

Abstract
Structures of G protein-coupled receptors (GPCRs) have a proven utility in the discovery of new antagonists and inverse agonists modulating signaling of this important family of clinical targets. Applicability of active-state GPCR structures to virtual screening and rational optimization of agonists, however, remains to be assessed. In this study of adenosine 5′ derivatives, we evaluated the performance of an agonist-bound A2A adenosine receptor (AR) structure in retrieval of known agonists and then employed the structure to screen for new fragments optimally fitting the corresponding subpocket. Biochemical and functional assays demonstrate high affinity of new derivatives that include polar heterocycles. The binding models also explain modest selectivity gain for some substituents toward the closely related A1AR subtype and the modified agonist efficacy of some of these ligands. The study suggests further applicability of in silico fragment screening to rational lead optimization in GPCRs.
INTRODUCTION
Adenosine receptors belong to the large family of seven-transmembrane (7TM) G protein-coupled receptors (GPCRs), the key sensors for a variety of extracellular signals that play a major role in human physiology and pathology.1,2 Receptors of the GPCR superfamily have been targeted by roughly 40% of all therapeutic drugs, and the number of indications and new targets steadily grows. Prior to the elucidation of detailed 3D information, structure-based drug discovery approaches to GPCRs were behind by more than 15 years as compared to other major drug families such as kinases and proteases; the situation is rapidly changing with the recent breakthroughs in GPCR crystallography.3 Over the past few years, high resolution crystal structures have been solved for a number of clinically relevant class A GPCRs in inactive forms bound to high affinity antagonists and inverse agonists.4–8 Structures of active, agonist-bound forms of rhodopsin,9 β-adrenergic,10,11 and adenosine receptor (AR) subtypes12,13 have also been solved recently, providing important insight into agonist-dependent activation mechanisms in GPCRs.
The elucidation of GPCR structures has already given rise to a number of benchmark studies assessing performance of docking and structure-based virtual ligand screening (VLS) approaches14–16 as well as prospective VLS campaigns demonstrating the high efficiency of these technologies in discovery of new antagonists and inverse agonists.17–21 At the same time, we are only starting to gain a first insight into the application of the active state structures to VLS and the rational design of agonists.22 Structure-based discovery of GPCR agonists has its own challenges, as in addition to high affinity recognition within the binding pocket, a newly designed agonist must be highly selective for active state conformational states as compared to inactive conformations.23,24 For many GPCRs that signal via recognition of small molecules as ligands, the proven and most successful strategy for discovery of new agonists is derivatization of the native ligand or ligand-like chemical scaffold as a lead compound, where design efforts are focused on fine-tuning the affinity and subtype selectivity as well as functional selectivity of these lead compounds.25 Rational drug design approaches that take advantage of the recently solved active state crystal structures are of particular interest, as they could streamline the optimization of agonist scaffolds toward specific properties related to drug efficacy and safety.
The AR subfamily26 is a perfect target for such a strategy of rational lead optimization, as each of the four closely related AR subtypes (A1, A2A, A2B, and A3) is considered as a potential therapeutic target, for example, in neurodegenerative,27,28 cardiovascular,29,30 immune, and inflammatory disorders31,32 and in cancer.33 AR antagonism is the principal mechanism of the most commonly used stimulatory drugs caffeine and theophylline,34 and a new generation A2AAR-selective agonist 2-{4-[(methylamino)carbonyl]-1H-pyrazol-1-yl}adenosine (regadenoson) has been approved for vasodilation in myocardial perfusion imaging.35 However, the clinical development of AR drug candidates for other indications is faced with various obstacles36,37 such as insufficient subtype selectivity of the ligands as well as off-target pathways involved in their actions.
The most prominent scaffold for the design of AR agonists has been provided by derivatization of the orthosteric agonist adenosine,38,39 and only a few other chemotypes40 have been found with agonist activity. Adenosine derivatives with various substitutions in the 2 or N6 positions of the adenine ring and 3′, 4′, or 5′ positions of the ribose ring37 have been developed as selective agonists for each of the four AR subtypes. Some of these studies attempted derivatization of the 5′ moiety of the adenosine ribose ring,41–45 as well as truncations that completely removed polar interaction in this site,46 and assessed their effect on AR selectivity and efficacy. However, only a limited set of substitutions were found to be compatible with binding to A2AAR or A1AR subtypes. Furthermore, many such compounds have been characterized pharmacologically only in nonhuman AR orthologues, which often have altered selectivity profiles. The recently solved high resolution crystal structures of the active-state human (h) A2AAR in complex with agonists 6-(2,2-diphenylethylamino)-9-((2R,3R,4S,5S)-5-(ethylcarbamoyl)-3,4-dihydroxytetrahydrofuran-2-yl)-N-(2-(3-(1-(pyridin-2-yl)piperidin-4-yl)ureido)ethyl)-9H-purine-2-carboxamide (2, UK-432097),12 adenosine, and 5′-N-ethylcarboxamidoadenosine (1)13 (Chart 1) give us a unique opportunity to put these previous findings in a 3D atomic context and use the knowledge of atomic interactions to predict new substituted ligands.
Chart 1.

Chemical Structures of a Nonselective Agonist 1 and A2AAR-Selective Agonist 2
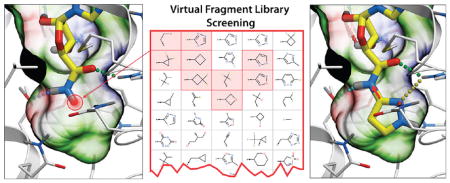
Here we employed a structure-based fragment design approach to comprehensively explore the ligand selectivity of the 5′ binding pocket. The designed compounds were synthesized and tested for their affinity at the ARs in comparison to some of the known 5′ derivatives. The observed high affinity of nucleoside analogues suggests a strong predictive power of the 3D structure-based virtual design approach in the selection of fragments compatible with agonist binding. While our results corroborate strict size limitations on adenosine 5′-carboxamide substitutions for A2AAR agonists, 45 we were able to find several new polar fragments that fit the binding pocket, some of them having unexpected selectivity for the A1AR subtype. The structural basis of agonist binding and selectivity suggests further approaches to the design of low molecular weight drug-like A1AR-selective agonists and partial agonists targeting this subpocket.
RESULTS AND DISCUSSION
Docking and VLS Performance with Known 5′ Adenosine Substituted A2AAR Agonists
To assess the suitability of crystal structure-based models of the A2AAR12 (PDB code 3QAK) for rational ligand design, we performed docking of known A2AAR agonists and benchmarked the VLS model performance in separating these ligands from decoy compounds. (Figure 1). High affinity A2AAR agonists with substitutions at the C5′ position of the ribose ring were selected from the ChEMBL database (Ki <1 μM), with total of 10 unique substituents (Supporting Information Table S1). Adenosine and its high-affinity derivatives were docked into VLS models of the A2AAR generated from the crystal structure of the receptor complex (PDB code 3QAK)12 with agonist 2, as described in the Experimental Section.47
Figure 1.

Performance of the A2AAR agonist binding model in docking and virtual screening. (A) Docking of high-affinity agonist 1 (shown with magenta carbons) into the binding site of 2 (yellow carbons) in an activated A2AAR structure (PDB code 3QAK). The binding pocket of 1 is shown by a semitransparent surface, colored by binding properties (green, hydrophobic; red, acceptor; blue, donor of hydrogen bonds). Hydrogen bonds are shown as dotted lines colored according to their calculated strength (from highest, green, to lowest, red). (B) cyclopropyl-NH analogue (compound 16). (C) Ethyl-tetrazole substituted agonist64 (see Supporting Information Table S1). (D) Cyclobutyl-NH analogue (compound 18). (E) Performance of virtual ligand screening for adenosine 5′ substitutions. ROC curves were calculated for three different models, including activated A2AAR structure (PDB code 3QAK, green), inactive antagonist-bound A2AAR structure (PDB code 3EML, red), and an agonist-optimized model described in ref 48; black line shows nondiscriminating model. AUC and normalized square root AUC (SQ_AUC) for the corresponding models are shown.
Reproducibly predicted docking poses for all 10 5′ adenosine derivatives showed the adenosine core conformations to be similar to that found for 2 in the crystal structure (rmsdcore <0.5 Å), confirming compatibility of these substituents with the active-state A2AAR pocket. All of the docked agonists reproduced the major interactions of the adenosine scaffold, including Phe168ECL2 stacking and Asn2536.55 hydrogen bonding for the adenine aromatic ring system, as well as polar interaction of the ribose OH groups with Ser2777.42 and His2787.43, as shown in Figure 1A. Binding poses for all four derivatives that contained an adenosine 5′-carboxamide moiety (N-methyl, N-ethyl, N-cyclopropyl, and N-cyclobutyl substituted at the 5′ position) also reproduced additional contacts and the hydrogen bonding pattern of the 5′-N-ethylcarbox-amido group of 2 with Thr883.36 and His2506.52 side chains (Figure 1 B, D), resulting in very high binding scores of these compounds (Score < −42 kJ/mol). Another polar substitution, 5′-(2-ethyl-tetrazol) shown in Figure 1C, also found polar contacts with Thr883.36, His2506.52, or Asn1815.42. At the same time, compounds with smaller (5′-hydroxyl, 5′-methyl-chloride) and charged (5′-methyl-amine) lacked interactions in the 5′ subpocket and showed reduced binding scores in the range of −26 to −30 kJ/mol. All these observations corroborate the importance of optimal 5′ subpocket interactions for high affinity of agonists.
The VLS performance of the crystal structure model was assessed by comparing binding scores of the 10 known adenosine substitutions with a decoy adenosine 5′ substitution set based on 200 randomly generated, small MW (<130) fragments from commercial compound libraries (Enamine and Chembridge). The green receiver operating characteristic (ROC) curve in Figure 1E suggested the active-state A2AAR model performed well with an area under curve (ROC_AUC) of about 93% and normalized square root AUC, NSQ_AUC = 78%.19 The four highest ranked compounds in this docking benchmark were true positives (hits). Note, that these VLS performance numbers were likely underestimated because many of the high-ranked “decoy” substitutions are very close analogues of true ligand substitutions and may have A2AAR affinity comparable to some of the true ligands. In addition to the active-state A2AAR model, performance in docking and VLS was assessed for another two conformationally distinct VLS models of the A2AAR based on: (i) the unmodified crystal structure of an inactive A2AAR with inverse agonist 4-[2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol (ZM241385)-bound (PDB code 3EML, red curve),6 and (ii) a side chain-optimized model of agonist binding derived from the inactive A2AAR structure (blue curve).48 The A2AAR inactive state model performance (ROC_AUC = 57%, NSQ_AUC = 8%) suggested that VLS with the unmodified inactive crystal structure did not provide any significant enrichment. The model based on agonist-guided side chain optimization with nonselective agonist adenosine-5′-N-ethyluronamide (1, NECA) described previously48 was found to yield significant enrichment (ROC_AUC = 79%, NSQ_AUC = 48%), though still inferior to the active-state A2AAR crystal structure model. Other activated, thermo-stabilized A2AAR complexes with adenosine and 1 have been crystallized recently.13 However, one of the stabilizing mutations in the receptor, Q89A, is located in the C5′ binding subpocket, substantially changing its shape and specificity, so these structures may not be suitable for the rational design of C5′ substitutions.
Rational Design and Fragment Screening for New Adenosine 5′-Carboxamide Derivatives
The crystal structure of the active state A2AAR (PDB code 3QAK) was used to rationally design new adenosine 5′-carboxamide derivatives with the 3D Ligand Editor tool in the ICM molecular modeling package,47 as described in the Experimental Section. Chemical structures of the substituted adenosine 5′-carboxamides (3–25) for which binding scores were predicted are shown in Table 1. Initially, the manual mode of the 3D Ligand Editor was employed to explore some of the sterically possible substitutions fitting in the binding pocket (Figure 2). This mode allows one to “grow” the ligand substitutions atom by atom, automatically optimizing the new atom positions, which is followed by redocking the modified flexible compound and calculating its binding score. This approach was used to computationally evaluate some simple modifications, including several previously tested derivatives 7, 9, 16, 18, and 22 and new halide substitutions of known compounds (compounds 3–6, 8, 17, 19, and 20). We also tested several positively and negatively charged substitutions, i.e. 11, 12, and 15, and these were predicted to have suboptimal binding energy due to a lack of ionic interactions in this subpocket.
Table 1.
Binding Affinities of a Series of Adenosine 5′-Carboxamide Derivatives at Three Subtypes of ARs (Human Unless Noted) and Relative Efficacy at hA1ARf
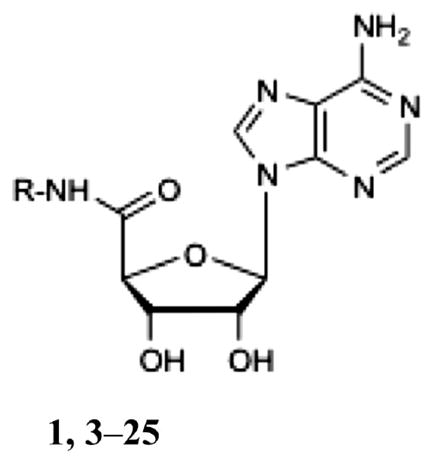 1,3–25 | |||||
|---|---|---|---|---|---|
| Compd | R = | Affinity Ki, nM or (% inhibition)a | Activation of A1, % of 1, 10 μM | ||
| A1 | A2A | A3 | |||
| 1* | CH3CH2 | 63 (rat),c 3.00±0.10 | 12 (rat),c 35.0±4.0 | 6.4,c 16.0±5.0 | 100 |
| 3# | CH2FCH2 | 3.40±1.10 | 140±60 | 240±60 | 77.9+14.1 |
| 4# | CHF2CH2 | 4.70±0.70 | 90±20 | 520±100 | 87.6±1.8 |
| 5* | CF3CH2 | 1190 (rat),c 15.0±2.0 | 405 rat,c 180±40 | 719,c 700±200 | 73.4±5.6 |
| 6* | CH2ClCH2 | 248 (rat),c | 410 (rat),c | 332,c | ND |
| 7* | CH3(CH2)2 | 1240 (rat),c 12.0±2.0 | 550 (rat),c 210±40 | 44,c 50.0±7.0 | 78.6±3.4 |
| 8# | F(CH2)3 | 8.90±1.70 | 690±70 | 150±30 | 59.5±6.2 |
| 9* | HO-CH2CH2 | 12.8b | 505b | 9450b | ND |
| 10# |
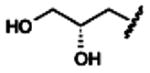
|
120±10 | 1570±370 | 1870±10 | 75.5±5.7 |
| 11* |
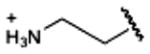
|
(>10%, rat)e | (>10%, rat)e 3120b 46,100d | 19,600e | ND |
| 12* |
|
>10,000b | >10,000b | >10,000b | ND |
| 13^ |
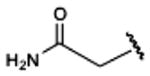
|
330±50 | 940±290 | (30%±5%) | 47.8±6.3 |
| 14^ |
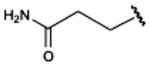
|
540±10 | (44%±4%) | (30%±5%) | 50.1±4.4 |
| 15# |
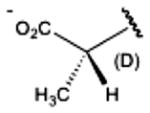
|
(13%) | (23%±3%) | (10%) | 9.4±6.4 |
| 16* |
|
6.4 (rat),b 63 (rat),c 1.90±0.60 | 13.4 (rat),b 12 (rat),c 50±10 | 1600 (rat),b 108,c 180±50 | 95.4±6.3 |
| 17^ |
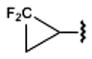
|
8.10±0.40 | 170±40 | 1790±250 | 80.9±7.3 |
| 18* |
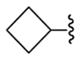
|
43 (rat),c 5.50±0.50 | 34.0 ±8.0 | 23,c 70.0±7.0 | 65.2±9.3 |
| 19^ |
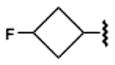
|
24.0±1.0 | 370±100 | 120±10 | 77.4±7.1 |
| 20^ |
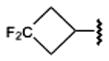
|
200±80 | 2470±450 | 230±40 | 63.4±5.4 |
| 21^ |
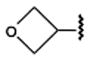
|
12.0±5.0 | 850±430 | 300±50 | 56.8±2.4 |
| 22* |
|
770 (rat),c 27.0±3.0 | 610 (rat),c 650±210 | 32,c 70±30 | 92.5±9.2 |
| 23^ |
|
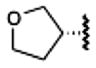
140±10 | 1710±460 | 210±20 | 77.9±0.9 |
| 24^ |
|
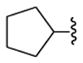
70±30 | 230±40 | 3070±730 | 91.9±9.2 |
| 25^ |
|
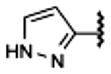
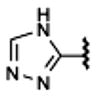
170±30 | 1720±400 | (40%±3%) | 50.0±3.8 |
Using CHO or HEK293 (A2A only) cells stably expressing a hAR; affinity was expressed as Ki value (n = 3–5) or percent inhibition of radioligand binding at 10 μM.
Values from de Zwaart et al.38
Values from Kim et al.40
Values from Jacobson et al.39
ND, not determined.
Previously known derivatives.
Suggested by manual structure-based design.
Predicted by fragment-based screening.
Figure 2.

Interactive design of 5′-carboxamino substitutions with the ICM 3D Ligand Editor.47 (A) A precalculated ligand pocket of A2AAR (mesh) is shown with the adenosine 5′-carboxamide (primary amide) scaffold. The pocket mesh guided the design by showing preferred placement of heavy atoms and by coloring according to binding properties (green, hydrophobic; red, acceptor; blue, donor of hydrogen bonds). The empty subpocket targeted here is highlighted by red circle). The terminal hydrogen in the scaffold can be manually replaced by a single atom or a by chemical group (a custom list shown in the left panel), and positions of the newly added atoms are automatically optimized on the fly. The designed compounds are then redocked into the binding cavity, and its VLS Score calculated without any bias to the initial pose of the scaffold. Hydrogen bonds are shown as dotted lines colored according to their calculated strength (from highest, green, to lowest, red). (B, C, D) Binding poses of compounds 3 (2,2,2-trifluoroethyl fragment), 13 (glycinamide fragment), and 15 (glycine fragment), respectively, and the interactions of the 5′ substituents are shown.
To further explore the range of chemical diversity to predict optimal adenosine 5′-carboxamide derivatives using the ICM Ligand Editor, we performed an automated fragment search in a nonredundant library of about 2000 small fragments (MW <150) compiled from Chembridge and Enamine building blocks and compound databases. Using this automated procedure, all the small fragments were connected to the adenosine carboxamide scaffold via the amide bond, and the resulting derivative compounds were energy optimized in the receptor binding pocket. For about 100 compounds with acceptable initial scores, the initial optimization was followed by full redocking and scoring steps, which were identical to those in the manual design. The screening resulted in 23 different fragments with predicted binding scores better than −42.0 kJ/mol. The predicted high-ranking substituents ranged from 2 to 7 heavy atoms in size (MW from 40 to 130) and included both hydrophobic and polar groups. The docking and screening results suggested very strict size limitations on C5′ substituents in the A2AAR, defined by the size of the corresponding subpocket buried deep in the binding cavity. The size limitations were also enforced by a highly conserved conformation of the adenosine 5′-carboxamide scaffold and high rigidity in the 5′ subpocket. These results were in good agreement with the known structure–activity relationship (SAR) for 5′-carboxamide derivatives at the A2AAR, as discussed above.
The examples of predictions by fragment screening for new derivatives 13 (glycinamide fragment), 21 (oxetane fragment), 23 (tetrahydrofuran fragment), and 24 (pyran fragment) are shown in Figure 3. (Docking poses and scores of all 23 compounds in Table 1 are shown in Supporting Information Figure S2 and Table S2). While all previously known high affinity compounds had hydrophobic C5′-carboxamide substituents (except for 2-hydroxyethyl derivative 9), some of the top ranked screening results suggested a variety of polar substituents. These fragments were predicted to form specific hydrogen bonds with Asn1815.42, Gln893.37, and His2506.52 side chains lining the subpocket. Most of the hydrogen bonds were formed with the side chain of Asn1815.42, which was also the only side chain in the subpocket with an unsatisfied H-bond donor group. Other potential hydrogen donors in the pocket were already involved in the intramolecular hydrogen bonding in the crystal structure and therefore were unlikely to make a significant contribution to the ligand binding energy.
Figure 3.
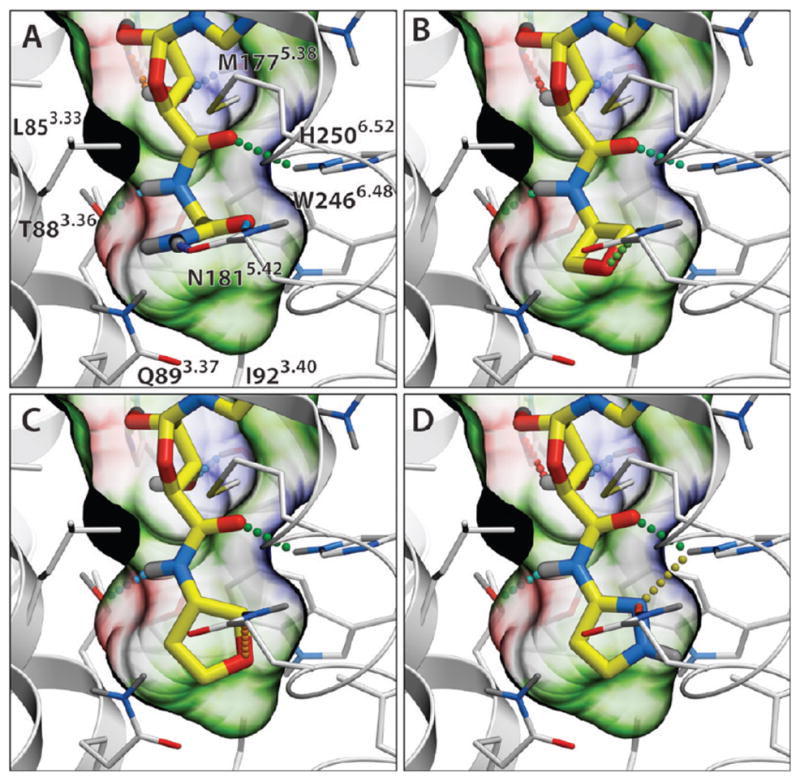
Examples of predicted A2AAR binding modes for novel AR 5′-carboxamide agonists (13, 21, 23, 24) selected by automated scanning of a diverse library of 2000 small fragments. Hydrogen bonds are shown as dotted lines colored according to their calculated strength (from highest, green, to lowest, red).
Chemical Synthesis and Biological Assay Results
We synthesized 16 rationally designed nucleosides, including both halogenated analogues and the top ranked adenosine 5′-carboxamide derivatives predicted by structure-based fragment screening. We also synthesized and tested at three subtypes of hARs eight compounds (5, 6, 7, 11, 12, 16, 18, and 22) whose binding was previously assayed only for rat (r) AR orthologues. The synthetic routes are shown in Scheme 1. A 2′,3′-isopropylidene protected 5′-carboxylic acid intermediate 26 was condensed with various amine moieties using the efficient coupling reagent 1-cyano-2-ethoxy-2-oxoethylidenaminooxy)-dimethylamino-morpholino-carbenium hexafluorophosphate (COMU).49 In initial attempts to deprotect 34 to provide β-alanine amide derivative 14 using 50%TFA in methanol at 70 °C, the N6-amino group was completely displaced by methoxy, which was not observed with other 2′,3′-isopropylidene intermediates. However, 1 N HCl in dioxane avoided this side reaction and gave the desired product, and these conditions were used generally for the deprotection. In one case, the product 40 was unstable in the acidic conditions needed to deprotect the isopropylidene group, and the unprotected adenosine 5′-carboxylic acid 45 was used instead as starting material for the oxetane derivative 21.
Scheme 1. Synthetic Routes to Adenosine 5′-Carboxamide Derivativesa.

aReagents and conditions: (i) RNH2 or salt, COMU, DIPEA, DMF, rt; (ii) oxetanamine, EDC, pyridine, rt; (iii) 1 N HCl, dioxane, rt. Compound 40 decomposed during the attempted deprotection, and the synthesis of 21 was accomplished from intermediate 45. Compounds 27–29, 35, 36, 39, and 42 were subjected to deprotection after rough purification.
The binding affinities of the nucleoside derivatives at the A1AR, A2AAR, and A3AR were measured in standard radio-ligand binding assays as described in the Experimental Section. We used membrane preparations from Chinese hamster ovary (CHO) cells stably expressing either the recombinant hA1 or hA3AR and human embryonic kidney (HEK) 293 cells stably expressing the hA2AAR.50–52 IC50 values were converted to Ki as described.53 Results of the binding assays are shown in Table 1.
For the majority of the new rationally designed adenosine 5′-carboxamide derivatives, the measured affinities were in the submicromolar range and comparable to affinity of the agonists adenosine and 1. In contrast, very weak or no affinity was observed for positively charged compounds (11, 12) or negatively charged compound (15), which agreed well with suboptimal binding scores predicted in the docking experiments. As shown previously,45 optimal interactions of the primary amines in this pocket require a “neoceptor” mutation of Gln893.37 to an acidic Glu side chain. Only one of the new compounds (14) with a high predicted binding score (Score <−42 kJ/mol) had an undefined Ki value at the A2AAR due to its weak affinity. The high hit rate for the 5′-carboxamide derivatives (13 out of 14 new compounds) suggested a predictive power of the structure-based model. The highest affinity at the A2AAR, however, was observed for known nonpolar 5′-carboxamide substituents in compounds 1, 16 (N-cyclopropyl), and 18 (N-cyclobutyl), suggesting that predicted polar interactions observed in the A2AAR model may be not sufficiently strong in the A2AAR to compensate for desolvation of the corresponding polar groups.
There was a substantial species dependence of the affinity at the A1AR, with affinity at the human orthologue generally more potent than at the rat A1AR. The most divergent examples of this dependence were compounds 5 and 7, with differences of approximately 100-fold between rat and human A1AR binding affinities.
Introduction of polar groups, including fluoroalkyls,54 in the adenosine 5′-carboxamide derivatives resulted in an improved A1AR over A2AAR subtype selectivity for some of the analogues of 1. For example, the N-propyl derivative 7 is 17.5-fold more selective for the hA1AR, while its N-3-fluoropropyl analogue 8 is 78-fold more selective for the hA1AR. However, the effect of fluorination is dependent on the alkyl moiety and the substitution position. For example, increasing fluorination at the 3-position of the cyclobutyl series 18–20 caused a general reduction of AR affinity. Replacement of the same position with an ether bridge in 21 increased A1AR selectivity, an effect not seen in the cyclopentyl series 22 and 23. It is noteworthy that the introduction of fluoroalkyl groups in the context of some alkyl- and cycloalkyl 5′-carboxamide moieties had an enhancing effect on A1AR selectivity. The carbon-fluoride derivatization is an important tool in lead optimization, as fluoride atoms can subtly change the steric fit in the subpocket and provide an alternative way to introduce polar interactions54 between the ligand and polar side chains.
We tested the adenosine derivatives in a functional assay at the A1AR consisting of inhibition of forskolin-stimulated cyclic AMP production in A1AR-expressing CHO cells.55,56 Table 1 summarizes the A1AR-mediated effects on cyclic AMP (single point determinations at 10 μM) expressed as a percent of the effect of full agonist 1 (= 100%). This high concentration was selected to be generally in great excess of the Ki value for each derivative to approximate the maximal effect. Most of the values that are considerably less than 100% represent less than full efficacy of these agonists, although for some compounds (for example 15) the weak binding precludes reliable conclusion about their efficacy level.
The impact of the 5′ substitutions on function of different AR subtypes was considered. Our analysis in Figure 4 suggests that variations of amino acid side chains between A1AR and A2AAR in proximity of the 5′ binding subpocket can lead to significant conformational and dynamic differences. In functional assays, the series of small cycloalkyl amides was compared in efficacy at two AR subtypes (Figure 5). The cyclopropyl 16 and cyclopentyl 22 derivatives were full agonists at the A1AR, but the cyclobutyl derivative 18 was a partial agonist with 65.2 ± 9.3% of the maximal efficacy. With an EC50 of 15.1 ± 1.97 nM, compound 18 was very potent, consistent with its binding affinity at the A1AR.
Figure 4.
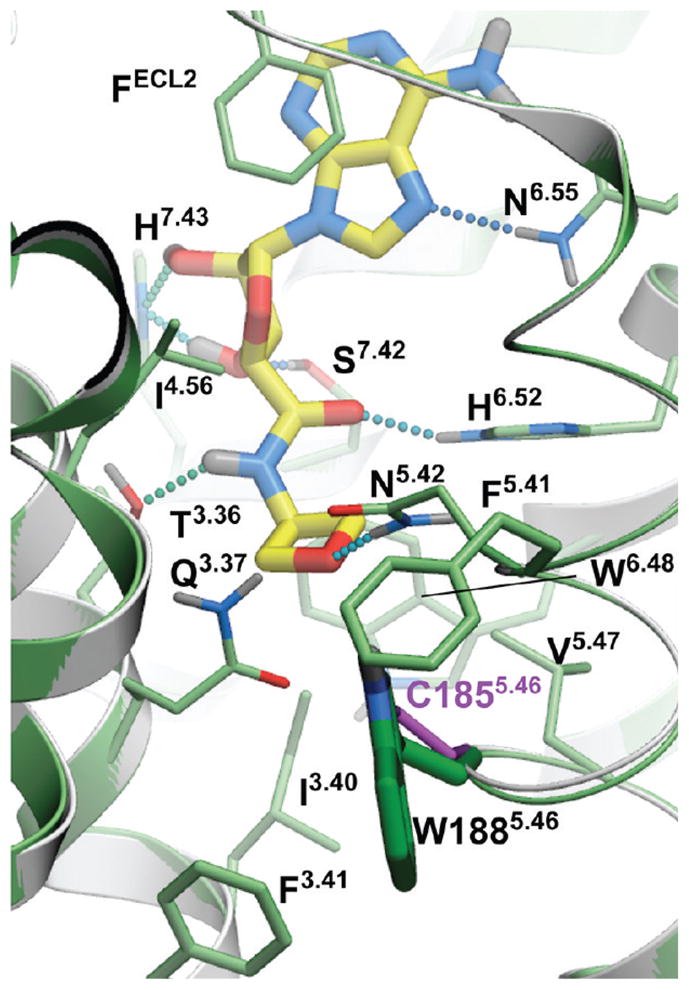
Subtype variations in proximity of the 5′ binding subpocket in the hA1AR model, generated from the hA2AAR crystal structure (PDB: 3QAK). Modification in position 5.46 from Cys1855.46 (purple carbons) in the hA2AAR to Trp1855.46 (bright-green carbons) in the hA1AR results in minor steric clashes with surrounding side chains and potentially leads to a slight (0.3–0.5 Å) increase of the distance between helices IV and V in this region. This can also impact some polar interactions in the pocket and conformational dynamics of ligand binding. Other residues of the hA1AR model are shown as sticks with light-green carbons, the position of docked compound 9 shown by sticks with yellow carbons.
Figure 5.

Concentration response curves for selected adenosine derivatives in a cyclic AMP accumulation assay. (A) Inhibition of cyclic AMP accumulation mediated by the hA1AR. EC50 value of compd 18 was found to be 15.1 ± 2.0 nM. (B) Stimuation of cyclic AMP accumulation mediated by the hA2AAR. EC50 values of homologous N-cycloalkyl compounds 16, 18, and 22 were found to be 22.9 ± 4.6, 26.8 ± 5.7, and 225 ± 39 nM, respectively.
Selected derivatives were examined in functional assays at the A2AAR consisting of stimulation of cyclic AMP production in A2AAR-expressing CHO cells to compare with its inhibition in the A1AR expressed in CHO cells. Curiously, at the A2AAR, compounds 16 and 18 were full agonists, while 22 displayed a slightly lower efficacy (~80%). Other clearly partial agonists at the hA1AR include mono- and trifluoro analogues of 1, i.e., 3 and 5, carboxamides 13 and 14, an oxetane 21, and a 1,2,4-triazole 25. Thus, subtle changes in the amide moiety had substantial effects on the ability to activate the ARs, and the pattern of effects at A1AR and A2AAR were different.
Structural Basis of A1AR Selectivity in the 5′ Subpocket
The introduction of polar moieties into the adenosine 5′-carboxamide derivatives resulted in modest improvement of A1AR over the A2AAR subtype selectivity as compared to compound 1, with maximum 8-fold gain for compounds 8 and 21. Analysis of the AR binding pockets shows that contact residues for 1 are fully conserved between human A2AAR and A1AR subtypes, with the only exception being a Ser2777.42 replacement to a similar Thr side chain in A1. Other sequence variations between these two subtypes are located at the extracellular entrance to the pocket,57 where they can have only indirect effects on the selectivity of bound 5′-carboxamide derivatives of 1.
The only variable residue in immediate proximity to the 5′ subpocket of hA2AAR is Cys1855.46, which is replaced by Trp in A1ARs (both hA1AR and rA1AR), or by Phe in the rA2AAR orthologue. Our modeling of the hA1AR based on the active-state structure of hA2AAR (PDB: 3QAK), illustrated in Figure 4, suggests that the Trp5.46 side chain in A1AR is too bulky to fit sterically in the model even after flexible side chain optimization of the receptor model. According to this model, introducing the C185W modification requires at least a minor adjustment in the protein backbone, pushing helix V approximately 0.3–0.5 Å away from helices IV and III in this region, thus slightly expanding the 5′ subpocket. This minor movement of the backbone can weaken polar interactions between Gln3.37 and Asn5.42 side chains in A1AR and make them more accessible for polar interactions with the ligand, which may explain why many of the newly designed polar 5′-carboxamide substitutions apparently confer some additional selectivity for the A1AR. Similarly, between A2AAR orthologues, modification of Cys5.46 in human to Phe5.46 in rat (rA2AAR) is likely to alter this subpocket conformation and dynamics, potentially making its selectivity profile more similar to the hA1AR rather than the hA2AAR. The results of docking for the series of hit compounds into the hA1AR model (Supporting Information Table S2) suggest slightly improved binding scores for some of the high-affinity derivatives with bulkier 5′ carboxamide substituents, for example, compound 21. In general, however, no significant correlation between the score and selectivity gain was observed due to a rather small range (<8-fold) of selectivity variations. Moreover, the C185W modification can also affect conformational dynamics of this region, which may contribute to specific aspects of the activation mechanisms operating in these receptor subtypes.
It is to be noted that the hA3AR subtype harbors much more dramatic differences compared to both hA1AR and hA2AAR among residues controlling the 5′ subpocket, including Q89H, N181S, and H250S substitutions. These changes, generally from larger to smaller polar side chains, apparently result in a more spacious subpocket in A3AR than in A2A and A1 subtypes, which is also in agreement with much bulkier 5′ substitutions being allowed for high-affinity A3AR selective agonists.58
Partial Agonism for 5′ Substitutions and Its Structural Basis
Functional activity of adenosine derivatives was tested in a cyclic AMP accumulation assay. All identified high-affinity binders in Table 1 were found to have significant agonistic activity at the hA1AR, ranging from 48–95% of the efficacy of full agonist 1. Although some of the agonists, e.g., compounds 16, 22, and 24, displayed more than 90% efficacy, a number of other compounds had significantly reduced activation capacity. We also observed some variation in the activation capacity of these new agonists between A1AR and A2AAR subtypes. For example, cyclic AMP activity curves for a series of homologous N-cycloalkyl compounds 16, 18, and 22 (from cyclopropyl to cyclopentyl) in Figure 5 shows that while compound 18 has a lower EC50 for the A1AR than for A2AAR, its maximum efficacy at the A1AR is only 65%, while it maintains full (99%) efficacy at the A2AAR subtype. On the other hand, compound 22 has somewhat higher efficacy at the A1AR (92%) than at the A2AAR subtype (75%).
The high sensitivity of the agonist efficacy to variations in adenosine 5′-carboxamide moieties can be explained by the fact that the 5′ subpocket undergoes dramatic conformational changes upon receptor activation (Figure 6). The most prominent changes include upward shifts towards the extracellular side of residues Gln893.37 and Ile923.40 lining the subpocket by more than 2 Å, as well as a shift by about 1.5 Å in a conserved Trp2466.48 residue, which is a key conserved aromatic residue involved in receptor activation in the A2AAR,12 rhodopsin,59 and potentially other GPCRs. The shifts in the residues lining the pocket result in a substantial change of its shape and reduction in size upon activation. Our models of agonist–receptor interactions show that bulkier 5′-carboxamide moieties can limit the range and dynamics of movement within the receptor, thus impacting the activation-related changes and ligand efficacy. Because partial agonism of A1AR-selective compounds can be therapeutically beneficial, for example, in treatment of clinical arrhythmias,44 rational control of this efficacy by 5′ modification may have clinical applications.
Figure 6.
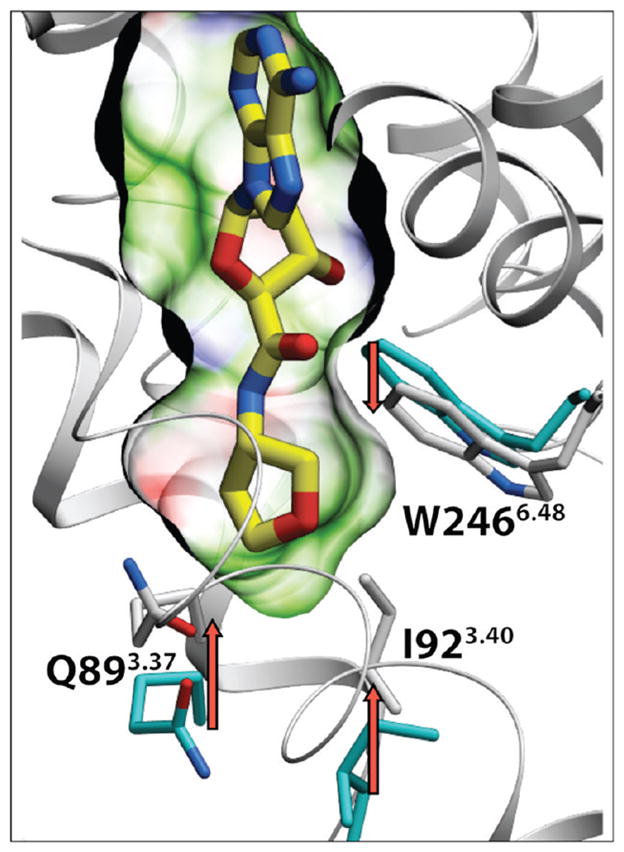
Conformational changes in the binding pocket upon receptor activation. Active state structure of hA2AAR (PDB code 3QAK, white ribbon) is shown here in predicted complex with a new partial agonist (with respect to the hA1AR), i.e., tetrahydrofuryl derivative 23 (yellow carbons). Residues lining the 5′ subpocket that display the most pronounced conformational changes upon receptor activation are shown as sticks, with white carbons for the active structure and cyan carbons for the superimposed inactive structure (PDB code 3EML). The binding pocket in the active structure is shown as a semitransparent surface colored according to its binding properties (green, hydrophobic; red, acceptor; blue, donor of hydrogen bonds).
CONCLUSIONS
Validated by benchmarking with known agonists, the crystal structure of the active-state A2AAR was employed in structure-based ligand screening and in the rational design of novel adenosine 5′-carboxamide derivatives. Out of 16 predicted high scoring candidates, including fluorinated, polar, and 5-membered ring aromatic compounds, 15 compounds had submicromolar affinity at the A2AAR, including several compounds with affinity that reached approximately 10 nM. Moreover, a number of predicted compounds had improved affinity and selectivity at the similar A1AR subtype, which has the same side chains lining the 5′ subpocket. A detailed analysis pointed to a change in the second layer of pocket residues, i.e., Cys1855.46 to Trp (in A1AR) or Phe (in the rat A2AAR), as a key variation in this region, which can contribute to ligand subtype selectivity and species selectivity. Comparison of conformational changes between inactive and active structures of the A2AAR also provides insight into the structural basis of partial agonism of some of the novel ligands, which has potential benefit in some therapeutic applications. Overall, the study shows the utility of a structure-based rational design approach in design of new agonist derivatives for ARs.
EXPERIMENTAL SECTION
Computational Procedures
Virtual Ligand Libraries for Optimization and Validation of A2AAR Models
High affinity A2AAR agonists with substitutions at the ribose C5′ position were selected from the ChEMBL database (Ki < 1 μM). This search yielded 10 unique substituents, as shown in Supporting Information Table S1. A set of decoy adenosine 5′ substituents was generated based on random 200 small (MW < 130) fragments from Enamine and Chembridge compound libraries. The benchmark compound library was generated by substituting the ethyl group in compound 1 by the selected moieties and by building all-atom molecular models from these 2D chemical representations.
Preparation of Adenosine Receptor Docking Models
The study employed three different docking models of the hA2AAR. The inactive hA2AAR (PDB: 3EML) and activated hA2AAR (PDB: 3QAK) were generated from the corresponding crystal structures by conversion into ICM internal coordinates object, removal of waters, automatic assignment, and optimization of hydrogens and Asn, Gln, and His side chain conformers. The previously published48 3EML-based model of activated hA2AAR was generated from the inactive crystal structure (PDB: 3EML) by docking compound 1 and co-optimization of the ligand with flexible side chains of the receptor binding pocket. The model of activated hA1AR was generated using ICM homology modeling with the hA2AAR crystal structure template (PDB: 3QAK), followed by energy-based optimization of the hA2AAR/compound 1 complex model with both side chain and backbone flexibility.
Ligand Docking and Small Scale VLS Benchmark
To use ICM fast docking and VLS procedures, the receptor all-atom models were converted into energy potential maps calculated on a fine 3D grid (0.5 Å cell).47 The grid potential maps account for van der Waals, hydrogen-bonding, hydrophobic, and electrostatic interactions between ligand and receptor.60,61 The ligand was represented by an all-atom model and considered fully flexible in the potential field of the receptor. The ligand and decoy compounds were automatically docked into the A2AAR models using the biased probability Monte Carlo (BPMC) global energy minimization procedure62 and sorted according to their ICM binding scores. To ensure convergence of the Monte Carlo optimization, three independent runs of the docking procedure were performed, and the best scoring pose per compound was kept. No distance restraints or any other experimentally derived information was used in the ligand docking procedure. The docking procedure takes about 30 s of Intel Xeon 2.8 GHz CPU time per compound and was performed using an 8-core Linux workstation.
Metrics for VLS Benchmark Performance
On the basis of the values of ICM binding scores for the docked compounds, we used several complementary metrics to assess VLS performance.63 ROC curves were plotted with True Positive rate (TP = Lf/Lt) on the Y axis vs False Positive rate (FP = Df/Dt) on the X axis for different fractions f of the data set. Area under ROC curve (AUC) was calculated along with Normalized Square Root AUC (NSQ_AUC).19 For NSQ_AUC, the area AUC* is calculated for the ROC curve plotted with X coordinate X = (FP)1/2 The NSQ_AUC value is then calculated as:
The value of NSQ_AUC is more sensitive to initial enrichment than the commonly used linear AUC. The NSQ_AUC measure returns the value of 100 for any perfect separation of signal from noise and values close to 0 for a random subset of noise.
Rational Design with ICM 3D Ligand Editor
ICM 3D Ligand Editor47 was employed to explore candidate adenosine 5′-carboxamide substitutions fitting in the corresponding subpocket (Figure 2). The initial setup included all atom flexible model of compound 1 docked into the crystal structure-based VLS model of A2AAR. The amide hydrogen specified as a terminal atom of the ligand scaffold can be replaced by any atom or synthetically feasible chemical group, followed by thorough energy optimization of the new derivative in the receptor model. In the interactive mode, the sequential design was guided by the 3D pocket mesh colored according to binding properties (green, hydrophobic; red, acceptor; blue, donor of hydrogen bonds), which visually suggest preferred placement of new heavy atoms of the derivative. This mode allows one to “grow” the ligand substitutions atom by atom, change bond types, and join fragments (or cycles) by linker bonds. The Ligand Editor performs on-the-fly energy-based optimization of position for each added atom or the whole fragment upon bond modification and calculates the new ligand binding score that guides design. In the automated fragment screening procedure, the terminal atom was replaced by a fragment from a predefined library, which in this case included 2000 diverse fragments with MW < 150 compiled from building blocks available from Chembridge and Enamine.
For all new compounds from interactive and automatic procedures with better than initial cutoff score (< −25 kJ/mol) were then independently redocked into the VLS model of A2AAR. Then 23 compounds with predicted binding score improvement over compound 1 (< −42 kJ/mol) were selected for synthesis.
Chemical Synthesis
General Chemical Methods
All reagents and solvents (regular and anhydrous) were of analytical grade and obtained from commercial suppliers and used without further purification. Compound 1 was obtained from R&D Systems-Tocris (Minneapolis, MN). Compounds 11 and 12 were synthesized previously.39,42 Compound 16 and adenosine 5′-carboxylic acid 45 were obtained from Sigma Aldrich (St. Louis, MO). Adenosine 5′-carboxylic acid 2′,3′-isopropylidene derivative 26 was obtained from Aberjona Laboratories, Inc. (Beverly, MA). Amine reagents as intermediates for the synthesis of adenosine 5′-carboxamides (as listed) were obtained from Sigma-Aldrich (3–5, 7, 8, 10, 13, 18, 22, 24, 25; St. Louis, MO), TCI America (14; Portland, OR), Milestone PharmaTech (21, 23; New Brunswick, NJ), Advanced ChemBlocks, Inc. (19, 20; Burlingame, CA), and Ryan Scientific (17; Mt. Pleasant, SC). Reactions were conducted under an atmosphere of nitrogen whenever anhydrous solvents were used. All reactions were monitored by thin-layer chromatography (TLC) using silica gel coated plates with a fluorescence indicator which were visualized: (a) under UV light, (b) by dipping in a mixture of anisaldehyde (2.5 mL)/concd H2SO4 (5 mL)/methanol (425 mL) or (c) by dipping the plate in a solution of ninhydrin (0.3 g in 100 mL EtOH, containing AcOH, 1.3 mL) followed by heating. Silica gel column chromatography was performed with silica gel (SiO2, 200–400 mesh, 60 Å) using moderate air pressure. Evaporation of solvents was carried out under reduced pressure at a temperature below 50 °C. After column chromatography, appropriate fractions were pooled, evaporated, and dried at high vacuum for at least 12 h to give the desired products in high purity (>95%). 1H NMR ascertained sample purity and spectra were recorded with a Bruker 400 MHz NMR spectrometer. Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane or using deuterated solvent as the internal standard (e.g., δH: DMSO-d6 2.50 ppm or CDCl3 7.26 ppm). For 19F NMR spectra, 19F signals were referenced to CFCl3 (internal standard, 0 ppm). ESI–high resolution mass spectroscopic (HRMS) measurements were performed on a proteomics optimized Q-TOF-2 (Micromass-Waters) using external calibration with polyalanine. Observed mass accuracies are those expected on the basis of known performance of the instrument as well as the trends in masses of standard compounds observed at intervals during the series of measurements. Reported masses are observed masses uncorrected for this time-dependent drift in mass accuracy.
General Synthetic Procedures for Compounds 3, 4, 5, 8, and 15
1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluoro-phosphate (COMU, 258 mg, 0.6 mmol), the amine, amine hydrochloride, or amino acid reactant (0.6 mmol), and 2′,3′-O-isopropylidene adenosine-5′-carboxylic acid 26 (129 mg, 0.4 mmol) were added to 1 mL of anhydrous DMF (dimethylformamide) under nitrogen. The resulting suspension (the acid is poorly soluble in DMF) was stirred for 1 h at room temperature. During this time, most of the acid dissolved to produce a slightly yellow opaque solution. DIPEA (N,N-diisopropylethylamine, 162 mg, 1.25 mmol for amine hydrochlorides and amino acids, or 84 mg, 0.65 mmol for amines) was added in one portion with stirring at room temperature, resulting in a bright-yellow turbid reaction mixture. The reaction mixture was stirred for additional 16 h and then diluted with 10 mL of water. Small amounts of insoluble materials were removed by filtration, and the clear solution was adjusted to pH = 8 with small amounts of sodium bicarbonate. The solution was extracted with 2 × 20 mL of ethyl acetate. The organic extract was washed with 3 × 40 mL of water, and the solvent was removed under reduced pressure. Then 1N HCl (1 mL) was added directly to the residue without further purification, and the reaction mixture was heated at 50 °C for 6–8 h. Dioxane (1 mL) was added to some reaction mixtures after 2 h of heating as required to improve the solubility of the starting material. Deprotection with 1 N HCl typically yielded a clear solution of the desired amide with a small amount of precipitate. The precipitate was removed by filtration, the filtrate was neutralized with several portions of solid sodium bicarbonate, and the solvent evaporated in a flow of dry nitrogen. Final purification was performed by HPLC using CH3CN–H2O gradient system (0–10% H2O). Acetic acid (5% in water) was used instead of water as a second solvent for the purification of adenosine-5′-carboxylic acid N-(D-alanyl)amide 15, which was further purified using HPLC (CH3CN–H2O gradient system, 0–25% H2O with 0.5% CF3COOH).
A general synthetic procedure for compound 10 that was also applied to compounds 13, 14, 18–20, 22, and 24, a shared procedure for compounds 17 and 23, and individual procedures for 21 and 25 are described in the Supporting Information.
Biological Assays
Cell Culture and Membrane Preparation
CHO cells50 stably expressing either the recombinant hA1 or hA3AR and HEK-293 cells stably expressing the human (h) A2AAR were cultured in DMEM and F12 (1:1), supplemented with 10% bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 μmol/mL glutamine. In addition, 800 μg/mL Geneticin or 500 μg/mL hygromycin were added to the A2A media or to the A1 and A3 media, respectively. After harvesting the cells, they were homogenized for 10 s with an electric homogenizer, centrifuged, and pipetted into 1 mL vials and then stored at −80 °C until binding experiments were conducted. The concentration of protein was determined using a BCA Protein Assay Kit from Pierce Biotechnology (Rockford, IL).
Radioligand Binding Studies
Radioligand binding assays at A1, A2A, and A3ARs were performed according to the procedures described previously.51,52 Each tube in the binding assay contained 100 μL of membrane suspension (20 μg of protein), 50 μL of a stock solution of agonist radioligand, and 50 μL of increasing concentrations of the test ligands in Tris-HCl buffer (50 mM, pH 7.5) containing 10 mM MgCl2. Nonspecific binding was determined using a final concentration of 10 μ M 1, a nonspecific agonist, diluted with the buffer.
The mixtures were incubated at 25 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under a reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburg, MD). Filters were washed three times with 5 mL of 50 mM ice-cold Tris-HCl buffer (pH 7.5). The radioactive agonists [3H]R-N6-(phenylisopropyl)adenosine (R-PIA) and [3H]2-[p-(2-carboxyethyl)-phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine (CGS21680) were used for the A1 and A2AAR assays, respectively, while [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (I-AB-MECA) was used for the A3AR assay. All of the filters were washed three times with Tris-HCl, pH 7.5. Filters for A1 and A2AAR binding were placed in scintillation vials containing 5 mL of Hydrofluor scintillation buffer and counted using a PerkinElmer Tricarb 2810TR liquid scintillation analyzer. Filters for A3AR binding were counted using a PerkinElmer Cobra II γ-counter. At least six concentrations of each compound were used for determination of Ki values, and results were repeated in at least three separate experiments.
Cyclic AMP Accumulation Assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method.55,56 CHO cells that expressed the recombinant hA1AR or hA2AAR were harvested by trypsinization. After centrifugation and resuspending in medium, cells were planted in 24-well plates in 1.0 mL medium. After 24 h, the medium was removed and cells were washed three times with 1 mL of DMEM containing 50 mM HEPES, pH 7.4. Cells were then treated with the agonist 1 (10 μM) and/or other test compound (1 or 10 μM) in the presence of rolipram (10 μM) and adenosine deaminase (3 U/mL) for 30 min. For the hA1AR functional assay, forskolin (10 μM) was added to the medium, and incubation was continued for an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20 °C. For determination of cyclic AMP production, a cyclic AMP kit (Sigma, St. Louis, MO) was used.
Data Analysis
Binding and functional parameters were calculated using Prism 5.0 software (GraphPAD, San Diego, CA, USA). IC50 values obtained from competition curves were converted to Ki values using the Cheng–Prusoff equation.53 Data were expressed as mean ± standard error.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases, and the NIH PSI:Biology grant (U54 GM094618) to R.C.S., R.A., and V.K.
ABBREVIATIONS USED
- AR
adenosine receptor
- VLS
virtual ligand screening
- ECL
extracellular loop
- GPCR
G protein-coupled receptor
- HPLC
high performance liquid chromatography
- PDB
Protein Data Bank
- ICM
internal coordinate mechanics
- NECA
5′-N-ethylcarboxamidoadenosine
- rmsd
root-mean-square deviation
- TM
transmembrane α-helix
- ROC
receiver operating characteristic
Footnotes
Notes
The authors declare no competing financial interest.
NOTE ADDED AFTER ASAP PUBLICATION
After this paper was published online April 30, 2012, several corrections were made. The corrected version was reposted May 1, 2012.
Known A2AAR agonists used in the initial docking model assessments. 3D binding models and scores for all ligands in Table 1. Synthetic procedures for the nucleoside derivatives and their characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nature Rev Drug Discovery. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 2.Tyndall JD, Sandilya R. GPCR agonists and antagonists in the clinic. Med Chem. 2005;1:405–421. doi: 10.2174/1573406054368675. [DOI] [PubMed] [Google Scholar]
- 3.Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends Pharmacol Sci. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shimamura T, Shiroishi M, Weyand S, Tsujimoto H, Winter G, Katritch V, Abagyan R, Cherezov V, Liu W, Han GW, Kobayashi T, Stevens RC, Iwata S. Structure of the human histamine H1 receptor complex with doxepin. Nature. 2011;475:65–70. doi: 10.1038/nature10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaakola VP, Griffth MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2. 6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human β2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a β1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choe HW, Park JH, Kim YJ, Ernst OP. Transmembrane signaling by GPCRs: insight from rhodopsin and opsin structures. Neuropharmacology. 2011;60:52–57. doi: 10.1016/j.neuropharm.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature. 2011;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao ZG, Cherezov V, Stevens RC. Structure of an Agonist-Bound Human A2A Adenosine Receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AG, Tate CG. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Topiol S, Sabio M. Use of the X-ray structure of the β2-adrenergic receptor for drug discovery. Bioorg Med Chem Lett. 2008;18:1598–1602. doi: 10.1016/j.bmcl.2008.01.063. [DOI] [PubMed] [Google Scholar]
- 15.Reynolds KA, Katritch V, Abagyan R. Identifying conformational changes of the β2 adrenoceptor that enable accurate prediction of ligand/receptor interactions and screening for GPCR modulators. J Comput-Aided Mol Des. 2009;23:273–288. doi: 10.1007/s10822-008-9257-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Congreve M, Langmead CJ, Mason JS, Marshall FH. Progress in structure based drug design for G protein-coupled receptors. J Med Chem. 2011;54:4283–4311. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabio M, Jones K, Topiol S. Use of the X-ray structure of the β 2-adrenergic receptor for drug discovery. Part 2: Identification of active compounds. Bioorg Med Chem Lett. 2008;18:5391–5395. doi: 10.1016/j.bmcl.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 18.Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of β2-adrenergic receptor ligands. Proc Natl Acad Sci USA. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katritch V, Jaakola VP, Lane JR, Lin J, Ijzerman AP, Yeager M, Kufareva I, Stevens RC, Abagyan R. Structure-based discovery of novel chemotypes for adenosine A2A receptor antagonists. J Med Chem. 2010;53:1799–1809. doi: 10.1021/jm901647p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, Iwata S, Shimamura T, Stevens RC, de Esch IJ, Leurs R. Crystal structure-based virtual screening for fragment-like ligands of the human histamine H1 receptor. J Med Chem. 2011;54:8195–8206. doi: 10.1021/jm2011589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nature Chem Biol. 2011;7:769–778. doi: 10.1038/nchembio.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deflorian F, Kumar TS, Phan K, Gao ZG, Xu F, Wu H, Katritch V, Stevens RC, Jacobson KA. Evaluation of Molecular Modeling of Agonist Binding in Light of the Crystallographic Structure of an Agonist-Bound A2A Adenosine Receptor. J Med Chem. 2012;55:538–552. doi: 10.1021/jm201461q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katritch V, Abagyan R. GPCR agonist binding revealed by modeling and crystallography. Trends Pharmacol Sci. 2011;32:637–643. doi: 10.1016/j.tips.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vilar S, Ferino G, Phatak SS, Berk B, Cavasotto CN, Costanzi S. Docking-based virtual screening for ligands of G protein-coupled receptors: not only crystal structures but also in silico models. J Mol Graphics Modell. 2011;29:614–623. doi: 10.1016/j.jmgm.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tautermann CS. The use of G-protein coupled receptor models in lead optimization. Future Med Chem. 2011;3:709–721. doi: 10.4155/fmc.11.24. [DOI] [PubMed] [Google Scholar]
- 26.Jacobson KA. Introduction to adenosine receptors as therapeutic targets. Handb Exp Pharmacol. 2009:1–24. doi: 10.1007/978-3-540-89615-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morelli M, Carta AR, Jenner P, Adenosine A. 2A Receptors and Parkinson’s Disease. Handb Exp Pharmacol. 2009:589–615. doi: 10.1007/978-3-540-89615-9_18. [DOI] [PubMed] [Google Scholar]
- 28.Sebastiao AM, Ribeiro JA. Adenosine receptors and the central nervous system. Handb Exp Pharmacol. 2009:471–534. doi: 10.1007/978-3-540-89615-9_16. [DOI] [PubMed] [Google Scholar]
- 29.Mustafa SJ, Morrison RR, Teng B, Pelleg A. Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharmacol. 2009:161–188. doi: 10.1007/978-3-540-89615-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Headrick JP, Lasley RD. Adenosine receptors and reperfusion injury of the heart. Handb Exp Pharmacol. 2009:189–214. doi: 10.1007/978-3-540-89615-9_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson CN, Nadeem A, Spina D, Brown R, Page CP, Mustafa SJ. Adenosine receptors and asthma. Handb Exp Pharmacol. 2009:329–362. doi: 10.1007/978-3-540-89615-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blackburn MR, Vance CO, Morschl E, Wilson CN. Adenosine receptors and inflammation. Handb Exp Pharmacol. 2009:215–269. doi: 10.1007/978-3-540-89615-9_8. [DOI] [PubMed] [Google Scholar]
- 33.Fishman P, Bar-Yehuda S, Synowitz M, Powell JD, Klotz KN, Gessi S, Borea PA. Adenosine receptors and cancer. Handb Exp Pharmacol. 2009:399–441. doi: 10.1007/978-3-540-89615-9_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultze-Werninghaus G, Meier-Sydow J. The clinical and pharmacological history of theophylline: first report on the bronchospasmolytic action in man by S. R. Hirsch in Frankfurt (Main) 1922. Clin Allergy. 1982;12:211–215. doi: 10.1111/j.1365-2222.1982.tb01641.x. [DOI] [PubMed] [Google Scholar]
- 35.Thomas GS, Thompson RC, Miyamoto MI, Ip TK, Rice DL, Milikien D, Lieu HD, Mathur VS. The RegEx trial: a randomized, double-blind, placebo- and active-controlled pilot study combining regadenoson, a selective A2A adenosine agonist, with low-level exercise, in patients undergoing myocardial perfusion imaging. J Nucl Cardiol. 2009;16:63–72. doi: 10.1007/s12350-008-9001-9. [DOI] [PubMed] [Google Scholar]
- 36.Yan L, Burbiel JC, Maass A, Muller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerging Drugs. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 37.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev Drug Discovery. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marone G, Plaut M, Lichtenstein LM. Characterization of a specific adenosine receptor on human lymphocytes. J Immunol. 1978;121:2153–2159. [PubMed] [Google Scholar]
- 39.Daly JW. Adenosine receptors: targets for future drugs. J Med Chem. 1982;25:197–207. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]
- 40.Beukers MW, Chang LCW, von Frijtag Drabbe Künzel JK, Mulder-Krieger T, Spanjersberg RF, Brussee J, IJzerman AP. New, non-adenosine, high-potency agonists for the human adenosine A2B receptor with an improved selectivity profile compared to the reference agonist N-ethylcarboxamidoadenosine. J Med Chem. 2004;47:3707–3709. doi: 10.1021/jm049947s. [DOI] [PubMed] [Google Scholar]
- 41.de Zwart M, Kourounakis A, Kooijman H, Spek AL, Link R, von Frijtag Drabbe Kunzel JK, APIJ 5′-N-Substituted carboxamidoadenosines as agonists for adenosine receptors. J Med Chem. 1999;42:1384–1392. doi: 10.1021/jm9804984. [DOI] [PubMed] [Google Scholar]
- 42.Jacobson KA, Gao ZG, Chen A, Barak D, Kim SA, Lee K, Link A, Rompaey PV, van Calenbergh S, Liang BT. Neoceptor concept based on molecular complementarity in GPCRs: a mutant adenosine A3 receptor with selectively enhanced affinity for amine-modified nucleosides. J Med Chem. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SK, Gao ZG, Van Rompaey P, Gross AS, Chen A, Van Calenbergh S, Jacobson KA. Modeling the adenosine receptors: comparison of the binding domains of A2A agonists and antagonists. J Med Chem. 2003;46:4847–4859. doi: 10.1021/jm0300431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zablocki JA, Wu L, Shryock J, Belardinelli L. Partial A1 adenosine receptor agonists from a molecular perspective and their potential use as chronic ventricular rate control agents during atrial fibrillation (AF) Curr Top Med Chem. 2004;4:839–854. doi: 10.2174/1568026043450998. [DOI] [PubMed] [Google Scholar]
- 45.Jacobson KA, Ohno M, Duong HT, Kim SK, Tchilibon S, Cesnek M, Holy A, Gao ZG. A neoceptor approach to unraveling microscopic interactions between the human A2A adenosine receptor and its agonists. Chem Biol. 2005;12:237–247. doi: 10.1016/j.chembiol.2004.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tosh DK, Phan K, Deflorian F, Wei Q, Gao ZG, Jacobson KA. Truncated (N)-Methanocarba Nucleosides as A1 Adenosine Receptor Agonists and Partial Agonists: Overcoming Lack of a Recognition Element. ACS Med Chem Lett. 2011;2:626–631. doi: 10.1021/ml200114q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abagyan RA, Orry A, Raush E, Budagyan L, Totrov M. ICM Manual, 3.0. MolSoft LLC; La Jolla, CA: 2011. [Google Scholar]
- 48.Jaakola VP, Lane JR, Lin JY, Katritch V, Ijzerman AP, Stevens RC. Ligand binding and subtype selectivity of the human A2A adenosine receptor: identification and characterization of essential amino acid residues. J Biol Chem. 2010;285:13032–13044. doi: 10.1074/jbc.M109.096974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.El-Faham A, Subiros Funosas R, Prohens R, Albericio F. COMU: a safer and more effective replacement for benzotriazole-based uronium coupling reagents. Chemistry. 2009;15:9404–9416. doi: 10.1002/chem.200900615. [DOI] [PubMed] [Google Scholar]
- 50.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes—characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 51.Kim YC, Ji X, Melman N, Linden J, Jacobson KA. Anilide derivatives of an 8-phenylxanthine carboxylic congener are highly potent and selective antagonists at human A2B adenosine receptors. J Med Chem. 2000;43:1165–1172. doi: 10.1021/jm990421v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gao ZG, Mamedova LK, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng Y, Prusoff WH. Relationship between Inhibition Constant (K1) and Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 54.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 55.Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 56.Post SR, Ostrom RS, Insel PA. Biochemical methods for detection and measurement of cyclic AMP and adenylyl cyclase activity. Methods Mol Biol. 2000;126:363–374. doi: 10.1385/1-59259-684-3:363. [DOI] [PubMed] [Google Scholar]
- 57.Katritch V, Kufareva I, Abagyan R. Structure based prediction of subtype selectivity for adenosine receptor antagonists. Neuropharmacology. 2011;60:108–115. doi: 10.1016/j.neuropharm.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morrison CF, Elzein E, Jiang B, Ibrahim PN, Marquart T, Palle V, Shenk KD, Varkhedkar V, Maa T, Wu L, Wu Y, Zeng D, Fong I, Lustig D, Leung K, Zablocki JA. Structure–affinity relationships of 5′-aromatic ethers and 5′-aromatic sulfides as partial A1 adenosine agonists, potential supraventricular anti-arrhythmic agents. Bioorg Med Chem Lett. 2004;14:3793–3797. doi: 10.1016/j.bmcl.2004.04.096. [DOI] [PubMed] [Google Scholar]
- 59.Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- 60.Totrov M, Abagyan R. Flexible protein–ligand docking by global energy optimization in internal coordinates. Proteins. 1997;(Suppl 1):215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 61.Totrov M. Accurate and efficient generalized born model based on solvent accessibility: derivation and application for LogP octanol/water prediction and flexible peptide docking. J Comput Chem. 2004;25:609–619. doi: 10.1002/jcc.10392. [DOI] [PubMed] [Google Scholar]
- 62.Abagyan R, Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J Mol Biol. 1994;235:983–1002. doi: 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- 63.Truchon JF, Bayly CI. Evaluating virtual screening methods: good and bad metrics for the “early recognition” problem. J Chem Inf Model. 2007;47:488–508. doi: 10.1021/ci600426e. [DOI] [PubMed] [Google Scholar]
- 64.Bosch MP, Campos F, Niubo I, Rosell G, Diaz JL, Brea J, Loza MI, Guerrero A. Synthesis and biological activity of new potential agonists for the human adenosine A2A receptor. J Med Chem. 2004;47:4041–4053. doi: 10.1021/jm031143+. [DOI] [PubMed] [Google Scholar]
- 65.Gallo-Rodriguez C, Ji XD, Melman N, Siegman BD, Sanders LH, Orlina J, Fischer B, Pu Q, Olah ME, van Galen PJ, et al. Structure–activity relationships of N6-benzyladenosine-5′-uronamides as A3-selective adenosine agonists. J Med Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.