

Abstract
The origin and timing of the diversification of modern birds remains controversial, primarily because phylogenetic relationships are incompletely resolved and uncertainty persists in molecular estimates of lineage ages. Here, we present a species tree for the major palaeognath lineages using 27 nuclear genes and 27 archaic retroposon insertions. We show that rheas are sister to the kiwis, emu and cassowaries, and confirm ratite paraphyly because tinamous are sister to moas. Divergence dating using 10 genes with broader taxon sampling, including emu, cassowary, ostrich, five kiwis, two rheas, three tinamous, three extinct moas and 15 neognath lineages, suggests that three vicariant events and possibly two dispersals are required to explain their historical biogeography. The age of crown group birds was estimated at 131 Ma (95% highest posterior density 122–138 Ma), similar to previous molecular estimates. Problems associated with gene tree discordance and incomplete lineage sorting in birds will require much larger gene sets to increase species tree accuracy and improve error in divergence times. The relatively rapid branching within neoaves pre-dates the extinction of dinosaurs, suggesting that the genesis of the radiation within this diverse clade of birds was not in response to the Cretaceous–Paleogene extinction event.
Keywords: vicariance biogeography, palaeognaths, phylogenetics, retroposon, bird origins, gene trees
1. Introduction
For over a century, the uncertainty surrounding the phylogenetic relationships among palaeognathous birds has made them one of the most controversial branches in the avian tree of life. Traditionally, this group has been subdivided into flighted tinamous of South and Central America and flightless ratites distributed across the southern continents. Ratites are a group of large flightless birds that lack a keel on their sternum, and include emu (Australia), cassowaries (Australia and New Guinea), rheas (South America), ostrich (Africa and formerly Asia), kiwis (New Zealand) and extinct moas (New Zealand) and elephant birds (Madagascar). Many studies using morphological and molecular data supported the monophyly of ratites and placed them sister to tinamous, although no consensus was achieved on relationships among ratites [1–5]. Their flightless condition and disjunct distribution on southern land masses led to a widely accepted biogeographic theory that their evolution and distribution was shaped by the break-up of Gondwana [1,3,5].
Recent analyses using multiple nuclear gene sequences of major clades of birds [6,7] have posited a seemingly radical phylogenetic hypothesis of ratite paraphyly because tinamous were found to be embedded within ratites. This view has also been suggested by a reanalysis of palaeognath mitochondrial genomes [8]. Contrary to the widely accepted view that the common ancestor of ratites was flightless, this hypothesis implies that the common ancestor was probably a flying bird. Additionally, these studies concluded that the branching pattern of the palaeognath tree does not match the separation of the land masses as Gondwana fragmented [6]. Instead, molecular dates estimated from mitochondrial genes suggest that many of the divergences post-date the break-up, leading to the prediction that the ancestors of different ratite lineages probably flew to different continents after Gondwana broke apart [8]. Flightlessness may have evolved much later, thus challenging the conventional biogeographic theory that their disjunct distribution in the southern hemisphere was shaped by plate tectonics.
Tests of these hypotheses have broader reach beyond the avian tree of life because they have general relevance to the relative roles of vicariance and dispersal in the southern biota, and the timing and potential causes of the flowering of crown group lineages near the end of the Cretaceous. Such tests must also address a major scientific issue in the dating of crown group divergence times. Most molecular estimates of the ages of the various nodes in the avian tree of life have relied largely on a single locus, the mitochondrial genome [9–11] (but see [12,13]), which yields divergence times considerably older than a strict interpretation of the fossil record [14].
A fundamental prerequisite in testing these hypotheses is the construction of a well-resolved and strongly supported phylogeny of avian clades. The tree produced from a multigene nuclear dataset [6,7] has the ostrich sister to a clade composed of the remaining extant palaeognaths, but fails to resolve the branching pattern between the rheas, kiwis, Casuariidae and tinamous with confidence [6]. A reanalysis of the mitochondrial genomes concluded that ‘beyond the sister-groupings of the tinamous and moa, and of emu and cassowaries, the relationships of the palaeognaths remain difficult to resolve’ ([8], p. 7). A striking feature of these tree topologies is the presence of successive short branches between major lineages deep within the tree, connected to long terminal branches. Rapid radiations causing this tree shape can pose problems in phylogenetic reconstruction owing to limits in the number of phylogenetically informative characters between closely spaced speciation events, which may be overwritten by subsequent mutations on long terminal branches. Rapid radiations can also lead to incomplete sorting of ancestral polymorphisms if population sizes were large [15,16]. In either case, this can produce gene tree discordance and failure to recover the underlying species tree with the common practice of concatenating gene sequences from multigene datasets [17,18]. Moreover, this will result in erroneous downstream conclusions about evolutionary processes when characters, life-history traits or geographical distributions are mapped onto incorrect tree topologies.
To further resolve palaeognath phylogenetic relationships and test different biogeographic hypotheses and the timing of key avian diversification events, we assembled two molecular datasets. The first consisted of 10 new nuclear gene sequences from a broad sampling of palaeognath species that were combined with 17 published nuclear genes to which we added new sequences from the extinct little bush moa (Anomalopteryx didiformis). To root the palaeognath tree and provide fossil calibration points for divergence dating of crown group birds, we also included 15 neognaths and two non-avian outgroup taxa. A second dataset of rare genomic changes was assembled to provide an independent test of palaeognath phylogenetic relationships. The CR1 non-LTR retrotransposon LINE family is ideally suited as a clade marker owing to its propagation properties and universality, as shown recently in several deeper-level avian phylogenetic studies [19–24]. Unlike DNA sequences, retroposon insertion analysis avoids problems of model misspecification, taxon sampling, long-branch attraction and base compositional biases. Both long-branch attraction and base compositional biases have been implicated in making phylogenetic analyses of the palaeognaths so problematic using mitochondrial genomes [3,8]. Although retroposon insertions avoid many of the pitfalls of DNA sequences, they are not exempt from incomplete lineage sorting. Conflict in rare genomic events can be useful as potential evidence of incomplete lineage sorting [22,23].
2. Material and methods
(a). DNA sequence datasets
(i). Ten nuclear genes with broad palaeognath taxonomic sampling
DNA sequences from 10 nuclear protein-coding genes were obtained from 16 species representing all major palaeognath lineages, including ostrich, five species of kiwi, emu, southern cassowary, both rhea species, three tinamou species and three species of extinct moa. Additionally, we sequenced these genes from 15 species of neognaths: blackish oystercatcher, painted snipe, wattled jacana, rifleman, zebra finch, common loon, Wilson's storm-petrel, saddle-billed stork, gentoo penguin, emperor penguin, chimney swift, ruby-throated hummingbird, chicken, greater white-fronted goose and magpie goose.
Sequences were also obtained for two non-avian outgroup taxa (alligator and caiman) to root the avian tree (further detail is available in the electronic supplementary material).
Total genomic DNA was isolated from all specimens and used to amplify and sequence BACH1, BDNF, BMP2, DNAH3, IRBP, NT3, PNN, PTPN12, RAG2 and TRAF6 using the polymerase chain reaction. Primers and amplification conditions are outlined in electronic supplementary material. All genes were translated and aligned by eye at the amino acid level, and nucleotides were made to conform to this alignment. The 10-gene dataset totalled 9909 base pairs (bp).
(ii). Twenty-seven nuclear genes with restricted palaeognath taxon sampling
To deal with possible gene tree heterogeneity we combined the 10 protein-coding genes with 17 of the 20 genes from Harshman et al. [6] and Hackett et al. [7]. One gene was common to both datasets, and two of the 20 genes could not be obtained from the little bush moa, so they were excluded (see the electronic supplementary material for details). Taxon sampling in the 17-gene dataset differed at the species level in four genera, and at the genera level for three taxa, so we made composite sequences with closely related species (see the electronic supplementary material). The 27-gene dataset totalled 31 685 bp.
(b). Model selection and phylogenetic analysis
For the 10 protein-coding genes, the best-fitting models for four potential partitioning schemes (one partition, partitioning by gene or codon position and partitioning by gene and codon position) were calculated with the program MrModeltest v. 2.3 [25] using the Akaike information criterion. Optimal partitioning was determined using Bayes factors with marginal likelihoods estimated by the stepping-stone (SS) [26] method implemented in PHYCAS v. 1.2 [27]. Partitioning by codon position was favoured, and GTR + I + Γ was estimated to be the most appropriate model for first and second codon positions, and GTR + Γ for third codon positions. In the 27-gene dataset, the additional 17 genes were partitioned by gene because most were non-coding.
Phylogenetic analyses on concatenated sequences of the 10- and 27-gene datasets were conducted using Bayesian inference in MrBayes v. 3.1.2 [28]. MrBayes was run on the CIPRES Science Gateway portal [29] multiple times with four chains set for 100 million generations and sampling every 2000 generations. All other run parameters were set as defaults, and convergence was examined using Tracer v. 1.4 [30] with a 25 per cent burn-in. Individual gene trees were estimated using maximum likelihood using the program RAxML [31]. Because gene tree discordance potentially can prevent recovery of the species tree with concatenated data [32], we used maximum pseudo-likelihood estimates of species trees (MP-EST) [33] to estimate the species tree from the set of gene trees. MP-EST has been shown to be statistically consistent in estimating the species tree even in the anomaly zone, and is computationally efficient for a moderate number of taxa, as in this study [33].
(c). Fossil calibrations and molecular dating
Because the 10-gene dataset had broader taxon sampling and very small amounts of missing sequence we used it to estimate divergence times among taxa. Node ages were estimated with the lognormal relaxed clock method in BEAST v. 1.6.2 [34], and were compared with relaxed clock estimates with correlated and uncorrelated rates in MCMCtree [35]. To calibrate molecular clocks we used seven fossil anchor points: crocodile/bird split 243–251 Ma [36], alligator/caiman split 66–71 Ma [36], emu/cassowary split 25–35 Ma [37], magpie goose/greater white-fronted goose split 66–68 Ma [38], penguin/petrel split 60–62 Ma [39], hummingbird/swift split 47–49 Ma with an upper limit of 53 Ma [12] and jacana/painted snipe split 30–32 Ma [40]. A uniform prior was placed on the origin of modern birds from 67 to 133 Ma for the relaxed clock methods. The lower limit was set by the oldest diagnostic neornithine fossil Vegavis [39]. The upper limit was set by mitochondrial estimates of modern bird origins [9]. Multiple independent runs of 100 million generations were performed in BEAST with sampling every 2000 generations. Dating in MCMCtree was performed with the available HKY model for 100 000 generations and sampling every 10 generations. Dates were averaged across the runs. Convergence of runs in both methods were checked using Tracer (ESS > 200).
(d). Retroposon identification from GenBank and library construction and screening
Genomic libraries were created for emu, ostrich, great spotted kiwi and lesser rhea, and were enriched for CR1 non-LTR retrotransposon insertions by adapting a primer walking protocol (www.biochem.uci.edu/steele/Splinkerette_page.html) and a CR1 consensus sequence (see the electronic supplementary material). Hundreds of CR1 insertion sites were recovered and identified using the RepeatMasker database [41]. Primers were designed in flanking regions to screen for presence or absence of CR1 inserts in homologous loci of other palaeognaths. Emu sequences from 19 BAC clones downloaded from GenBank were also examined to identify additional CR1 inserts, which were screened against the other palaeognaths (see the electronic supplementary material). All CR1 inserts had flanking domains used in BLASTn searches (www.ensembl.org) against chicken and zebra finch genomes to determine whether inserts were also present in neognaths. CR1 insertions were also identified in the avian nuclear sequence dataset of Hackett et al. [7], kindly provided by Rebecca Kimball. The GenBank accession numbers for all the sequences collected here are JX532994–JX533444.
3. Results and discussion
(a). Gene sequence analysis
Bayesian analyses of the concatenated dataset of 10 nuclear protein-coding genes reaffirms the fundamental split in birds between the Palaeognathae and Neognathae, and the split between the Galloanserae and Neoaves within the Neognathae (figure 1). Within the Palaeognathae, there is strong support (posterior probability, PP = 1) for ostrich as sister to the remaining palaeognaths, in agreement with studies using multiple nuclear loci [6,7]. The tinamous are embedded within the ratites, rendering ratites paraphyletic, and are strongly supported as sister to the moas as in the reanalysis of mitochondrial genomes [8]. The moa–tinamou clade is sister to rheas, kiwis, emu and cassowaries. In contrast, the nuclear gene trees of Harshman et al. [6] placed tinamous as either sister to emu, cassowaries and kiwis or as sister to the rheas, but both were poorly supported at these nodes. MtDNA genomes recovered the first of these topologies, but again with low nodal support [8].
Figure 1.
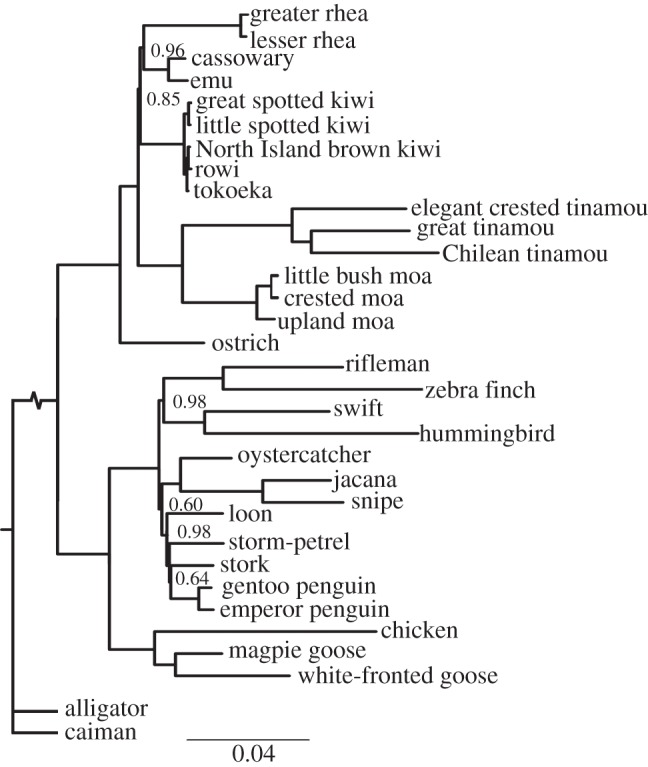
Phylogenetic analysis using the 10-gene concatenated dataset in MrBayes. Bayesian posterior probabilities are shown at nodes with less than maximal support.
In our tree, emu and cassowary (Casuariidae) are strongly supported as sister taxa (PP = 1), which in turn are sister to rheas (PP = 0.96). This topology differs from previous mitochondrial [3,5,8] and nuclear gene phylogenies [6,7], which strongly supported kiwis as sister to emu and cassowaries (figure 1). However, internal branches at this node are very short, increasing the potential for incomplete lineage sorting or polytomy in gene trees owing to lack of phylogenetic signal.
Individual consensus gene trees estimated in RAxML are discordant and have low bootstrap support at nodes with short internal branches (see electronic supplementary material, figure S1). However, all gene trees except BMP2 have strong bootstrap support (1.0) for the sister-group relationships of neognaths and palaeognaths and of Galloanserae and Neoaves (0.85), and place moa sister to tinamous (0.86). Five gene trees place ostrich sister to the other palaeognaths (0.93). There is only weak support from three genes for either rheas or kiwis as sister to emu and cassowary (0.53 and 0.47, respectively).
As statistical consistency in estimating the species tree from discordant gene trees is likely to increase with more genes [33], we analysed the 27-gene dataset with MrBayes. The tree is strongly supported (figure 2), and relationships among the palaeognaths are mostly congruent with the tree from the 10 genes. The exceptions in the 27-gene phylogeny are that kiwis are sister to Casuariidae instead of rheas (PP = 1.0), rheas are instead sister to this clade (PP = 1.0), and Chilean tinamou is sister to elegant crested tinamou rather than to great/white-throated tinamou. Among Neoaves, petrel is sister to penguins, rather than to stork, as recovered in the 10-gene dataset.
Figure 2.
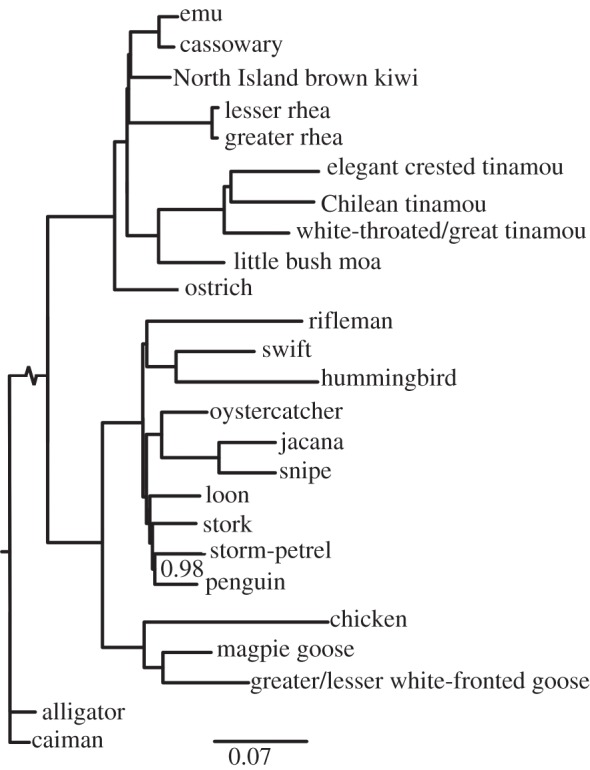
Phylogenetic analysis using the 27-gene concatenated dataset in MrBayes. Bayesian posterior probabilities are shown at nodes with less than maximal support.
The bootstrapped gene trees were then used to estimate the species consensus tree using MP-EST. Because MP-EST needs rooted trees, we restricted the estimation of the species trees from the 10- and 27-gene datasets to palaeognaths rooted with chicken sequences, because alignments to crocodilian introns were problematic. The topology of the palaeognath species tree in MP-EST for the 10-gene dataset closely matched the tree from the concatenated dataset under MrBayes. The exception is the relationships among tinamous, which changed to match the topology of the 27-gene dataset concatenated tree. MP-EST for 27 genes is identical to that from the 27 genes under concatenation (figure 3).
Figure 3.
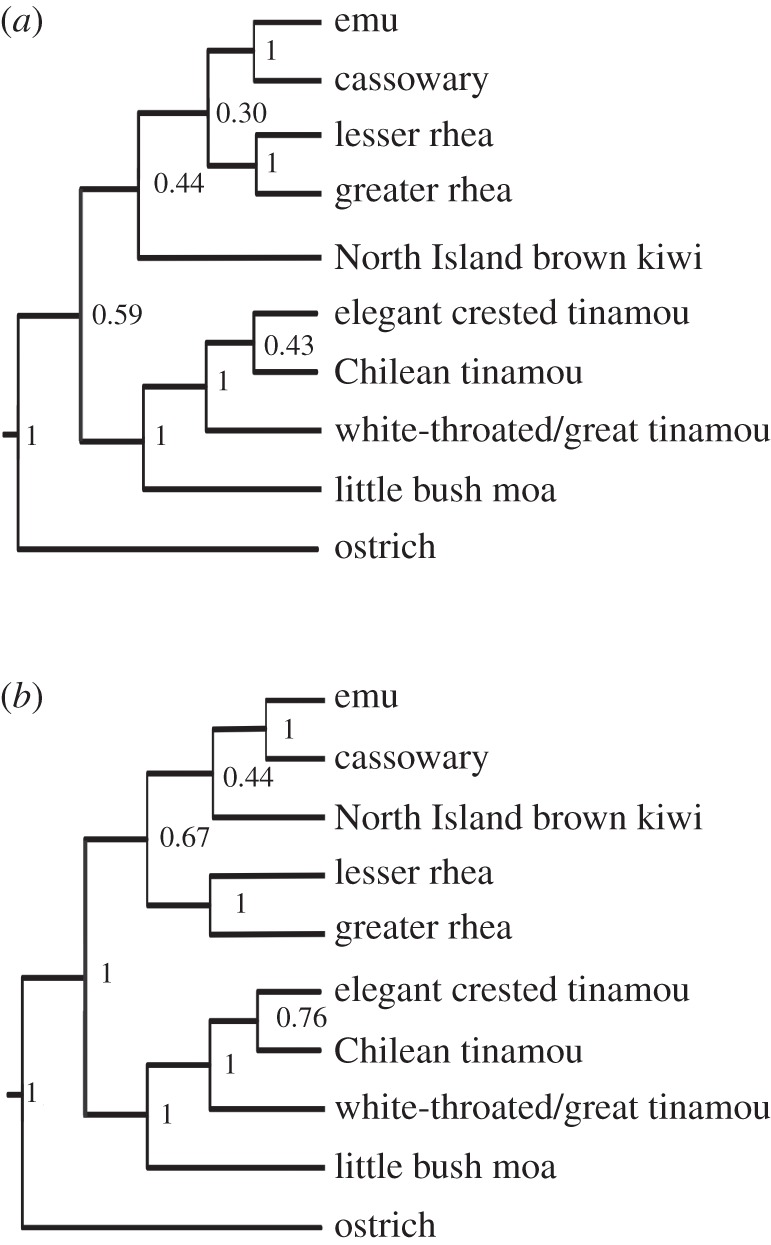
Consensus trees for the (a) 10- and (b) 27-gene datasets constructed from the species trees estimated by MP-EST with 1000 bootstrap datasets and majority-rule-extension in CONSENSUS from the PHYLIP package [42].
However, bootstrap support for both MP-EST trees is much lower than the posterior probabilities found for their respective concatenated trees. These low bootstrap numbers probably reflect the level of uncertainty of clades within and among gene trees [33], and imply that the Bayesian concatenation method may have overestimated the posterior probabilities. Increasing the number of loci from 10 to 27 did increase the bootstrap support at the difficult nodes with very short internal branches, as expected from the results of simulations by Liu et al. [33]. However, it is clear that many more genes will be required to resolve these nodes and estimate strongly supported species trees of the palaeognaths and of the Neoaves, where similarly short branches occur at internal nodes.
(b). Retroposon insertion analysis and tree topology
To independently test competing tree topologies inferred from sequences analysed here and the published nuclear [6,7] and mitochondrial gene sequences [8], we compiled a dataset of CR1 retroposon insertions from emu sequences available in GenBank and from four palaeognath genomic libraries. Among the hundreds of retroposon insertions screened, 22 were phylogenetically informative (figure 4; electronic supplementary material, table S2). Six are common to all the palaeognaths and not present in homologous loci in chicken or zebra finch genomes. By contrast, one insert was common to all neognaths but absent in the palaeognaths. These insertions support the fundamental split in the modern bird tree between the Palaeognathae and Neognathae.
Figure 4.

Chronogram based on the multilocus species tree with retroposon insertions mapped onto their respective nodes. Grey horizontal bars correspond to 95% highest posterior density of the Bayesian posterior distribution of molecular time estimates. Land mass provenance for each palaeognath follows species labels. A red clock marks each of the seven fossil calibration points. Simplified maps below the time scale show the fragmentation of Gondwana. The separations of Africa, New Zealand and Australia from Gondwana have their estimated fragmentation times mapped as coloured vertical bars. Gondwana map: Af, Africa; Ant, Antarctica; Aus, Australia; NZ, New Zealand; SA, South America. Bird drawings are modified from del Hoyo et al. [43], with permission from Lynx Edicions, Barcelona, Spain.
Within the Palaeognathae, five insertion sites are common to tinamous and all extant ratites with the exception of ostrich. As retroposon insertions are self-polarizing, these five shared insertions identify all palaeognaths minus ostrich as a monophyletic group, reaffirming ratite paraphyly detected in sequence trees. Also in agreement with the 10- and 27-gene trees are two retroposon insertions exclusive to kiwis, rheas, emu and cassowary that support the monophyly of this clade. These two insertions cannot be mapped parsimoniously on the trees estimated from the published multigene nuclear datasets [6,7] and the reanalysis of the mitochondrial genomes [8]. However, three insertions are estimated to be required at a node for statistical significance at p < 0.05 [24], but the two we found are consistent with the species tree estimated with MP-EST and with concatenation in MrBayes. The monophyly of the kiwis, emu and cassowaries is supported by two insertion sites they share, but which are absent in the rheas, ostrich, tinamous and moa (only one of the two loci amplified for the moa ancient DNA).
A single retroposon insertion unique to emu and cassowary supports the monophyly of this clade. Two insertions were unique to the kiwis (one common to all kiwis and the other only in the spotted kiwis). Three insertions were unique to rheas. All retroposon insertions were mapped on the 27 gene tree without incongruence. No conflicting insertions were found, and thus there is no evidence for incomplete lineage sorting in these retro-elements, but sample size for some nodes is small.
Within the Neognathae, five phylogenetically informative CR1 insertion sites identified from the nuclear sequence dataset of Hackett et al. [7] were mapped onto our sequence tree (figure 4). Two of these support jacana as sister to painted snipe (Scolopaci), two are common to all passerines and one was common to all penguins (Sphenisciformes).
(c). Molecular dating and palaeognath biogeography
Divergence times estimated across the avian tree are pivotal in understanding the roles of historical events such as plate tectonics and the large-scale mass extinction at the end of the Cretaceous in shaping the modern avian radiation. A likelihood ratio test performed in DAMBE [44] rejected a strict clock with p < 0.0001. Divergence times were therefore estimated with relaxed clock models in BEAST and MCMCtree, with the 10-gene dataset constrained to the 27-gene and retroposon-supported species tree topology. Estimates of node ages using uncorrelated and correlated rates among lineages in MCMCtree were very similar, so we only present the former to compare with those from BEAST (table 1). Mean node ages were similar using both methods, but among the paleognaths the 95 per cent highest posterior density (HPD) estimates were markedly broader in BEAST. For this reason, we focus on MCMCtree estimates, because the 95 per cent HPDs are conservative in comparing divergence times with the dates of fragmentation of Gondwana.
Table 1.
Divergence times and 95% HPD estimated with uncorrelated rates in MCMCtree and BEAST using seven fossil constraints.
| node/divergence | MCMCtree |
BEAST |
||
|---|---|---|---|---|
| node age | 95% HPD | node age | 95% HPD | |
| 1. Neognathae/Palaeognathae | 131 | 122–138 | 127 | 114–133 |
| 2. ostrich/other palaeognaths | 97 | 89–108 | 97 | 73–119 |
| 3. (moa, tinamou)/other palaeognaths | 84 | 77–91 | 81 | 59–105 |
| 4. rheas/(emu, cassowary, kiwis) | 81 | 74–84 | 76 | 52–101 |
| 5. kiwis/(emu, cassowary) | 78 | 71–83 | 73 | 50–100 |
| 6. moa/tinamous | 69 | 63–76 | 65 | 47–87 |
| 7. moa | 19 | 12–27 | 21 | 8–39 |
| 8. little bush moa/crested moa | 9 | 5.2–14 | 8.9 | 2–20 |
| 9. tinamous | 45 | 37–54 | 41 | 24–61 |
| 10. erect crested tinamou/Chilean tinamou | 36 | 26–45 | 30 | 15–48 |
| 11. greater rhea/lesser rhea | 4.0 | 1.9–7.4 | 5.8 | 1–15 |
| 12. emu/cassowary | 27 | 25–31 | 29 | 25–34 |
| 13. kiwis | 4.9 | 3.0–7.9 | 6.3 | 2.3–14 |
| 14. great spotted kiwi/little spotted kiwi | 1.4 | 0.6–2.7 | 1.85 | 0.3–4.9 |
| 15. (North Island brown kiwi, rowi)/tokoeka | 1.9 | 1.0–3.2 | 2.5 | 0.7–5.6 |
| 16. North Island brown kiwi/rowi | 1.2 | 0.6–2.2 | 1.4 | 0.3–3.3 |
| 17. Neoaves/Galloanserae | 107 | 97–118 | 106 | 91–121 |
| 18. chicken/(white-fronted goose, magpie goose) | 83 | 74–94 | 83 | 71–98 |
| 19. white-fronted goose/magpie goose | 70 | 65–79 | 68 | 67–71 |
| 20. Neoaves | 81 | 75–89 | 83 | 71–98 |
| 21. passerines/(swifts, hummingbirds) | 76 | 69–84 | 76 | 62–93 |
| 22. rifleman/zebra finch | 51 | 40–67 | 49 | 26–69 |
| 23. swift/hummingbird | 51 | 47–53 | 50 | 47–53 |
| 24. shorebirds/waterbirds | 77 | 72–85 | 79 | 69–92 |
| 25. shorebirds | 68 | 59–75 | 63 | 46–80 |
| 26. jacana/snipe | 36 | 32–43 | 33 | 32–37 |
| 27. loon/other waterbirds | 72 | 66–80 | 72 | 65–83 |
| 28. stork/(penguins, petrel) | 69 | 63–77 | 67 | 63–74 |
| 29. petrel/penguins | 67 | 62–76 | 65 | 62–70 |
| 30. penguins | 13 | 7.0–22 | 14 | 2.4–34 |
The age of crown group birds was estimated to be 131 Ma with 95 per cent HPD 122–138 Ma, consistent with earlier mitochondrial clock estimates [9–11]. This is almost twice the age of the oldest modern bird fossil Vegavis (66–68 Ma), but this fossil taxon is highly derived and placed in a modern order [38]. Despite the use of widely accepted fossil calibrations, a gap persists between the age of the oldest crown group fossils and molecular estimates of the age of their most recent common ancestor (MRCA). However, Neornithes have been posited to have originated in the Southern Hemisphere [45], a region less sampled for fossils than the Northern Hemisphere, and perhaps where exposures with fossils of greater antiquity may yet be discovered to bridge the gap. Additionally, a recent assessment of the poor quality and patchy regional sampling of the fossil record in the Late Cretaceous suggested that the presence of neornithine lineages at this time cannot be refuted based on current evidence [46].
The MRCA of Palaeognathae was estimated at about 97 Ma (95% HPD 89–108 Ma) when the lineage leading to ostrich diverged. Earlier studies placed ostrich in a more derived position in the palaeognath tree, which translated to a molecular divergence time of 65 Ma. This was long after Africa separated from Gondwana around 100 Ma [47,48]. A mixture of dispersal and continental drift were invoked to explain the ostrich's presence in Africa and Asia [3,5]. By contrast, the much older ostrich divergence time we derived borders on the time of rifting of Africa from Gondwana, and thus dispersal need not be invoked.
The rheas were estimated to branch off from the lineage leading to kiwis, emu and cassowaries around 81 Ma (95% HPD 74–84 Ma). At this time, South America was connected to Australia via Antarctica, well before separation of Australia at approximately 55 Ma. The vicariance hypothesis predicts that the isolation of the rhea lineage in the Antarctic/South America land mass would therefore have occurred around the time Australia rifted away. Although this continental fragmentation time overlaps the lower 95 per cent HPD (52–101 Ma) for rhea divergence estimated in BEAST, a vicariant origin for this split seems unlikely given the much older mean divergence times estimated by both dating methods. Instead, a more ancient dispersal event is inferred in the Campanian.
The branching point between the kiwi lineage and Casuariidae was estimated at 78 Ma (95% HPD 71–83 Ma), close to the 80–82 Ma period when New Zealand began to separate from Gondwana [49], and thus vicariance cannot be rejected. The early divergence time of about 84 Ma (95% HPD 77–91 Ma) estimated for the lineage leading to moas and tinamous is intriguing because it too coincides with the rifting of New Zealand. If the MRCA of these lineages was on the New Zealand land mass at this time, only one dispersal to South America is required to explain the occurrence of tinamous in South America. Dispersal may have occurred when the two lineages diverged about 69 Ma (95% HPD 63–76 Ma). In this scenario, palaeognath biogeography requires possibly two dispersals, and three vicariance events.
Geological forces may have influenced diversifications of palaeognath lineages even after the fragmentation of Gondwana. The MRCA of the extant kiwis was estimated to occur around 5 Ma (95% HPD 3–8 Ma). The timing of the diversification of the kiwis into the five recognized species [50] coincides with an extensive mountain-building period in New Zealand and cycles of glaciation that probably facilitated allopatric speciation through habitat fragmentation. The three moa species sampled in this study included taxa from the two most divergent moa branches. This allowed estimation of the divergence time of their MRCA to about 19 Ma (95% HPD 12–27 Ma), consistent with previous dating by Baker et al. [51]. This timing follows closely after the Oligocene ‘drowning’ of New Zealand, a period when most of New Zealand became inundated by the sea [52]. The upper limit of our estimate coincides with the rebound and re-emergence of the New Zealand land mass around 23 Ma. This is much earlier than the 5.8 Ma proposed by Bunce et al. [53] based on mitochondrial sequences. The timing of the moa radiation is consistent with a terrestrial biota that has undergone a severe bottleneck owing to habitat loss followed by a subsequent diversification as the environment expanded.
The MRCA of Neognathae was estimated to have occurred around 107 Ma (95% HPD 97–118 Ma). Chicken and the two geese lineages were estimated to diverge around 83 Ma (95% HPD 74–94 Ma), similar to times obtained with mitochondrial sequences [9–11]. Within the Neoaves, the split of the clade of waterbirds (loon, stork, petrel, penguins) plus shorebirds (oystercatcher, jacana, snipe) from the Passeriformes (rifleman and zebra finch) plus Apodiformes (swift and hummingbird) was dated at around 81 Ma (95% HPD 75–89 Ma). These divergence times of major clades in the Neoaves predate the mass extinction at the end of the Cretaceous, and instead occur in the Campanian age (70–83 Ma), when the fossil record shows a coincident pulse of diversification within non-avian dinosaurs [54].
Conversely, in our tree, two nodes were significantly younger than had been estimated in earlier molecular studies. The MRCA of extant penguins was estimated at approximately 13 Ma (95% HPD 7–22 Ma), as opposed to the 40 Ma estimate derived from mostly mitochondrial genes and relaxed molecular clocks [55]. The New Zealand endemic rifleman was calculated to have diverged from the zebra finch around 51 Ma (95% HPD 40–67 Ma), which makes the origin of the Passeriformes much younger than earlier estimates [56], and similar in age to the oldest passerine fossil found in Australia [57].
4. Conclusions
In summary, the use of nuclear multigene datasets to resolve the relationships among the palaeognaths [6,7] and improved methods of modelling base substitutions in mitochondrial genomes [8] have contributed to our understanding of the phylogenetic relationships among these birds. However, the persistence of poorly resolved nodes within these trees limits the conclusions that can be made about their evolution and biogeography. By improving upon taxon sampling, adding more gene sequences and introducing a large number of rare genomic changes, we have produced the most robust estimate to date of a multigene species tree of the palaeognaths that takes into account gene tree uncertainty. Nevertheless, support at some key branches in the species tree is still low, clearly indicating the need for a phylogenomics approach with a much larger gene set in future studies. The use of multiple fossil calibrations in molecular dating of the multilocus species tree produced estimates of divergence times that imply that both continental drift and dispersal are required to explain palaeognath biogeography. This is in contrast to dating of gene trees where divergence times can be overestimated, because there is no correction for genetic divergence prior to speciation [58]. We conclude that the problems encountered in resolving phylogenetic relationships among the Palaeognathae and a limited sampling of the Neognathae are likely to be common across the sweep of modern bird lineages. Rapid radiations leading to incomplete lineage sorting and discordant or unresolved gene trees can be anticipated across short internal branches, which are a striking feature at the base of the bird tree. This will probably require coalescent methods combined with evidence from rare genomic events to help resolve the branching order of the major clades in the avian tree of life.
Acknowledgements
We thank the American Museum of Natural History, the Academy of Natural Sciences of Philadelphia, Louisiana State University and Carol Ritland for blood and tissue samples. This work was supported by grants to A.J.B. from the Natural Sciences and Engineering Research Council (Canada), the Royal Ontario Museum Foundation and Gloria Chen.
References
- 1.Cracraft J. 1973. Continental drift, paleoclimatology, and the evolution and biogeography of birds. J. Zool. 169, 455–545 10.1111/j.1469-7998.1973.tb03122.x (doi:10.1111/j.1469-7998.1973.tb03122.x) [DOI] [Google Scholar]
- 2.Sibley C. G., Ahlquist J. E. 1990. Phylogeny and classification of birds: a study in molecular evolution. New Haven, CT: Yale University Press [Google Scholar]
- 3.Haddrath O., Baker A. J. 2001. Complete mitochondrial DNA genome sequences of extinct birds: ratite phylogenetics and the vicariance biogeography hypothesis. Proc. R. Soc. Lond. B 268, 939–945 10.1098/rspb.2001.15 (doi:10.1098/rspb.2001.15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee K., Feinstein J., Cracraft J. 1997. The phylogeny of ratite birds: resolving conflicts between molecular and morphological data sets. In Avian molecular evolution and systematics (ed. Mindell D. P.), pp. 173–211 San Diego, CA: Academic Press [Google Scholar]
- 5.Cooper A., Lalueza-Fox C., Anderson S., Rambaut A., Austin J., Ward R. 2001. Complete mitochondrial genome sequences of two extinct moas clarify ratite evolution. Nature 409, 704–707 10.1038/35055536 (doi:10.1038/35055536) [DOI] [PubMed] [Google Scholar]
- 6.Harshman J., et al. 2008. Phylogenomic evidence for multiple losses of flight in ratite birds. Proc. Natl Acad. Sci. USA 105, 13 462–13 467 10.1073/pnas.0803242105 (doi:10.1073/pnas.0803242105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hackett S. J., et al. 2008. A phylogenomic study of birds reveals their evolutionary history. Science 320, 1763–1768 10.1126/science.1157704 (doi:10.1126/science.1157704) [DOI] [PubMed] [Google Scholar]
- 8.Phillips M. J., Gibb G. C., Crimp E. A., Penny D. 2009. Tinamous and moa flock together: mitochondrial genome sequence analysis reveal independent losses of flight among ratites. Syst. Biol. 59, 1–18 10.1093/sysbio/syp079 (doi:10.1093/sysbio/syp079) [DOI] [PubMed] [Google Scholar]
- 9.Brown J. W., Rest J. S., García-Moreno J., Sorenson M. D., Mindell D. P. 2008. Strong mitochondrial DNA support for a Cretaceous origin of modern avian lineages. BMC Biol. 6, 6–24 10.1186/1741-7007-6-6 (doi:10.1186/1741-7007-6-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pereira S. L., Baker A. J. 2006. A mitogenomics timescale for birds detects variable phylogenetic rates of molecular evolution and refutes the standard molecular clock. Mol. Biol. Evol. 23, 1731–1740 10.1098/rspb.2005.3260 (doi:10.1098/rspb.2005.3260) [DOI] [PubMed] [Google Scholar]
- 11.Paton T., Haddrath O., Baker A. J. 2002. Complete mitochondrial DNA genome sequences show that modern birds are not descended from transitional shorebirds. Proc. R. Soc. Lond. B 269, 839–846 10.1098/rspb.2002.1961 (doi:10.1098/rspb.2002.1961) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ericson P. G. P., et al. 2006. Diversification of neoaves: integration of molecular sequence data and fossils. Biol. Lett. 2, 543–547 10.1098/rsbl.2006.0523 (doi:10.1098/rsbl.2006.0523) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper A., Penny D. 1997. Mass survival of birds across the Cretaceous–Tertiary boundary: molecular evidence. Science 275, 1109–1113 10.1126/science.275.5303.1109 (doi:10.1126/science.275.5303.1109) [DOI] [PubMed] [Google Scholar]
- 14.Fountaine T. M. R., Benton M. J., Dyke G. J., Nudds R. L. 2005. The quality of the fossil record of Mesozoic birds. Proc. R. Soc. B 272, 289–294 10.1098/rspb.2004.2923 (doi:10.1098/rspb.2004.2923) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rokas A., Carroll S. B. 2006. Bushes in the tree of life. PLoS Biol. 4, 1899–1904 10.1371/journal.pbio.0040352 (doi:10.1371/journal.pbio.0040352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards S. V. 2009. Is a new and general theory of molecular systematics emerging? Evolution 63, 1–19 10.1111/j.1558-5646.2008.00549.x (doi:10.1111/j.1558-5646.2008.00549.x) [DOI] [PubMed] [Google Scholar]
- 17.Degnan J. H., Rosenberg N. A. 2006. Discordance of species trees with their most likely gene trees. PLoS Genet. 2, 762–768 10.1371/journal.pgen.0020068 (doi:10.1371/journal.pgen.0020068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards S. V., Liu L., Pearl D. K. 2007. High-resolution species trees without concatenation. Proc. Natl Acad. Sci. USA 104, 5936–5941 10.1073/pnas.0607004104 (doi:10.1073/pnas.0607004104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Treplin S., Tiedemann R. 2007. Specific chicken repeat 1 (CR1) retrotransposon insertion suggests phylogenetic affinity of rockfowls (genus Picathartes) to crows and ravens (Corvidae). Mol. Phylogenet. Evol. 43, 328–337 10.1016/j.ympev.2006.10.020 (doi:10.1016/j.ympev.2006.10.020) [DOI] [PubMed] [Google Scholar]
- 20.Kaiser V., van Tuinen M., Ellegren H. 2007. Insertion events of CR1 retrotransposable elements elucidate the phylogenetic branching order in galliform birds. Mol. Biol. Evol. 24, 338–347 10.1093/molbev/msl164 (doi:10.1093/molbev/msl164) [DOI] [PubMed] [Google Scholar]
- 21.Watanabe M., Nikaido M., Tsuda T. T., Inoko H., Mindell D. P., Murata K., Okada N. 2006. The rise and fall of the CR1 subfamily in the lineage leading to penguins. Gene 365, 57–66 10.1016/j.gene.2005.09.042 (doi:10.1016/j.gene.2005.09.042) [DOI] [PubMed] [Google Scholar]
- 22.Kriegs J. O., Churakov G., Kiefmann M., Jordan U., Brosius J., Schmitz J. 2006. Retroposed elements as archives for the evolutionary history of placental mammals. PLoS Biol. 4, 537–544 10.1371/journal.pbio.0040091 (doi:10.1371/journal.pbio.0040091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishihara H., Maruyama S., Okada N. 2009. Retroposon analysis and recent geological data suggest near-simultaneous divergence of the three superorders of mammals. Proc. Natl Acad. Sci. USA 106, 5235–5240 10.1073/pnas.0809297106 (doi:10.1073/pnas.0809297106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suh A., Paus M., Kiefmann M., Churakjov G., Franke F. A., Brosius J., Kriegs J. O., Schmitz J. 2011. Mesozoic retroposons reveals parrots as the closest living relatives of the passerine birds. Nat. Commun. 2, 443. 10.1038/ncomms1448 (doi:10.1038/ncomms1448) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nylander J. A. 2004. MrModeltest v. 2.0. Program distributed by the author. Uppsala, Sweden: Evolutionary Biology Centre, Uppsala University [Google Scholar]
- 26.Lewis P. O., Holder M. T., Swofford D. L. 2008. Phycas: software for phylogenetic analysis. Storrs, CT: University of Connecticut. See www.phycas.org. [Google Scholar]
- 27.Fan Y., Chen M., Kuo L., Lewis P. O. 2011. Choosing among partition models in Bayesian phylogenetics. Mol. Biol. Evol. 28, 523–532 10.1093/molbev/msq224 (doi:10.1093/molbev/msq224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huelsenbeck J. P., Ronquist F. 2001. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755 10.1093/bioinformatics/17.8.754 (doi:10.1093/bioinformatics/17.8.754) [DOI] [PubMed] [Google Scholar]
- 29.Miller M. A., Pfeiffer W., Schwartz T. 2010. Creating the CIPRES science gateway for inference of large phylogenetic trees. In Proc. Gateway Computing Environments Workshop (GCE), 14 November 2010, New Orleans, LA, pp. 1–8 Piscataway Township, NJ: Institute of Electrical and Electronics Engineers. [Google Scholar]
- 30.Rambaut A., Drummond A. J. 2007. Tracer v. 1.5. See http://beast.bio.ed.ac.uk/Tracer.
- 31.Silvestro D., Michalak I. 2011. RAXMLGUI: a graphical front-end for RAXML. Organ. Divers. Evol. (doi:10.1007/s13127-011-0056-0) [Google Scholar]
- 32.Heled J., Drummond A. 2010. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580 10.1093/molbev/msp274 (doi:10.1093/molbev/msp274) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L., Yu L., Edwards S. V. 2010. A maximum pseudo-likelihood approach for estimating species trees under the coalescent model. BMC Evol. Biol. 10, 302. 10.1186/1471-2148-10-302 (doi:10.1186/1471-2148-10-302) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drummond A. J., Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214. 10.1186/1471-2148-7-214 (doi:10.1186/1471-2148-7-214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z., Rannala B. 2006. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 23, 212–226 10.1093/molbev/msj024 (doi:10.1093/molbev/msj024) [DOI] [PubMed] [Google Scholar]
- 36.Müller J., Reisz R. R. 2005. Four well-constrained calibration points from the vertebrate fossil record for molecular clock estimates. BioEssays 27, 1069–1075 10.1002/bies.20286 (doi:10.1002/bies.20286) [DOI] [PubMed] [Google Scholar]
- 37.Boles W. E. 1992. Revision of Dromaius gidju Patterson and Rich 1987 from Riversleigh, northwestern Queensland, Australia, with a reassessment of its generic position. Nat. Hist. LA County Sci. Ser. 36, 195–208 [Google Scholar]
- 38.Clarke J. A., Tambussi C. P., Noriega J. I., Erickson G. M., Ketcham R. A. 2005. Definitive fossil evidence for the extant avian radiation in the Cretaceous. Nature 433, 305–306 10.1038/nature03150 (doi:10.1038/nature03150) [DOI] [PubMed] [Google Scholar]
- 39.Slack K. E., Jones C. M., Ando T., Harrison G. L., Fordyce R. E., Arnason U., Penny D. 2006. Early penguin fossils, plus mitochondrial genomes, calibrate avian evolution. Mol. Biol. Evol. 23, 1144–1155 10.1093/molbev/msj124 (doi:10.1093/molbev/msj124) [DOI] [PubMed] [Google Scholar]
- 40.Rasmussen D. T., Olson S. L., Simmons E. L. 1987. Fossil birds from the Oligocene Jebel Qatrani Formation, Fayum Province, Egypt. Smithsonian Contrib. Paleobiol. 62, 1–20 10.5479/si.00810266.62.1 (doi:10.5479/si.00810266.62.1) [DOI] [Google Scholar]
- 41.Smit A. F. A., Hubley R., Green P. 1996–2010. RepeatMasker Open-3.0. See http://www.repeatmasker.org. [Google Scholar]
- 42.Felsenstein J. 2005. PHYLIP v. 3.6. Seattle, WA: Department of Genome Science, University of Washington [Google Scholar]
- 43.del Hoyo J., Elliot A., Sargatal J. 1992. Handbook of the birds of the world. Barcelona, Spain: Lynx Edicions [Google Scholar]
- 44.Xia X., Xie Z. 2001. DAMBE: data analysis in molecular biology and evolution. J. Hered. 92, 371–373 10.1093/jhered/92.4.371 (doi:10.1093/jhered/92.4.371) [DOI] [PubMed] [Google Scholar]
- 45.Cracraft J. 2001. Avian evolution, Gondwana biogeography and the Cretaceous–Tertiary mass extinction event. Proc. R. Soc. Lond. B 268, 459–469 10.1098/rspb.2000.1368 (doi:10.1098/rspb.2000.1368) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brocklehurst N., Upchurch P., Mannion P. D., O'Connor J. 2012. The completeness of the fossil record of Mesozoic birds: implications for early avian evolution. PLoS ONE 7, e39056. 10.1371/journal.pone.0039056 (doi:10.1371/journal.pone.0039056) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krause D. W., O'Connor P. M., Rogers K. C., Sampson S. D., Buckley G. A., Rogers R. R. 2006. Late Cretaceous terrestrial vertebrates from Madagascar: implications for Latin American biogeography. Ann. Mon. Bot. Gard. 93, 178–208 10.3417/0026-6493(2006)93[178:LCTVFM]2.0.CO2 (doi:10.3417/0026-6493(2006)93[178:LCTVFM]2.0.CO2) [DOI] [Google Scholar]
- 48.Sereno P. C., Wilson J. A., Conrad J. L. 2004. New dinosaurs link southern landmasses in the Mid-Cretaceous. Proc. R. Soc. Lond. B 271, 1325–1330 10.1098/rspb.2004.2692 (doi:10.1098/rspb.2004.2692) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cooper R. A., Millener P. 1993. The New Zealand biota: historical background and new research. Trends Ecol. Evol. 8, 429–433 10.1016/0169-5347(93)90004-9 (doi:10.1016/0169-5347(93)90004-9) [DOI] [PubMed] [Google Scholar]
- 50.Burbidge M. L., Colbourne R. M., Roberston H. A., Baker A. J. 2003. Molecular and other biological evidence supports the recognitions of at least three species of brown kiwi. Conserv. Genet. 4, 167–177 10.1023/A:1023386506067 (doi:10.1023/A:1023386506067) [DOI] [Google Scholar]
- 51.Baker A. J., Huynen L. J., Haddrath O., Millar C. D., Lambert D. M. 2005. Reconstructing the tempo and mode of evolution in an extinct clade of birds with ancient DNA: the giant moas of New Zealand. Proc. Natl Acad. Sci. USA. 102, 8257–8262 10.1073/pnas.0409435102 (doi:10.1073/pnas.0409435102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Landis C. A., Campbell H. J., Begg J. G., Mildenhall D. C., Paterson A. M., Trewick S. A. 2008. The Waipounamu erosion surface: questioning the antiquity of the New Zealand land surface and terrestrial fauna and flora. Geol. Mag. 145, 173–197 10.1017/S0016756807004268 (doi:10.1017/S0016756807004268) [DOI] [Google Scholar]
- 53.Bunce M., et al. 2009. The evolutionary history of the extinct ratite moa and New Zealand Neogene paleogeography. Proc. Natl Acad. Sci. USA 106, 20 646–20 651 10.1073/pnas.0906660106 (doi:10.1073/pnas.0906660106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barrett P. M., McGowan A. J., Page V. 2009. Dinosaur diversity and the rock record. Proc. R. Soc. B 276, 2667–2674 10.1098/rspb.2009.0352 (doi:10.1098/rspb.2009.0352) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baker A. J., Pereira S. L., Haddrath O., Edge K. A. 2006. Multiple gene evidence for expansion of extant penguins out of Antarctica due to global cooling. Proc. R. Soc. B 273, 11–17 10.1098/rspb.2005.3260 (doi:10.1098/rspb.2005.3260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barker F. K., Cibois A., Schikler P., Feinstein J., Cracraft J. 2004. Phylogeny and diversification of the largest avian radiation. Proc. Natl Acad. Sci. USA 101, 11 040–11 045 10.1073/pnas.0401892101 (doi:10.1073/pnas.0401892101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boles W. E. 1995. The world's oldest songbird. Nature 374, 21–22 10.1038/374021b0 (doi:10.1038/374021b0) [DOI] [Google Scholar]
- 58.McCormack J. E., Heled J., Delaney K. S., Peterson A. T., Knowles L. L. 2011. Calibrating divergence times on species trees versus gene trees: implications for speciation history of Aphelocoma jays. Evolution 65, 184–202 10.1111/j.1558-5646.2010.01097.x (doi:10.1111/j.1558-5646.2010.01097.x) [DOI] [PubMed] [Google Scholar]