

Abstract
After feeding microbes with a defined 13C substrate, unique isotopic patterns (isotopic fingerprints) can be formed in their metabolic products. Such labelling information not only can provide novel insights into functional pathways but also can determine absolute carbon fluxes through the metabolic network via metabolic modelling approaches. This technique has been used for finding pathways that may have been mis-annotated in the past, elucidating new enzyme functions, and investigating cell metabolisms in microbial communities. In this review paper, we summarize the applications of 13C approaches to analyse novel cell metabolisms for the past 3 years. The isotopic fingerprints (defined as unique isotopomers useful for pathway identifications) have revealed the operations of the Entner–Doudoroff pathway, the reverse tricarboxylic acid cycle, new enzymes for biosynthesis of central metabolites, diverse respiration routes in phototrophic metabolism, co-metabolism of carbon nutrients and novel CO2 fixation pathways. This review also discusses new isotopic methods to map carbon fluxes in global metabolisms, as well as potential factors influencing the metabolic flux quantification (e.g. metabolite channelling, the isotopic purity of 13C substrates and the isotopic effect). Although 13C labelling is not applicable to all biological systems (e.g. microbial communities), recent studies have shown that this method has a significant value in functional characterization of poorly understood micro-organisms, including species relevant for biotechnology and human health.
Keywords: carbon fluxes, isotopic effect, isotopomers, channelling, novel enzymes
1. Introduction
13C labelling is a powerful tool for elucidating in vivo enzyme activities in micro-organisms. After a particular 13C-labelled substrate is fed to microbes, the label is transferred to the downstream metabolites through metabolic pathways. As a consequence, some metabolic products (i.e. the analysed compound) comprise mixtures of 13C isotopomers with specific labelling patterns, which are termed isotopic fingerprints. Via unique isotopologue profiles, researchers can trace the paths of reactants to products in the metabolic network and thus reveal functional enzymes and metabolic pathways. 13C labelling can not only accurately define the structure of a complete pathway but also quantify global enzymatic reactions (i.e. carbon flux) in the metabolic network by incorporating 13C fingerprints into metabolic modelling (13C metabolic flux analysis (13C-MFA)) [1–3]. Owing to post-transcriptional/post-translational regulation mechanisms, genetic analysis sometimes cannot reflect the final metabolic outputs; however, the metabolic fluxes can provide experimental validation of the results obtained from genomics and transcriptomics studies. The 13C technique is widely applied in poorly understood microbial species and has filled in gaps between genome sequencing and functional characterization. Meanwhile, the 13C technique is being improved to speed up screening and exploitation of native metabolic features of non-model microbes for biotechnology applications. Figure 1 shows a protocol describing the 13C technique for probing microbial metabolism, which includes two key procedures: measuring isotopic labelling patterns in metabolites and examining metabolic pathways [1,4,5].
Figure 1.

Procedures for metabolism analysis through GC-MS-based isotopic labelling.
In the past 3 years, numerous papers have reported novel metabolic insights from 13C fingerprints. Other papers have described different new methods to obtain metabolic labelling fingerprints and quantify the fluxome in the cell metabolism, such as the extraction of targeted metabolites in microbial communities, new isotopomer analysis approaches (gas chromatography–combustion–isotope ratio mass spectrometry (GC-C-IRMS) and liquid chromatography–mass spectrometry (LC-MS)) and high-performance fluxomics algorithms/software. Because 13C isotopic labelling is extensively used in cell metabolism analyses, it is difficult to provide a complete and balanced view in this short review paper. Therefore, we highlight only a small portion of recent studies (after 2009) on environmental micro-organisms.
2. Pathway elucidation via 13C fingerprints
For many micro-organisms, genetic analysis tools (such as genome annotation and microarrays) cannot precisely reveal the topology of the functional metabolic network. 14C labelling has historically played a key role in the discovery of functional metabolic pathways. Although 14C methods are sensitive owing to the radioactivity, stable 13C is instrumentally safer and easier for researchers to use high-performance nuclear magnetic resonance (NMR) or mass spectrometry to detect metabolic pathways [6]. 13C-labelled metabolites (isotopic fingerprints) can be used for investigating carbon metabolism. For example, by examining whether the 13C fingerprints in key metabolites are consistent with the predicted labelling patterns from the annotated pathway, we can verify annotated enzymatic reactions. In parallel to the 13C technique, we can elucidate the functional pathway by conducting a BLAST search for potential genes that have been reported in other micro-organisms, perform transcription analysis of gene candidates, detect intermediate metabolites, and overexpress or knock out the target genes. Table 1 highlights several pathways that have been examined via 13C fingerprints in diverse micro-organisms since 2009.
Table 1.
Examples of recent pathway discoveries via 13C tracing.
| pathway identification | microbes | labelled substrate | fingerprint metabolite | 13C fingerprint description | reference |
|---|---|---|---|---|---|
| ED pathway | Roseobacter denitrificans | [1-13C]glucose | serine and alanine | serine labelling is significantly lower than alanine labelling | [7] |
| non-phosphorylative ED pathway for C5 sugar metabolism | Sulfolobus solfataricus and acidocaldarius | [1-13C]xylose | serine, Asp and alanine | less than 4% serine and Asp are labelled, while (approx. 50%) alanine is labelled on its first position | [8] |
| incomplete TCA cycle | Heliobacterium modesticaldum | [1-13C]PYR | Asp and Glu | Asp is labelled with two carbons; Glu is labelled with one carbon | [9] |
| reductive branch of TCA pathway, OAA → SUC | Mycobacterium tuberculosis | [1,4-13C]Asp or [1,4-13C] OAA | SUC | SUC retains both 13C carbons (doubly labelled) | [10] |
| oxidative branch of TCA pathway, OAA → CIT → SUC | Mycobacterium tuberculosis | [1,4-13C]Asp or [1,4-13C]OAA | SUC | SUC is not labelled | [10] |
| PEPCK activity, OAA → PEP + CO2 | Mycobacterium tuberculosis | [U-13C]acetate | Asp, MAL, PYR, serine, PEP, G3P | 13C from OAA (reflected by Asp) is moved to glycolytic derived intermediates (PEP and serine) | [11] |
| co-catabolism of multiple carbon sources | Mycobacterium tuberculosis | only one of the substrates is labelled | central metabolites (e.g. PYR, SUC) | if the 13C substrate is co-used, the 13C can enter central metabolites | [12] |
| complete reverse TCA cycle | Chlorobaculum tepidum | [3-13C]PYR and 12CO2 | Asp, Glu and histidine | 13C in Asp and Glu (derived from TCA cycle) is diluted by 12CO2, while 13C in histidine (derived from R5P) is apparently higher than 13C in Asp and Glu | [13] |
| citramalate pathway | Chlorobaculum tepidum | [2-13C]PYR or [1-13C] acetate | leucine and isoleucine | leucine and isoleucine labelling are identical owing to same precursors | [13] |
| (Re)-CIT synthase (anaerobic bacteria) | Dehalococcoides ethenogenes | [1-13C]acetate | Glu | glutamate is labelled in α-carboxyl group | [14] |
| Wood–Ljungdahl pathway (mixotrophic) | Dehalococcoides ethenogenes | [1-13C]acetate and 12CO2 | alanine and leucine | alanine and leucine labelling would be diluted by 12C | [14] |
| utilization of exogenous amino acids | Dehalococcoides ethenogenes | 13C substrates with 12C amino acids | proteinogenic amino acids | if the cells cannot de novo synthesize amino acids, but use supplemented 12C amino acids, significant 12C amino acids are present in proteinogenic amino acids | [15] |
| oxidative PP pathway | Thermoanaerobacter | [1-13C]glucose | alanine | the 13C is lost as CO2 in this pathway, and thus non-labelled alanine will be more than 50% | [16] |
| photorespiratory: RuBP → G3P + phosphoglycolate | Synechocystis 6803 | 13CO2 (dynamic labelling approach) | glycerate, G3P and PEP | 13C INST-MFA reveals the photorespiratory flux | [17] |
| EMCP | Methylobacterium extorquens | [1-13C]acetate (via dynamic labelling) | CoA intermediate | 13C is subsequently found in acetyl-CoA, 3-hydroxybutyryl-CoA, ethylmalonyl-CoA, methylsuccinyl-CoA, mesaconyl-CoA, β-mesaconyl-CoA (C5-CoA) and propionyl-CoA (13C % in the C5-CoA is similar) | [18] |
| serine cycle associated with EMCP | Methylobacterium extorquens | [13C]methanol and 12CO2 | glycine and serine | 3rd carbon of serine is labelled, 2nd carbon of glycine is highly labelled | [18] |
| glyoxylate shunt | Methylobacterium extorquens | [1,2-13C]glyoxylate and 12C ethylamine | MAL | glyoxylate shunt through the malyl-CoA route produces doubly labelled MAL, if acetyl CoA is not labelled | [19] |
| alternative glycine and serine synthesis glyoxylate → glycine glycine + C1 → serine | Methylobacterium extorquens | [1,2-13C]glyoxylate and 12C ethylamine | glycine and serine | glycine is doubly labelled, serine can be doubly labelled (with 12C1) or triply labelled (with 13C1) | [19] |
| quantify mixotrophic CO2 fixation (Calvin–Benson cycle) | Cyanothece 51142 | 12CO2 with labelled carbon substrates | histidine (precursor R5P) and serine (precursor G3P) | serine and histidine labelling are significantly diluted owing to 12CO2 fixation | [20] |
| anaplerotic pathway: PEP + CO2 → OAA | Cyanothece 51142 | 13CO2 and 12C glycerol | Asp | Asp labelling will be enriched in its fourth position | [20] |
| 3-hydroxypropionate bi-cycle for glyoxylate assimilation | Chloroflexus aurantiacus | [1,2,3-13C]propionyl-CoA (from labelled propionate) | PYR and C5 CoA- intermediate (e.g. mesaconyl-CoA and citramalyl-CoA) | PYR will be fully labelled, while C5 CoA-intermediates are labelled with three carbons | [21] |
2.1. Central metabolic pathways for carbon substrate catabolism
13C fingerprints have provided new insights into the central pathways in poorly characterized micro-organisms (figure 2). First, the Entner–Doudoroff pathway (ED pathway) is an alternative pathway to glycolysis that catabolizes glucose to pyruvate (PYR) via two key enzymes—6-phosphogluconate (6PG) dehydratase and 2-keto-3-deoxyphosphogluconate aldolase. This pathway has been reported in several micro-organisms, including Pseudomonas, Agrobacterium, Rhodobacter, Zymomonas, Azotobacter and Sinorhizobium meliloti [22–24]. A recent report shows that Roseobacter denitrificans (an aerobic marine proteobacterium) exclusively uses the ED pathway for glucose photoheterotrophic metabolism, because the Embden–Meyerhof–Parnas pathway (glycolysis) is not active owing to the lack of the enzyme 6-phosphofructokinase [7]. On the other hand, in some hyperthermophilic archaeal species (such as Sulfolobus solfataricus), a non-phosphorylative ED pathway metabolizes glucose to PYR and glyceraldehyde. This non-phosphorylative ED pathway is also responsible for metabolizing C5 sugars (d-xylose and l-arabinose) to PYR and glycolaldehyde (the latter can be subsequently converted to glycolate and glyoxylate) [8].
Figure 2.

Metabolic pathways revealed by 13C fingerprints. DHAP, dihydroxyacetone phosphate; E4P, erythrose-4-phosphate; F6P, fructose-6-phosphate; S7P, sedoheptulose-7-phosphate; ICT, isocitrate.
Second, the oxidative pentose phosphate pathway (PP pathway) (glucose-6-phosphate (G6P) → ribose-5-phosphate, ribulose-5-phosphate, ribulose-1,5-bisphosphate, xylulose-5-phosphate (X5P) + CO2 + 2NADPH) is an important resource for NADPH synthesis. Thermoanaerobacter sp. strain X514 (a thermophilic ethanol producer) has been studied via 13C experiments with [1-13C]glucose. On the basis of an isotopic analysis of PYR (reflected by alanine) labelling, the oxidative PP pathway for glucose metabolism shows low activity in strain X514. This result indicates that X514 may contain an alternative route for NADPH production [16].
Third, phosphoenolpyruvate carboxykinase (PEPCK) redirects the carbon flux from the tricarboxylic acid (TCA) cycle metabolites to gluconeogenesis (oxaloacetate (OAA) + GTP (or ATP) → phosphoenolpyruvate (PEP) + CO2 + GDP (or ADP)). This reaction offers microbes an alternative route (PEP–glyoxylate cycle: OAA → PEP → acetyl-coenzyme A (AcCoA) → citrate (CIT) → glyoxylate → OAA) to oxidize intermediates in the TCA cycle to CO2 [25]. This enzyme is also essential for Mycobacterium tuberculosis growth on fatty acids [11].
Fourth, the TCA cycle in most microbes is often branched under oxygen-limited conditions. In M. tuberculosis, Watanabe et al. [10] have proved that the branched TCA cycle is active for succinate (SUC) accumulation under hypoxia. Their finding is based on the observation that SUC has a labelling pattern similar to that of aspartate (Asp) after feeding the cells with [1, 4-13C]Asp. As another example, a study of Plasmodium falciparum [26] determined the major carbon source contributing to TCA intermediates by culturing parasite-infected red blood cells in a 13C- and 15N-labelled medium ([U-13C]glucose, [U-13C-15N]Asp or [U-13C-15N]glutamine). After separation of P. falciparum from red blood cells, an LC-MS analysis of its central metabolites showed that this parasite relies on glucose fermentation for energy, but its branched TCA cycle is disconnected from glycolysis (i.e. its TCA cycle metabolites are synthesized from glutamate/glutamine). Furthermore, heliobacteria (a phylum of anaerobic anoxygenic phototrophic bacteria) were considered to have an incomplete reverse TCA cycle owing to their lack of a conventional CIT synthase. Recently, Tang et al. [9] reported that the oxidative TCA route (OAA → CIT → α-ketoglutarate) is operative via a novel (Re)-CIT synthase in contrast to the conventional (Si)-CIT synthase. The stereo-specific enzyme reaction of (Re)-CIT synthase has been revealed by tracing 13C transitions from OAA (reflected by Asp) to α-ketoglutarate (reflected by glutamate). The in vivo activity of (Re)-CIT synthase has also been found in other anaerobic bacteria via 13C tracing along with the heterologous gene expression method. The (Re)-CIT synthases from Clostridium and Desulfovibrio are phylogenetically related to isopropylmalate synthase [27–29]. Meanwhile, a new (Re)-CIT synthase (previously annotated as homoCIT synthase) is reported to be responsible for the TCA pathway function in Dehalococcoides [30].
2.2. CO2 fixation pathways
The Calvin–Benson cycle is the common autotrophic CO2 assimilation pathway in microalgae. Using labelled carbon substrates and unlabelled CO2, Feng et al. [20] have studied the mixotrophic metabolism in a hydrogen-producing cyanobacterium Cyanothece 51142. They found that the CO2 fixation rate is dependent on both the nitrogen source and the carbon substrate. Under favourable conditions, with glycerol and nitrate, Cyanothece 51142 can switch its mixotrophic metabolism to photoheterotrophic metabolism to promote biomass growth. Under such conditions, Cyanothece still uses the non-autotrophic CO2 fixation pathway via PEP carboxylase.
Furthermore, AcCoA-related carbon fixation pathways have been recently reported in different micro-organisms. These pathways include the reverse TCA cycle, the Wood–Ljungdahl pathway, dicarboxylate/4-hydroxybutyrate cycle, 3-hydroxypropionate/4-hydroxybutyrate cycle, the 3-hydroxypropionate bi-cycle and the ethylmalonyl-CoA pathway (EMCP). These alternative autotrophic pathways may lead to new insights into the global carbon cycle, the early evolution of metabolism and interpretations of geological records [31]. Feng et al. [13] have studied a green sulphur bacterium, Chlorobaculum tepidum, for acetate and PYR metabolism, in which they reported three routes for CO2 fixation: the reverse TCA cycle, AcCoA + CO2 → PYR and PEP + CO2 → OAA. Moreover, Dehalococcoides ethenogenes 195 has been speculated to use the Wood–Ljungdahl pathway because its genome contains several key enzymes in this pathway. However, the isotopomer data disproved the complete function of this pathway when strain 195 was grown with H2, trichloroethylene (TCE) and acetate [14]. The EMCP has been proposed in the photosynthetic proteobacterium Rhodobacter sphaeroides [32]. This pathway starts from AcCoA, and forms intermediate metabolites, including ethylmalonyl-CoA, methylsuccinyl-CoA, methylmalyl-CoA and propionyl-CoA. The propionyl-CoA can then be converted to malate (MAL) in the TCA cycle. The net reaction of EMCP is: AcCoA + 2CO2 → 2 glyoxylate + CoA. Peyraud et al. [18] applied [1-13C]acetate and 13C metabolomics to determine the order of biochemical reactions starting from AcCoA during growth of Methylobacterium extorquens AM1 on methanol. During short-term 13C-labelling experiments, an LC-MS analysis of CoA-based intermediate metabolites tracked 13C percolating through cell metabolism. Meanwhile, they used 13C-labelled methanol in combination with NMR so as to provide final demonstration of the methanol utilization pathway by measurement of proteinogenic glycine labelling. This study not only discovered the operation of the EMCP, but also identified a serine cycle to assimilate methanol and convert the EMCP product glyoxylate to central metabolites. In the serine cycle, glyoxylate produces glycine, and glycine assimilates methanol through a 5,10-methylene-THF (C1) unit (N5, N10-methylene-tetrahydrofolate) to form serine. Serine can be converted to 2-phosphoglycerate that enters glycolysis and the TCA cycle. The complete serine cycle fixes both CO2 and methanol (or methane). Furthermore, Okubo et al. [19] reported that M. extorquens AM1 is able to condense glyoxylate and AcCoA to MAL (the glyoxylate shunt) via a novel malyl-CoA/β-methylmalyl-CoA lyase. This finding indicates an alternative route responsible for glyoxylate consumption, besides the serine cycle. Later on, Schneider et al. [33] used LC-MS-based 13C-MFA to profile the carbon fluxes through three metabolic cycles for acetate metabolism in M. extorquens (metabolic activities: the TCA cycle > the EMCP > the serine cycle).
The filamentous anoxygenic phototrophic bacterium Chloroflexus aurantiacus uses the 3-hydroxypropionate bi-cycle for CO2 fixation [21]. Similar to the EMCP, the first cycle starts from AcCoA and forms malonyl-CoA by fixing one CO2. The malonyl-CoA is the precursor of 3-hydroxypropionate. Via multiple enzymatic steps, 3-hydroxypropionate is converted to propionyl-CoA, and subsequently to malyl-CoA by fixing another CO2. The malyl-CoA splits to glyoxylate and AcCoA as the end products from the first cycle of the 3-hydroxypropionate bi-cycle. The second cycle consumes the glyoxylate from the first cycle of the 3-hydroxypropionate bi-cycle. Glyoxylate and propionyl-CoA form β-methylmalyl-CoA, followed by three reactions to generate citramalyl-CoA; the citramalyl-CoA is then cleaved into AcCoA and PYR to form the second cycle.
Finally, two additional CO2 fixation cycles (dicarboxylate/4-hydroxybutyrate cycle and 3-hydroxypropionate/4-hydroxybutyrate cycle) have been discovered via 13C and 14C isotopic labelling. Both cycles contain a key metabolic route (i.e. 4-hydroxybutyrate pathway: succinyl-CoA (SucCoA) → 4-hydroxybutyrate → acetoacetyl-CoA → AcCoA). A recent review has a thorough discussion of the two CO2 fixation cycles in various environmental micro-organisms, including enzyme steps, energy (cofactor) demand and thermodynamic equilibrium of each reaction [31].
2.3. Unconventional biosynthesis pathways
13C experiments are useful tools to check annotated biosynthesis pathways. A recent report confirms diverse amino acid synthesis routes in the pathogen Streptococcus pneumoniae, using labelled glucose and glycine [34]. It is the first 13C study of this pathogen that revealed multiple carbon usage (i.e. identifying, that some amino acids were unlabelled and taken from the medium, whereas others were made de novo). In some non-model micro-organisms, 13C experiments may find unconventional biosynthesis pathways. The citramalate pathway is an alternate pathway for isoleucine synthesis in some anaerobic bacteria (such as Geobacter species and Thermoanaerobacter species) and methanogenic Archaea [16,35–37]. Recently, this threonine-independent pathway (using PYR and AcCoA as the precursors) has also been discovered in photosynthetic bacteria, including Rsb. denitrificans [7], Hbt. modesticaldum [9], Cba. tepidum [13] and Cyanothece 51142 [38]. The citramalate synthase can be used to engineer the carbon flux to 2-ketobutyrate (a precursor of butanol), and thus promote alcohol biosynthesis [39]. Moreover, glyoxylate can be an alternative precursor instead of glycerate 3-phosphate (G3P) for synthesis of glycine and serine. For example, Mycobacterium smegmatis may employ glycine dehydrogenase to convert glyoxylate to glycine and consume excess NADH under oxygen-limited growth conditions [40]. This amino acid synthesis route forms an initial step to remove glyoxylate from the EMCP in M. extorquens, generating serine and subsequently metabolites for glycolysis [18].
The 13C technique has been applied to investigate the biosynthesis of the building blocks of products. Tao et al. [41] applied 13C-NMR to study Aspergillus glaucus (a filamentous fungus) for the biosynthesis of aspergiolide A (a novel anti-tumour compound). Through tracer experiments using singly or fully labelled 13C-labelled sodium acetate in a nutrient-rich medium followed by 13C-NMR spectroscopic investigation, they found that acetate is the only carbon source for the polyketide pathway for aspergiolide A synthesis, and 12 carbon atoms of the final product are derived from the carboxylic group of acetate. Another example is the investigation of the carbon sources for ergosterol biosynthesis in Trypanosoma brucei (a parasite that causes African trypanosomiasis) [42]. Via 13C-labelled lanosterol, acetate, leucine and glucose labelling, this study analysed the incorporation of 13C into sterols and investigated the operation of the sterol metabolic network in the parasite. The results demonstrate sets of coordinated pathways to yield distinct sterol profiles, including glucose metabolism in the glycosome, AcCoA synthesis and leucine catabolism in the mitochondria, the acetate mevalonate pathway and the isopentenyl pyrophosphate pathway (regulated in the cytosol), and ergosterol synthesis reactions.
2.4. Carbon nutrient utilization
13C-labelled experiments are used extensively to study the co-utilization of carbon nutrients by micro-organisms. In such experiments, one of the carbon substrates is 13C labelled, while other substrates are naturally labelled. By measuring the labelling enrichment in central metabolites, we can quantify the co-catabolism of carbon nutrients. For example, Feng et al. [20] have examined the uptake of 13C-labelled glucose by Cyanothece 51142 under CO2 photoautotrophic growth conditions. The results showed that Cyanothece 51142 cannot use glucose efficiently owing to the lack of a specific glucose transporter. Furthermore, by tracing 13C enrichment of the TCA cycle metabolites using a bis(trimethylsilyl)trifluoroacetamide-based GC-MS method, Tang et al. [40] have reported that M. smegmatis upregulates Tween 80 utilization once it enters hypoxia. Meanwhile, 13C-based metabolomics [12] revealed that M. tuberculosis catabolizes multiple carbon sources (glucose, acetate and glycerol) simultaneously to achieve optimal biomass growth. Finally, a 13C experiment can optimize nutritional conditions for slow-growing micro-organisms. To differentiate selective utilization of exogenous amino acids by D. ethenogenes 195, 13C-labelled acetate and non-labelled amino acids have been used in the culture medium [15]. After five subcultures in the rich medium with labelled acetate, strain 195 showed significant improvement of biomass growth and TCE degradation. The analysis of 13C enrichment in proteinogenic amino acids from those subcultures implied that phenylalanine, isoleucine, leucine and methionine are most highly imported from the culture medium, while strain 195 cannot efficiently uptake other amino acids to promote biomass synthesis. The cultivation experiments showed that the four highly used amino acids can enhance biomass growth and dechlorination activities to levels similar to those observed in a rich medium.
3. Metabolic flux profiling in non-model micro-organisms via 13C fingerprints
13C-MFA uses 13C fingerprints to calculate intracellular metabolic fluxes. 13C-MFA has been extensively applied to study model micro-organisms and their mutants, such as Escherichia coli, Bacillus subtilis and Saccharomyces cerevisiae. Here, 13C-MFA has also been shown to be a powerful tool to answer unresolved metabolic questions in non-model microbial species, including cofactor generation/consumption, regulation of pathways under different growth conditions, and operation of complicated metabolic cycles.
The Calvin–Benson cycle is considered to be indispensable for photoheterotrophic metabolisms. McKinlay et al. [43] studied a photoheterotrophic bacterium (Rhodopseudomonas palustris). They found that cells oxidize 13C-labelled acetate to CO2 via the glyoxylate shunt and the partially functioning TCA cycle, and then fix 68 per cent of this CO2 into biomass via the Calvin–Benson cycle. The Calvin–Benson cycle in R. palustris actually plays a major role in cofactor recycling, which can reoxidize nearly half of the reduced cofactors (NADH) generated during conversion of acetate to biomass. Meanwhile, in a nitrogenase active mutant, CO2 fixation by the Calvin–Benson cycle is downregulated, because R. palustris channels NADH to produce H2. By further analysing R. palustris growth with different 13C substrates (fumarate, SUC, acetate or butyrate), the same authors [44] determined the factors that influence the H2 yield of R. palustris. Their 13C-MFA results indicate that the Calvin–Benson cycle and H2 production are two key pathways to compete for the reducing equivalents generated during the oxidation of organic carbon sources. Thus, inhibition of the Calvin–Benson cycle will promote H2 production.
13C-MFA can be used to examine the mixotrophic metabolisms. For example, it is well known that the green sulphur bacteria use the reverse TCA cycle for CO2 fixation, but not much was known about its electron-balancing function [45]. 13C-MFA has been used in quantitative analysis of reducing equivalents in Cba. tepidum under mixotrophic growth conditions [13]. The fluxomics not only estimated the absolute CO2 fixation by the reverse TCA cycle during assimilation of acetate or PYR, but also revealed the reducing equivalents harvested from phototrophic reactions.
Young et al. [17] applied dynamic isotopic labelling analysis to decipher the autotrophic metabolism of Synechocystis sp. PCC6803. They built an isotopically non-stationary MFA (INST-MFA) to quantify the carbon flux from CO2 to the overall central metabolism. Their results indicated that a significant amount of the fixed carbon is lost via the oxidative PP pathway, which had been previously considered to be minimal in phototrophic metabolisms. Their INST-MFA also indicated that the glyoxylate shunt is absent, the measurable glyoxylate flux is redirected to the photorespiratory pathway and the malic enzyme flux (MAL → PYR) is the rate-limiting step of biomass synthesis. A recent paper [46] reported two new enzymes that perform the function of the missing alpha-ketoglutarate dehydrogenase in cyanobacteria to close the TCA cycle. However, INST-MFA indicated no measurable flux from α-ketoglutarate to SUC.
13C-MFA has wide applications in microbes important for bioremediation, disease control and bioenergy. Chavarría et al. [47] used 13C-MFA to analyse the phosphoenolpyruvate phosphotransferase system (PTS) in Pseudomonas putida, a soil bacterium capable of degrading polycyclic aromatic hydrocarbons. By comparing glucose and fructose metabolisms in both wild-type and PTS mutant strains, they showed that a PTS mutant strain lacking PTSNtr (N-related PTS that controls cellular functions unrelated to the transport of carbohydrates) displays significantly higher fluxes in the PYR shunt (MAL → PYR), which indicates that PTSNtr regulates P. putida sugar metabolism by mediating its TCA cycle. 13C-MFA was also used to probe the metabolism of the pathogen Mycobacterium bovis. The results showed that carbon fluxes through a complete TCA cycle are minimal, while the PYR is metabolized via the glyoxylate shunt coupled with the anaplerotic reactions [48]. In the bioenergy field, 13C-MFA has recently been applied to study oleaginous Chlorella protothecoides for oil production [49]. Under heterotrophic conditions, the isotopic fingerprints in proteinogenic amino acids were used to study the fluxes through glycolysis, the PP pathway and the TCA cycle. The results indicated that the PP pathway is upregulated, while global flux distribution remains relatively stable after the culture was switched from a nitrogen-sufficient condition to a nitrogen-limited condition.
4. Recent advances in 13C techniques
Paralleling the application of 13C techniques to identify functional pathways and quantify metabolic fluxes, good progress has been recently made in improving 13C techniques.
4.1. Dynamic flux analysis
Traditionally, 13C-MFA is inferred from steady-state distributions of carbon–carbon bonds of metabolites from different metabolic pathways. However, INST-MFA can speed up the experimentation for industrially relevant processes (e.g. non-growing cells or fed batch cultures) with moderately changing process conditions [50]. Such non-stationary 13C-MFA depends on dynamic carbon-labelling experiments for metabolic steady-state cultures (after medium replacement with 13C substrate, measuring multiple labelling samples during the isotopically transient time course) [51]. Proof-of-concept studies have developed algorithms to decipher fluxes based on measured metabolite pool sizes and isotopomer concentrations [51–53], and to speed up INST-MFA calculation [54]. A recent milestone INST-MFA paper on environmental microbes resolved photoautotrophic metabolism in cyanobacteria [17]. A steady-state culture using single carbon CO2 as the sole carbon source cannot provide any constraints for solving the flux distribution [55]. To overcome this problem, this pioneering study investigated the photoautotrophic metabolism by examining the change of isotopomer distributions in fast-turnover metabolites immediately after step-switching the carbon source from 12CO2 to 13CO2 [17]. Such INST-MFA estimates both intracellular metabolic fluxes and metabolite pool sizes, on the basis of the measured isotope labelling dynamics of 16 metabolites. The tracking of free metabolites uses both LC-MS and GC-MS. As expected, INST-MFA requires solving nonlinear differential equations to track the labelling of metabolites as a function of time. To reduce the computational complexity of INST-MFA, the model employs an elementary metabolite unit (EMU) approach to simplify simulation of isotopomer distributions throughout the metabolic network [56].
Exploratory studies have also been performed to analyse metabolic unsteady-state bioprocesses to obtain global enzyme kinetic parameters [57]. The Sauer group reported the use of dynamic amino acid labelling to obtain time-dependent fluxomics results from increasing glucose limitation in a B. subtilis culture overproducing riboflavin [58]. Because metabolic transition in the culture is slow, dynamic 13C-MFA decomposes the culture process into numerous quasi-steady states (time interval = 30 ∼ 60 min). Then steady-state 13C-MFA can be used to resolve the fluxomics and estimate the cofactor balance at different quasi-steady time intervals (mini-13C-MFA). Through snapshots of the metabolism at various time points, the dynamic metabolism of B. subtilis was revealed. Another milestone dynamic MFA paper reported a new approach to profile E. coli metabolism in a fed-batch process [59]. Via a rigorous statistical analysis, inflection time points can be determined so that the MFA model divides the time domain of the entire fed batch culture into several intervals where the fluxes have linear change in each interval. Then the algebraic solutions to time-dependent metabolite mass balance equations can be obtained to facilitate the calculation of dynamic fluxes in each time interval.
4.2. Microbial community studies
In microbial communities, species evolve cross-feeding interactions (one species obtains nutrients from another). Microbial communities play important roles in ecology, bioremediation, biofuel production and biomedical research [60]. However, these communities present great modelling challenges because flux models for communities have to simulate both the metabolism in individual species and the exchange of metabolites among species. In the past, an isotope-based microbial community study usually treated the community as a single metaorganism without considering the metabolic interactions between individual subspecies. For example, 13C-MFA was applied for gut microflora to review the overall nutrient metabolism in intestinal bacteria [61]. By linking MFA with the other systems-biology technologies, researchers can have insights into the metabolic regulations involved in microbe–host mutualism and their relevance for health. Besides, communities are also characterized in terms of a subcompartment-based flux balance analysis [60,62], where the intracellular flux in subspecies and the metabolite interactions between species are predicted using objective functions (e.g. optimal biomass growth). Such an analysis (without using isotopic labelling) is not precise because true metabolisms in the communities are often suboptimal. Therefore, it is beneficial to employ novel 13C-MFA to facilitate understanding the links between the physiology of individual species and their community characteristics. However, traditional 13C-MFA has not been readily applicable to mixed cultures because it is difficult to obtain 13C-labelling patterns of metabolites from a given population within a mixed culture. The targeted species have to be separated using centrifugation or fluorescence-assisted cell sorting. Unfortunately, centrifugation separates only species with sufficient density differences, while fluorescence-assisted sorting takes too long to obtain enough cells for a reliable 13C-labelling pattern analysis. To overcome this problem, the Sauer group [63] invented a proof-of-concept 13C-MFA of a microbial community. In their approach, a reporter protein is used to assist the subpopulation-specific MFA of E. coli mutants in a mixed culture of E. coli strains. Specifically, a reporter protein that has been synthesized in only one species of the consortium can be separated via protein purification, and thus 13C patterns of proteinogenic amino acids from the reporter protein can be used to resolve the fluxes in this single species. This method (i.e. labelling patterns of amino acids from purified proteins can be used to infer metabolic fluxes of targeted organisms in a mixed culture) may eventually extend 13C-MFA to study more complex community systems [64].
4.3. Investigation on host–pathogen systems
13C-labelling approaches (such as MS-based amino acid analysis) have been used to examine the metabolic interactions between a microbial pathogen and its host [65]. The investigation of host–pathogen systems not only provides understanding of the key central pathways for survival of pathogens (as described in §2.1, [26]), but also reveals host responses to pathogen infections. Combined with other experimental techniques, 13C isotopic labelling gives important medical insights into the treatment of pathogens. For example, Listeria monocytogenes is a food-borne pathogen that can cause invasive infection in susceptible humans. For proliferation within hosts, this facultative intracellular pathogen uses specific metabolic pathways (e.g. an incomplete TCA cycle owing to a lack of 2-ketoglutarate dehydrogenase) for nutrient utilization during infection [66]. A 13C/14C-labelling study on extracellular L. monocytogenes cultures demonstrated that the ATP-dependent PYR carboxylase (pycA) plays an important role in its central carbon metabolism. Later on, the virulence of the pycA defective mutant was shown to be highly attenuated in mouse infection experiments [67]. To decipher the metabolism of pathogens inside host cells during the infection phase, Götz et al. [68] supplied [U-13C]glucose to the enteroinvasive E. coli and Salmonella enterica replicating in epithelial colorectal adenocarcinoma cells. To separate the pathogens and infected host cells after 13C-labelling cultivation, they lysed host cells to release bacteria first; then removed the host cell debris from bacteria by repeated centrifugations. Such a separation protocol minimized the contamination of the host cell protein fraction with bacterial proteins (less than 10%), and thus the proteinogenic amino acids from host cells or bacteria could be isolated for GC-MS measurements. By comparing the labelling patterns in bacterial amino acids from wild-type and mutant strains (e.g. devoid of sugar uptake systems), they revealed the carbon sources (e.g. glucose, glucose phosphate, glycerol, lactate, PYR, amino acids) used by pathogens as well as bacterial metabolic pathways (e.g. anaplerotic reactions and the ED pathway) during the infections. However, the pathogen infections seem not to affect the carbon metabolism of the used Caco-2 host cells, as judged by 13C incorporation into host cell amino acids.
4.4. Metabolism profiling using integrated approaches
The 13C technique is often applied with other approaches, including genome-scale reconstruction, enzyme assay, metabolite profiling, genetic manipulation (i.e. knockout of key enzymes) and transcription expression measurements. First, mis-annotations can be easily generated from genome sequences and propagated through databases. 13C metabolism analysis is useful for the development and validation of new methods for accurate gene predictions. For example, 13C-labelled leucine experiments have identified the leucine degradation pathway in B. subtilis, and facilitated the development of a new computational algorithm (an automatic policing method) to detect biochemical mis-annotations [69]. Second, unknown genes may be revealed by integrating the 13C technique with genomic analyses. For example, Veit et al. [70] analysed co-metabolism of acetate and glucose in Corynebacterium glutamicum. Via 13C labelling, genetic mutation, enzyme and microarray analysis, they not only profiled the fluxomics during co-utilization of acetate and glucose, but also discovered a redundant gene encoding CoA transferase that can activate acetate and propionate in the presence of glucose. Third, for genome-scale metabolic reconstruction, integration of metabolic models with 13C-MFA can unravel the topology of cell metabolism. For example, recent work combined in silico metabolic network reconstruction with 13C fluxomics and found that methanol assimilation pathways in M. extorquens operate as a unique process, which is tightly connected by several major metabolic cycles (serine cycle, EMCP, TCA cycle and anaplerotic processes) [71]. Moreover, 13C-MFA can measure gene regulation by correlating fluxomics (functional output) with multiple-level genomics data (such as transcription and protein profiles). In a global investigation of B. subtilis, it has determined the influence of the ferric uptake regulator (Fur) on the TCA cycle fluxes [72]. The Sauer group has also examined metabolic adaptation across multiple levels of regulation in B. subtilis, including transcriptomics, proteomics, promoter activities, metabolomics, fluxomics and biomass growth. Such analyses reveal controlling genes responsible for metabolic adaptation and post-transcriptional regulatory mechanisms [73].
4.5. New analytical and computational tools for 13C-metabolic flux analysis
The scope and the resolution of 13C-MFA can be improved by metabolomics tools (GC-MS and LC-MS) to obtain extensive 13C fingerprints. Rühl et al. [74] applied LC-MS/MS to measure both intact and fragmented central metabolites, obtaining complete 13C fingerprints in each metabolite. Because collisional fragmentation of metabolites may partially give 13C positional information for certain isotopomers, LC-MS/MS-based 13C-MFA improved estimations of fluxes in the PP pathway, glycolysis and the TCA cycle. Moreover, GC-C-IRMS is used for analysis of isotopomers when 13C enrichment in the metabolites is low (well below 10%). The GC-C-IRMS isotopomer data allow 13C-MFA to provide good flux resolution even when the tracer experiments are performed using very low 13C concentration substrates in order to reduce the cost of 13C substrates [75].
NMR is a powerful tool to probe 13C in organic chemicals [76]. NMR has been extensively used for collecting isotopic data and has provided valuable metabolic insights into a broad range of biological systems [23]. Although recent studies on environmental microbes (after 2009) have relied more on MS techniques because of low cost/high sensitivity of metabolite measurement, NMR still plays an important role in metabolism studies. For example, by measuring time courses for 13C enrichment in metabolites using NMR, a kinetic model quantified the fluxes through human gut bacterial metabolism [61]. In general, NMR identifies the positional labelling information in metabolites, while MS measures the total labelling enrichment in the metabolites. So combining NMR with MS techniques can provide complementary isotopic labelling information to reveal novel metabolic insights [18,77].
Computational tools for high-performance 13C-MFA have been continuously published in the past 3 years. User-friendly software (such as 13C-FLUX [78]) allows scientists with little programming knowledge to perform 13C fluxomics studies. A 13C-MFA toolbox, OpenFLUX, is a computationally efficient software package [79]. The software incorporates a high-performance EMU framework (a decomposition method identifying the minimum amount of information needed to simulate isotopic labelling) for calculation of isotopomer balances within a reaction network [56]. For a more detailed introduction to computational tools, a recent review paper summarized major computational models and software [80], covering areas of both metabolic reconstruction tools and 13C flux analysis platforms. Finally, new mathematical algorithms are proposed to reformulate 13C-MFA problems and simplify the flux calculation. Recently, a new set of variables (called ‘fluxomers’) has been invented by combining both fluxes and isotopomer abundances [81]. The fluxomers represent the rate of a metabolic reaction transferring labelling from substrate isotopomers to product isotopomers. Such variables provide efficient computation of metabolic fluxes and show robustness to measurement noise and initial conditions.
5. Influential factors in 13C metabolism analysis
Several factors that may affect 13C metabolism analysis have been reported recently. First, metabolite channelling resulting from the formation of multi-enzyme complexes is common in some eukaryotes [82]. In this process, metabolites in metabolic pathways are transferred from enzyme to enzyme without being released into the bulk phase of the cell. The channelling can change isotopomer fingerprints because it may prevent the randomization of orientation for symmetric metabolites [83]. For example, the orientations of the TCA cycle metabolites (SUC and MAL) can be conserved in S. cerevisiae in the pathway from [4-13C]α-ketoglutarate to OAA [84]. Because of the channelling of SUC/fumarate formation enzymes (SucCoA synthetase, SUC dehydrogenase and fumarase), the 13C fractions in the second and third carbon in OAA resulting from [4-13C]α-ketoglutarate are not completely randomized (i.e. 13C2/13C3 ≠ 1). Because 13C-MFA assumes a homogeneous condition for all intracellular metabolites, channelling may affect 13C-MFA-based flux results. In mammalian cells, isotopic experiments (3H and 13C) have been used to investigate metabolite channelling and compartmentation in cytosolic and mitochondrial metabolite pools [82,85]. In plant cells, channelled fluxes can be detected in glycolysis and the TCA cycle. Therefore, it is suggested that 13C-MFA of eukaryotes may require correction coefficients to account for the impact of channelling on the redistribution of 13C in key metabolites [83].
Moreover, metabolic channelling has also been discovered in bacterial species. For example, the channelling phenomenon in E. coli has been investigated by feeding cells with 14C-labelled glucose and 14C-labelled glycolytic intermediates [86]. By measuring the evolved 14CO2 production, the authors concluded that fructose 1,6-bisphosphate (FBP) was strongly channelled all the way to CO2, whereas fructose-6-phosphate (F6P) was not. Metabolic channelling may provide an alternative route for the labelling of pathway intermediates, leading to a change in the isotopic fingerprints (figure 3). Similarly, a 13C INST-MFA of autotrophic metabolism finds metabolic channelling in Synechocystis 6803 [17]. Specifically, PEP showed higher 13C enrichments than other metabolites right after the cells were fed with 13CO2, while the Calvin–Benson cycle intermediates (e.g. F6P, glyceraldehyde-3-phosphate (GAP) and ribose-5-phosphate (R5P)) were significantly less labelled than their downstream products, such as ribulose-5-phosphate (Ru5P) and ribulose-1,5-bisphosphate (RuBP). To correct for this effect, the flux model has to include ‘dilution parameters’ for these Calvin–Benson cycle intermediates. In other words, 13C INST-MFA becomes a new tool for investigating the occurrence and extent of enzymatic channelling in microbial species.
Figure 3.
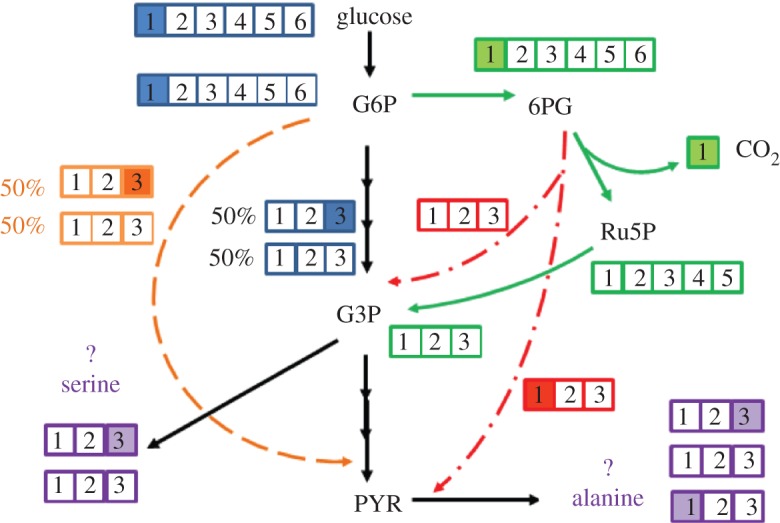
Example of glycolysis channelling. It provides an alternative route for the labelling of PYR. The filled boxes represent labelled carbon. When the first position glucose is used as the carbon source, the oxidative PP pathway (green line) generates only non-labelled G3P and PYR. Both the ED pathway (red line) and glycolysis channelling (orange line) increase the labelled PYR fractions (measured from alanine), but reduce labelled G3P fractions (measured from serine).
Isotopic fractionation is present if 13C and 12C substrates are both in the medium. Enzymes show subtle selectivity in the uptake of their 12C and 13C substrates. It has been well established that, by using the CO2-fixing enzyme RuBisCO, photosynthetic micro-organisms favour 12CO2 over 13CO2. The reported isotopic fractionation, δ13C (a measure of the relative ratio of 13C : 12C, reported in parts per thousand), of biological processes (Cyanobacteria or Calvin–Benson cycle plants) can be over −20 [87]. Feng and Tang [88] examined the effect of isotopic fractionation on flux analysis using 20 per cent fully labelled glucose and 80 per cent naturally labelled glucose. The measured δ13C (based on several key amino acids) is around −40, which is just above the GC-MS measurement errors (instrumental bias may also contribute to δ13C ≈ −20 or higher). The study also pointed out that commercial fully labelled glucose from the market often contains 3–4% of five-carbon labelled glucose, and such isotopic impurity in 13C substrates causes a much greater impact on flux calculation than does isotopic fractionation. Therefore, isotopic fractionation, though evident, has a smaller effect on conventional 13C-MFA than other experimental noise sources. If tracer experiments employ a very low labelling percentage of 13C substrates (well below 10%) to save experimental cost, the isotope effect on 13C-MFA is reported to be higher [75]. In such tracer experiments, 13C enrichment data for metabolites (measured by sensitive GC-C-IRMS) can be corrected using a mathematical approach before using the isotopomer data for flux calculations.
6. Conclusions
Metabolism analysis through 13C isotopic fingerprints is a powerful tool to map microbial ultimate phenotypic outcomes, both qualitatively and quantitatively. New methods of 13C metabolism analysis are being developed and greatly facilitate the analytic resolution and scope, and provide complementary knowledge to results obtained from other ‘omics’ approaches. 13C-based metabolism analyses have often been used to profile industrial microbial metabolisms in order to improve their product yields, but few successful cases for significant improvements of product yields through 13C-MFA have been demonstrated so far. On the other hand, recent studies show the significant value of 13C metabolism analyses of poorly characterized environmental micro-organisms as well as human pathogens. Many non-model microbes play important roles in the processes involved in ecology, disease, biofuels and bioremediation, and therefore the 13C technique can be widely applied to speed up their screening and provide us with novel metabolic insights and benefits there from.
Acknowledgements
This study was supported in part by an NSF career grant (MCB0954016) to Y.J.T. and a NASA exobiology grant (NNX12AD85G) to R.E.B. J.K.H.T. was supported by start-up funds from Clark University. We are thankful for Dr Danny Kohl's useful discussion on channelling, and Dr Xueyang Feng's useful comments on 13C-MFA.
References
- 1.Zamboni N., Fendt S. M., Rühl M., Sauer U. 2009. 13C-based metabolic flux analysis. Nat. Protoc. 4, 878–892 10.1038/nprot.2009.58 (doi:10.1038/nprot.2009.58) [DOI] [PubMed] [Google Scholar]
- 2.Zamboni N., Sauer U. 2009. Novel biological insights through metabolomics and 13C-flux analysis. Curr. Opin. Microbiol. 12, 553–558 10.1016/j.mib.2009.08.003 (doi:10.1016/j.mib.2009.08.003) [DOI] [PubMed] [Google Scholar]
- 3.Wiechert W. 2001. 13C metabolic flux analysis. Metab. Eng. 3, 195–206 10.1006/mben.2001.0187 (doi:10.1006/mben.2001.0187) [DOI] [PubMed] [Google Scholar]
- 4.Wittmann C. 2007. Fluxome analysis using GC-MS. Microb. Cell Factories 6, 6. 10.1186/1475-2859-6-6 (doi:10.1186/1475-2859-6-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wittmann C. 2002. Metabolic flux analysis using mass spectrometry. Adv. Biochem. Eng./Biotechnol. 74, 39–64 10.1007/3-540-45736-4_3 (doi:10.1007/3-540-45736-4_3) [DOI] [PubMed] [Google Scholar]
- 6.Tang Y. J., Martin H. G., Myers S., Rodriguez S., Baidoo E. K., Keasling J. D. 2009. Advances in analysis of microbial metabolic fluxes via 13C isotopic labeling. Mass Spectrom. Rev. 28, 362–375 10.1002/mas.20191 (doi:10.1002/mas.20191) [DOI] [PubMed] [Google Scholar]
- 7.Tang K.-H., Feng X., Tang Y. J., Blankenship R. E. 2009. Carbohydrate metabolism and carbon fixation in Roseobacter denitrificans OCh114. PLoS ONE 4, e7233. 10.1371/journal.pone.0007233 (doi:10.1371/journal.pone.0007233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nunn C. E., Johnsen U., Schonheit P., Fuhrer T., Sauer U., Hough D. W., Danson M. J. 2010. Metabolism of pentose sugars in the hyperthermophilic archaea Sulfolobus solfataricus and Sulfolobus acidocaldarius. J. Biol. Chem. 285, 33 701–33 709 10.1074/jbc.M110.146332 (doi:10.1074/jbc.M110.146332) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang K.-H., Feng X., Zhuang W.-Q., Alvarez-Cohen L., Blankenship R. E., Tang Y. J. 2010. Carbon flow of Heliobacterium modesticaldum is more related to Clostridia than to the green sulfur bacteria. J. Biol. Chem. 285, 35 104–35 112 10.1074/jbc.M110.163303 (doi:10.1074/jbc.M110.163303) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe S., Zimmermann M., Goodwin M. B., Sauer U., Barry C. E., III, Boshoff H. I. 2011. Fumarate reductase activity maintains an energized membrane in anaerobic mycobacterium tuberculosis. PLoS Pathog. 7, e1002287. 10.1371/journal.ppat.1002287 (doi:10.1371/journal.ppat.1002287) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marrero J., Rhee K. Y., Schnappinger D., Pethe K., Ehrt S. 2010. Gluconeogenic carbon flow of tricarboxylic acid cycle intermediates is critical for Mycobacterium tuberculosis to establish and maintain infection. Proc. Natl Acad. Sci. USA 107, 9819–9824 10.1073/pnas.1000715107 (doi:10.1073/pnas.1000715107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Carvalho L. P., Fischer S. M., Marrero J., Nathan C., Ehrt S., Rhee K. Y. 2010. Metabolomics of Mycobacterium tuberculosis reveals compartmentalized co-catabolism of carbon substrates. Chem. Biol. 17, 1122–1131 10.1016/j.chembiol.2010.08.009 (doi:10.1016/j.chembiol.2010.08.009) [DOI] [PubMed] [Google Scholar]
- 13.Feng X., Tang K.-H., Blankenship R. E., Tang Y. J. 2010. Metabolic flux analysis of the mixotrophic metabolisms in the green sulfur bacterium Chlorobaculum tepidum. J. Biol. Chem. 285, 39 544–39 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Y. J., Yi S., Zhuang W., Zinder S. H., Keasling J. D., Alvarez-Cohen L. 2009. Investigation of carbon metabolism in ‘Dehalococcoides ethenogenes’ strain 195 via isotopic and transcriptomic analysis. J. Bacteriol. 191, 5224–5231 10.1128/JB.00085-09 (doi:10.1128/JB.00085-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuang W. Q., Yi S., Feng X., Zinder S. H., Tang Y. J., Alvarez-Cohen L. 2011. Selective utilization of exogenous amino acids by Dehalococcoides ethenogenes strain 195 and its effects on growth and dechloronation activity. Appl. Environ. Microbiol. 77, 7797–7803 10.1128/AEM.05676-11 (doi:10.1128/AEM.05676-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng X., et al. 2009. Characterization of the central metabolic pathways in Thermoanaerobacter sp. x514 via isotopomer-assisted metabolite analysis. Appl. Environ. Microbiol. 75, 5001–5008 10.1128/AEM.00715-09 (doi:10.1128/AEM.00715-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Young J. D., Shastri A. A., Stephanopoulos G., Morgan J. A. 2011. Mapping photoautotrophic metabolism with isotopically nonstationary 13C flux analysis. Metab. Eng. 13, 656–665 10.1016/j.ymben.2011.08.002 (doi:10.1016/j.ymben.2011.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peyraud R., Kiefer P., Christen P., Massou S., Portais J.-C., Vorholt J. A. 2009. Demonstration of the ethylmalonyl-CoA pathway by using 13C metabolomics. Proc. Natl Acad. Sci. USA 106, 4846–4851 10.1073/pnas.0810932106 (doi:10.1073/pnas.0810932106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okubo Y., Yang S., Chistoserdova L., Lidstrom M. E. 2010. Alternative route for glyoxylate consumption during growth on two-carbon compounds by Methylobacterium extorquens AM1. J. Bacteriol. 192, 1813–1823 10.1128/JB.01166-09 (doi:10.1128/JB.01166-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng X., Banerjee A., Berla B., Page L., Wu B., Pakrasi H. B., Tang Y. J. 2010. Mixotrophic and photoheterotrophic metabolisms in Cyanothece sp. ATCC 51142 under continuous light. Microbiology 156, 2566–2574 10.1099/mic.0.038232-0 (doi:10.1099/mic.0.038232-0) [DOI] [PubMed] [Google Scholar]
- 21.Zarzycki J., Brecht V., Müller M., Fuchs G. 2009. Identifying the missing steps of the autotrophic 3-hydroxypropionate CO2 fixation cycle in Chloroflexus aurantiacus. Proc. Natl Acad. Sci. USA 106, 21 317–21 322 10.1073/pnas.0908356106 (doi:10.1073/pnas.0908356106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuhrer T., Fischer E., Sauer U. 2005. Experimental identification and quantification of glucose metabolism in seven bacterial species. J. Bacteriol. 187, 1581–1590 10.1128/JB.187.5.1581-1590.2005 (doi:10.1128/JB.187.5.1581-1590.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beale J. M., Foster J. L. 1996. Carbohydrate fluxes into alginate biosynthesis in Azotobacter vinelandii NCIB 8789: NMR investigations of the triose pools. Biochemistry 35, 4492–4501 10.1021/bi951922v (doi:10.1021/bi951922v) [DOI] [PubMed] [Google Scholar]
- 24.Portais J.-C., Tavernier P., Gosselin I., Barbotin J.-N. 1999. Cyclic organization of the carbohydrate metabolism in Sinorhizobium meliloti. Eur. J. Biochem. 265, 473–480 10.1046/j.1432-1327.1999.00778.x (doi:10.1046/j.1432-1327.1999.00778.x) [DOI] [PubMed] [Google Scholar]
- 25.Fischer E., Sauer U. 2003. A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli. J. Biol. Chem. 278, 46 446–46 451 10.1074/jbc.M307968200 (doi:10.1074/jbc.M307968200) [DOI] [PubMed] [Google Scholar]
- 26.Olszewski K. L., Mather M. W., Morrisey J. M., Garcia B. A., Vaidya A. B., Rabinowitz J. D., Llinas M. 2010. Branched tricarboxylic acid metabolism in Plasmodium falciparum. Nature 466, 774–778 10.1038/nature09301 (doi:10.1038/nature09301) [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Tang Y., Pingitore F., Mukhopadhyay A., Phan R., Hazen T. C., Keasling J. D. 2007. Pathway confirmation and flux analysis of central metabolic pathways in Desulfovibrio vulgaris hildenborough using gas chromatography-mass spectrometry and Fourier transform-ion cyclotron resonance mass spectrometry. J. Bacteriol. 189, 940–949 10.1128/JB.00948-06 (doi:10.1128/JB.00948-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crown S. B., Indurthi D. C., Ahn W. S., Choi J., Papoutsakis E. T., Antoniewicz M. R. 2011. Resolving the TCA cycle and pentose-phosphate pathway of Clostridium acetobutylicum ATCC 824: isotopomer analysis, in vitro activities and expression analysis. Biotechnol. J. 6, 300–305 10.1002/biot.201000282 (doi:10.1002/biot.201000282) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li F., Hagemeier C. H., Seedorf H., Gottschalk G., Thauer R. K. 2007. Re-Citrate synthase from Clostridium kluyveri is phylogenetically related to homocitrate synthase and isopropylmalate synthase rather than to Si-citrate synthase. J. Bacteriol. 189, 4299–4304 10.1128/JB.00198-07 (doi:10.1128/JB.00198-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marco-Urrea E., Paul S., Khodaverdi V., Seifert J., von Bergen M., Kretzschmar U., Adrian L. 2011. Identification and characterization of a Re-citrate synthase in Dehalococcoides strain CBDB1. J. Bacteriol. 193, 5171–5178 10.1128/JB.05120-11 (doi:10.1128/JB.05120-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuchs G. 2011. Alternative pathways of carbon dioxide fixation: insights into the early evolution of life? Annu. Rev. Microbiol. 65, 631–658 10.1146/annurev-micro-090110-102801 (doi:10.1146/annurev-micro-090110-102801) [DOI] [PubMed] [Google Scholar]
- 32.Erb T. J., Berg I. A., Brecht V., Müller M., Fuchs G., Alber B. E. 2007. Synthesis of C5-dicarboxylic acids from C2-units involving crotonyl-CoA carboxylase/reductase: the ethylmalonyl-CoA pathway. Proc. Natl Acad. Sci. USA 104, 10 631–10 636 10.1073/pnas.0702791104 (doi:10.1073/pnas.0702791104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider K., Peyraud R., Kiefer P., Christen P., Delmotte N., Massou S., Portais J.-C., Vorholt J. A. 2012. The ethylmalonyl-CoA pathway is used in place of the glyoxylate cycle by Methylobacterium extorquens AM1 during growth on acetate. J. Biol. Chem. 287, 757–766 10.1074/jbc.M111.305219 (doi:10.1074/jbc.M111.305219) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Härtel T., Eylert E., Schulz C., Petruschka L., Gierok P., Grubmüller S., Lalk M., Eisenreich W., Hammerschmidt S. 2012. Characterization of central carbon metabolism of Streptococcus pneumoniae by isotopologue profiling. J. Biol. Chem. 287, 4260–4274 10.1074/jbc.M111.304311 (doi:10.1074/jbc.M111.304311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risso C., Van Dien S. J., Orloff A., Lovley D. R., Coppi M. V. 2008. Elucidation of an alternate isoleucine biosynthesis pathway in Geobacter sulfurreducens. J. Bacteriol. 190, 2266–2274 10.1128/JB.01841-07 (doi:10.1128/JB.01841-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu H., Zhang Y. Z., Guo X. K., Ren S. X., Staempfli A. A., Chiao J. S., Jiang W. H., Zhao G. P. 2004. Isoleucine biosynthesis in Leptospira interrogans serotype lai strain 56601 proceeds via a threonine-independent pathway. J. Bacteriol. 186, 5400–5409 10.1128/JB.186.16.5400-5409.2004 (doi:10.1128/JB.186.16.5400-5409.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howell D. M., Xu H., White R. H. 1999. (Re)-citramalate synthase in methanogenic archaea. J. Bacteriol. 181, 331–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu B., Zhang B., Feng X., Rubens J. R., Huang R., Hicks L. M., Pakrasi H. B., Tang Y. J. 2010. Alternative isoleucine synthesis pathway in cyanobacterial species. Microbiology 156, 596–602 10.1099/mic.0.031799-0 (doi:10.1099/mic.0.031799-0) [DOI] [PubMed] [Google Scholar]
- 39.Atsumi S., Liao J. C. 2008. Directed evolution of Methanococcus jannaschii citramalate synthase for biosynthesis of 1-propanol and 1-butanol by Escherichia coli. Appl. Environ. Microbiol. 74, 7802–7808 10.1128/AEM.02046-08 (doi:10.1128/AEM.02046-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang Y. J., Shui W. Q., Myers S., Feng X., Bertozzi C., Keasling J. D. 2009. Central metabolism in Mycobacterium smegmatis during the transition from O2-rich to O2-poor conditions as studied by isotopomer-assisted metabolite analysis. Biotechnol. Lett. 31, 1233–1240 10.1007/s10529-009-9991-7 (doi:10.1007/s10529-009-9991-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao K., Du L., Sun X., Cai M., Zhu T., Zhou X., Gu Q., Zhang Y. 2009. Biosynthesis of aspergiolide A, a novel antitumor compound by a marine-derived fungus Aspergillus glaucus via the polyketide pathway. Tetrahedron Lett. 50, 1082–1085 10.1016/j.tetlet.2008.12.094 (doi:10.1016/j.tetlet.2008.12.094) [DOI] [Google Scholar]
- 42.Nes C. R., Singha U. K., Liu J., Ganapathy K., Villalta F., Waterman M. R., Lepesheva G. I., Chaudhuri M., Nes W. D. 2012. Novel sterol metabolic network of Trypanosoma brucei procyclic and bloodstream forms. Biochem. J. 443, 267–277 10.1042/BJ20111849 (doi:10.1042/BJ20111849) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKinlay J. B., Harwood C. S. 2010. Carbon dioxide fixation as a central redox cofactor recycling mechanism in bacteria. Proc. Natl Acad. Sci. USA 107, 11 669–11 675 . 10.1073/pnas.1006175107 (doi:10.1073/pnas.1006175107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKinlay J. B., Harwood C. S. 2011. Calvin cycle flux, pathway constraints, and substrate oxidation state together determine the H2 biofuel yield in photoheterotrophic bacteria. MBio 2, e00323-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang K.-H., Tang Y. J., Blankenship R. E. 2011. Carbon metabolic pathways in phototrophic bacteria and their broader evolutionary implications. Front. Microbiol. 2, 165. 10.3389/fmicb.2011.00165 (doi:10.3389/fmicb.2011.00165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang S., Bryant D. A. 2011. The tricarboxylic acid cycle in cyanobacteria. Science 334, 1551–1553 10.1126/science.1210858 (doi:10.1126/science.1210858) [DOI] [PubMed] [Google Scholar]
- 47.Chavarría M., Kleijn R. J., Sauer U., Pflüger-Grau K., de Lorenzo V. 2012. Regulatory tasks of the phosphoenolpyruvate-phosphotransferase system of Pseudomonas putida in central carbon metabolism. MBio 3, e00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beste D. J. V., et al. 2011. 13C metabolic flux analysis identifies an unusual route for pyruvate dissimilation in mycobacteria which requires isocitrate lyase and carbon dioxide fixation. PLoS Pathog. 7, e1002091. 10.1371/journal.ppat.1002091 (doi:10.1371/journal.ppat.1002091) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiong W., Liu L., Wu C., Yang C., Wu Q. 2010. 13C-tracer and gas chromatography-mass spectrometry analyses reveal metabolic flux distribution in the oleaginous microalga Chlorella protothecoides. Plant Physiol. 154, 1001–1011 10.1104/pp.110.158956 (doi:10.1104/pp.110.158956) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiechert W., Nöh K. 2005. From stationary to instationary metabolic flux analysis. Adv. Biochem. Eng. Biotechnol. 92, 145–172 [DOI] [PubMed] [Google Scholar]
- 51.Nöh K., Wahl A., Wiechert W. 2006. Computational tools for isotopically instationary 13C labeling experiments under metabolic steady state conditions. Metab. Eng. 8, 554–577 10.1016/j.ymben.2006.05.006 (doi:10.1016/j.ymben.2006.05.006) [DOI] [PubMed] [Google Scholar]
- 52.Schaub J., Mauch K., Reuss M. 2008. Metabolic flux analysis in Escherichia coli by integrating isotopic dynamic and isotopic stationary 13C labeling data. Biotechnol. Bioeng. 99, 1170–1185 10.1002/bit.21675 (doi:10.1002/bit.21675) [DOI] [PubMed] [Google Scholar]
- 53.Nöh K., Grönke K., Luo B., Takors R., Oldiges M., Wiechert W. 2007. Metabolic flux analysis at ultra short time scale: isotopically non-stationary 13C labeling experiments. J. Biotechnol. 129, 249–267 10.1016/j.jbiotec.2006.11.015 (doi:10.1016/j.jbiotec.2006.11.015) [DOI] [PubMed] [Google Scholar]
- 54.Young J. D., Walther J. L., Antoniewicz M. R., Yoo H., Stephanopoulos G. 2008. An elementary metabolite unit (EMU) based method of isotopically nonstationary flux analysis. Biotechnol. Bioeng. 99, 686–699 10.1002/bit.21632 (doi:10.1002/bit.21632) [DOI] [PubMed] [Google Scholar]
- 55.Shastri A. A., Morgan J. A. 2007. A transient isotopic labeling methodology for 13C metabolic flux analysis of photoautotrophic microorganisms. Phytochemistry 68, 2302–2312 10.1016/j.phytochem.2007.03.042 (doi:10.1016/j.phytochem.2007.03.042) [DOI] [PubMed] [Google Scholar]
- 56.Antoniewicz M. R., Kelleher J. K., Stephanopoulos G. 2007. Elementary metabolite units (EMU): a novel framework for modeling isotopic distributions. Metab. Eng. 9, 68–86 10.1016/j.ymben.2006.09.001 (doi:10.1016/j.ymben.2006.09.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wahl S., Noh K., Wiechert W. 2008. 13C labeling experiments at metabolic nonstationary conditions: an exploratory study. BMC Bioinform. 9, 152. 10.1186/1471-2105-9-152 (doi:10.1186/1471-2105-9-152) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rühl M., Zamboni N., Sauer U. 2010. Dynamic flux responses in riboflavin overproducing Bacillus subtilis to increasing glucose limitation in fed-batch culture. Biotechnol. Bioeng. 105, 795–804 [DOI] [PubMed] [Google Scholar]
- 59.Leighty R. W., Antoniewicz M. R. 2011. Dynamic metabolic flux analysis (DMFA): a framework for determining fluxes at metabolic non-steady state. Metab. Eng. 13, 745–755 10.1016/j.ymben.2011.09.010 (doi:10.1016/j.ymben.2011.09.010) [DOI] [PubMed] [Google Scholar]
- 60.Stolyar S., Van Dien S., Hillesland K. L., Pinel N., Lie T. J., Leigh J. A., Stahl D. A. 2007. Metabolic modeling of a mutualistic microbial community. Mol. Syst. Biol. 3, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Graaf A. A., Maathuis A., de Waard P., Deutz N. E. P., Dijkema C., de Vos W. M., Venema K. 2010. Profiling human gut bacterial metabolism and its kinetics using [U-13C]glucose and NMR. NMR Biomed. 23, 2–12 10.1002/nbm.1418 (doi:10.1002/nbm.1418) [DOI] [PubMed] [Google Scholar]
- 62.Salimi F., Zhuang K., Mahadevan R. 2010. Genome-scale metabolic modeling of a clostridial co-culture for consolidated bioprocessing. Biotechnol. J. 5, 726–738 10.1002/biot.201000159 (doi:10.1002/biot.201000159) [DOI] [PubMed] [Google Scholar]
- 63.Rühl M., Hardt W. D., Sauer U. 2011. Subpopulation-specific metabolic pathway usage in mixed cultures as revealed by reporter protein-based 13C analysis. Appl. Environ. Microbiol. 77, 1816–1821 10.1128/AEM.02696-10 (doi:10.1128/AEM.02696-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shaikh A. S., Tang Y. J., Mukhopadhyay A., Keasling J. D. 2008. Isotopomer distributions in amino acids from a highly expressed protein as a proxy for those from total protein. Anal. Chem. 80, 886–890 10.1021/ac071445+ (doi:10.1021/ac071445+) [DOI] [PubMed] [Google Scholar]
- 65.Eisenreich W., Dandekar T., Heesemann J., Goebel W. 2010. Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol. 8, 401–412 10.1038/nrmicro2351 (doi:10.1038/nrmicro2351) [DOI] [PubMed] [Google Scholar]
- 66.Fuchs T. M., Eisenreich W., Kern T., Dandekar T. 2012. Towards a systemic understanding of Listeria monocytogenes metabolism during infection. Front. Microbiol. 3, 23. 10.3389/fmicb.2012.00023 (doi:10.3389/fmicb.2012.00023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schär J., Stoll R., Schauer K., Loeffler D. I. M., Eylert E., Joseph B., Eisenreich W., Fuchs T. M., Goebel W. 2010. Pyruvate carboxylase plays a crucial role in carbon metabolism of extra- and intracellularly replicating Listeria monocytogenes. J. Bacteriol. 192, 1774–1784 10.1128/JB.01132-09 (doi:10.1128/JB.01132-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Götz A., Eylert E., Eisenreich W., Goebel W. 2010. Carbon metabolism of enterobacterial human pathogens growing in epithelial colorectal adenocarcinoma (Caco-2) cells. PLoS ONE 5, e10586. 10.1371/journal.pone.0010586 (doi:10.1371/journal.pone.0010586) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsiao T.-L., Revelles O., Chen L., Sauer U., Vitkup D. 2010. Automatic policing of biochemical annotations using genomic correlations. Nat. Chem. Biol. 6, 34–40 10.1038/nchembio.266 (doi:10.1038/nchembio.266) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Veit A., Rittmann D., Georgi T., Youn J.-W., Eikmanns B. J., Wendisch V. F. 2009. Pathway identification combining metabolic flux and functional genomics analyses: acetate and propionate activation by Corynebacterium glutamicum. J. Biotechnol. 140, 75–83 10.1016/j.jbiotec.2008.12.014 (doi:10.1016/j.jbiotec.2008.12.014) [DOI] [PubMed] [Google Scholar]
- 71.Peyraud R., Schneider K., Kiefer P., Massou S., Vorholt J., Portais J.-C. 2011. Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 5, 189. 10.1186/1752-0509-5-189 (doi:10.1186/1752-0509-5-189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smaldone G. T., Revelles O., Gaballa A., Sauer U., Antelmann H., Helmann J. D. 2012. A global investigation of the Bacillus subtilis iron-sparing response identifies major changes in metabolism. J. Bacteriol. 194, 2594–605 10.1128/JB.05990-11 (doi:10.1128/JB.05990-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buescher J. M., et al. 2012. Global network reorganization during dynamic adaptations of Bacillus subtilis metabolism. Science 335, 1099–1103 10.1126/science.1206871 (doi:10.1126/science.1206871) [DOI] [PubMed] [Google Scholar]
- 74.Rühl M., Rupp B., Nöh K., Wiechert W., Sauer U., Zamboni N. 2012. Collisional fragmentation of central carbon metabolites in LC-MS/MS increases precision of 13C metabolic flux analysis. Biotechnol. Bioeng. 109, 763–771 10.1002/bit.24344 (doi:10.1002/bit.24344) [DOI] [PubMed] [Google Scholar]
- 75.Yuan Y., Yang T. H., Heinzle E. 2010. 13C metabolic flux analysis for larger scale cultivation using gas chromatography-combustion-isotope ratio mass spectrometry. Metab. Eng. 12, 392–400 10.1016/j.ymben.2010.02.001 (doi:10.1016/j.ymben.2010.02.001) [DOI] [PubMed] [Google Scholar]
- 76.Caytan E., Remaud G. S., Tenailleau E., Akoka S. 2007. Precise and accurate quantitative 13C NMR with reduced experimental time. Talanta 71, 1016–1021 10.1016/j.talanta.2006.05.075 (doi:10.1016/j.talanta.2006.05.075) [DOI] [PubMed] [Google Scholar]
- 77.Tang Y. J., Hwang J. S., Wemmer D., Keasling J. D. 2007. The Shewanella oneidensis MR-1 fluxome under various oxygen conditions. Appl. Environ. Microbiol. 73, 718–729 10.1128/AEM.01532-06 (doi:10.1128/AEM.01532-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wiechert W., Möllney M., Petersen S., de Graaf A. A. 2001. A universal framework for 13C metabolic flux analysis. Metab. Eng. 3, 265–283 10.1006/mben.2001.0188 (doi:10.1006/mben.2001.0188) [DOI] [PubMed] [Google Scholar]
- 79.Quek L.-E., Wittmann C., Nielsen L. K., Krömer J. O. 2009. OpenFLUX: efficient modelling software for 13C-based metabolic flux analysis. Microb. Cell Factories 8, 25. 10.1186/1475-2859-8-25 (doi:10.1186/1475-2859-8-25) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Copeland W. B., Bartley B. A., Chandran D., Galdzicki M., Kim K. H., Sleight S. C., Maranas C. D., Sauro H. M. 2012. Computational tools for metabolic engineering. Metab. Eng. 14, 270–280 10.1016/j.ymben.2012.03.001 (doi:10.1016/j.ymben.2012.03.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Srour O., Young J. D., Eldar Y. C. 2011. Fluxomers: a new approach for 13C metabolic flux analysis. BMC Syst. Biol. 5, 129. 10.1186/1752-0509-5-129 (doi:10.1186/1752-0509-5-129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Malaisse W. J., Zhang Y., Jijakli H., Courtois P., Sener A. 2004. Enzyme-to-enzyme channelling in the early steps of glycolysis in rat pancreatic islets. Int. J. Biochem. Cell Biol. 36, 1510–1520 10.1016/j.biocel.2003.12.013 (doi:10.1016/j.biocel.2003.12.013) [DOI] [PubMed] [Google Scholar]
- 83.Williams T. C. R., Sweetlove L. J., Ratcliffe R. G. 2011. Capturing metabolite channeling in metabolic flux phenotypes. Plant Physiol. 157, 981–984 10.1104/pp.111.184887 (doi:10.1104/pp.111.184887) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sumegi B., Sherry A. D., Malloy C. R., Srere P. A. 1993. Evidence for orientation-conserved transfer in the TCA cycle in Saccharomyces cerevisiae: carbon-13 NMR studies. Biochemistry 32, 12 725–12 729 10.1021/bi00210a022 (doi:10.1021/bi00210a022) [DOI] [PubMed] [Google Scholar]
- 85.Niklas J., Sandig V., Heinzle E. 2011. Metabolite channeling and compartmentation in the human cell line AGE1.HN determined by 13C labeling experiments and 13C metabolic flux analysis. J. Biosci. Bioeng. 112, 616–623 10.1016/j.jbiosc.2011.07.021 (doi:10.1016/j.jbiosc.2011.07.021) [DOI] [PubMed] [Google Scholar]
- 86.Shearer G., Lee J. C., Koo J. A., Kohl D. H. 2005. Quantitative estimation of channeling from early glycolytic intermediates to CO2 in intact Escherichia coli. FEBS J. 272, 3260–3269 10.1111/j.1742-4658.2005.04712.x (doi:10.1111/j.1742-4658.2005.04712.x) [DOI] [PubMed] [Google Scholar]
- 87.Madsen E. L. 2008. Environmental microbiology: from genomes to biogeochemistry. Malden, MA: Blackwell Publishing [Google Scholar]
- 88.Feng X., Tang Y. J. 2011. Evaluation of isotope discrimination in 13C-based metabolic flux analysis. Anal. Biochem. 417, 295–297 10.1016/j.ab.2011.06.022 (doi:10.1016/j.ab.2011.06.022) [DOI] [PubMed] [Google Scholar]