

Abstract
Seminal fluid proteins (Sfps) alter female behaviour and physiology and can mediate sexual conflict. In Drosophila melanogaster, a single Sfp, the sex peptide (SP), triggers remarkable post-mating responses in females, including altered fecundity, feeding, immunity and sexual receptivity. These effects can favour the evolutionary interests of males while generating costs in females. We tested the hypothesis that SP is an upstream master-regulator able to induce diverse phenotypes through efficient induction of widespread transcriptional changes in females. We profiled mRNA responses to SP in adult female abdomen (Abd) and head+thorax (HT) tissues using microarrays at 3 and 6 h following mating. SP elicited a rich, subtle signature of temporally and spatially controlled mRNAs. There were significant alterations to genes linked to egg development, early embryogenesis, immunity, nutrient sensing, behaviour and, unexpectedly, phototransduction. There was substantially more variation in the direction of differential expression across time points in the HT versus Abd. The results support the idea that SP is an important regulator of gene expression in females. The expression of many genes in one sex can therefore be under the influence of a regulator expressed in the other. This could influence the extent of sexual conflict both within and between loci.
Keywords: seminal fluid proteins, Acp, transcription, transcriptome, sexual conflict
1. Introduction
The interplay between the sexes over reproduction generates substantial phenotypic variability upon which selection can act [1–3]. To understand the pace, dynamics and trajectory of coevolution arising from this interplay, it is necessary to understand the molecular interactions between males and females [4]. Costs and benefits of reproductive interactions will determine whether coevolution is antagonistic [5–7]. A mechanistic understanding of costs should prove particularly informative in revealing the nature and extent of the potential manipulation of gene expression in one sex by the other. This would allow an assessment of the potential ease with which females could evolve resistance to manipulative adaptations [4] and reveal any constraints arising from the genetic architecture of the pathways involved.
Insights into the molecular interplay between males and females have been gained through economic studies of single genes [2,8,9], studies of fitness effects of interactions between different male and female genotypes [1,10], the characterization of male and female reproductive proteins [3,11] and the measurement of genome-wide responses to mating [12–17]. Of these methods, only genome-wide studies can identify the overall extent to which male and female transcriptomes are altered when the sexes interact, and they offer a valuable opportunity to probe the underlying basis of the interacting pathways involved. Much of the mechanistic work has used the powerful genetic resources of the fruitfly Drosophila melanogaster. Particularly well studied are the functions and fitness effects of seminal fluid proteins (Sfps) in males [2,18], which cause a substantial reprogramming of female behaviour and physiology [2]. The genome-wide profiles of gene expression changes in response to courtship and mating [12,13,16,17] and to the receipt of Sfps [12,15,19] have been investigated. Here, we aimed to capture an in-depth genomic profile of gene expression changes resulting from the transfer of a single Sfp with striking fitness effects [20–26], the sex peptide (SP).
Sex peptide can produce a startling array of female responses, e.g. reduced female receptivity, increased egg laying, feeding and antimicrobial peptide production, and altered sleep patterns and water balance [20–26]. It also adheres to sperm and influences the rate of sperm release from storage [27,28]. The receptor for SP [29] is expressed in the female brain, nervous system and reproductive system, and responses to SP depend upon a circuit of neurons that express the sex determination genes doublesex [30] and fruitless [29]. Insight into the extent of the influence of SP in females is of interest because of the finding that SP can mediate sexual conflict. Females exposed to high levels of SP have reduced fitness [18], the magnitude of which is dependent upon female nutritional status [6]. Males, on the other hand, benefit from SP transfer [31], an effect that is also nutrition dependent [32]. These findings show that costs arising from sexual conflict are context dependent, and highlight the role of nutritional status in determining their magnitude. They are also consistent with the hypothesis that males can manipulate female reproductive processes by altering the sensitivity of nutrient-sensing pathways. Our aim in this study was to reveal the full extent of genomic responses of female D. melanogaster to SP, to measure at the genomic level the extent of male manipulation and to predict potential evolutionary genomic responses in females.
2. Methods
(a). Rationale
To examine differences in mRNA expression in females following SP receipt, we mated wild-type females to males lacking SP (null, SP0) or to control, SP-producing (SP+), males [21]. We assayed mRNAs regulated by SP at 3 and 6 h after mating, to coincide with phenotypic responses observed at 3 h [20,21,23,33–35] and through 6 h and beyond [14,20]. We conducted fourfold independent biological replication. We profiled abdomen (Abd) and head and thorax (HT) tissues separately. This was to minimize the loss of mRNA signal arising from tissue-specific expression [36] and to capture distinct signals arising from the reproductive tissue (Abd) versus central nervous system (HT). Our goal was to identify the full range of biological processes affected by SP receipt. Hence, we opted for a gene category enrichment analysis (Catmap [37]), rather than defining lists of differentially expressed genes with relatively low fold changes based on a cutoff. All expressed genes were ranked based on significance (Bay-t statistic, see below), and this list was then analysed for over-represented functional annotation terms. This Catmap approach integrated information from all expressed genes, giving sensitive detection of, and well-supported biological insight into, process-level changes induced by receipt of SP.
(b). Fly stocks and husbandry
Females were from an outbred, laboratory-adapted Dahomey stock maintained in population cages with overlapping generations. Control and SP null males were derived as in [21]. SP0 bears a non-functional SP gene opposite Δ130, a deletion that covers SP [21]. The SP+ genetically matched control gives wild-type SP expression [21], but contains both the mutant and wild-type SP genes. We backcrossed the deletion and mutant alleles into the Dahomey genetic background (as in [6]). Flies were kept at 25°C in non-humidified rooms on a 12 L : 12 D cycle and supplied throughout with sugar yeast (SY) food (as in [31]) supplemented with excess live yeast granules.
(c). Matings with SP0 and SP+ males
Parents of experimental flies were 4 days old when their eggs were sampled. All flies were grown at a low standard density (18 µl eggs/70 ml food [38]). Virgin Dahomey females were collected on ice within 6 h of eclosion and stored in single sex groups of 10. SP0 and SP+ males were collected within 24 h of eclosion using ice or CO2 anaesthesia, and stored in single sex groups of 10. Mating assays were performed when females were 6 and males were 5–6 days old. The day before matings, two males were aspirated into each mating vial (Nunc cryotube, 4.5 ml with starfoot, containing SY food and a standard-size, live yeast grain). On the day of the assay, a single female was aspirated into each vial (the first female into an SP0 vial, the second into a SP+, the third into an SP0 vial and so on). Females that mated for less than 10 min were discarded, as complete ejaculate transfer was unlikely [39]. Vials containing once-mated females were formed into matched pairs, in which SP0 and SP+ matings began within 5 min of each other. Following matings, males were removed. Profiling time points were 3 h and 6 h after the start of mating (ASM). The first pair of females was allocated to the 3 h profile, the second to the 6 h, the third to the 3 h and so on. At 3 and 6 h ASM, the females were frozen in their cryotubes in liquid N2. After freezing but before RNA extraction, Abd tissues were separated from HT without defrosting. Body parts (HT or Abd) of females within each SP treatment (SP0 or SP+) within each time point were combined into a single microcentrifuge tube (eight tubes in total). The experiment was replicated, using exactly the same procedure, four times. Thus, a total of 32 tubes were produced (i.e. two tissue types (HT, Abd) × 2 treatments (SP+, SP0) × 2 time points (3 h, 6 h) × 4 replicates = 32). The final sample size of body parts within each tube was N = 61–74. To verify the phenotype of backcrossed SP0 and SP+ lines, we performed a bioassay. During replicate 2, we performed 20 additional matings for each treatment. Females were transferred individually to fresh vials after mating and allowed to oviposit for 24 h. They were then introduced to vials containing two wild-type males, and the number of females that remated within 30 min was recorded. As expected, females mated to SP0 males laid significantly fewer eggs than controls (F1,38 = 14.28, p = 0.0006) and were significantly more receptive (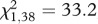 , p < 0.0001, data not shown).
, p < 0.0001, data not shown).
(d). RNA extractions, labelling, hybridizations
Total RNA (TRNA) was isolated using the mirVana kit (Ambion) following the manufacturer's protocol. Quality and quantity of the isolated RNA were verified using spectrophotometry and gel electrophoresis. The TRNA sample was used for mRNA expression analysis and the remainder stored at −80°C for qRT-PCR validation. mRNA array assays were conducted at the Glasgow Polyomics Facility (University of Glasgow). Biotinylated cRNA probes were generated with the one-cycle Genechip target labelling protocol and hybridized to high-density oligonucleotide Affymetrix microarrays (Drosophila Genome 2.0 Array), as described in the Affymetrix GeneChip Expression Analysis Technical Manual (2000).
(e). Gene expression (mRNA) data analyses
Microarray data are lodged in ArrayExpress (www.ebi.ac.uk/arrayexpress; accession nos. E-MEXP-2070 and E-MEXP-2074 for the HT and Abd datasets, respectively). With the exception of Catmap perl program for gene category analysis, all analyses were done using R v. 2.6.2 [40] and BioConductor [41]. Preliminary quality control identified one HT outlier microarray, which was excluded from further analyses. Hence, a total of 16 arrays were analysed for the Abd and 15 for the HT. Hierarchical clustering among samples and ANOVA analysis indicated a batching effect correlated with the ‘biological replicate’ random variable. ANOVA revealed that variance among time-points was heterogeneous, suggesting that time-points should be treated separately.
The numbers of present/expressed genes in the HT and Abd datasets were determined using MAS5 present/absent calls, based on GC-RMA-transformed PM and MM values, as previously implemented in R [42]. A transcript was considered present if the p-value was <0.111 at either time-point/genotype. Normalizations were performed using GC-RMA on all probes [43], followed by Loess [44] on present probe sets. We included the biological replication batching structure in global statistical analysis by conducting paired comparisons. The Cyber-T [45] Bayesian statistic was used to perform paired t-tests between SP+ and SP0 strains for each time-point. False discovery rates were estimated using the Storey method [46–48], as implemented in R (Qvalue package).
To identify significant differential expression of functionally related categories of genes, we used Catmap analysis [37]. This program was populated with functional annotations for the genome of D. melanogaster, using universal vocabularies from GeneOntology (GO) 1.107 [49], Interpro [50] and KEGG v. 44 [51–53], as well as links to PubMed identifiers for published genes retrieved from Flybase [54]. An additional functional category comprised groups of genes with known tissue-specific expression [36]. This allowed us to test for overlap of particular categories of genes with clusters of genes expressed in specific tissues—hence to discover with greater resolution in which tissues the Abd and HT genes were differentially expressed. Raw data (.CEL) files for 15 fly tissues from FlyAtlas [36] (GEO accession no. GSE7763) were used together with an additional dataset (N. Alic, M. E. Giannakou & L. Partridge 2009, unpublished data) corresponding to adult cuticle and associated fat body in white Dahomey S1106/+ flies [55]. The number of present genes in these datasets was determined as described above [42], and normalizations performed on present genes using both GC-RMA [56] with a Loess [44] option, and MAS v. 5.0 [57] with a quantiles [58] option. Soft clustering of these FlyAtlas-expressed genes was achieved using the mfuzz program [59,60] to incorporate the resulting clusters.
For each time-point and body part, Catmap analysis was conducted on two lists of genes ranked based on the Bayes t statistic from Cyber-T. One was based on ranking all expressed genes from the most significantly up- to downregulated, and the second on the reverse. As a category input file, the same list of present genes mapped to functional categories was supplied. As Catmap p-values are positively biased towards categories with many genes under the gene permutation null hypothesis [37], categories comprising less than five genes were discarded. The one-sided Wilcoxon rank sum was used to generate a score based on the sum of the rankings of present genes with a particular functional annotation. The significance of that score (the p-value) was calculated analytically based on a random gene rank distribution. Adjusted p-values (correction for multiple testing) were calculated based on 1000 random permutations of gene rankings. Following identification of enriched functional categories by Catmap, we tested the significance of functional overlaps between tissue-specific FlyAtlas clusters and other functional categories (such as GO, KEGG, etc.) that showed similar regulation profiles, using Fisher exact tests.
(f). Quantitative RT-PCR
To confirm the up- and downregulation of gene categories, individual genes were selected for qRT-PCR. We chose 10 genes from 3 h in the HT, and 10 from 6 h in the Abd. We used the same samples for qRT-PCR validation as for the microarrays. The choice was based on the degree of up- or downregulation (expression fold change) and statistical confidence (ranked p-values from the Bayes t statistic [45] and the q-value after Storey multiple testing correction [47,48]). RNA was quantified using a NanoDrop ND1000 spectrophotometer (LabTech, Ringmer UK). One microgram of TRNA was reverse-transcribed using 200 ng Random Primers and Supercript ll reverse transcriptase (Invitrogen, Paisley, UK), according to the manufacturer's instructions. qRT-PCR reactions were performed using the Taqman ABI 7900, with 12.5 µl of SYBR Green PCR Master Mix (Applied Biosystems, Warrington), 200 nM of each primer and 5 ng of cDNA in 25 µl total volume. Tests with control genes gave extremely low variability, and technical replicates were not used. Dissociation curves were run following each reaction to ensure single product amplification. No contamination in the melting curves was found for any primers used (for sequences, see the electronic supplementary material, table S1). To determine relative RNA levels, standard curve analysis was used. qRT-PCR reactions were prepared by using the cDNA from one sample and making 2-fold serial dilutions covering the range equivalent to 100 ng to 3.125 ng of RNA. αtubulin84β (FBgn0003884) was used as an endogeneous control, as this was more stable than 18S rRNA.
3. Results and discussion
Receipt of SP led to widespread but subtle changes in mRNAs (see figure 1 and the electronic supplementary material, figures S1–S9 and table S2). Throughout, upregulated refers to functional categories (groups) of genes with higher expression following receipt of SP, and downregulated to lowered expression induced by SP, in comparison with SP-lacking controls.
Figure 1.

Summary heat maps of adjusted p-values from Catmap analyses performed on (a) the head and thorax (HT) and (b) on the abdomen (Abd) datasets (full outputs are given in electronic supplementary material, table S2 and figures S1–S6). The colour key gives significance of p-values for significantly up (red) or down (blue) regulated gene categories. p-Values were log10-transformed in a positive and negative scale for up- and downregulated categories, respectively. The plotted categories include annotations for protein domains (six-digit InterPro identifiers), pathways (five-digit Kegg identifiers), Biological Process/BP terms (seven-digit GO identifiers) and results from the intersection with FlyAtlas tissue-specific gene clustering. Only categories with an adjusted p-value < 0.001 in at least one of the two time points (3, 6 h) are shown. Further editing of these categories was carried out in order to omit redundant terms and omit Flyatlas clusters that did not show evidence for specificity (expression peaks identified in more than four tissue types). For the Abd GO BP terms, comprising 218 categories with an adjusted p-value < 0.001, we selected 78 categories with a p-value < 10–14 prior to omission of redundant terms.
(a). mRNA responses: global patterns
Changes in gene expression were globally small in expression ratios (see the electronic supplementary material, figures S8, S9 and table S3), consistent with previous studies [12,13]. We assessed both quantitative (correlation) and qualitative (direction) validation of the microarray data by qRT-PCR. This was consistent overall with the differential expression detected by microarrays (see the electronic supplementary material, figures S8, S9 and table S3). Quantitative validation was provided by the significant positive correlation (r = 0.668, p = 0.002, n = 19) between fold changes from Affymetrix arrays versus qRT-PCR (see the electronic supplementary material, figure S9). Of the 20 genes tested by qRT-PCR, 18 showed directional agreement with the array data (see the electronic supplementary material, table S3 and figure S8; binomial probability p = 0.0004). However, the small fold changes observed render statistical (e.g. t-test) analysis of the qRT-PCR data insensitive (particularly for fold changes less than 1.4 [61]). Consistent with this, few genes were confirmed by t-tests as significantly regulated (see the electronic supplementary material, table S3 and figure S8). Exact regulation patterns for genes subjected to qRT-PCR should therefore be treated with caution. Indeed, this was a motivation for exploring expression patterns through the process-level Catmap analysis, instead of focusing on individual genes drawn from gene lists.
A summary of Catmap analysis of all expressed genes in the HT (8274 genes) and Abd (7626 genes) (figure 1; full results in the electronic supplementary material, table S2) illustrates enriched categories from gene functional annotations [62]. In the Abd, we observed subtle but widespread downregulation of mRNAs at 3 h, a pattern that persisted but was less marked at 6 h. Functional enrichment analysis illustrated that 452 and 199 gene categories were downregulated at 3 and 6 h, respectively (see figure 1 and electronic supplementary material, figures S1–S3). Only a few categories showed significant upregulation, i.e. 68 and 26 categories at 3 and 6 h. In contrast, the HT showed a much more dynamic pattern. At 3 h, 97 categories were up- and 134 downregulated; at 6 h, 73 categories were up- and only 19 downregulated. It was common in the HT profile to observe significant enrichment within a category at only one time point or even opposite profiles between 3 and 6 h (see figure 1 and the electronic supplementary material, figures S4–S6).
mRNA responses to SP in the HT encompassed diverse biological processes, and the direction of differential expression varied markedly within and across timepoints (figure 1a). In contrast, Abd profiles exhibited a more consistent pattern of downregulation, with changes in genes related to a narrower set of potential functions. There were several specific examples of tight regulation across time. For example, differential expression of genes regulated by TOR (target of rapamycin), a nutrient-sensitive regulator of cell growth, proliferation, dietary choices and lifespan [63], was significantly downregulated at 3 h in the HT, but showed an opposite expression profile at 6 h (see the electronic supplementary material, figure S5). This finding suggests precise transcriptional control over short time-scales. There were also examples of apparent systemic coordination of classes of genes across different body parts. For example, Jonah genes and peptidases showed time- and tissue-specific activation and were significantly upregulated at 3 h in the Abd and downregulated in the HT at 6 h (see figure 1 and the electronic supplementary material, figures S2, S5). We hypothesize that these proteins could have a role in digestion, or play a role in defense in the midgut.
(b). mRNA responses: potential functions of differentially expressed mRNAs
In the HT, there was significant differential expression in gene categories related to signalling pathways, behaviour, cell communication and development (figure 1). Novel and unanticipated signatures included the upregulation of genes involved in phototransduction [64], highly time-specific regulation of genes regulated by TOR and downregulation of chorion genes (see the electronic supplementary material, figure S5) [65]. In marked contrast to the HT, Abd profiles showed changes in genes related to fewer potential functions. We describe major mRNA signatures below.
(i). Phototransduction
In the HT profile at 3 h post-mating, there was significant upregulation of processes related to phototransduction (figure 1a). Related components, such as the G-signalling pathway, transmembrane receptor and molecular transducer activities were all differentially expressed, as well as genes involved in cell communication, brain and behaviour, and synaptic transmissions (see figure 1 and the electronic supplementary material, figures S4–S6). Lysosomal tetraspanin, a key protein related to phototransduction, was also significantly upregulated at 3 h (see the electronic supplementary material, figure S5). To determine the specific tissues in which these sets of genes were expressed, we cross-referenced to gene clusters built from the FlyAtlas data [36]. Two clusters upregulated at 3 h (11 and 19, comprising genes expressed in the brain and in the thoracic ganglion, respectively), contained genes with significant functional overlap with genes upregulated in response to the SP at 3 h (see figure 1a and the electronic supplementary material, figure S7a; Fisher test p-value < 10–7). The overlapping functional enrichment was for phototransduction, G-signalling, neuropeptide function and synapse structure (cluster 19), as well as for neurological processes, cell surface and molecular transducers (cluster 11). This result suggests that visual perception is altered following the receipt of SP, perhaps to facilitate the search for suitable oviposition sites.
(ii). TOR pathway
The amino acid sensing TOR pathway was downregulated by SP in the HT at 3 h post mating, along with related categories in energy metabolism and energy biogenesis (see the electronic supplementary material, figure S5). This suggests a link between the receipt of SP and the regulation of nutrient sensing [66,67], which could mediate the known role of SP in feeding and nutrient balancing [24,68]. This is consistent with the finding that the effects of Sfps, and the costs of SP receipt, are significantly altered by dietary manipulations [6,32]. Notably, the TOR pathway showed the opposite profile (upregulation) at 6 h, indicating that SP induces rapid changes in nutrient sensing. Signalling related to increased feeding to support egg production is potentially induced relatively late (from 6 h).
(iii). Ovary and development genes
The majority of categories differentially expressed in the Abd were involved in diverse aspects of egg and early embryo development (see figure 1 and the electronic supplementary material, figure S2). Examples include cell motility and migration, cytoskeleton genes, signal transduction (e.g. JAK/STAT signalling), regulation of transcription and RNA localization, regulation of divisions/cell cycle and cell size and gamete formation (see figure 1 and the electronic supplementary material, figures S1–S3). These signals reflect the role of SP in activating egg development (e.g. cytoskeleton genes) and also the increased rate of production of eggs loaded with translationally inactive maternal mRNAs that direct early embryonic development [69,70]. Early development genes could therefore be differentially expressed in females because of transcription of maternally loaded mRNAs, and/or from early zygotic transcription in retained fertilized eggs within the female reproductive tract. However, the majority of functional categories were significantly down- rather than upregulated in the Abd, in contrast to the expectation given that receipt of SP significantly elevates egg production [20,21]. A plausible explanation is the loss of maternal mRNAs [69,70] in the significantly increased numbers of eggs laid. An additional and unexpected signature was downregulation in the HT of ‘ovary-related’ (e.g. chorion) genes (see figure 1 and the electronic supplementary material, figures S4–S6). These did not show significant overlap with the Abd profiles noted earlier. Significant downregulation was observed for the maternal gene oskar, various cyclins, and genes involved in mRNA translation, splicing and nucleic acid binding via Myc-Max-Mad/Mnt transcription factors (see figure 1 and the electronic supplementary material, figure S5). Such changes related to the control of growth and proliferation are predicted for egg development and maturation [71], but are expected for Abd and not for HT profiles. By cross-referencing to clusters of tissue-specific expression, we found a significant overlap with a set of genes expressed in the cuticle-fat body as well as in the ovaries (cluster 15, figure 1a; electronic supplementary material, figure S7a). The enrichment of these genes in the fat body potentially resolves the paradox of ‘ovary genes’ in the HT (also observed previously by others [19]). Consistent with this, reduced expression of CG1478 (Chorion protein 36 or Cp36) in the HT was supported by the qRT-PCR (see table 1 and the electronic supplementary material, figure S8a).
Table 1.
Known SP phenotypes, their potential genomic signatures detected by the microarrays, and the opportunity for sexual conflict.
| phenotype | possible underlying genomic signatures observed, as detected by microarray (see figure 1 and the electronic supplementary material, figures S1–S6) | opportunity for selection arising from sexual conflict |
|---|---|---|
| increased egg production [20,21] | Cytoskeleton organization+biogenesis; Jak-STAT signalling; cell cycle; chorion genes; germ plasm assembly; TOR pathway; oocyte determination; maternal transcription and egg activation. | Increased fecundity following SP receipt is likely favoured by both sexes up to a point. However, males may favour a higher egg production rate overall, because they gain but do not bear the costs of elevated fecundity. Therefore, via the actions of SP, males may try to increase a female's fecundity investment. |
| increased production of juvenile hormone (JH) [72] | Neuropeptide hormone activity; molecular transducer activity; signal transduction activity. | Following the reasoning above, males should favour high levels of JH production (leading to high levels of fecundity) in females. |
| decreased receptivity [20, 21] | Neurological processes; central nervous system development; G-protein-coupled receptor protein pathway; transmission of nerve impulse; response to external stimulus; synaptic transmission; phototransduction; cell–cell signalling; behaviour. | There is conflict over optimum remating rate (lower for females than for males) arising from intra- and inter-sexual competition. However, as well as being selected to mate often (over-riding low female receptivity), males should favour reductions in female receptivity (to prevent their mates from mating again). Females should gain by effective reduction of receptivity to minimize costly frequent matings. |
| increased production of antimicrobial peptides [23] | Turandot pathway; Jonah genes; immunoglobulin genes; Toll and Imd pathways. | There are potential costs of mounting an immune response for females and as yet undetermined benefits. Mating males may suffer reduced immunity [73]; however, it is not known whether this is related to SP. |
| increased feeding and nutrient balancing [24,66] | TOR pathway; signal transduction activity; neuropeptide hormone activity; molecular transducer activity. | Both sexes may gain by quantitative and qualitative modification by females of nutrient intake to support increased reproduction. However, there are potential conflicts because of longer-term deleterious effects of high nutrient intake/signalling on female survival, costs not incurred by the current mating male. |
| siesta sleep [25] | Neurological processes; central nervous system development; G-protein-coupled receptor protein pathway; transmission of nerve impulse; response to external stimulus; synaptic transmission; phototransduction; cell–cell signalling; behaviour; circadian genes. | Females receiving SP experience reduced levels of siesta sleep, and higher activity levels. Females that do not receive SP have male-like activity levels. Elevated levels of activity may incur costs, or may benefit females through increased oviposition seeking or resistance to mating behaviours. There is potential for conflict over optimum female activity levels. |
| water balance [26] | Cell communication; intracellular cascade; ion transmembrane activity. | Females that do not receive SP have fluid-retention profiles comparable to virgin females. Conflict may occur if female responses to SP to avoid costs result, as a side effect, in suboptimal water homeostasis. |
| adhesion of SP to sperm [27] | Epithelium morphogenesis; cell surface receptor linked signal transduction. | Males gain from the adhesion of SP to sperm through extended SP phenotypes. Conflict is likely over optimum adhesion times depending on the extent to which adhered SP mediates other phenotypes. |
| release of sperm from storage [28] | Cell communication; epithelium morphogenesis; cell surface receptor linked signal transduction. | Depletion of sperm from female storage sites is significantly slower in the absence of SP. Likely to be the subject of strong competition between males, with potential female costs. If fertility is not maintained. |
(iv). Immune genes
As expected [12,13,19], immune genes formed part of SP responses. They were one of the few categories to be significantly upregulated in the Abd. At 3 h, there was evidence for differential expression in peptidases, proteases/endopeptidases, hydrolases, in Toll-lmd-mediated immune responses and defence-related Jonah genes (e.g. electronic supplementary material, figures S2, S5). There was significant overlap with genes specifically expressed in the midgut (cluster 7, electronic supplementary material, figure S7b) and serine-type proteases, immune response and Jonah genes (Fisher test p ≤ 5×10–10). Part of the immune response was also mediated via a cluster of genes upregulated in the fat body (cluster 15, figure 1b; electronic supplementary material, figure S7b), significantly correlated with Toll-lmd pathway, oxidoreductases and serine proteases (p ≤ 7×10–4). A strong association with Toll-lmd pathway, immune response (p ≤ 3×10–21) and endopeptidase genes (p = 8.8×10–10) was also found in differentially expressed spermathecae genes (cluster 10, figure 1b; electronic supplementary material, figure S7b). This cluster displayed a continuous pattern of upregulation at both 3 and 6 h, suggesting a sustained activation of immune responses in mated females. Novel aspects of the immune response therefore became apparent by the cross-referencing to Flyatlas [36]. First, SP induced a defence response in genes expressed in the gut (e.g. Jonah genes). Second, SP triggered a systemic immune response involving Toll-Imd, as noted previously [23]. Third, evidence that immune genes were also expressed in tissues such as the spermathecae suggests that there was also a local immune response [74]. These data therefore reveal a multi-faceted immune response to SP.
(c). Signatures of sexual conflict
The extent of female transcriptional responses suggests that SP is an upstream master regulator of many post-mating processes. This reveals multiple routes by which females might alter their responses to SP. It also suggests that global, evolutionary responses by females to SP are likely to be complex. Such responses could be constrained through pleiotropic effects on essential processes [9]; for example, SP signalling depends upon neural circuits that express sex-determination genes [29]. Hence, SP is plugged into fundamental biological processes of the female, and dampening down global SP responses might not be feasible. Opportunities for antagonistic selection depend upon the magnitude of the difference in the optimum value of reproductive traits across the sexes, and mechanisms available in each sex to enforce their interests [75]. It has been suggested that the evolution of sex-limited expression can release genes from intra-locus sexual conflict by removing the tug of war over optimum expression levels [5]. However, such loci can then quickly/simultaneously become influenced by other loci (e.g. such as SP) and become subject instead to inter-locus conflict. Hence, the evolution of sex limitation per se does not represent evidence of conflict resolution in general (whether within or between loci). SP can also influence the expression of loci in females that are both sex-limited and not sex-limited and could, in principle, simultaneously influence the extent of conflict between loci subject to both intra- and inter-locus sexual conflict. Table 1 summarizes expression signatures from this study consistent with traits affected by SP, and predicts opportunities for antagonistic selection. It is possible that traits over which there is conflict could be typified by contrasting expression profiles between virgins and mated females that do and do not receive SP. The transient expression profiles we observed for TOR signalling along the time course of mating (see the electronic supplementary material, figure S5) could represent such an example, but expression profiles of virgins are needed to confirm this. Many changes in response to SP were in genes expressed in both sexes that have contrasting (often essential) roles at different times (table 1). To further explore how the processes in table 1 might reflect sexual conflict at the transcriptional level, one could compare and manipulate the expression of such genes in both sexes at different life-history stages.
4. Conclusions
Receipt of SP caused significant differential expression in mRNAs. The rich mRNA signatures reflected known and new responses in previously unknown pathways (e.g. phototransduction). Responses in the HT were varied across different gene categories and time. Complexity was evident in several examples of tight temporal and spatial control of mRNA expression. The results show the widespread and complex nature of SP-induced alteration of the female transcriptome. Hence, females that are under selection to alter their responses to the amount and/or potency of SP may have a formidable set of ‘obstacles’ in their way, as would be predicted, given the sexually antagonistic effects of SP [6,18].
Acknowledgements
We thank Andy Barnes, James Boone, Su Davies, Yasmine Driege, Sebastian Grönke and Giovanna Vinti for assisting with the experiments. We thank Daniela Wieser for help with data analysis. Funding was provided by the NERC, BBSRC, The Royal Society, The Lloyd's Tercentenary Foundation and the Wellcome Trust.
References
- 1.Clark A. G., Begun D. J., Prout T. 1999. Female × male interactions in Drosophila sperm competition. Science 283, 217–220 10.1126/science.283.5399.217 (doi:10.1126/science.283.5399.217) [DOI] [PubMed] [Google Scholar]
- 2.Ram K. R., Wolfner M. F. 2007. Seminal influences: Drosophila Acps and the molecular interplay between males and females during reproduction. Int. Comp. Biol. 47, 427–445 10.1093/icb/icm046 (doi:10.1093/icb/icm046) [DOI] [PubMed] [Google Scholar]
- 3.Clark N. L., Gasper J., Sekino M., Springer S. A., Aquadro C. F., Swanson W. J. 2009. Coevolution of interacting fertilization proteins. PLoS Genet. 5, e1000570. 10.1371/journal.pgen.1000570 (doi:10.1371/journal.pgen.1000570) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowe L., Cameron E., Day T. 2005. Escalation, retreat, and female indifference as alternative outcomes of sexually antagonistic coevolution. Am. Nat. 165, S5–S18 10.1086/429395 (doi:10.1086/429395) [DOI] [PubMed] [Google Scholar]
- 5.Arnqvist G., Rowe L. 2005. Sexual conflict (eds Krebs J. R., Clutton-Brock T.). Princeton, NJ: Princeton University Press [Google Scholar]
- 6.Fricke C., Bretman A., Chapman T. 2009. Female nutritional status determines the magnitude and sign of responses to a male ejaculate signal in Drosophila melanogaster. J. Evol. Biol. 23, 157–165 10.1111/j.1420-9101.2009.01882.x (doi:10.1111/j.1420-9101.2009.01882.x) [DOI] [PubMed] [Google Scholar]
- 7.Brommer J. E., Fricke C., Edward D. A., Chapman T. 2011. Interactions between genotype and sexual conflict environment influence transgenerational fitness in Drosophila melanogaster. Evolution 66, 517–531 10.1111/j.1558-5646.2011.01449.x (doi:10.1111/j.1558-5646.2011.01449.x) [DOI] [PubMed] [Google Scholar]
- 8.Chapman T., Davies S. J. 2004. Functions and analysis of the seminal fluid proteins of male Drosophila melanogaster fruit flies. Peptides 25, 1477–1490 10.1016/j.peptides.2003.10.023 (doi:10.1016/j.peptides.2003.10.023) [DOI] [PubMed] [Google Scholar]
- 9.Khila A., Abouheif E., Rowe L. 2012. Function, developmental genetics, and fitness consequences of a sexually antagonistic trait. Science 336, 585–589 10.1126/science.1217258 (doi:10.1126/science.1217258) [DOI] [PubMed] [Google Scholar]
- 10.Chow C. Y., Wolfner M. F., Clark A. G. 2010. The genetic basis for male × female interactions underlying variation in reproductive phenotypes of Drosophila. Genetics 186, 1355–1365 10.1534/genetics.110.123174 (doi:10.1534/genetics.110.123174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swanson W. J., Clark A. G., Waldrip-Dail H. M., Wolfner M. F., Aquadro C. F. 2001. Evolutionary EST analysis identifies rapidly evolving male reproductive proteins in Drosophila. Proc. Natl Acad. Sci. USA 98, 7375–7379 10.1073/pnas.131568198 (doi:10.1073/pnas.131568198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McGraw L. A., Gibson G., Clark A. G., Wolfner M. F. 2004. Genes regulated by mating, sperm, or seminal proteins in mated female Drosophila melanogaster. Curr. Biol. 14, 1509–1514 10.1016/j.cub.2004.08.028 (doi:10.1016/j.cub.2004.08.028) [DOI] [PubMed] [Google Scholar]
- 13.Lawniczak M. K., Begun D. J. 2004. A genome-wide analysis of courting and mating responses in Drosophila melanogaster females. Genome 47, 900–910 10.1139/g04-050 (doi:10.1139/g04-050) [DOI] [PubMed] [Google Scholar]
- 14.Mack P. D., Kapelnikov A., Heifetz Y., Bender M. 2006. Mating-responsive genes in reproductive tissues of female Drosophila melanogaster. Proc. Natl Acad. Sci. USA 103, 10 358–10 363 10.1073/pnas.0604046103 (doi:10.1073/pnas.0604046103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGraw L. A., Clark A. G., Wolfner M. F. 2008. Post-mating gene expression profiles of female Drosophila melanogaster in response to time and to four male accessory gland proteins. Genetics 179, 1395–1408 10.1534/genetics.108.086934 (doi:10.1534/genetics.108.086934) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dalton J. E., Kacheria T. S., Knott S. R. V., Lebo M. S., Nishitani A., Sanders L. E., Sitirling E. J., Winbush A., Arbeitman M. N. 2010. Dynamic, mating-induced gene expression changes in females head and brain tissues of Drosophila melanogaster. BMC Genomics 11, 541. 10.1186/1471-2164-11-541 (doi:10.1186/1471-2164-11-541) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis L. L., Carney G. E. 2010. Mating alters gene expression patterns in Drosophila melanogaster male heads. BMC Genomics 11, 558. 10.1186/1471-2164-11-558 (doi:10.1186/1471-2164-11-558) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wigby S., Chapman T. 2005. Sex peptide causes mating costs in female Drosophila melanogaster. Curr. Biol. 15, 316–321 10.1016/j.cub.2005.01.051 (doi:10.1016/j.cub.2005.01.051) [DOI] [PubMed] [Google Scholar]
- 19.Domanitskaya E. V., Liu H., Chen S., Kubli E. 2007. The hydroxyproline motif of male sex peptide elicits the innate immune response in Drosophila females. FEBS J. 274, 5659–5668 10.1111/j.1742-4658.2007.06088.x (doi:10.1111/j.1742-4658.2007.06088.x) [DOI] [PubMed] [Google Scholar]
- 20.Chapman T., Bangham J., Vinti G., Seifried B., Lung O., Wolfner M. F., Smith H. K., Partridge L. 2003. The sex peptide of Drosophila melanogaster: female post-mating responses analyzed by using RNA interference. Proc. Natl Acad. Sci. USA 100, 9923–9928 10.1073/pnas.1631635100 (doi:10.1073/pnas.1631635100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H., Kubli E. 2003. Sex-peptide is the molecular basis of the sperm effect in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 100, 9929–9933 10.1073/pnas.1631700100 (doi:10.1073/pnas.1631700100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubli E. 2003. Sex peptides: seminal peptides of the Drosophila male. Cell. Mol. Life Sci. 60, 1689–1704 10.1007/s00018-003-3052 (doi:10.1007/s00018-003-3052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng J., Zipperlen P., Kubli E. 2005. Drosophila sex-peptide stimulates female innate immune system after mating via the Toll and Imd pathways. Curr. Biol. 15, 1690–1694 10.1016/j.cub.2005.08.048 (doi:10.1016/j.cub.2005.08.048) [DOI] [PubMed] [Google Scholar]
- 24.Ribeiro C., Dickson B. J. 2010. Sex peptide receptor and neuronal TOR/S6K signaling modulate nutrient balancing in Drosophila. Curr. Biol. 20, 1000–1005 10.1016/j.cub.2010.03.061 (doi:10.1016/j.cub.2010.03.061) [DOI] [PubMed] [Google Scholar]
- 25.Isaac R. E., Li C., Leedale A. E., Shirras A. D. 2010. Drosophila male sex peotide inhibits siesta sleep and promotes locomotor activity in the post-mated female. Proc. R. Soc. B 277, 65–70 10.1098/rspb.2009.1236 (doi:10.1098/rspb.2009.1236) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cognigni P., Bailey A. P., Miguel-Aliaga I. 2011. Enteric neurons and systemic signals couple nutritional and reproductive status with intestinal homeostasis. Cell Metab. 13, 92–104 10.1016/j.cmet.2010.12.010 (doi:10.1016/j.cmet.2010.12.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng J., Chen S., Busser S., Liu H., Honegger T., Kubli E. 2005. Gradual release of sperm bound sex-peptide controls female postmating behavior in Drosophila. Curr. Biol. 15, 207–213 10.1016/j.cub.2005.01.034 (doi:10.1016/j.cub.2005.01.034) [DOI] [PubMed] [Google Scholar]
- 28.Avila F. W., Ram K. R., Qazi M. C. B., Wolfner M. F. 2010. Sex peptide is required for the efficient release of stored sperm in mated Drosophila females. Genetics 186, 595–600 10.1534/genetics.110.119735 (doi:10.1534/genetics.110.119735) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yapici N., Kim Y. J., Ribeiro C., Dickson B. J. 2008. A receptor that mediates the post-mating switch in Drosophila reproductive behaviour. Nature 451, 33–37 10.1038/nature06483 (doi:10.1038/nature06483) [DOI] [PubMed] [Google Scholar]
- 30.Rezaval C., Pavlou H. J., Dornan A. J., Chan Y.-B., Kravitz E. A., Goodwin S. F. 2012. Neural circuitry underlying Drosophila female postmating behavioral responses. Curr. Biol. 22, 1155–1165 10.1016/j.cub.2012.04.062 (doi:10.1016/j.cub.2012.04.062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fricke C., Wigby S., Hobbs R., Chapman T. 2009. The benefits of male ejaculate sex peptide transfer in Drosophila melanogaster. J. Evol. Biol. 22, 275–286 10.1111/j.1420-9101.2008.01638.x (doi:10.1111/j.1420-9101.2008.01638.x) [DOI] [PubMed] [Google Scholar]
- 32.Fricke C., Bretman A., Chapman T. 2008. Adult male nutrition and reproductive success in Drosophila melanogaster. Evolution 62, 3170–3177 10.1111/j.1558-5646.2008.00515.x (doi:10.1111/j.1558-5646.2008.00515.x) [DOI] [PubMed] [Google Scholar]
- 33.Aigaki T., Fleischmann I., Chen P. S., Kubli E. 1991. Ectopic expression of sex peptide alters reproductive behavior of female D. melanogaster. Neuron 7, 557–563 10.1016/0896-6273(91)90368-A (doi:10.1016/0896-6273(91)90368-A) [DOI] [PubMed] [Google Scholar]
- 34.Heifetz Y., Lung O., Frongillo E. A., Jr, Wolfner M. F. 2000. The Drosophila seminal fluid protein Acp26Aa stimulates release of oocytes by the ovary. Curr. Biol. 10, 99–102 10.1016/S0960-9822(00)00288-8 (doi:10.1016/S0960-9822(00)00288-8) [DOI] [PubMed] [Google Scholar]
- 35.Soller M., Bownes M., Kubli E. 1997. Mating and sex peptide stimulate the accumulation of yolk in oocytes of Drosophila melanogaster. Eur. J. Biochem. 243, 732–738 10.1111/j.1432-1033.1997.00732.x (doi:10.1111/j.1432-1033.1997.00732.x) [DOI] [PubMed] [Google Scholar]
- 36.Chintapalli V. R., Wang J., Dow J. A. 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39, 715–20 10.1038/ng2049 (doi:10.1038/ng2049) [DOI] [PubMed] [Google Scholar]
- 37.Breslin T., Eden P., Krogh M. 2004. Comparing functional annotation analyses with Catmap. BMC Bioinform. 5, 193. 10.1186/1471-2105-5-193 (doi:10.1186/1471-2105-5-193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clancy D. J., Kennington W. J. 2001. A simple method to achieve consistent larval density in bottle cultures. Dros. Inf. Serv. 84, 168–169 [Google Scholar]
- 39.Gilchrist A. S., Partridge L. 2000. Why it is difficult to model sperm displacement in Drosophila melanogaster: the relation between sperm transfer and copulation duration. Evolution 54, 534–542 [DOI] [PubMed] [Google Scholar]
- 40.Ihaka R., Gentleman R. 1996. The R project for statistical computing. J. Comp. Graph. Stat. 5, 299–314 10.2307/1390807 (doi:10.2307/1390807) [DOI] [Google Scholar]
- 41.Gentleman R. C., et al. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. 10.1186/gb-2004-5-10-r80 (doi:10.1186/gb-2004-5-10-r80) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuster E. F., Blanc E., Partridge L., Thornton J. M. 2007. Correcting for sequence biases in present/absent calls. Genome Biol. 8, R125. 10.1186/gb-2007-8-6-r125 (doi:10.1186/gb-2007-8-6-r125) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Z., Irizarry R. A., Gentleman R., Martinez Murillo F., Spencer F. 2003. A model based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 99, 907–917 [Google Scholar]
- 44.Yang Y. H., Dudoit S., Luu P., Lin D. M., Peng V., Ngai J., Speed T. P. 2002. Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucl. Acids Res. 30, e15. 10.1093/nar/30.4.e15 (doi:10.1093/nar/30.4.e15) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldi P., Long A. D. 2001. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17, 509–519 10.1093/bioinformatics/17.6.509 (doi:10.1093/bioinformatics/17.6.509) [DOI] [PubMed] [Google Scholar]
- 46.Storey J. D. 2002. A direct approach to false discovery rates. J. R. Stat. Soc. 64, 479–498 10.1111/1467-9868.00346 (doi:10.1111/1467-9868.00346) [DOI] [Google Scholar]
- 47.Storey J. D. 2003. The positive false discovery rate: a Bayesian interpretation and the q-value. Ann. Stat. 31, 2013–2035 10.1214/aos/1074290335 (doi:10.1214/aos/1074290335) [DOI] [Google Scholar]
- 48.Storey J. D., Tibshirani R. 2003. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA 100, 9440–9445 10.1073/pnas.1530509100 (doi:10.1073/pnas.1530509100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashburner M., et al. 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 10.1038/75556 (doi:10.1038/75556) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulder N. J., et al. 2003. The InterPro Database, 2003 brings increased coverage and new features. Nucl. Acids Res. 31, 315–318 10.1093/nar/gkg046 (doi:10.1093/nar/gkg046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanehisa M., et al. 2008. KEGG for linking genomes to life and the environment. Nucl. Acids Res. 36, D480–D484 10.1093/nar/gkm882 (doi:10.1093/nar/gkm882) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanehisa M., Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28, 27–30 10.1093/nar/28.1.27 (doi:10.1093/nar/28.1.27) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanehisa M., Goto S., Hattori M., Aoki-Kinoshita K. F., Itoh M., Kawashima S., Katayama T., Araki M., Hirakawa M. 2006. From genomics to chemical genomics: new developments in KEGG. Nucl. Acids Res. 34, D354–D357 10.1093/nar/gkj102 (doi:10.1093/nar/gkj102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tweedie S., et al. 2009. FlyBase: enhancing Drosophila gene ontology annotations. Nucl. Acids Res. 37, D555–D559 10.1093/nar/gkn788 (doi:10.1093/nar/gkn788) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giannakou M. E., Goss M., Junger M. A., Hafen E., Leevers S. J., Partridge L. 2004. Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science 305, 361. 10.1126/science.1098219 (doi:10.1126/science.1098219) [DOI] [PubMed] [Google Scholar]
- 56.Wu Z., Irizarry R. A. 2005. Stochastic models inspired by hybridization theory for short oligonucleotide arrays. J. Comput. Biol. 12, 882–893 10.1089/cmb.2005.12.882 (doi:10.1089/cmb.2005.12.882) [DOI] [PubMed] [Google Scholar]
- 57.Affymetrix 2002. Statistical Algorithms Description Document. http://www.affymetrix.com/support/technical/whitepapers/sadd_whitepaper.pdf [Google Scholar]
- 58.Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., Speed T. P. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 10.1093/biostatistics/4.2.249 (doi:10.1093/biostatistics/4.2.249) [DOI] [PubMed] [Google Scholar]
- 59.Futschik M. E., Carlisle B. 2005. Noise-robust soft clustering of gene expression time-course data. J. Bioinform. Comput. Biol. 3, 965–988 10.1142/S0219720005001375 (doi:10.1142/S0219720005001375) [DOI] [PubMed] [Google Scholar]
- 60.Kumar L., Futschik M. E. 2007. Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 10.6026/97320630002005 (doi:10.6026/97320630002005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morey J. S., Ryan J. C., Van Dolah F. M. 2006. Microarray validation: factors influencing correlation between oligonuceotide microarrays and real-time PCR. Biol. Proc. Online 8, 175–193 10.1251/bpo126 (doi:10.1251/bpo126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hunter S., et al. 2009. InterPro: the integrative protein signature database. Nucl. Acids Res. 37, D224–D228 10.1093/nar/gkn833 (doi:10.1093/nar/gkn833) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oldham S., Hafen E. 2003. Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol. 13, 79–85 10.1016/S0962-8924(02)00042-9 (doi:10.1016/S0962-8924(02)00042-9) [DOI] [PubMed] [Google Scholar]
- 64.Katz B., Minke B. 2009. Drosophila photoreceptors and signaling mechanisms. Front. Cell. Neurosci. 3, 2. 10.3389/neuro.03.002.2009 (doi:10.3389/neuro.03.002.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cavaliere V., Bernardi F., Romani P., Duchi S., Gargiulo G. 2008. Building up the Drosophila eggshell: first of all the eggshell genes must be transcribed. Dev. Dyn. 237, 2061–2072 10.1002/dvdy.21625 (doi:10.1002/dvdy.21625) [DOI] [PubMed] [Google Scholar]
- 66.Carvalho G. B., Kapahi P., Anderson D. J., Benzer S. 2006. Allocrine modulation of feeding behavior by the sex peptide of Drosophila. Curr. Biol. 16, 692–696 10.1016/j.cub.2006.02.064 (doi:10.1016/j.cub.2006.02.064) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barnes A. I., Boone J. M., Partridge L., Chapman T. 2007. A functioning ovary is not required for sex peptide to reduce receptivity to mating in D. melanogaster. J. Insect Physiol. 53, 343–348 10.1016/j.jinsphys.2006.12.008 (doi:10.1016/j.jinsphys.2006.12.008) [DOI] [PubMed] [Google Scholar]
- 68.Vargas M. A., Luo N. G., Yamaguchi A., Kapahi P. 2010. A Role for S6 Kinase and serotonin in postmating dietary switch and balance of nutrients in D. melanogaster. Curr. Biol. 20, 1006–1011 10.1016/j.cub.2010.04.009 (doi:10.1016/j.cub.2010.04.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hooper S. D., Boué S., Krause R., Jensen L. J., Mason C. E., Ghanim M., White K. P., Furlong E. M., Bork P. 2007. Identification of tightly regulated groups of genes during Drosophila melanogaster embryogenesis. Mol. Syst. Biol. 3, 72. 10.1038/msb4100112 (doi:10.1038/msb4100112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Baker D. A., Russell S. 2009. Gene expression during Drosophila melanogaster egg development before and after reproductive diapause. BMC Genomics 10, 242. 10.1186/1471-2164-10-242 (doi:10.1186/1471-2164-10-242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gallant P. 2006. Myc/Max/Mad in invertebrates: the evolution of the max network. Myc/max/mad Transcript. Fact. Network 302, 235–253 10.1007/3-540-32952-8_9 (doi:10.1007/3-540-32952-8_9) [DOI] [PubMed] [Google Scholar]
- 72.Moshitzky P., Fleischmann I., Chaimov N., Saudan P., Klauser S., Kubli E., Applebaum S. W. 1996. Sex-peptide activates juvenile hormone biosynthesis in the Drosophila melanogaster corpus allatum. Arch. Insect. Biochem. Physiol. 32, 363–74 (doi:10.1002/(SICI)1520-6327(1996)32:3/4<363::AID-ARCH9>3.0.CO;2-T) [DOI] [PubMed] [Google Scholar]
- 73.McKean K. A., Nunney L. 2001. Increased sexual activity reduces male immune function in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 98, 7904–7909 10.1073/pnas.131216398 (doi:10.1073/pnas.131216398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kapelnikov A., Zelinger E., Gottlieb Y., Rhrissorrakrai K., Gunsalus K. C., Heifetz Y. 2008. Mating induces an immune response and developmental switch in the Drosophila oviduct. Proc. Natl Acad. Sci. USA 105, 13 912–13 917 10.1073/pnas.0710997105 (doi:10.1073/pnas.0710997105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chapman T. 2006. Evolutionary conflicts of interest between males and females. Curr. Biol. 16, 744–754 10.1016/j.cub.2006.08.020 (doi:10.1016/j.cub.2006.08.020) [DOI] [PubMed] [Google Scholar]