

Abstract
Castleman disease is a rare lymphoproliferative lesion that is predominantly found in the mediastinum. Retroperitoneal and pararenal localizations are very rare. We describe a 36-year-old man with a hyaline vascular type of Castleman disease involving renal parenchyma and a paraaortic lymph node. Most reported renal Castleman disease was plasma cell type with systemic symptoms. Herein, we report the first Korean case of the hyaline vascular type of Castleman disease involving the renal parenchyma and the paraaortic lymph node simultaneously.
Keywords: Kidney; Hyaline vascular type, Multicentric; Giant lymph node hyperplasia
Castleman disease (CD) is a rare non-neoplastic lymphoproliferative disorder that has been subclassified on histologic forms: hyaline vascular type (HV) and plasma cell type (PC).1 The cause is unknown to date, but recently, some reports suggest that HVCD is a benign neoplasm of follicular dendritic cells, because these cells have aberrant expressions of adhesion molecules and may show cytogenetic abnormalities common among benign mesenchymal tumors.2 In addition, some reports propose that PCCD is related to some viral infections.3
CD is predominantly found in mediastinal lymph nodes.1 Some extrathoracic cases involving nodal and extranodal locations have been reported, however, CD of the kidney is extremely rare.4 We report a very rare case of HVCD that involved the renal parenchyma and a regional lymph node simultaneously. Two renal cases of CD have been reported in Korea but these cases were PCCD. Renal HVCD with simultaneous involvement of regional lymph node has not been reported before.
CASE REPORT
A 36-year-old man was hospitalized due to abdominal discomfort with fatigue. His family history states that his mother died of liver cirrhosis associated with the hepatitis B virus (HBV). Physical examination indicated no abnormal findings. All laboratory findings were normal, but serum hepatitis B surface antigen, hepatitis B e antigen, and HBV DNA were positive. The human immunodeficiency virus (HIV) antibody tests were all negative. The abdominal computed tomography did not show any abnormality in the liver, but a 2.4 cm sized enhancing renal mass in the left upper pole was incidentally found. An enlarged lymph node was seen in the left paraaortic area (Fig. 1). A whole body isotope scan to assess for metastasis did not show any abnormal uptake.
Fig. 1.
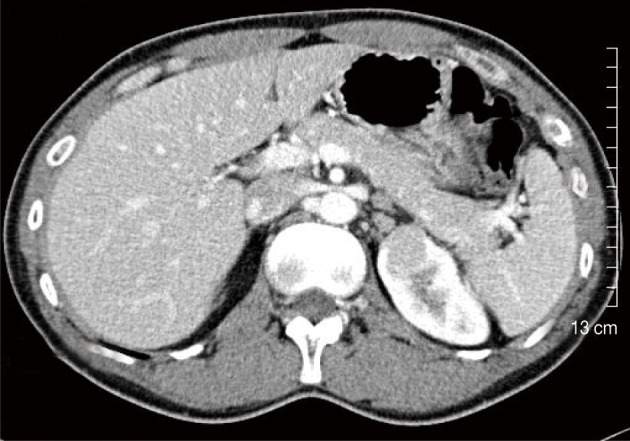
Abdominal computed tomography scan reveals an enhanced mass in the upper pole of the left kidney and an enlarged paraaortic lymph node.
The renal mass and the enlarged lymph node were excised by robotic-assisted laparoscopic partial nephrectomy based on the preoperative impression of renal cell carcinoma, renal oncocytoma, or adenoma. Microscopic pathologic examination was performed intraoperatively, and lymphoproliferative lesion with negative margins was demonstrated by a frozen section.
The wedge resected specimen was 5×3×2.5 cm in size and 20 g in weight. The cut surface showed a well demarcated, white tan and solid mass measuring 2.2×1.8 cm in the renal parenchyma (Fig. 2). The paraaortic lymph node was 1 cm in its greatest dimension. The histopathological examination revealed the features of the HVCD. There were many lymphoid follicles with hyalinized germinal centers and broad mantle zones. Blood vessels were prominent in the germinal centers and the interfollicular areas. These findings were seen in the renal cortex, medulla, and the paraaortic lymph node (Fig. 3).
Fig. 2.
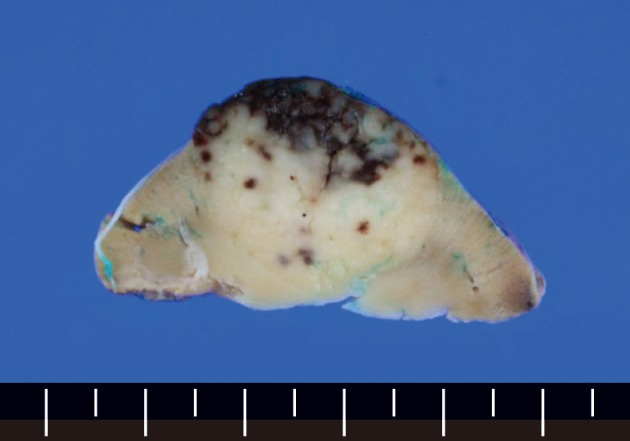
The cut surface of the partial nephrectomy specimen shows an oval white tan solid mass with focal hemorrhage involving the renal parenchyma.
Fig. 3.

Lower power-view shows extensive lymphoid infiltration forming germinal centers in the renal cortex and medulla (A) and several small regressively transformed germinal centers surrounded by a broad mantle zone in the paraaortic lymph node (B). High power-view shows a hyalinized lymphoid follicle and many blood vessels in and around the follicles (C).
Both renal tissue and lymph node tested negative for human herpsevirus 8 (HHV-8) and Epstein-Barr virus infection. The levels of interleukin 6 (IL-6) and erythrocyte sedimentation rate (ESR) were not assessed.
DISCUSSION
CD is an unusual non-neoplastic lymphoproliferative disorder which was first described as a disease entity in 1956.1 It has been called angiofollicular lymphoid hyperplasia, giant lymph node hyperplasia and angiomatous lymphoid hyperplasia in the past.5 The cause is undetermined but is most likely related to abnormal follicular dendritic cells.
CD arises primarily in the mediastinum, and about 70% of the reported cases tend to be of mediastinal origin, followed by lymph nodes in the abdomen, neck, and other sites.3 Retroperitoneal and pararenal localizations are very rare and CD of the kidney is extremely rare.4 To the best of our knowledge, fewer than 5 cases of CD involving the kidney that are of the localized HV type have been published in the English and Japanese literature.4,6,7 From the 1980s to 2010s, CD has been reported in Korea for more than 80 times. Of these, fewer than 15 occurred in the retroperitoneal area and only 2 in the kidney. All renal cases of CD were PC type.8,9 Our case is a HVCD involving renal parenchyma and a paraaortic lymph node simultaneously.
CD has been generally subclassified based on histologic features into HV, PC, and mixed type. More recently, a plasmablastic variant has been described as a fourth subtype.3 However, its clinical significance is determined by another classification of extent that is localized or multicentric.5
Nearly all cases of HV type (about 80%) and a minority of PC types (about 20%) are localized, which implies that the disorder affects a single anatomic site or a single group of lymph nodes. A localized HV type usually occurs in young people and is asymptomatic and associated with a benign clinical course. It is characterized by giant lymphoid follicles with small, regressively transformed hyalinized germinal centers and interfollicular vascular proliferation. Localized PC type usually affects mediastinal or intra-abdominal lymph nodes. This type has sheets of mature plasma cells in the interfollicular area and associated symptoms and laboratory abnormalities, such as anemia, fever, weight loss, night sweats, hypergammaglobulinemia, hypoalbuminemia and elevated ESR. Following excision of the mass, the abnormalities disappear, and the patients tend to do well overall.3
By contrast, multicentric CD (MCD) is a systemic disease with lesions of two or more separate anatomic sites.3 MCD was first proposed in 1983 by Frizzera et al.10 Recently, MCD has been defined as "a systemic disease with multiple peripheral lymphadenopathy or multiorgan involvement."10 However, another recent studies suggested that MCD is often composed of several disease entities, including idiopathic MCD and secondary MCD due to HIV infection, autoimmune disease-associated lymphadenopathy, POEMS (polyneuropathy, organomegaly, endocrinopathy, anasarca, M-proteins and skin lesions) syndrome, and non-Hodgkin lymphomas.11
Multicentric forms usually have histologic features of the PC type and only rarely of the HV type. Systemic symptoms tend to be more severe than those in localized PC type. The deregulated overproduction of IL-6 is responsible for symptoms. The levels of IL-6 appear to correlate with the systemic inflammatory manifestations.3 Recent reports suggested that HHV-8 has a viral homologue of human IL-6 in its genomic DNA, and HHV-8 infection stimulates B lymphocytes to induce IL-6 production. Moreover, other exogenous or endogenous factors may induce IL-6 secretion from B lymphocytes in HHV-8 negative CD.12 Nearly all HIV-positive patients and about half of HIV-negative patients with MCD have evidence of HHV-8 infection.3 However, Suda et al.13 reported that HHV-8 appears to be unrelated to the etiology of idiopathic MCD in Japan. Idiopathic MCD in Japan usually exhibits a chronic disease course, and appears to be related to a negative outcome for HHV-8 infection.
As above, MCD lacks a clear definition to date and there are various applications. In this regard, although our case had no significant systemic symptoms, we think it can be considered as MCD, at least from an anatomic point of view.
In summary, we report a very rare case of the multicentric HVCD involving the renal parenchyma and the paraaortic lymph node at the same time. The patient presented with a renal mass without evidence of HHV-8 infection and systemic symptoms. A few cases of multicentric HVCD have been reported before,14,15 but the renal location with involvement of regional lymph node has not been reported before in Korea.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9:822–830. doi: 10.1002/1097-0142(195607/08)9:4<822::aid-cncr2820090430>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 2.Cokelaere K, Debiec-Rychter M, De Wolf-Peeters C, Hagemeijer A, Sciot R. Hyaline vascular Castleman's disease with HMGIC rearrangement in follicular dendritic cells: molecular evidence of mesenchymal tumorigenesis. Am J Surg Pathol. 2002;26:662–669. doi: 10.1097/00000478-200205000-00013. [DOI] [PubMed] [Google Scholar]
- 3.Hsi ED. Hematopathology. Philadelphia: Churchill Livingstone Elsevier; 2007. [Google Scholar]
- 4.Hatano K, Fujita S, Tsujimoto Y, et al. Rare case of the hyaline vascular type of Castleman's disease of the kidney. Int J Urol. 2007;14:1098–1100. doi: 10.1111/j.1442-2042.2007.01900.x. [DOI] [PubMed] [Google Scholar]
- 5.Peterson BA, Frizzera G. Multicentric Castleman's disease. Semin Oncol. 1993;20:636–647. [PubMed] [Google Scholar]
- 6.Kaneko T, Ogushi T, Asakage Y, Kitamura T. Hyaline vascular type of Castleman's disease confined to the kidney. Nihon Hinyokika Gakkai Zasshi. 2008;99:597–600. doi: 10.5980/jpnjurol1989.99.597. [DOI] [PubMed] [Google Scholar]
- 7.Mah NA, Peretsman SJ, Teigland CM, Banks PM. Castleman disease of the hyaline-vascular type confined to the kidney. Am J Clin Pathol. 2007;127:465–468. doi: 10.1309/51U9EWBTPQAM61TT. [DOI] [PubMed] [Google Scholar]
- 8.Ryu JH, Oh JW, Kim KH, et al. Castleman disease misdiagnosed as a neoplasm of the kidney. Korean J Urol. 2009;50:413–416. [Google Scholar]
- 9.Won JE, Jeong SJ, Cho JH, et al. A case of Castleman's disease with kidney involvement. Korean J Nephrol. 2007;26:767–771. [Google Scholar]
- 10.Frizzera G, Banks PM, Massarelli G, Rosai J. A systemic lymphoproliferative disorder with morphologic features of Castleman's disease: pathological findings in 15 patients. Am J Surg Pathol. 1983;7:211–231. doi: 10.1097/00000478-198304000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Kojima M, Nakamura N, Tsukamoto N, et al. Clinical implications of idiopathic multicentric castleman disease among Japanese: a report of 28 cases. Int J Surg Pathol. 2008;16:391–398. doi: 10.1177/1066896908315812. [DOI] [PubMed] [Google Scholar]
- 12.Yoshizaki K, Matsuda T, Nishimoto N, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood. 1989;74:1360–1367. [PubMed] [Google Scholar]
- 13.Suda T, Katano H, Delsol G, et al. HHV-8 infection status of AIDS-unrelated and AIDS-associated multicentric Castleman's disease. Pathol Int. 2001;51:671–679. doi: 10.1046/j.1440-1827.2001.01266.x. [DOI] [PubMed] [Google Scholar]
- 14.Park JB, Hwang JH, Kim H, et al. Castleman disease presenting with jaundice: a case with the multicentric hyaline vascular variant. Korean J Intern Med. 2007;22:113–117. doi: 10.3904/kjim.2007.22.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JE, Kim CJ, Park IA, et al. Clinicopathologic study of Castleman's disease in Korea. J Korean Med Sci. 2000;15:393–398. doi: 10.3346/jkms.2000.15.4.393. [DOI] [PMC free article] [PubMed] [Google Scholar]