and centrifuged (700 rcf, 10 min RT). The supernatant was discarded and the PRP-pellet was resuspended in HEPES-Tyrode buffer (145 mM NaCl, 5 mM KCl, 0.5 mM Na2HPO4, 1 mM MgSO4, 10 mM HEPES, 5 mM glucose, 0.3 U/ml apyrase, pH 7.4), and pelleted again. The supernatant was removed and the pellet was resuspended to give 1.65 × 108 cells/ml in modified HEPES-Tyrode buffer (145 mM NaCl, 3 mM KCl, 0.5 mM Na2HPO4, 1 mM MgSO4, 10 mM HEPES, 5 mM glucose, 0.6 U/ml apyrase, 1 mM IBMX – phosphatase inhibitor, pH 7.4). 0.2 × 108 platelets were incubated for 15 min at 37°C before addition of stimulants. After 10 min platelets were pelleted by centrifugation at 800 rcf for 5 min. The platelets were lysed in 100 μl of 0.1 M HCl and pelleted again (1,200 rpm, 5 min, 4°C). The supernatant was stored in −80°C until analysis. For measuring the stimulated platelets a Cyclic AMP EIA assay (Cayman Chemical) was used following the manufacturer’s instructions. The readout of the results from the cAMP assay was performed in a Victor3 spectrophotometer, 1420 Multilabel Counter (Perkin Elmer).
Platelet [35S]GTPγS binding
PRP was prepared from ACD-treated whole blood through centrifugation (150 rcf, 20 min, RT) and passed carefully through a Pall Autostop™ Leukocyte removal filter (Pall Medical, Port Washington, NY, USA). The filtrated PRP was centrifuged (17,000 rcf, 15 min, RT). The pellet was resuspended in a room tempered membrane-buffer (20 mM HEPES, 20 mM EDTA, 150 mM NaCl, pH 7.4, RT) and the centrifugation was repeated once. The pellet was first resuspended into 3 ml ice-cold membrane-buffer (20 mM HEPES, 5 mM EDTA, pH 7.4) then homogenised for 30 s in a Polytron (setting 6), and diluted to 30 ml with ice-cold membrane-buffer (20 mM HEPES, 5 mM EDTA, pH 7.4). The platelet homogenate was pelleted twice (35,000 rcf, 15 min, 4°C). The final pellet was resuspended in ice-cold membrane buffer (20 mM HEPES, 0.6 mM EDTA, 5 mM MgCl2, 0.1 mM PMSF, pH 7.4) and stored at −80°C. Protein concentration was measured with a modified Lowry protein assay kit (Thermo Scientific, Rockford, IL, USA).
The binding assay was performed essentially as described (10). Briefly, the final volume of the assay was 200 μl and contained 20 μg platelet membranes in binding buffer [20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 10 μg/ml saponin, pH 7.4 + 4 nM [35S]GTPγS, 20 μM guanosine diphosphate (GDP)]. The mixture was incubated (30 min, 30°C) and then filtered through a glass fibre filter (GF/C, Whatman, Brentford, UK). The filters were washed with 3 × 500 μl ice cold binding buffer and then counted using OptiPhase Hisafe 3 (PerkinElmer) in a Liquid Scintillation Counter (Beckman Coulter).
[35S]GTPγS binding on P2Y12-transfected CHO-K1 cell membranes
pcDNA3 vector with cDNA for the human P2Y12 receptor (Missouri S&T cDNA Resource Center, Rolla, MO, USA) was transfected into Chinese Hamster Ovary Cells-K1 (CHO-K1) by using TransIT®-CHO Transfection kit (Mirus Bio, Madison, WI, USA). [35S]GTPγS binding studies on competent P2Y12-transfected CHO-K1 cell membrane were performed essentially as described (11).
Membrane preparations from Sf9 cells expressing the human P2Y12 receptor
The human P2Y12 receptor was expressed in Sf9 insect cells using the baculovirus expression system (Alsdorf B, Schiedel A, Müller CE, unpublished data) by analogy to a previously described method (12). Cells were disrupted and homogenised with 20 strokes in a Dounce homogeniser in 50 mM Tris-HCl buffer, pH 7.4, containing 1 mM phenylmethylsulfonyl fluoride. The resulting suspension was centrifuged for 10 min at 200 g and 4°C. The supernatant was then centrifuged at 48,000 g for 60 min at 4°C, and the resulting cell membrane pellet was resuspended in 50 mM Tris-HCl buffer, pH 7.4. The protein concentration was determined. Membranes were kept frozen at −80°C until use.
Radioligand binding experiments
The competition assays were carried out using [3H]PSB-0413 (13), at a concentration of 5 nM. The non-specific binding was determined using either 10 mM ADP or 100 μM 2-methylthio-ADP, with both ligands giving the same results. Different dilutions of test compound were prepared in assay buffer consisting of Tris-HCl (50 mM), NaCl (100 mM), and MgCl2 (5 mM). The assays were performed in a final volume of 500 μl in duplicate in test tubes containing incubation buffer. After addition of the membrane preparation (100–150 μg of protein/vial) samples were incubated for 60 min at RT. The incubation was stopped by vacuum filtration through Whatman GF/B glass fiber filters using a 48-channel cell harvester (Brandel, Gaithersburg, MD, USA). The vials were rinsed three times with 1 ml aliquots each of Tris-HCl buffer, 50 mM, pH 7.4.
Molecular modelling
The crystallographic structure of the human chemokine receptor type 4 (CXCR4) (14) was retrieved from the Protein Data Base (15), (PDB ID: 3ODU) and used as structural template for the construction of the P2Y12 receptor model by means of the Molecular Operating Environment software (MOE 2009.10, Chemical Computing Group Inc., Montreal, QC, Canada). The protein was cleaned from water molecules and ions and hydrogen atoms were added to the protein. All the ligands structures were sketched in Maestro and minimised with the Polak-Ribiere Conjugate Gradient (PRCG) minimisation consisting of 1,000 steps of CG until a gradient of 0.01 kJ/(molÅ2).
The molecular docking of the ligands was performed with Glide (16). The docking site was defined according to the site-directed mutagenesis data (17, 18) and previous modelling results (19–21). For each ligand, the best ranked docking conformations were retained and subjected to conformational analysis by means of Monte Carlo Multiple Minima (MCMM) for further structural optimisation and energy minimisation of the complex. During the conformational search full flexibility was granted to the ligand and the residues within a radius of 4 Å from the ligand. All the other residues of the receptor were considered conformationally frozen during the calculations. A total of 1,000 steps of MCMM were performed with the MMFF force field and the GB/SA model for water as implicit solvent. The PRCG minimisation method with a convergence threshold on the gradient of 0.05 kJ/mol·Å was used. No constraints were used during any of the docking calculations. The top scored model for each ligand was chosen as a final binding conformation.
Statistics
Calculations and statistics were performed using GraphPad Prism 4.0 software. Statistical significance was accepted when p < 0.05, using one-way ANOVA or Student’s t-test to analyse the data. Values are presented as mean ± standard error of the mean (SEM) (n = number of tests).
Results
FPP potentiates LPA-stimulated platelet shape change but has no effect on LPA-stimulated aggregation
FPP has previously been reported to stimulate platelet shape change. However while our results show that 30 μM FPP clearly potentiated LPA-stimulated shape change (*p=0.017, n=10), we were unable to show an effect of FPP alone (Fig. 1A). While FPP affected LPA stimulated shape change it had no effect on LPA stimulated aggregation (Fig. 1B). That was also the case for N-arachidonyl glycine (NAG) which, like FPP, has been reported to activate LPA5 and LPA4 (22) and (4).
Figure 1. LTA with PRP.
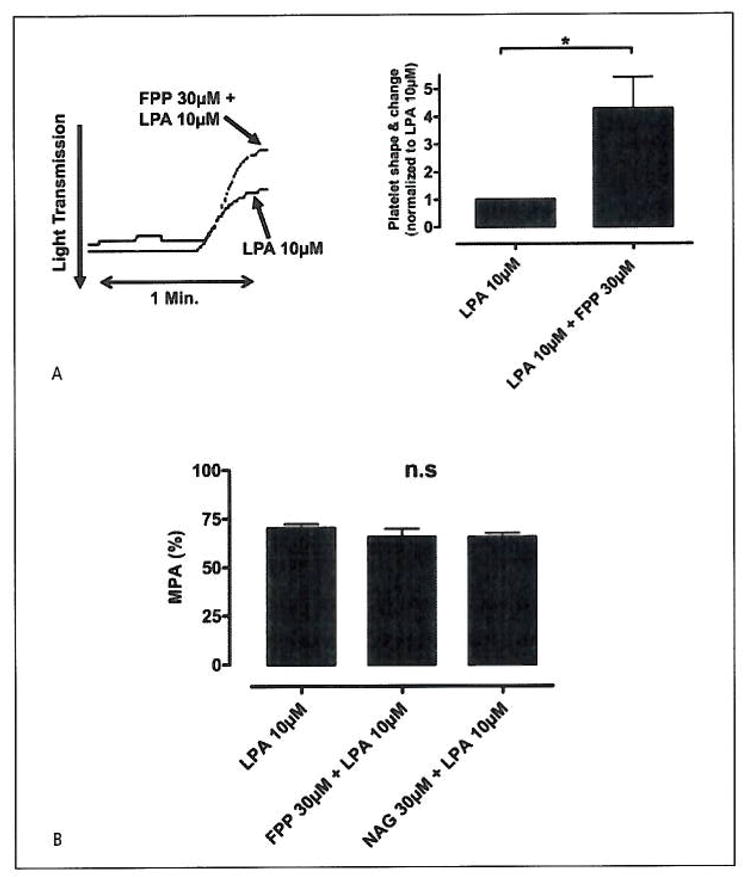
A) 30 μM FPP has an additive effect on 10 μM LPA-mediated platelet shape/change. (Student’s t-test: *p=0.017, n=10). Representative shape/change curves from platelets treated with LPA 10 μM and platelets first pre-treated with FPP, 30 μM for 5 min and further treated with LPA 10 μM. B) LTA on platelets aggregation with LPA (10 μM) alone or with platelets pre-incubated with FPP (30 μM) or NAG (30 μM). No statistically significant inhibitory effect was seen on the platelet aggregation between platelets treated with LPA vs. platelets pre-treated with FPP or NAG (ANOVA: n.sp=0.13, n=8). The values from the turbometric LTA PRP tests are presented as mean platelet aggregation (MPA), where a mean value is calculated from the sum of all observations divided with the number of observation, and presented in %.
FPP antagonises 2-MeSADP-stimulated platelet aggregation
While confirming the results of Williams et al. (4), we noticed that FPP was also exerting an inhibitory effect on platelet aggregation in addition to the documented effect on shape change. In order to investigate this effect, we stimulated platelets with 30 nM 2-MeS-ADP in the presence of either 30 μM FPP or 30 μM NAG. The results revealed a clear, concentration-dependent inhibition of aggregation for FPP while NAG was without effect (Student’s t-test: **p<0.01, n=15) (Fig. 2A). Interestingly, the inhibitory effect was specific for ADP-mediated aggregation, as aggregation stimulated by both TRAP (15 μM), (Student’s t-test: n.sp=0.077, n=7) and epinephrine (1 μM), (Student’s t-test: n.sp=0.063, n=10) was unaffected by 30 μM FPP (Fig. 2B, C). In the next experiment, we tested the other intermediates of the mevalonate pathway for their ability to inhibit ADP-stimulated aggregation, but FPP was the only member with this activity (ANOVA: **p=0.01, n=6) (Fig. 3A). The FPP-mediated inhibition of aggregation was dose-dependent resulting in 40 ± 5% inhibition at 10 μM (**p=0.01, n=15) (Fig. 3B The dose-dependency of FPP was further demonstrated by performing 2-MeSADP dose response curves in the presence of increasing concentrations of FPP. It was found that increasing the FPP concentration successively displaced the dose response curves to the right while, at the same time, depressing the maximal signal (Fig 3C) (ANOVA:*p=0.0122, n=6).
Figure 2. LTA with PRP.
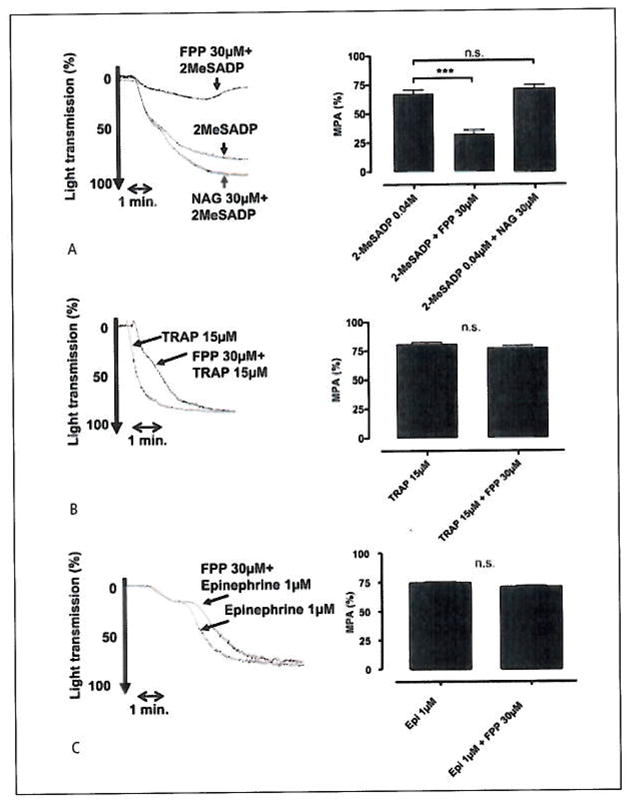
A) (30 μM) FPP shows an inhibitory effect on (0.03 μM) 2-MeS-ADP mediated platelet activation (Student’s t-test: **p<0.01, n=15). B) (30 μM) FPP has no inhibitory effect on (10 μM) TRAP-mediated platelet activation (Student’s t-test: n.sp=0.077, n=7). C) (30 μM) FPP has no inhibitory effect on (1 μM) epinephrine-mediated platelet activation, (Student’s t-test: n.sp=0.063, n=15). A representing tracer accompanies each of the bar graphs.
Figure 3. FPP inhibits-ADP-induced platelet aggregation.
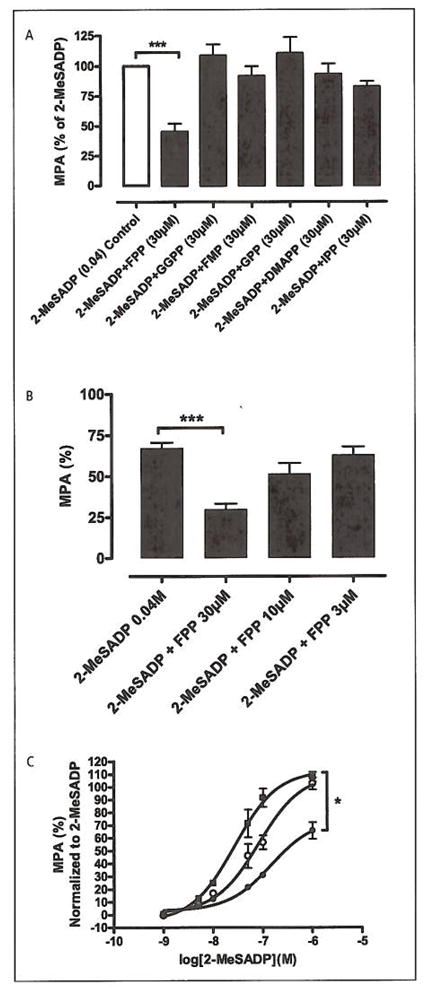
A) LTA study on washed platelets. The different intermediates of the mevalonate pathway were screened for antagonistic activity on platelets stimulated by 2-MeSADP (0.04 μM). 2-MeSADP were also used as control, from which MPA where calculated. Farnesyl pyrophosphate (FPP), Geranylgeranyl pyrophosphate (GGPP), Farnesyl mono-phosphate (FMP), Geranyl pyrophosphate (GPP), Dimethylallyl pyrophosphate (DMAPP), Isopentenyl-5-pyrophosphate (IPP). Intermediates (30 μM) vs. 2-MeSADP (0.04 μM). Only FPP displayed a statistically significant effect (ANOVA: **p<0.01, n=6). The values from each of the observations with the intermediates where normalised vs. its control (2-MeSADP). B) LTA on PRP. Bar graphs representing the effect of increasing FPP concentration on 2-MeSADP stimulated platelet aggregation (ANOVA:**p<0.01, n=15). C) Dose-response curves with LTA on platelets stimulated with increasing concentrations of 2-MeSADP (■) EC50= 27 nM and pre-incubated with [FPP] 15 μM (○) EC50 = 82 nM and FPP 30 μM (●) EC50= 0.15 μM. LTA results are normalised to [2-MeSADP]. (ANOVA:*p=0.012, n=6).
FPP decreases ADP-stimulated activation of GPIIb/IIIa and P-selectin presentation on platelets and inhibits ADP-mediated attenuation of cAMP production, but has no effect on calcium mobilisation
To assess the effect of FPP on the platelet activation markers glycoprotein (GP)IIb/IlIa and p-selectin, dose-response experiments were performed on platelets stimulated with ADP or 2-MeSADP in the presence or absence of 30 μM FPP (n=4). As expected, ADP and 2-MeSADP dose-dependently increased the proportion of PAC-1+/p-selectin+ platelets as measured by flow cytometry (Fig 4A). FPP caused a significant right shift of the dose-response curve for both ADP and 2MeS-ADP. The EC50 value for ADP alone was 16 μM and 59 μM in the presence of FPP (p=0.01) whereas the EC50 for 2MeS-ADP was 2.9 nM and 7.8 nM in the presence of FPP (p=0.04). A statistically significant decrease in the maximal response to both ADP and 2MeS-ADP was also observed in the presence of 30 μM FPP (P=0.01 and 0.03, respectively).
Figure 4. Platelet effects of FFP on PAC-1, p-selectin, cAMP and calcium flux.
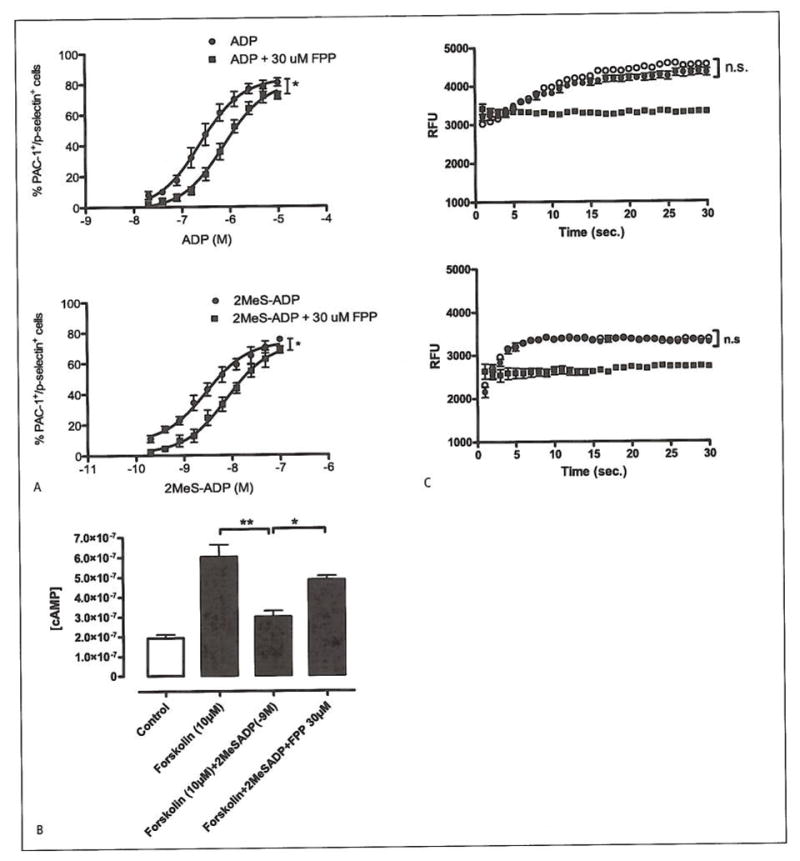
A) Dose-response experiments with ADP or 2MeS-ADP in the presence of FPP (n=4). The proportion of PAC-1+/p-selectin+ platelets was measured in the presence of increasing ADP (EC50=16 μM) or 2MeS-ADP (EC50=3 nM) concentrations, either alone or pre-incubated with 30 μM FPP (EC50=59 μM and 8 nM, respectively). The maximal response of both ADP and 2MeS-ADP was significantly decreased in the presence of 30 μM FPP (p=0.01 and 0.03, respectively). *p<0.05. B) cAMP measurements in platelets. Statistical significance was observed between (10 μM) forskolin-treated platelets vs. platelets incubated with (1*10−9M) 2-MeS-ADP, (ANOVA: **p<0.01). A significant difference in cAMP production was also seen between platelets treated with 2-MeSADP and forskolin vs. platelets also pre-treated with (30 μM) FPP, (ANOVA:*p<0.05, n=4). Untreated platelets were used as control. C) Calcium flux in platelets. Trace 1.2-MeSADP-treated platelets (?) vs. platelets pre-treated with (30 μM) FPP (●), (Student’s t-test: n.sP=0.24, n=6). Trace 2. (0.5 U/ml) thrombin (○) vs. platelets, pre-treated with (30 μM) FPP (●), (Student’s t-test: n.sp=0.46, n=6). Calcium flux is expressed in values of relative fluorescence units (RFU). Unstimulated platelets were used as control (■).
However, when we loaded platelets with Fluo-4AM and tested the effect of FPP on calcium signalling, stimulated by ADP and by thrombin, the results revealed no statistical significance between platelets stimulated either with 5 μM ADP (n.sp =0.24, n=6) (Fig. 4C:1) or with thrombin (0.5 U/ml) (n.sp=0.46, n=6) in the presence or absence of 30 μM FPP (Fig. 4C:2). The absence of a typical activation peak is likely caused by the employed technique of injecting the platelets, resulting in the platelets being partially in an activated state.
2MeSADP-stimulated [35S]GTPγS binding in platelets is antagonised by FPP
As the preceding results indicated the likely target to be the P2Y12 receptor, we isolated platelet plasma membranes and investigated the ability of FPP to interfere with 2-MeSADP-stimulated [35S]GTPγS binding. 2-MeSADP-stimulated [35S]GTPγS binding displayed a slightly bell-shaped dose-response curve a property of the P2Y12 receptor that has previously been reported by van Giezen et al. (11). The presence of 60 nM of the P2Y12-specific antagonist ticagrelor right-shifted the dose response curve, thus confirming the identity of the target as the P2Y12 receptor (2-MeSADP; EC50=17 nM vs. 2-MeSADP + 60 nM ticagrelor; EC50=0.42 μM) (Fig. 5A). FPP 1 M alone generated a small, but significant, increase in [35S]GTPγS binding, an effect that was readily blocked by 10 μM ticagrelor (*p=0.05, **p=0.05, n=4) (Fig. 5). However, when increasing the concentration of FPP, the antagonist properties became more obvious by simultaneously decreasing the efficacy and right-shifting the concentration-response curve of 2-MeSADP (Fig. 5C). EC50 values were calculated and analysed, (EC50(Control): 2.6 nM, EC50([FPP] 1*10 −5M): 7.8 nM, EC50([FPP] 5*10 −5M): 22 nM). The inhibitory effect mediated by FPP vs. 2-MeSADP 1 μM yielded an IC50 value of 45 μM.
Figure 5. Effects of FFP on ADP-induced [35S]GTPγS binding.

A) Upper panel: 2-MeSADP stimulated [35S]GTPγS binding in isolated platelet membranes. Adding the P2Y12-receptor antagonist ticagrelor, shifted the concentration-response curve to the right. Platelet membranes treated with increasing concentrations of 2-MeSADP (○) display an EC50 value of 17 nM, while in platelet membranes pre-incubated with ticagrelor (60 nM) (●) the EC50 value is increased to 0.4 μM. Lower panel: The bar graphs represents the effect of ticagrelor (1 and 10 μM, respectively) on 2-MeSADP-mediated P2Y12 receptor in platelet membranes (ANOVA: **p<0.01, ***p<0.001, n=4). Untreated platelet membranes where used as control. The number of disintegrations per minute (dpm) from basal and fully stimulated cells (signal-to-noise) in the assay was 3,191 ± 239 and 6,507 ± 324 dpm (mean ± SEM), respectively. B) Bar graph representing the stimulating effect of 1 μM FPP on platelet [35S]GTPγS binding. This effect was suppressed by 10 μM of the P2Y12 receptor antagonist ticagrelor, which indicates that FPP interacts with the P2Y12 receptor. (ANOVA: **p<0.01, ***p<0.001, n=4). Untreated platelet membranes where used as control. C) Upper panel: 2-MeSADP stimulated [35S]GTPγS binding in isolated platelet membranes. Dose response curves were generated in the presence of increasing concentrations of FPP. 2-MeSADP (control), EC50=2.6 nM (■), 2-MeSADP with; [FPP] 10 μM, EC50=7.8 nM (□), [FPP] 50 μM, EC50=22 nM (●), and [FPP] 100 μM, EC50=n.d. (○). Increasing the FPP concentration depressed the maximal signal and shifted the concentration-response curve to the right. The radioactivity counts from basal and fully stimulated cells (signal-to-noise) in the assay were 3,217 ± 70 and 6,727 ± 199 dpm (mean ± SEM), respectively. Lower panel: 2-MeS-ADP-stimulated [35S]GTPS binding in isolated platelet membranes. Platelet membranes were stimulated with 2-MeSADP (1 μM) in the presence of increasing concentrations of FPP. The IC50 value was calculated to be 45 μM.
The backbone structure of FPP is similar to that of ADP since both ligands consist of a hydrophobic and a hydrophilic region. While the hydrophobic part of FPP is made up of a 15-carbon chain, the corresponding part on the ADP nucleotide is composed of a ribose and adenine ring. However, the hydrophilic parts of the two ligands are similar in that they both contain a negatively charged pyrophosphate group
FPP is a ligand to the P2Y12 receptor
We next repeated the [35S]GTPγS binding experiment using CHO cells stably expressing the P2Y12 receptor (Fig. 6). The results clearly confirm the identity of the P2Y12 receptor as the target. This conclusion is based on the dose-dependent inhibition pattern of 2-MeSADP-stimulated [35S]GTPγS binding that is very similar to the one obtained with platelet membranes. The higher maximal response at 100 μM FPP is likely due to the higher receptor density of the CHO cells in combination with the partial agonist nature of FPP. This was confirmed by the P2Y12-CHO cells having an transcription level about 20 times higher than platelets (58 copies/ng RNA vs 2.6 copies/ng RNA). We repeated the calculation and analysis of EC50 values from the CHOK1 binding assay, (EC50(2-MeS-ADP): 3.7 nM, EC50([FPP] 1*10−5M):10.3 nM, EC50([FPP] 5*10−5M): 29.4 nM, EC50([FPP] 1*10−4M): 21 nM), and the results were similar, i.e. increasing the concentration of FPP decreased the potency of 2-MeSADP to about the same extent.
Figure 6. 2-MeSADP-stimulated [35S]GTPS binding in membranes isolated from CHOK1-cells expressing the human P2Y12 receptor.
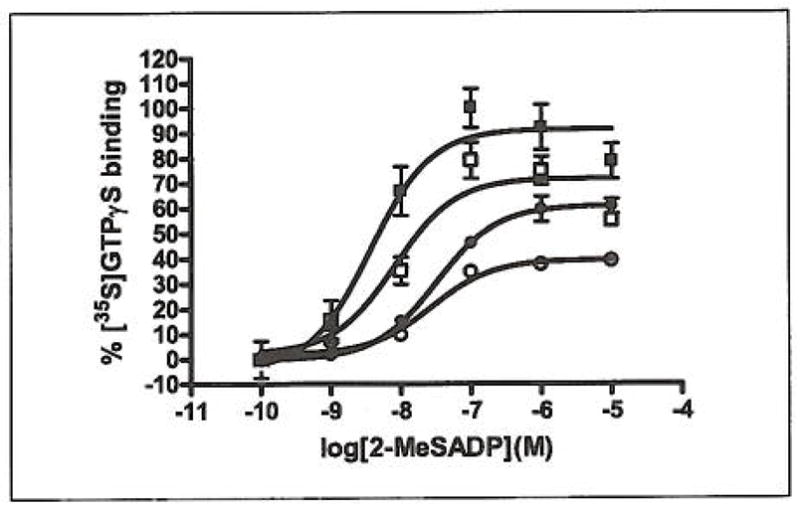
Dose response curves were generated in the presence of increasing concentrations of FPP. 2-MeSADP (control), EC50=3.7 nM (■), 2-MeSADP with: [FPP] 10 μM, EC50=10.3 nM (□), [FPP] 50 μM, EC50=29.4 nM (●), and [FPP] 100 μM, EC50=21 nM (○). As with platelet membranes, increasing the FPP concentration resulted in a depressed maximal signal and a right-shifted concentration-response curve. The radioactivity counts from basal and fully stimulated cells (signal-to-noise) in the assay were 8,180 ± 398 and 12,913 ± 561 dpm (mean ± SEM), respectively.
Radioligand competition binding assays were carried out using the P2Y12-selective nucleotide antagonist radioligand [3H]PSB-04132 (5 nM) in membrane preparations from Sf9 insect cells recombinantly expressing the human P2Y12 receptor. Non-specific binding was determined using 10 mM ADP or 100 μM 2-methylthio-ADP, both of which gave the same results.
We showed that [3H]PSB-0413 exhibited high-affinity binding to Sf9 cell membranes expressing the P2Y12 receptor, but not to membrane preparations of non-transfected cells. The labelled binding sites showed the following rank order of potency typical for P2Y12 receptors: the nonnucleotide PSB-0739 (21, 22). >2-methylthio-ADP > ADP (Abdelrahman A, Alsdorf BB, El-Tayeb A, Müller CE, unpublished data). IC50 values were about 10-fold higher as compared to previously published values for these compounds obtained in radioligand binding studies at human platelet membrane preparations. This may be explained by a different environment of the receptors within the Sf9 insect cell membrane as compared to platelet membranes, and/or by different posttranslational modifications in insect versus mammalian cells.
FPP caused a concentration-dependent inhibition of [3H]PSB-0413 binding with an IC50 value of 65.8 μM (Fig. 7). While the P2Y12 agonists ADP and 2methylthio-ADP as well as the antagonist PSB-0739 led to a complete inhibition of binding of the ATP analogue [3H]PSB-0413, FPP only resulted in a partial inhibition of radioligand binding even at very high concentrations. Maximal inhibition by FPP amounted to about 30–40%.
Figure 7. Concentration-dependent inhibition of [3H]PSB-0413 binding (5 nM) to human P2Y12 receptors recombinantly expressed in Sf9 insect cells (membrane preparations) by FPP.
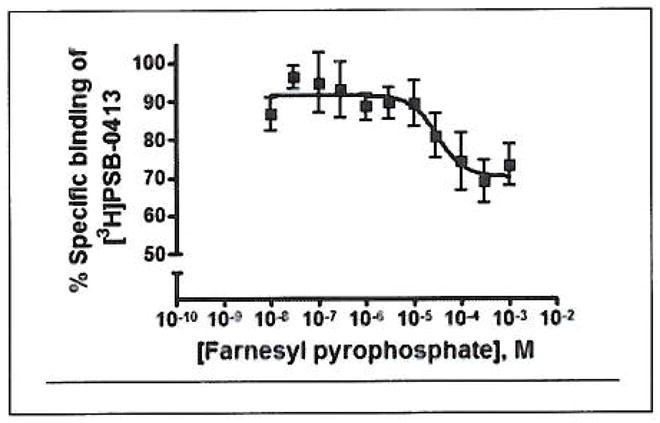
An IC50 value of 65.8 ± 27.1 μM was determined (n=4).
Docking of FPP to a P2Y12 receptor model
FPP contains a hydrophobic chain and a negatively charged pyrophosphate group. It presents a very different scaffold from ADP or 2-MeSADP, typical agonists of the P2Y12 receptor, but it shares a negatively charged phosphate moiety with ADP and 2-MeSADP. In order to propose a hypothetical binding mode of FPP and 2-MeS-ADP within the same receptor and to find a common rational basis for these modes, a molecular modelling study was conducted. A three-dimensional model of the P2Y12 receptor was built (21), based on the recently released X-ray structure of the chemokine CXCR4 receptor (PDB code: 3ODU) (14), and the binding modes of 2-MeS-ADP and FPP were studied by means of flexible molecular docking.
The putative binding site of the P2Y12 receptor was identified, based on the available site-directed mutagenesis data (16, 17) and on previously published modelling studies by Costanzi et al. (19). The recently published mutagenesis data confirmed the key role in the ligand binding of residues including Arg2566.55 and Tyr2596.58 in the sixth transmembrane domain (TM6) and Lys2807.35 in TM7, as also suggested by the docking model proposed by Costanzi et al. In our P2Y12 receptor model with docked agonists, these residues are directly involved in H-bond interactions with the ligands. The binding pocket in our model is located in a pocket formed by residues of TM1, TM2, TM3, TM6, and TM7. In addition to the hydrophilic and charged residues such as Arg2566.55 and Lys2807.35, several aromatic residues including Tyr321.39, Phe1043.32, Tyr1053.33, Tyr2596.58, Phe2777.32 and Phe177 (EL2), hydrophobic residues including Met1083.36 and Leu2847.39, and polar residues including Thr1003.28 and Ser1013.29 delineate the binding pocket of the P2Y12 receptor.
From the docking results of 2-MeSADP in the P2Y12 receptor binding pocket, there is a crucial role of Arg2566.55 and Lys2807.35 in anchoring the diphosphate group of 2-MeSADP to the binding cavity. In addition, the hydroxyl group of Tyr2596.58 stabilises the molecule from above with another H-bond interaction with the distal phosphate group. The adenine ring of 2-MeSADP is involved in a π-π stacking with the aromatic side chain of Phe177 in EL2, while the amino group at the N6 position interacts with the carboxyl group of the Asp842.63 side chain. Other favourable interactions occur between 2-MeSADP and the residues Phe1043.32 and Leu2847.39 at the bottom of the pocket, Tyr321.39 and the carbon chain of Lys802.60, as shown in Figure 8B.
Figure 8. Binding modes of FPP and 2-MeSADP after docking in the human P2Y12 receptor putative binding site.
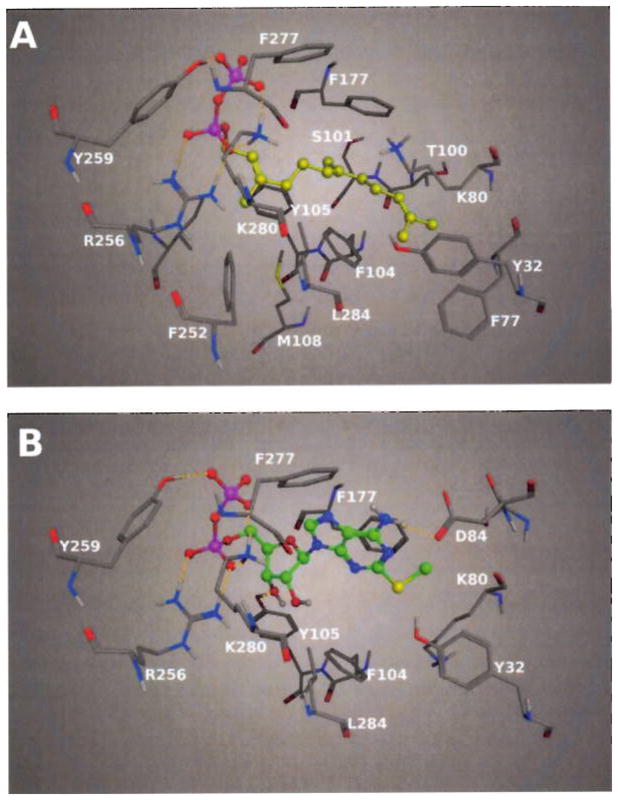
A) Calculated docking pose of FPP in the binding cavity of the P2Y12 receptor model. The FPP (depicted as ball and stick with yellow carbon atoms) is anchored in the pocket by strong electrostatic interactions between the charged and hydrophilic side chains and the negatively charged diphosphate group. The isoprenoid chain of FPP is favourably embedded by aromatic and hydrophobic residues of the P2Y12 receptor. B) 2-MeSADP in a ball and stick representation with green carbon atoms. The diphosphate group of 2-MeSADP has the same interactions with the residues of the P2Y12 receptor shown for FPP in panel A. Differently from the hydrophobic chain of FPP, the more hydrophilic adenosine group of 2-MeSADP is anchored in the pocket by other electrostatic interactions with the residues in the pocket.
From the docking results of FPP in the P2Y12 receptor binding site, the pyrophosphate group binds, as expected, in a similar way to the diphosphate moiety of 2-MeSADP (Fig. 8A). Strong electrostatic interactions occur between the diphosphate moiety of FPP and the side chains groups of Arg2566.55, Tyr2596.58 and Lys2807.35. The binding mode of FPP showed a conformation with an extended orientation of the flexible chain of FPP. The hydrophobic nature of the isoprenoid chain of FPP would be expected to lead to a more folded conformation. The 15-carbon flexible farnesyl group can assume either a trans or gauche disposition about the C4-C5 bond of the chain. The trans conformation leads to an extended form of the chain, while the gauche conformation leads to a folded form of FPP. From the docking results of FPP in the P2Y12 receptor binding pocket, the compound adopts a trans conformation about the C4-C5 bond with an extended isoprenoid chain. An extended conformation of the farnesyl chain of FPP both in solution and in a protein-bound conformation was already observed in several other experimental and computational studies on isoprenoid derivatives (23–25). The diphosphate group of FPP in the docking pose is located between TM6 and TM7, anchored by strong H-bond interactions, while the long hydrocarbon chain is oriented toward TM2 and TM3, where the aromatic and hydrophobic side chains of residues such as Phe792.59, Phe1043.32 and Thr1003.28 make optimal contacts creating a favourable environment to embed the FPP. Other residues that are close to the isoprenoid chain of FPP are Phe177 in EL2 and Leu2847.39 in TM7 that lie, respectively, above and below the compound in the pocket.
Some of the residues in the pocket arrange their side chains in different ways in the two complexes, adjusting the pocket to optimise the interactions with each ligand. The aromatic ring of Phe177 in EL2, for example, in the 2MeSADP-P2Y12 complex, is parallel to the adenine ring to form π-π stacking with the compound. In the FPP-P2Y12 complex, the side chain of Phe177 is instead above the isoprenoid chain of the ligand. In TM3, Tyr105 acts as an H-bond acceptor from the 3′-hydroxyl group of 2MeSADP, while in the FPP-P2Y12 complex the Tyr105 ring is slightly rotated with the hydroxyl group farther from the hydrophobic chain of the ligand, rendering the pocket less polar. Another residue that moves its side chain in order to make more favourable contacts with the ligands is Lys802.60. In both complexes the charged group of Lys80 makes no direct contact with the ligands, but the long carbon chain is oriented in different ways. In the 2MeSADP-P2Y12 complex, Lys80 is oriented deep in the pocket exposing the carbon chain to the 2-methylthio group of the ligand. In the complex with FPP, Lys80 is instead pointing up to interact with Glu2817.36, to open a space in the pocket to accommodate the terminal portion of the isoprenoid chain.
Figure 9 illustrates a superposition of the best docking poses of FPP and 2-MeSADP to show the overlap between the two molecules. The diphosphate group of FPP is superimposed on the diphosphate group of 2-MeSADP, and the diphosphate groups of both compounds share the same strong electrostatic interactions with the residues in the top part of the P2Y12 receptor binding pocket.
Figure 9. Calculated superposition of the docking poses of FPP (in yellow) and 2-MeSADP (in green) in the binding pocket of the human P2Y12 receptor.
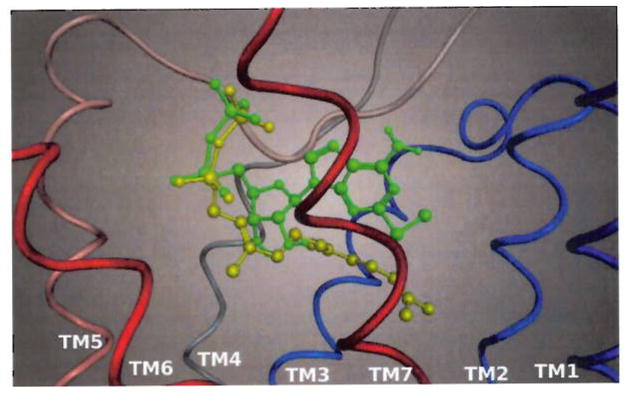
Only the ribbon representing the backbone of the receptor is shown, sequentially coloured from blue (TM1) to red (TM7).
Discussion
In the present study we demonstrate that FPP, apart from being an intermediate in the mevalonate pathway, is an endogenous antagonist of the P2Y12 receptor acting as an inhibitor of ADP-induced platelet aggregation.
The mevalonate pathway is a metabolic pathway of great importance to all eukaryotic cells. Although the isoprenoid metabolites of this pathway, FPP, GGPP, DMPP and IPP are mainly known as precursors in the biosynthetic route to cholesterol or to mediate the membrane association of certain GTPases (1), there have been several recent reports that indicate alternative functions. FPP was reported to serve as an antagonist at the classical LPA2 and LPA3 receptors (26, 27) while being an agonist on the atypical LPA4, LPA5 and LPA6 (28). Furthermore, FPP has also been described as an agonist of the pain producing TRPV3 channel (29), while GGPP is known to antagonise the liver X receptor (LXR) (30). In two previous studies, the message of the recently deorphanised LPA receptors LPA4 and LPA5 were identified in human platelet mRNA (5). As these receptors both have been described as receptors for FPP by Williams et al. (4), we tested the effect of this compound on platelet activation. Although we were able to confirm the findings of Williams et al. who reported an effect of FPP on platelet shape change mediated by LPA5, we noticed that the major effect was antagonistic rather than agonistic and specifically antagonistic to ADP-stimulated aggregation, as aggregation triggered by either TRAP, epinephrine or LPA was unaffected. Furthermore, NAG was without effect on both LPA and 2-MeSADP stimulated aggregation. Since NAG which, like FPP has been reported as an agonist to LPA4–LPA6, was without effect, this rules out the involvement of LPA receptor mechanisms. FPP had no effect on 2-MeSADP-stimulated calcium mobilisation in platelets, therefore eliminating the other pro-aggregatory ADP receptor, P2Y1, as a target. Moreover, dose response curves, for 2-MeSADP and ADP stimulated P-selectin/ GPIIb/IIIa activation, were both displaced to the right in the presence of FPP. These results correlate well with results from the cAMP measurement, which showed that FPP repressed an ADP-induced decrease in cAMP generation. When we studied the effect of FPP on 2-MeSADP-stimulated [35S]GTPγS binding in platelet membranes, we found that the action of FPP at concentrations above 10 μM was clearly antagonistic affecting both efficacy and potency, i.e. indicative of the typical hallmarks of noncompetitive antagonism. At concentrations below 6 μM, FPP independently stimulated [35S]GTPγS binding rather than blocking it. This effect could be completely reversed by ticagrelor to identify the P2Y12 receptor as the main target. In order to confirm the interaction with this receptor, we tested the ability of FPP to compete with 2-MeSADP on P2Y12 receptors expressed in CHO cells. The results substantiated the platelet results as FPP was able to antagonise the effects of 2-MeSADP stimulated [35S]GTPγS binding also in a recombinant system. The antagonistic response to FPP in P2Y12-CHO cells was slightly less than for platelet membranes, and the most likely explanation for this difference is that FPP, being a partial agonist, is more dependent on receptor density. Thus, in a recombinant cell system with a high receptor density an affinity-driven partial agonist will activate a higher proportion of receptors. Binding studies using the P2Y12-selective antagonist radioligand [3H]PSB-0413 demonstrated that although FPP does compete with [3H]PSB-0413 only 30–40 % of the radioligand is displaced. These results indicates that FPP binds to the human P2Y12 receptor; however, its binding site appears to be – at least partly – different from that of the physiological agonist ADP and its competitive inhibitors ATP and PSB-0739.
The affinity of FPP for the P2Y12 receptor is understandable, partly because of the structural similarity between ADP and FPP, but also because the LPA5 and LPA6 receptors, both phylogenetically related to P2Y12, have previously been demonstrated to bind FPP. As LPA5, LPA6 and P2Y12 receptors all belong to the purinergic cluster of GPCRs (31) and share a high degree of general structural similarity, they are also likely to have similarities in the receptor binding pocket. Indeed, a chemogenomic evaluation of the trans-membrane ligand binding cavity of GPCRs placed LPA5 (GPR92), LPA6 (P2Y5) and P2Y12 in the same group (32).
To further explore a possible structural basis for the observed nucleotide-competitive component of FPP binding, we evaluated the ability of FPP to dock into the binding cavity of the human P2Y12 receptor. From the docking poses of 2-MeSADP and FPP, we could deduce that the positively charged residues in the upper part of the binding cavity of the P2Y12 receptor are essential for the binding of the negatively charged diphosphate groups. The docking modes of FPP and 2-MeSADP closely superpose, with the side chain at die C2 position of 2-MeSADP and the terminal part of the farnesyl chain of FPP pointing both toward a region of the P2Y12 receptor binding pocket formed by residues of TM2 and TM3. The aromatic and hydrophobic residues in the P2Y12 receptor cavity create a favourable environment for both compounds. The side chains of the residues in the binding pocket are able to adapt well around the different structural components of the ligands. To summarise, as the docking modes of FPP and ADP are super-imposable, there is reason to expect that these coincident docking poses also reflect function, i.e., it is likely that FPP would display properties as a partial agonist or antagonist. We cannot fully explain with molecular modelling the complex interaction of FPP with the P2Y12 receptor, and the possibility of binding to multiple sites remains. While the docking study cannot distinguish between an allosteric and an orthosteric modulator, it shows that FPP can bind to the binding site of the P2Y12 receptor with good interaction with the residues of the pocket.
FPP is probably best described as an antagonist with an insurmountable component since it both produces a parallel rightward shift of the agonist concentration-response curve and depresses the maximal response. Insurmountable antagonists are usually classified as either allosteric modulators that interact with alternative binding sites or antagonists with a slow dissociation rate. While the radioligand binding studies indicates a complex binding mode involving orthosteric as well as allosteric interactions, it also supports the view of FPP as an insurmountable antagonist of the first type characterised by allosteric modulation of receptor function. The presence of alternative binding sites on P2Y12 capable of allosteric modulation agonist function has previously been reported by van Giezen et al. (33). It could be speculated that the orthosteric FPP binding site, supported by both the docking and the binding study, and a putative allosteric interaction with an alternative site could have different functional effects on the receptor protein that might explain the partial agonism observed.
Although FPP has been reported to be present in circulation (2), only a single report describes physiological circumstances affecting FPP plasma levels, showing that fasting and feeding cause different diurnal variations in the FPP plasma levels (3). To understand a possible physiological role for FPP as platelet regulator, we need to know the plasma concentrations under different physiological and pathophysiological situations, such as coronary ischaemia and inside a thrombus. At low concentrations, FPP could have a prothrombotic effect both by stimulating LPA-induced shape change via LPA receptors or by partial agonism at the P2Y12 receptor. However, at higher concentrations (micromolar), FPP is anti-thrombotic by blocking the fundamentally important positive feedback loop of ADP stimulating the P2Y12 receptor. P2Y12 blockers are among the most widely used drugs for the treatment of acute coronary syndromes. Clopidogrel has a variable effect, and patients with diabetes or CYP2C19 variants are commonly poor responders. The more potent P2Y12 antagonists prasugrel and ticagrelor have been shown to be superior to clopidogrel in preventing major adverse cardiac events (32, 34). Another interesting aspect relates to the action of the widely used statins, which are inhibitors of the mevalonate pathway and thereby FPP synthesis. Could this be one explanation for the pleiotropic effects of statins? In the MIRACLE and ARMYDA-ACS studies, the incidence of myocardial infarction was reduced very early, and it has been argued that this is too short a time for cholesterol lowering to reduce plaque burden or even to stabilise the plaque (34, 35). Based on this, pleiotropic effects have been suggested to explain the early effects of statins (36). Statins have been shown to reduce platelet aggregation. Whether changes in FPP synthesis could contribute to these effects needs to be examined.
In conclusion, FPP is mainly an antagonist of ADP-induced P2Y12 receptor-mediated platelet aggregation with a complex mode of interaction with the receptor giving rise to an insurmountable component and partial agonism. Further insights into the structural basis for these interactions would require the use of site-directed mutagenesis of the receptor. It could be an endogenous antithrombotic factor modulating the strong platelet aggregating effects of ADP in a manner similar to the use of clopidogrel, prasugrel or ticagrelor in the treatment of ischaemic heart disease.
What is known about this topic?
The ADP-receptor P2Y12 is of fundamental importance for platelet function.
Pharmaceutical therapies acting as P2Y12-receptor antagonist drugs are widely used, reduce the risk of myocardial infarction and increase survival.
What does this paper add?
The paper demonstrates that farnesyl pyrophosphate, which is an intermediate in cholesterol biosynthesis, is an insurmountable antagonist of ADP-induced platelet aggregation mediated by the P2Y12 receptor.
The effect of farnesyl pyrophosphate is demonstrated on platelet function testing, flow cytometry, intracellular signalling, binding experiments and with receptor modelling.
It could possibly act as an endogenous antithrombotic factor.
Acknowledgments
The study was supported by grants from the Swedish Scientific Research Counsil (521–2009–2276) and the Swedish Heart-Lung Foundation. KAJ and FD thank the NIDDK, NIH Intramural Research Program.
Abbreviations
- ACD
acid citrate dextrose
- FPP
3,7,11-trimethyl-2,6,10-dodecatrien-1-yl pyrophosphate
- FMP
3,7,11-trimethyl-2,6,10-dodecatrien-1-yl monophosphate
- GGPP
all trans-3,7,11,15-tetra-methyl-2,6,10,14-hexadecatetraenyl pyrophosphate
- GPP
trans-3,7-dimethyl-2,6-octadienyl pyrophosphate
- IPP
isopentenyl pyrophosphate
- DMPP
γ,γ-dimethylallyl pyrophosphate triammonium salt
- LPA
lysophosphatidic acid
- LTA
light transmission aggregation
- MeSADP
2-methylthioadenosine-5′-diphosphate
- MCMM
Monte Carlo Multiple Minima
- PRP
platelet rich plasma
- PSB-0413
2-propylthioadenosine-5′-adenylic acid (1,1-dichloro-1-phosphonomethyl-1-phosphonyl) anhydride
- PSB-0739
1-amino-4-[4-phenylamino-sulfophenylamino]-9,10-dioxo-9,10-dihydroanthracene-2-sulfonic acid disodium salt
- TRAP
Thrombin Receptor Activator Peptide
- NAG
N-arachidonoylglycine
Footnotes
Conflicts of interest
None declared.
References
- 1.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 2.McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63:255–267. doi: 10.1007/s00018-005-5298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saisho Y, Morimoto A, Umeda T. Determination of farnesyl pyrophosphate in dog and human plasma by high-performance liquid chromatography with fluorescence detection. Analyt Biochem. 1997;252:89–95. doi: 10.1006/abio.1997.2314. [DOI] [PubMed] [Google Scholar]
- 4.Williams JR, Khandoga AL, Goyal P, et al. Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J Biol Cheni. 2009;284:17304–17319. doi: 10.1074/jbc.M109.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amisten S, Braun OO, Bengtsson A, et al. Gene expression profiling for the identification of G-protein coupled receptors in human platelets. Thromb Res. 2008;122:47–57. doi: 10.1016/j.thromres.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 6.Khandoga AL, Fujiwara Y, Goyal P, et al. Lysophosphatidic acid-induced platelet shape change revealed through LPA (1–5) receptor-selective probes and albumin. Platelets. 2008;19:415–427. doi: 10.1080/09537100802220468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hogberg C, Erlinge D, Braun OO. Mild hypothermia does not attenuate platelet aggregation and may even increase ADP-stimulated platelet aggregation after clopidogrel treatment. Thromb J. 2009;7:2. doi: 10.1186/1477-9560-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cazenave JP, Ohlmann P, Cassel D, et al. Preparation of washed platelet suspensions from human and rodent blood. Methods Mol Biol. 2004;272:13–28. doi: 10.1385/1-59259-782-3:013. [DOI] [PubMed] [Google Scholar]
- 9.Liu EC, Abell LM. Development and validation of a platelet calcium flux assay using a fluorescent imaging plate reader. Analyt Biochem. 2006;357:216–224. doi: 10.1016/j.ab.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 10.Vasiljev KS, Uri A, Laitinen JT. 2-Alkylthio-substituted platelet P2Y12 receptor antagonists reveal pharmacological identity between the rat brain Gi-linked ADP receptors and P2Y12. Neuropharmacol. 2003;45:145–154. doi: 10.1016/s0028-3908(03)00142-4. [DOI] [PubMed] [Google Scholar]
- 11.van Giezen JJ, Nilsson L, Berntsson P, et al. Ticagrelor binds to human P2Y(12) independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation. J Thromb Haemost. 2009;7:1556–1565. doi: 10.1111/j.1538-7836.2009.03527.x. [DOI] [PubMed] [Google Scholar]
- 12.von Kugelgen I, Schiedel AC, Hoffmann K, et al. Cloning and functional expression of a novel Gi protein-coupled receptor for adenine from mouse brain. Mol Pharmacol. 2008;73:469–477. doi: 10.1124/mol.107.037069. [DOI] [PubMed] [Google Scholar]
- 13.El-Tayeb A, Griessmeier KJ, Muller CE. Synthesis and preliminary evaluation of [3H]PSB-0413, a selective antagonist radioligand for platelet P2Y12 receptors. Bioorg Med Chem Lett. 2005;15:5450–5452. doi: 10.1016/j.bmcl.2005.08.104. [DOI] [PubMed] [Google Scholar]
- 14.Jaakola VP, Griffith MT, Hanson MA, et al. The 2. 6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sherman W, Day T, Jacobson MP, et al. Novel procedure for modeling ligand/receptor induced fit effects. I Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 17.Cattaneo M, Zighetti ML, Lombardi R, et al. Molecular bases of defective signal transduction in the platelet P2Y12 receptor of a patient with congenital bleeding. Proc Natl Acad Sci USA. 2003;100:1978–1983. doi: 10.1073/pnas.0437879100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffmann K, Sixel U, Di Pasquale F, et al. Involvement of basic amino acid residues in transmembrane regions 6 and 7 in agonist and antagonist recognition of the human platelet P2Y(12)-receptor. Biochem Pharmacol. 2008;76:1201–1213. doi: 10.1016/j.bcp.2008.08.029. [DOI] [PubMed] [Google Scholar]
- 19.Costanzi S, Mamedova L, Gao ZG, et al. Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem. 2004;47:5393–5404. doi: 10.1021/jm049914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov AA, Costanzi S, Jacobson KA. Defining the nucleotide binding sites of P2Y receptors using rhodopsin-based homology modeling. J Comp Aid Mol Design. 2006;20:417–426. doi: 10.1007/s10822-006-9054-2. [DOI] [PubMed] [Google Scholar]
- 21.Deflorian F, Jacobson KA. Comparison of three GPCR structural templates for modeling of the P2Y12 nucleotide receptor. J Comput Aided Mol Des. 2011;25:329–338. doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh DY, Yoon JM, Moon MJ, et al. Identification of farnesyl pyrophosphate and N-arachidonylglycine as endogenous ligands for GPR92. J Biol Chem. 2008;283:21054–21064. doi: 10.1074/jbc.M708908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui G, Merz KM., Jr Computational studies of the farnesyltransferase ternary complex part II: the conformational activation of farnesyldiphosphate. Biochemistry. 2007;46:12375–12381. doi: 10.1021/bi701324t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zahn TJ, Ksebati MB, Gibbs RA. Synthesis and conformational analysis of dil3C-labeled farnesyl diphosphate analogs. Tetrahedron Lett. 1998;39:3991–3994. [Google Scholar]
- 25.Zahn TJ, Eilers M. Evaluation of Isoprenoid Conformation in Solution and in the Active Site of Protein-Farnesyl. J Am Chem Soc. 2000;122:7153–7165. [Google Scholar]
- 26.Lec M, Choi S, Hallden G, et al. P2Y5 is a G{alpha}i, G{alpha} 12/13 G Protein Coupled Receptor Activated by Lysophosphatidic Acid that Reduces Intestinal Cell Adhesion. Am J Physiol. 2009;297:G641–654. doi: 10.1152/ajpgi.00191.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liliom K, Tsukahara T, Tsukahara R, et al. Farnesyl phosphates are endogenous ligands of lysophosphatidic acid receptors: inhibition of LPA GPCR and activation of PPARs. Biochim Biophys Acta. 2006;1761:1506–1514. doi: 10.1016/j.bbalip.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bang S, Yoo S, Yang TJ, et al. Farnesyl pyrophosphate is a novel pain-producing molecule via specific activation of TRPV3. J Biol Chem. 2010;285:19362–19371. doi: 10.1074/jbc.M109.087742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gan X, Kaplan R, Menke JG, et al. Dual mechanisms of ABCA1 regulation by geranylgeranyl pyrophosphate. J Biol Chem. 2001;276:48702–48708. doi: 10.1074/jbc.M109402200. [DOI] [PubMed] [Google Scholar]
- 30.Fredriksson R, Lagerstrom MC, Lundin LG, et al. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 31.Surgand JS, Rodrigo J, Kellenberger E, et al. A chemogenomic analysis of the transmembrane binding cavity of human G-prolein-coupled receptors. Proteins. 2006;62:509–538. doi: 10.1002/prot.20768. [DOI] [PubMed] [Google Scholar]
- 32.Schwartz GG, Olsson AG, Ezekowitz MD, et al. Effects of atorvastatin on early recurrent ischemic events in acute coronary syndromes: the MIRACL study: a randomized controlled trial. J Am Med Assoc. 2001;285:1711–1718. doi: 10.1001/jama.285.13.1711. [DOI] [PubMed] [Google Scholar]
- 33.Patti G, Pasceri V, Colonna G, et al. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: results of the ARMYDA-ACS randomized trial. J Am Coll Cardiol. 2007;49:1272–1278. doi: 10.1016/j.jacc.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 34.Labios M, Martinez M, Gabriel F, et al. Effect of atorvastatin upon platelet activation in hypercholesterolemia, evaluated by flow cytometry. Thromb Res. 2005;115:263–270. doi: 10.1016/j.thromres.2004.08.016. [DOI] [PubMed] [Google Scholar]