

Abstract
Hemolytic transfusion reactions represent one of the most common causes of transfusion-related mortality. Although many factors influence hemolytic transfusion reactions, complement activation represents one of the most common features associated with fatality. In this paper we will focus on the role of complement in initiating and regulating hemolytic transfusion reactions and will discuss potential strategies aimed at mitigating or favorably modulating complement during incompatible red blood cell transfusions.
1. Introduction
As pathogens began to evolve elaborate antigenic structures to avoid innate immunity, vertebrates evolved an equally impressive mechanism of combating antigenic diversity among pathogens [1]. Indeed, adaptive immunity appears to possess the capacity to respond to a nearly infinite number of antigens, enabling immunological protection against a wide variety of potential pathogens [1, 2]. However, not all foreign antigens represent a pathogenic threat. Although tolerance mechanisms exist that reduce the likelihood of developing antibodies against innocuous antigens, individuals can possess significant antibodies against antigenic polymorphisms on human tissue [2]. Indeed, hemolytic transfusion reactions typically reflect the engagement of antibodies directed against antithetical antigens on donor red blood cells (RBCs).
The earliest example of human donor rejection occurred following transfusion of ABO (H) incompatible RBCs [3]. Although ABO(H) represent the first RBC polymorphic antigens described, many other carbohydrate and protein antigenic differences became apparent as transfusion practices increased [4]. Interestingly, these immune-mediated discoveries provided the first example of significant polymorphisms within the human population long before DNA was recognized as the molecular basis of inheritance [5–8]. As hemolytic transfusion reactions (HTRs) can occur following transfusion of incompatible RBCs or following transfer of antibodies present in donor units, such as platelets or plasma, significant testing occurs prior to transfusion to insure utilization of antigen compatible blood products [9, 10]. Unfortunately, these procedures occasionally fail. In addition, some patients fail to demonstrate detectable antibodies but exhibit amnestic antibody responses to previously exposed RBC antigens following transfusion [11]. Under these circumstances, cellular rejection in the form of a hemolytic transfusion reaction may occur.
Hemolytic transfusion reactions may not only cause significant morbidity and compromise the therapeutic efficacy of transfusion, but ultimately these reactions can prove fatal. Indeed, hemolytic transfusion reactions represent one of the most common causes of transfusion-related mortality. Furthermore, in highly immunized patients, securing antigen compatible blood can be difficult, if not impossible, preventing appropriate and timely life-saving intervention [12]. As a result, a greater understanding of the factors that may influence hemolytic transfusion reactions is needed. Although many factors influence hemolytic transfusion reactions, in this paper we will focus on the potential role of complement in initiating and regulating hemolytic transfusion reactions, with a particular focus on potential strategies aimed at mitigating or favorably modulating complement during incompatible RBC transfusions.
2. Early Transfusion Reactions
While many diseases reflect complement dysregulation [13], perhaps the earliest and most potent example of complement-mediated mortality predates the discovery of microbes and immunity. In 1667, Dr. Jean-Babtiste Deny transfused several patients multiple times with either sheep or calf blood. Although the patients appeared to initially tolerate transfusion, repeated transfusions uniformly resulted in patient death [4, 14]. Subsequent attempts nearly two centuries later utilizing human donors for transfusion resulted in more favorable outcomes; however, patients receiving transfusions from human donors occasionally experience similar fatalities despite many attempts to predict favorable responses to transfusion [4].
Prompted by previous work suggesting that antigenic differences on RBCs occur between different mammalian species, Karl Landsteiner sought to determine whether similar differences may account for incompatible transfusions using human donors [3]. In 1900, Landsteiner published his seminal work demonstrating that sera isolated from patients could differentially agglutinate donor RBCs [5]. Within the next decade, the discovery of A, B, and C (O) antigens enabled accurate prediction of immunological compatibility between donor and recipient, for which Landsteiner was awarded the Nobel prize in physiology and medicine in 1930 [3].
While the factors responsible for fatal outcomes during an incompatible transfusion remained unknown for many years, naturally occurring antibodies directed against carbohydrate xenoantigens on animal RBCs or ABO(H) antigens on human RBCs likely mediated activation of complement [15]. Robust complement activation not only results in significant intravascular hemolysis, but complement products also independently induce significant physiological changes. Indeed, early transfusion reactions likely reflected significant complement-mediated hemolysis and systemic alterations that ultimately resulted in fatal outcomes [15–18].
3. Complement: A Brief History
The identification of microbes as a potential cause of human illness drove intense research by numerous investigators to understand host factors that may inhibit microbial invasion. Early studies by Pastuer and others demonstrated that inoculation of animals with microbes could induce a form of host resistance to further infection [19]. Although the specific players responsible for acquired host immunity remained unknown for many years, both cellular and serological factors appeared to possess the ability to protect animals against reinfection [20].
Early studies focused on characterizing serological factors demonstrated that blood possesses intrinsic bactericidal activity following inoculation of a specific microbe. Subsequent studies demonstrated that heating serum at 55°C effectively eliminated this bactericidal activity in vitro. However, infusion of heat inactivated serum isolated from previously inoculated animals protected recipients from infection. These results suggested that serum contains heat labile and heat stable components. Paul Ehrlich subsequently coined the term complement to describe the heat labile component of immunity, which he postulated worked in concert with a heat stable “amboceptor” that provided target specificity [21]. Subsequent studies demonstrate that a complex pathway of complement activation and regulation occurs following “amboceptor” (antibody) engagement of antigen.
4. Complement Initiation and Regulation: An Overview
Complement activation can be divided into three primary cascades of activation, classical (antibody), lectin, and alternative (Figure 1) [22, 23]. While the lectin and alternative pathways of complement activation play a key role in immunity [24, 25], antibodies provide the primary initiating activity of complement activation during a hemolytic transfusion [26]. Regardless of the initiating stimulus, each pathway converges on the formation of an enzyme complex capable of converting complement component C3 into active products, C3a and C3b. C3b, working in concert with additional complement components, ultimately propagates a cascade that terminates in the formation of a membrane attack complex and eventual target lysis [27].
Figure 1.

Antibody-mediated complement activation. IgM antibodies primarily exist as planar molecules. However, upon engagement of antigen, IgM antibodies undergo significant conformation changes that result in favorable exposure of multiple C1q binding domains. As a result, a single IgM molecule can initiate significant complement activation. In contrast, two separate IgG antibodies must engage antigen within close enough proximity to simultaneously engage the C1q fix complement.
Following transfusion of incompatible blood, antigen-antibody interactions facilitate engagement of the first component of the classical pathway, C1q. Once bound, C1q induces conformational changes in the serine protease, C1r, which allows C1r to cleave C1s, resulting in an active C1s protease [28]. This complex then cleaves C2 and C4, generating target bound C2b and C4b and the release of soluble C2a and C4a (Figure 1). Bound C2b and C4b form a complex, the C3 convertase, which cleaves C3 to form C3a and C3b [27]. C3a provides a soluble complement regulator of variety of biological pathways, including activation of mast cells, endothelial cells, and phagocytes in addition to intrinsically possessing antimicrobial activity [29, 30]. In contrast, C3b covalently attaches to the target membrane through a highly reactive thioester and thereby facilitates continuation of the complement cascade [23, 27]. Bound C3b, in complex with C2b and an additional C3b molecule, then facilitates conversion of C5 to C5a and C5b. Similar to C3a, C5a regulates a wide variety of systemic factors in immunity [31]. Bound C5b recruits additional complement factors, including C6, C7, and C8, which together facilitate the insertion and polymerization of C9 in the target membrane [32]. Ultimately, C9-mediated pore formation results in osmotic lysis of the target [32].
In contrast to the antibody-mediated pathway, the lectin and alternative pathways do not require adaptive immunity to target pathogen. For example, the lectin pathway utilizes a series of innate immune lectins, such as mannan binding lectin (MBL), which target microbes by recognizing common pathogen-associated molecular motifs [25]. Upon pathogen recognition, MBL engages adapter proteins, MASP-1 and MASP-2, which cleave C4 and C2 similar to C1s in the classical pathway [25, 28]. In contrast to the classic and lectin pathways, the alternative pathway does not actively engage pathogens through a target molecule. Instead, the alternative pathway relies on spontaneous activation of C3 to form C3 (H2O). C3 (H2O) then binds an additional complement factor, factor B, which renders factor B sensitive to cleavage by constitutively active factor D [24]. A complex of factor C3 (H2O) and Bb, stabilized by a factor P, then forms the C3 convertase of the alternative pathway [33, 34]. Importantly, C3b generated during antibody or lectin-induced activation can also directly facilitate D-mediated factor B activation, amplifying complement activation even when antibody alone can provide an initiating stimulus [25]. While antibody-antigen complexes primarily utilize the classic pathway, several studies demonstrate that antibody can also directly activate the alternative pathway [35]. As a result, although antibody-induced activation likely provides the primary driving force, each pathway likely contributes to the overall effect of complement-mediated transfusion reactions.
In addition to facilitating complement activation, complement factors, such as C3b, can serve as opsonins on the target membrane for a series of complement receptors [36]. Each complement receptor appears to be expressed on distinct leukocyte populations and tissue [37]. Furthermore, engagement of complement receptors on different cells can result in distinct immunological outcomes. For example, several complement receptors, including CR1 (CD35), CR3 (CD11bCD18), CR4 (CD11cCD18), and CRIg, typically facilitate phagocytosis of opsonized targets following C3b ligation [38–43]. In contrast, CR2 (CD21) engagement of complement can influence B cell activation and tolerance [44–46]. Additional complement receptors, which recognize soluble complement components, C3a and C5a, also regulate a wide variety of biological pathways following ligation [27, 31, 47]. Although not functionally important in complement activation, several complement factors actually possess unique antigenic structures used in blood group classification, such as the Cromer, Chido, and Rodgers blood groups, and therefore could also theoretically facilitate RBC removal as unique target antigens [48, 49]. Taken together, complement activation results in the production of a variety of highly active immunoregulatory components capable of influencing a broad range of biological processes.
As with all immune effector functions, unregulated activation of complement can result in significant pathological sequelae. Indeed, many disease states are defined by genetic or acquired dysregulation of complement [50]. Given the evolutionary ancient history of complement, many inhibitory pathways exist that facilitate complement regulation. For example, constitutively expressed cell surface proteins, such as CD35 (CR1), CD46 (MCP), CD55, or CD59, provide various inhibitory activities to inhibit off-target complement activation [40, 51–53]. Importantly, each of these factors displays unique regulatory activities. For example, CD35 and CD46 bind C3b and C4b preventing complex formation with Bb and 2b, respectively, [40, 51, 54], thereby inhibiting active C3 convertase formation. Similarly, CD55 (decay accelerating factor or DAF) along with CD35 actively dissociates C3 convertase complexes once formed by displacing Bb and 2b [51, 53, 55]. Furthermore, CD59 (protectin) binds the C5, C6, C7, and C8 complex and inhibits C9 binding and formation of the membrane attack complex [56–58]. In concert, all of these mammalian, membrane bound receptors inhibit off-target complement deposition from actively destroying an individual's own cells.
In addition to cell surface factors, several soluble factors also aid in complement regulation. Similar to the inhibitory activities of CD35, CD46, and CD55, C4 binding protein (C4BP) can inactivate C4b by preventing binding to C2b [59], thereby also limiting C3 convertase formation [55]. In contrast, other soluble factors provide additional checkpoints in complement regulation. For example, although activated fragments of complement will readily hydrolyze in the absence of target attachment [60, 61], inhibitory factor H binds soluble C3b to further reduce off-target effects [62]. C1 inhibitor (C1INH) actually dissociates C1r and C1s from C1q, thereby directly targeting the effector arm of antibody-mediated complement activation [63]. Patients with hereditary angioneurotic edema illustrate the importance of C1INH in complement regulation.
Complement degradation also plays a key role in complement regulation. For example, factor I cleaves surface bound C3b and C4b. Although formation of the first cleavage product of C3b, iC3b retains the ability to bind complement receptors, iC3b no longer facilitates complement activation [27]. iC3b can be additionally cleaved by factor I to form C3dg and C3d which are no longer recognized by phagocytosis-inducing complement receptors (Figure 2). Indeed, CD21 serves as the primary receptor for C3dg, where CD21 ligation significantly enhances antibody production against opsonized antigen [45, 64]. Importantly, factor I requires coactivation by several other factors, including factor H, CD35, CD46, and CD55, allowing soluble and membrane inhibitory factors to work in concert to regulate complement activation [55, 65], while also directing complement inactivation toward an individual's own cells. Importantly, several studies recently described additional soluble and membrane bound regulators of complement, adding to the complexity of complement homeostasis [66, 67]. Thus, complement activation reflects a highly regulated pathway responsible for differentially targeting pathogens while protecting an individual's own cells.
Figure 2.
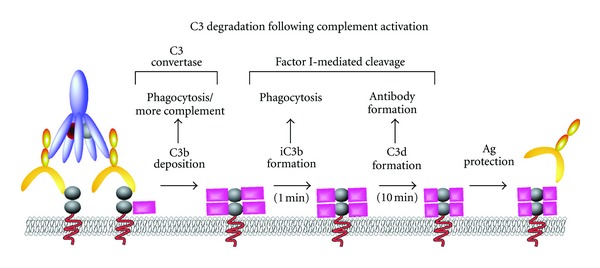
C3 degradation following complement activation. Conversion of C3 to C3b by C3 convertase complex results in rapid deposition of highly reactive C3b on the target surface. Degradation by inhibitory factor I, in concert with other regulatory molecules, results in conversion of C3b to iC3b. Although iC3b retains the capacity of C3b to engage complement receptors as an opsonin, it no longer participates in the production of additional complement. Additional cleavage of iC3b by factor I results in the formation of C3d, which no longer fixes complement or serves as an opsonin, although it may engage B cells and facilitate antibody formation. Decoration of antigen with C3d may result in protection of the antigen from additional antibody binding and subsequent effector function.
5. Factors Influencing Complement Activation
While complement can result in significant pathophysiology during a hemolytic transfusion reaction, not all HTRs result in complement activation [26]. Although the precise factors responsible for dictating whether complement activation will occur remain enigmatic, several factors appears to play a role. Of these factors, the type of antibody and antigen mediating the HTR appears to play the most significant roles in determining the outcome of an incompatible transfusion.
Although different isotypes of antibody fix complement, not all antibodies possess an equivalent capacity to engage this pathway. Appropriate engagement of C1q requires accommodation of two separate CH2 domains on antibody Ig heavy chains [68]. As a result, CH2 availability can influence antibody proclivity for complement fixation. For example, while a single pentameric IgM antibody possesses five separate CH2 domains, an IgG molecule only contains a single domain capable of C1q engagement. In solution, IgM exists as a planar molecule that fails to bind significant C1q, preventing IgM-mediated complement activation in the absence of antigen. However, IgM engagement of antigen induces a conformation change that enables a single IgM to engage C1q and activate complement [69] (Figure 1). In contrast, two IgG molecules must engage distinct antigenic epitopes within 20–30 nm of each other in order to adequately engage C1q and initiate complement [70] (Figure 1). As a result, fewer IgM antibodies must bind antigen to fix equivalent levels of complement as IgG [71]. Equally important, different IgG subclasses possess distinct hinge regions that can differentially impact the conformational flexibility of the heavy chain, thereby also potentially impacting the ability of the CH2 domain to engage complement [72, 73]. Intrinsic differences between subtypes in the CH2 domain may also directly impact C1q engagement [74]. In contrast to IgG and IgM, while IgA may induce complement activation through engagement of the lectin pathway [75], the CH2 domains of IgA, IgD, and IgE possess little affinity for C1q [73]. As a result, IgM and IgG antibodies provide the primary stimulus for classical complement activation.
In addition to intrinsic differences in the ability of Ig isotypes to induce complement activation, a variety of enzymatic and nonenzymatic posttranslational modifications can impact antibody-induced complement activation. For example, posttranslational glycosylation of the Fc domain can also impact the ability of IgG molecules to engage C1q or activate the lectin pathway of complement [76, 77]. Furthermore, nonenzymatic formation of advanced glycation end-products can also affect the ability of antibodies to induce complement activation [78]. Importantly, none of these factors in isolation predicts the ability of an antibody-antigen interaction to result in complement deposition [35]. As a result, the influence of an antibody on complement activation reflects a variety of distinct factors. As polyclonal antibodies typically mediate HTR, the unique composition, affinity, and potential modifications of antibodies within a recipient will impact the likelihood of complement activation during an incompatible transfusion.
In addition to the antibody isotype, the antigen itself appears to independently predict the likelihood of complement activation. For example, ABO(H) HTRs result in robust complement activation and rapid intravascular hemolysis [79]. Similar studies suggest that IgG antibodies against other RBC antigens, including the Kidd antigens (Jka and JkB) and Fya, appear to induce complement-mediated hemolysis [79, 80]. Other antigens, such as Kell, appear to result in mixed forms of clearance following antibody engagement, utilizing both complement and Fc receptors [81]. It should be noted that many of these studies employed complement deposition, clearance kinetics, or intravascular hemolysis as a surrogate for complement-mediated destruction [79]. Although intravascular hemolysis typically occurs in the presence of complement deposition, it remains to be definitively tested using mammalian animal models whether complement may be required in each of these settings. For example, while some results suggest that complement may be required for both IgM and IgG-induced hemolysis in an animal model of a glycophorin A-mediated HTR [82], a recent study using the same model system suggests that hemolysis occurs independent of Fc receptors or complement [83]. As a result, HTRs reflect complex immunological reactions that need additional investigation in genetically defined animal models to be fully elucidated.
In contrast to ABO(H) HTRs, the Rh(D) antigen, the second most common antigen implicated in HTR, appears to mediate RBC clearance independent of complement. For example, Rh(D) alloimmunized patients who receive incompatible blood display little complement activation, despite several studies demonstrating that similar IgG subclasses facilitate complement activation in other settings [84, 85]. Similarly, administration of Rh immune globulin (RhIG) results in complement-independent hemolysis, likely through Fc-receptor-dependent phagocytosis [86]. Although the Rh antigen complex provides the most compelling example of complement-independent hemolysis, several other antigens, including S and M, also appear to predominately induce HTR independent of complement [79, 87].
While the exact mechanism whereby distinct antigens dictate the mode of clearance remains unknown, the density of the antigen likely plays a significant role. Consistent with this, A and B antigens not only represent the most common complement-mediated HTR, but also display the highest antigen density, typically ranging from 700,000 to over a million antigens per cell [88, 89]. In contrast, the Rh(D) antigen typically occurs at approximately 20,000 sites per cell [90]. As a result, the lower density of Rh likely reduces the probability of successful engagement of C1q [73], especially when the antibody composition mediating an HTR primarily reflects the IgG isotype. However, additional antigen factors likely influence this process. For example, although Rh(D) and Kell both reflect low-density antigens [91, 92], Rh mediates complement-independent hemolysis while Kell can engage complement and Fc receptor effector responses [79, 81]. With low-density antigens, antibody-induced clustering likely influences the efficiency of C1q engagement and complement activation [70]. As a result, lateral mobility and location of different antigens within the RBC membrane likely play an important role. Consistent with this, recent studies demonstrate that the cytoplasmic domains of Rh antigens engage RBC cytoskeletal proteins [93], likely limiting the lateral mobility needed to effectively engage C1q. In contrast, Kell possesses a very small cytoplasmic domain with little cytoskeletal attachment [94], thereby potentially enhancing lateral mobility and antibody-induced cluster formation.
As antithetical antigens occasionally only differ at discrete binding sites, the unique orientation and presentation of antigens may also influence the ability of antibody-antigen complexes to mediate complement fixation. Indeed, several antithetical antigens responsible for HTRs only differ at a single nucleotide polymorphism [95]. As a result, effective antibody-mediated crosslinking and cluster formation may be compromised if each individual molecule only possesses a single epitope. Although some antigens may exist in higher order complexes [96], providing additional epitopes, the availability of the antigenic sites may also be sterically constrained, thus, reducing the overall ability of antibodies to engage antigen. Conversely, carbohydrate A and B antigens display high levels of expression on highly mobile glycolipids and exist as multiple copies on glycoproteins [97], directly facilitating optimal antibody engagement for complement fixation. Taken together, a variety of biochemical features at the antigen level appear to possess the ability to impact antibody-induced complement activation.
Although the type of antibody and antigen can independently impact the probability of a complement-mediated HTR, the type of antigen itself partially dictates the type of antibody generated in response to a particular antigen. Although it remains to be formally examined, previous studies suggest that bacterial flora in the gut that expresses A- and B-like antigens stimulates anti-A and anti-B antibody formation required for an ABO(H) HTR [98, 99]. As these antigens reside on O antigen LPS molecules, they do not typically possess the T cell epitopes required for significant antibody class switching [15]. As a result, anti-A and anti-B antibodies form naturally by the fourth month of life without prior exposure secondary to transfusion and predominately reflect IgM antibodies [15, 100].
In contrast to ABO(H) antigens, most antibodies mediating HTR to protein antigens, such as Rh(D), Kell, and Duffy, do not form without prior antigen exposure. Although IgM development during a primary exposure to antigen can actually shorten the lifespan of transfused blood [101, 102], primary antibody responses do not typically result in a routinely detectable HTR. However, during this primary antigen exposure, T cell-mediated class switching results in significant production of IgG. As a result, RBCs transfused into an individual previously immunized against a protein antigen will likely encounter IgG antibodies [103]. Taken together, antibody isotype, antigen density and location, and the modes and mechanisms of antibody stimulation can significantly impact whether complement mediates a HTR.
6. Consequences of Complement Activation
Given optimal antigen and antibody conditions, incompatible transfusion can result in robust activation of complement. Complement activation may not only result in intravascular hemolysis, but complement split products, in particular C5a, can directly impact vascular permeability by activating endothelial cells and inducing significant cytokine production [27, 47]. Thus, complement can directly impact hemodynamic stability following an HTR. Free hemoglobin (Hb) released following complement-mediated hemolysis also induces significant alterations in vascular tone. In addition, Hb may also induce cytokine production, result in significant coagulopathy, and can be directly nephrotoxic [104–108]. As a result, exuberant complement activation following exposure to a large bolus of incompatible RBCs can induce significant pathophysiological changes that may ultimately result in a fatal outcome.
Although rapid complement activation can result in significant mortality, complement-mediated HTRs do not uniformly result in patient death. While several factors, including the volume of incompatible blood and titer of recipient antibody, likely influence patient prognosis following complement-mediated HTR [10, 109], RBC intrinsic factors may also play a role. For example, while ABO(H) HTR may saturate complement inhibitory factors, lower density antigens, such as Kell antigens, likely fix insufficient complement to adequately overcome regulatory pathways, causing arrest of complement activation prior to complete intravascular hemolysis [92]. Indeed, many non-ABO(H) HTRs arrest at the C3b stage of the complement cascade [92]. Although C3b-mediated phagocytic removal may increase cytokine production in these situations [110, 111], limited activation of C5a and reduced release of intravascular Hb also likely limit systemic consequences of complement-mediated HTR. These differences likely contribute to the significantly mortality rate associated with ABO(H) HTR [16].
Regulation of complement activation may also influence the rate and magnitude of RBC clearance during a complement-mediated HTR. For example, while C3 retains opsonizing activity following initial factor I-mediated cleavage [27], additional degradation to C3dg and CD3d renders RBCs relatively resistant to C3-mediated removal [112–114] (Figure 2). Consistent with this, complement-mediated HTRs characteristically result in rapid clearance during the first 10 minutes followed by an abrupt change in clearance kinetics that mirror the degradation rate of C3 to C3dg [79, 115, 116]. These results strongly suggest that alterations in clearance at least partially reflect the kinetics of factor I-mediated degradation. Interestingly, once cells accumulate C3dg, they become relatively transparent to the immune system and may circulate with C3dg virtually undetected by phagocytic cells [115–117] (Figure 2). In addition, ongoing degradation of complement may actually result in release of phagocyte-engaged cells, as several studies demonstrate that RBC can reappear hours following a complement-mediated HTR [110, 118].
A variety of additional variables can influence the extent and consequences of complement activation following an HTR. For example, although stochastic competition between activating and inhibitory stimuli likely dictates which cells will resist complement-mediated lysis, recent studies suggest that the activity of complement inhibitory proteins may be altered as RBCs age. Indeed, accumulation of advanced glycation end products on complement inhibitory proteins appears to directly diminish their function, strongly suggesting that older RBCs may be more sensitive to complement-mediated hemolysis [119]. Furthermore, the age of the patient may also impact the extent of complement-mediated hemolysis, as pediatric patients possess a less mature complement inhibitory apparatus than adults [120]. Thus a variety of cell-specific factors may also independently regulate the consequences of complement activation.
While many modifications or alterations in the expression of complement inhibitor proteins can impact RBC sensitivity to complement-mediated hemolysis, additional alterations affecting general RBC physiology may also impact complement sensitivity. In addition to hemolyzing transfused cells during HTR, occasionally patients will hemolyze their own cells, a phenomenon called bystander hemolysis [121]. Although bystander hemolysis may theoretically occur in any patient [122], individuals with sickle cell disease appear to be particularly prone [121]. Interestingly, several studies suggest that sickle cells may be intrinsically sensitive to complement-mediated lysis, suggesting that complement may in part be responsible [123, 124]. However, as several antigens that may not activate complement, such as Duffy, can also induce bystander hemolysis [125], the actual role of complement in the pathophysiology of this process remains largely unknown. In addition, complement activation may also be involved in non-hemolytic transfusion reactions, such as transfusion-related acute lung injury (TRALI) [126], another leading cause of transfusion-related mortality [127]. Indeed, recent studies suggest that, in addition to anti-HLA antibodies, anti-neutrophil antibodies, and other factors [127], differences in complement may also contribute to the gender influence of donor plasma on the development of TRALI [128].
7. Possible Complement-Targeted Therapeutic Interventions
Once an HTR is suspected, immediate intervention takes place in the form of stopping the transfusion and initiating intravenous fluids in an effort to achieve hemodynamic stability and protect the kidneys from the nephrotoxic effects of hemoglobin. However, no intervention in routine clinical practice specifically targets the HTR itself. As some patients, particularly patients with paroxysmal nocturnal hemoglobinuria, suffer from chronic hemolysis secondary to a deficiency in CD55 and CD59, a number of complement inhibitors have been developed in an effort to treat this and other complement-related disorders. While many inhibitors exist [129–135], only a few inhibitors have been evaluated using animal models of HTRs and no inhibitors have been formally examined clinically in an HTR setting. Injection of a soluble form of CR1 significantly attenuates complement activation following transfusion and increases posttransfusion survival of incompatible blood. Similar results were obtained using small molecule inhibitors of complement or smaller fragments of CR1 [136, 137]. Specific targeting of complement inhibition to RBCs through antibody-DAF chimeras may also inhibit complement-induced hemolysis [138]. Additional experimental models also suggest that intravenous immune globulin (IVIG) can inhibit complement-mediated HTRs [139, 140]. Although many of these inhibitors demonstrate promise in animal models, other complement inhibitors, including the C5 inhibitor, eculizumab [141], which are currently available for clinical use in other settings, need to be formally examined in the setting of HTR. As a result, future studies need to explore the potential role of complement inhibition as a prophylactic agent in the event that antigen-compatible blood may not be available or as therapy following an unfortunate HTR.
Although reengagement of antibody with antigen would be expected to provide a continuous supply of active complement, RBCs represent the only cells in the human body that do not resynthesize protein following maturation. As a result, whatever antigen exists on an RBC represents the only antigen that RBC will possess for the duration of its lifetime. While this may not alter basic antibody-antigen interactions, as C3 molecules attach to target membrane through covalent association, previous studies suggest that deposition of complement on antigens can sterically or directly mask targets from additional antibody binding [115, 116, 142–144] (Figure 2). As a result, once antigen masking and complement degradation are complete, cells become relatively transparent to the effector mechanism of hemolysis. These studies suggest that deposition and inactivation of complement in vitro may allow complement-mediated masking of antigen prior to transfusion in an effort to generate compatible RBCs for highly immunized patients (Figure 2). While these possibilities are intriguing, more studies are needed utilizing defined animal models to determine whether complement-mediated RBC manipulation might enable transfusion across immunological barriers.
8. Conclusions
HTRs historically provided significant insight into the role of complement as an antibody effector system [79]. Fortunately for patients, extreme vigilance exercised by transfusion medicine services has drastically reduced the rate of hemolytic transfusions [9]. As a result, most detailed studies examining HTRs predate the exponential increase in genetically manipulated animal models designed to appreciate subtle, yet critical, factors responsible for complement regulation in vivo [145–147]. Furthermore, until recently, no mammalian models existed to study clinically relevant RBC antigens in the setting of HTR [82, 148–151]. Indeed, despite Landsteiner's discovery of ABO(H) antigens over a century ago, no animal model currently exits to formally examine the role of complement or other factors in ABO(H) HTRs. Despite the relatively decreased incidence of mistransfusion and HTRs, the risk still remains. In 2011, HTR represented the second leading cause of transfusion-associated mortality reported to the FDA [152]. Thus, additional models need to be developed with a renewed focus on the mechanisms responsible for complement-dependent and complement-independent mechanisms of RBC hemolysis. These future studies possess the potential to provide rational approaches to not only develop therapeutic interventions in the unfortunate event that an HTR occurs, but to also extend the scope of immunological compatibility by manipulating complement to render otherwise incompatible transfusions compatible. Future work will likely provide significant insight into complement activation and regulation with the potential to significantly impact patient care.
References
- 1.Litman GW, Rast JP, Fugmann SD. The origins of vertebrate adaptive immunity. Nature Reviews Immunology. 2010;10(8):543–553. doi: 10.1038/nri2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cannon JP, Haire RN, Rast JP, Litman GW. The phylogenetic origins of the antigen-binding receptors and somatic diversification mechanisms. Immunological Reviews. 2004;200:12–22. doi: 10.1111/j.0105-2896.2004.00166.x. [DOI] [PubMed] [Google Scholar]
- 3.Schwarz HP, Dorner F. Karl Landsteiner and his his major contributions to haematology. British Journal of Haematology. 2003;121(4):556–565. doi: 10.1046/j.1365-2141.2003.04295.x. [DOI] [PubMed] [Google Scholar]
- 4.Hajdu SI. Blood transfusion from antiquity to the discovery of the Rh factor. Annals of Clinical and Laboratory Science. 2003;33(4):471–473. [PubMed] [Google Scholar]
- 5.Landsteiner K. Zur Kenntnis der antifermativen, lytischen und agglutinierenden Wirkungendes Blutserums und der Lymphe. Zentralbl Bakteriol Parasitenkd. 1900;27:357–363. [Google Scholar]
- 6.Landsteiner K, Wiener AS. Studies on an agglutinogen (Rh) in human blood reacting with anti-rhesus sera and with human isoantibodies. The Journal of Experimental Medicine. 1941;74:309–320. doi: 10.1084/jem.74.4.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avery OT, MacLeod CM, McCarty M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types. Induction of transformation by a desoxyribonucleic acid fraction isolated from Pneumococcus type III. Journal of Experimental Medicine. 1944;79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watson JD, Crick FHC. Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 9.Sazama K. Transfusion errors: scope of the problem, consequences, and solutions. Current Hematology Reports. 2003;2:518–521. [PubMed] [Google Scholar]
- 10.Josephson CD, Castillejo MI, Grima K, Hillyer CD. ABO-mismatched platelet transfusions: strategies to mitigate patient exposure to naturally occurring hemolytic antibodies. Transfusion and Apheresis Science. 2010;42(1):83–88. doi: 10.1016/j.transci.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 11.Salama A, Mueller-Eckhardt C. Delayed hemolytic transfusion reactions. Evidence for complement activation involving allogeneic and autologous red cells. Transfusion. 1984;24(3):188–193. doi: 10.1046/j.1537-2995.1984.24384225018.x. [DOI] [PubMed] [Google Scholar]
- 12.Flickinger C. In search of red blood cells for alloimmunized patients with sickle cell disease. Immunohematology. 2006;22(3):136–142. [PubMed] [Google Scholar]
- 13.Holers VM. The spectrum of complement alternative pathway-mediated diseases. Immunological Reviews. 2008;223(1):300–316. doi: 10.1111/j.1600-065X.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- 14.Myhre BA. The first recorded blood transfusions: 1656 to 1668. Transfusion. 1990;30(4):358–362. doi: 10.1046/j.1537-2995.1990.30490273445.x. [DOI] [PubMed] [Google Scholar]
- 15.Galili U. Xenotransplantation and ABO incompatible transplantation: the similarities they share. Transfusion and Apheresis Science. 2006;35(1):45–58. doi: 10.1016/j.transci.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 16.Sazama K. Reports of 355 transfusion-associated deaths: 1976 through 1985. Transfusion. 1990;30(7):583–590. doi: 10.1046/j.1537-2995.1990.30790385515.x. [DOI] [PubMed] [Google Scholar]
- 17.Janatpour KA, Kalmin ND, Jensen HM, Holland PV. Clinical outcomes of ABO-incompatible RBC transfusions. American Journal of Clinical Pathology. 2008;129(2):276–281. doi: 10.1309/VXY1ULAFUY6E6JT3. [DOI] [PubMed] [Google Scholar]
- 18.Bacon JM, Young IF. ABO incompatible blood transfusion. Pathology. 1989;21(3):181–184. doi: 10.3109/00313028909061055. [DOI] [PubMed] [Google Scholar]
- 19.Hendriksen CF. A short history of the use of animals in vaccine development and quality control. Developments in Biological Standardization. 1996;86:3–10. [PubMed] [Google Scholar]
- 20.Silverstein AM, Bialasiewicz AA. History of immunology. A history of theories of acquired immunity. Cellular Immunology. 1980;51(1):151–167. doi: 10.1016/0008-8749(80)90245-2. [DOI] [PubMed] [Google Scholar]
- 21.Enrlich P. Zur Theorie der Lysin Wirkung. Berliner Klinische Wochenschrift. 1899;1:6–9. [Google Scholar]
- 22.Walport MJ. Advances in immunology. Complement (second of two parts) The New England Journal of Medicine. 2001;344(15):1140–1144. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 23.Walport MJ. Advances in immunology. Complement (first of two parts) The New England Journal of Medicine. 2001;344(14):1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 24.Pangburn MK, Muller-Eberhard HJ. The alternative pathway of complement. Springer Seminars in Immunopathology. 1984;7(2-3):163–192. doi: 10.1007/BF01893019. [DOI] [PubMed] [Google Scholar]
- 25.Fujita T. Evolution of the lectin —complement pathway and its role in innate immunity. Nature Reviews Immunology. 2002;2(5):346–353. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- 26.Engelfriet CP. The immune destruction of red cells. Transfusion Medicine. 1992;2(1):1–6. doi: 10.1111/j.1365-3148.1992.tb00128.x. [DOI] [PubMed] [Google Scholar]
- 27.Lambris JD. The multifunctional role of C3, the third component of complement. Immunology Today. 1988;9(12):387–393. doi: 10.1016/0167-5699(88)91240-6. [DOI] [PubMed] [Google Scholar]
- 28.Gál P, Barna L, Kocsis A, Závodszky P. Serine proteases of the classical and lectin pathways: similarities and differences. Immunobiology. 2007;212(4-5):267–277. doi: 10.1016/j.imbio.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 29.Kang YS, Do Y, Lee HK, et al. A Dominant Complement Fixation Pathway for Pneumococcal Polysaccharides Initiated by SIGN-R1 Interacting with C1q. Cell. 2006;125(1):47–58. doi: 10.1016/j.cell.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 30.Nordahl EA, Rydengård V, Nyberg P, et al. Activation of the complement system generates antibacterial peptides. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(48):16879–16884. doi: 10.1073/pnas.0406678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward PA. Functions of C5a receptors. Journal of Molecular Medicine. 2009;87(4):375–378. doi: 10.1007/s00109-009-0442-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Müller-Eberhard HJ. The membrane attack complex of complement. Annual Review of Immunology. 1986;4:503–528. doi: 10.1146/annurev.iy.04.040186.002443. [DOI] [PubMed] [Google Scholar]
- 33.Smith CA, Pangburn MK, Vogel CW, Muller-Eberhard HJ. Molecular architecture of human properdin, a positive regulator of the alternative pathway of complement. Journal of Biological Chemistry. 1984;259(7):4582–4588. [PubMed] [Google Scholar]
- 34.Fishelson Z, Pangburn MK, Muller-Eberhard HJ. Characterization of the initial C3 convertase of the alternative pathway of human complement. Journal of Immunology. 1984;132(3):1430–1434. [PubMed] [Google Scholar]
- 35.Seino J, Eveleigh P, Warnaar S, Van Haarlem LJM, Van Es LA, Daha MR. Activation of human complement by mouse and mouse/human chimeric monoclonal antibodies. Clinical and Experimental Immunology. 1993;94(2):291–296. doi: 10.1111/j.1365-2249.1993.tb03446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Enriquez-Rincon F, Andrew E, Parkhouse RME, Klaus GGB. Suppression of follicular trapping of antigen-antibody complexes in mice treated with anti-IgM or anti-IgD antibodies from birth. Immunology. 1984;53(4):713–719. [PMC free article] [PubMed] [Google Scholar]
- 37.Carroll MC. The role of complement and complement receptors in induction and regulation of immunity. Annual Review of Immunology. 1998;16:545–568. doi: 10.1146/annurev.immunol.16.1.545. [DOI] [PubMed] [Google Scholar]
- 38.Yan J, Větvička V, Xia Y, Hanikýřová M, Mayadas TN, Ross GD. Critical role of Kupffer cell CR3 (CD11b/CD18) in the clearance of IgM-opsonized erythrocytes or soluble β-glucan. Immunopharmacology. 2000;46(1):39–54. doi: 10.1016/s0162-3109(99)00157-5. [DOI] [PubMed] [Google Scholar]
- 39.Helmy KY, Katschke KJ, Jr., Gorgani NN, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124(5):915–927. doi: 10.1016/j.cell.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 40.Khera R, Das N. Complement Receptor 1: disease associations and therapeutic implications. Molecular Immunology. 2009;46(5):761–772. doi: 10.1016/j.molimm.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham IL, Anderson DC, Holers VM, Brown EJ. Complement receptor 3 (CR3, Mac-1, integrin α(M)β2, CD11b/CD18) is required for tyrosine phosphorylation of paxillin in adherent and nonadherent neutrophils. Journal of Cell Biology. 1994;127(4):1139–1147. doi: 10.1083/jcb.127.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taborda CP, Casadevall A. CR3 (CD11b/CD18) and CR4 (CD11c/CD18) are involved in complement-independent antibody-mediated phagocytosis of Cryptococcus neoformans . Immunity. 2002;16(6):791–802. doi: 10.1016/s1074-7613(02)00328-x. [DOI] [PubMed] [Google Scholar]
- 43.Brown EJ. Complement receptors and phagocytosis. Current Opinion in Immunology. 1991;3:76–82. doi: 10.1016/0952-7915(91)90081-b. [DOI] [PubMed] [Google Scholar]
- 44.Prodeus AP, Goerg S, Shen LM, et al. A critical role for complement in maintenance of self-tolerance. Immunity. 1998;9(5):721–731. doi: 10.1016/s1074-7613(00)80669-x. [DOI] [PubMed] [Google Scholar]
- 45.Dempsey PW, Allison MED, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science. 1996;271(5247):348–350. doi: 10.1126/science.271.5247.348. [DOI] [PubMed] [Google Scholar]
- 46.Roozendaal R, Carroll MC. Complement receptors CD21 and CD35 in humoral immunity. Immunological Reviews. 2007;219(1):157–166. doi: 10.1111/j.1600-065X.2007.00556.x. [DOI] [PubMed] [Google Scholar]
- 47.Rittirsch D, Flierl MA, Nadeau BA, et al. Functional roles for C5a receptors in sepsis. Nature Medicine. 2008;14(5):551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Neill GJ, Yang SY, Tegoli J. Chido and Rodgers blood groups are distinct antigenic components of human complement C4. Nature. 1978;273(5664):668–670. doi: 10.1038/273668a0. [DOI] [PubMed] [Google Scholar]
- 49.Lublin DM, Kompelli S, Storry JR, Reid ME. Molecular basis of Cromer blood group antigens. Transfusion. 2000;40(2):208–213. doi: 10.1046/j.1537-2995.2000.40020208.x. [DOI] [PubMed] [Google Scholar]
- 50.Botto M, Kirschfink M, Macor P, Pickering MC, Würzner R, Tedesco F. Complement in human diseases: lessons from complement deficiencies. Molecular Immunology. 2009;46(14):2774–2783. doi: 10.1016/j.molimm.2009.04.029. [DOI] [PubMed] [Google Scholar]
- 51.Seya T, Atkinson JP. Functional properties of membrane cofactor protein of complement. Biochemical Journal. 1989;264(2):581–588. doi: 10.1042/bj2640581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicholson-Weller A. Decay accelerating factor (CD55) Current Topics in Microbiology and Immunology. 1992;178:7–30. doi: 10.1007/978-3-642-77014-2_2. [DOI] [PubMed] [Google Scholar]
- 53.Wilcox LA, Ezzell JL, Bernshaw NJ, Parker CJ. Molecular basis of the enhanced susceptibility of the erythrocytes of paroxysmal nocturnal hemoglobinuria to hemolysis in acidified serum. Blood. 1991;78(3):820–829. [PubMed] [Google Scholar]
- 54.Paccaud JP, Carpentier JL, Schifferli JA. Direct evidence for the clustered nature of complement receptors type 1 on the erythrocyte membrane. Journal of Immunology. 1988;141(11):3889–3894. [PubMed] [Google Scholar]
- 55.Kim DD, Song WC. Membrane complement regulatory proteins. Clinical Immunology. 2006;118(2-3):127–136. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 56.Huang Y, Fedarovich A, Tomlinson S, Davies C. Crystal structure of CD59: implications for molecular recognition of the complement proteins C8 and C9 in the membrane-attack complex. Acta Crystallographica D. 2007;63(6):714–721. doi: 10.1107/S0907444907015557. [DOI] [PubMed] [Google Scholar]
- 57.Farkas I, Baranyi L, Ishikawa Y, et al. CD59 blocks not only the insertion of C9 into MAC but inhibits ion channel formation by homologous C5b-8 as well as C5b-9. Journal of Physiology. 2002;539(2):537–545. doi: 10.1113/jphysiol.2001.013381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rollins SA, Sims PJ. The complement-inhibitory activity of CD59 resides in its capacity to block incorporation of C9 into membrane C5b-9. Journal of Immunology. 1990;144(9):3478–3483. [PubMed] [Google Scholar]
- 59.Fujita T, Gigli I, Nussenzweig V. Human C4-binding protein. II. Role in proteolysis of C4b by C3b-inactivator. Journal of Experimental Medicine. 1978;148(4):1044–1051. doi: 10.1084/jem.148.4.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dodds AW, Ren XD, Willis AC, Law SKA. The reaction mechanism of the internal thioester in the human complement component C4. Nature. 1996;379(6561):177–179. doi: 10.1038/379177a0. [DOI] [PubMed] [Google Scholar]
- 61.Gadjeva M, Dodds AW, Taniguchi-Sidle A, Willis AC, Isenman DE, Law SKA. The covalent binding reaction of complement component C3. Journal of Immunology. 1998;161(2):985–990. [PubMed] [Google Scholar]
- 62.Ferreira VP, Pangburn MK. Factor H-mediated cell surface protection from complement is critical for the survival of PNH erythrocytes. Blood. 2007;110(6):2190–2192. doi: 10.1182/blood-2007-04-083170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davis AE, III, Mejia P, Lu F. Biological activities of C1 inhibitor. Molecular Immunology. 2008;45(16):4057–4063. doi: 10.1016/j.molimm.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dempsey PW, Fearon DT. Complement: instructing the acquired immune system through the CD21/CD19 complex. Research in Immunology. 1996;147(2):71–75. doi: 10.1016/0923-2494(96)87176-8. [DOI] [PubMed] [Google Scholar]
- 65.Ollert MW, David K, Bredehorst R, Vogel CW. Classical complement pathway activation on nucleated cells: role of factor H in the control of deposited C3b. Journal of Immunology. 1995;155(10):4955–4962. [PubMed] [Google Scholar]
- 66.Heinen S, Hartmann A, Lauer N, et al. Factor H-related protein 1 (CFHR-1) inhibits complement C5 convertase activity and terminal complex formation. Blood. 2009;114(12):2439–2447. doi: 10.1182/blood-2009-02-205641. [DOI] [PubMed] [Google Scholar]
- 67.He JQ, Wiesmann C, van Lookeren Campagne M. A role of macrophage complement receptor CRIg in immune clearance and inflammation. Molecular Immunology. 2008;45(16):4041–4047. doi: 10.1016/j.molimm.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 68.Duncan AR, Winter G. The binding site for C1q on IgG. Nature. 1988;332(6166):738–740. doi: 10.1038/332738a0. [DOI] [PubMed] [Google Scholar]
- 69.Weiner EM. On the interaction of the first complement component C1 and its subunit C1q with solid-phase IgM immune complexes. Scandinavian Journal of Immunology. 1988;28(4):425–430. doi: 10.1111/j.1365-3083.1988.tb01471.x. [DOI] [PubMed] [Google Scholar]
- 70.Swanson SM, Dombrink-Kurtzman MA, Voss EW., Jr. C1q binding by a high affinity anti-fluorescein murine monoclonal IgM antibody and monomeric subunits. Molecular Immunology. 1988;25(6):545–554. doi: 10.1016/0161-5890(88)90076-4. [DOI] [PubMed] [Google Scholar]
- 71.Schreiber AD, Frank MM. Role of antibody and complement in the immune clearance and destruction of erythrocytes. II. Molecular nature of IgG and IgM complement-fixing sites and effects of their interaction with serum. Journal of Clinical Investigation. 1972;51(3):583–589. doi: 10.1172/JCI106847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jing QF, Mozdzanowska K, Gerhard W. Complement component C1q enhances the biological activity of influenza virus hemagglutinin-specific antibodies depending on their fine antigen specificity and heavy-chain isotype. Journal of Virology. 2002;76(3):1369–1378. doi: 10.1128/JVI.76.3.1369-1378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valim YML, Lachmann PJ. The effect of antibody isotype and antigenic epitope density on the complement-fixing activity of immune complexes: a systematic study using chimaeric anti-NIP antibodies with human Fc regions. Clinical and Experimental Immunology. 1991;84(1):1–8. doi: 10.1111/j.1365-2249.1991.tb08115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tao MH, Canfield SM, Morrison SL. The differential ability of human IgG1 and IgG4 to activate complement is determined by the COOH-terminal sequence of the C(H)2 domain. Journal of Experimental Medicine. 1991;173(4):1025–1028. doi: 10.1084/jem.173.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roos A, Bouwman LH, Van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the Mannan-Binding lectin pathway. Journal of Immunology. 2001;167(5):2861–2868. doi: 10.4049/jimmunol.167.5.2861. [DOI] [PubMed] [Google Scholar]
- 76.Wright A, Morrison SL. Effect of C2-associated carbohydrate structure on Ig effector function: studies with chimeric mouse-human IgG1 antibodies in glycosylation mutants of Chinese hamster ovary cells. Journal of Immunology. 1998;160(7):3393–3402. [PubMed] [Google Scholar]
- 77.Malhotra R, Wormald MR, Rudd PM, Fischer PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nature Medicine. 1995;1(3):237–243. doi: 10.1038/nm0395-237. [DOI] [PubMed] [Google Scholar]
- 78.Hennessey PJ, Black CT, Andrassy RJ. Nonenzymatic glycosylation of immunoglobulin G impairs complement fixation. Journal of Parenteral and Enteral Nutrition. 1991;15(1):60–64. doi: 10.1177/014860719101500160. [DOI] [PubMed] [Google Scholar]
- 79.Cutbush M, Mollison PL. Relation between characteristics of blood-group antibodies in vitro and associated patterns of redcell destruction in vivo. British Journal of Haematology. 1958;4(2):115–137. doi: 10.1111/j.1365-2141.1958.tb03843.x. [DOI] [PubMed] [Google Scholar]
- 80.Howard JE, Winn LC, Gottlieb CE. Clinical significance of the anti-complement component of antiglobulin antisera. Transfusion. 1982;22(4):269–272. doi: 10.1046/j.1537-2995.1982.22482251204.x. [DOI] [PubMed] [Google Scholar]
- 81.Jones NC, Mollison PL, Veall N. Removal of incompatible red cells by the spleen. British Journal of Haematology. 1957;3(2):125–133. doi: 10.1111/j.1365-2141.1957.tb05779.x. [DOI] [PubMed] [Google Scholar]
- 82.Schirmer DA, Song SC, Baliff JP, et al. Mouse models of IgG- and IgM-mediated hemolysis. Blood. 2007;109(7):3099–3107. doi: 10.1182/blood-2006-08-040139. [DOI] [PubMed] [Google Scholar]
- 83.Liepkalns JS, Hod EA, Stowell SR, Cadwell CM, Spitalnik SL, Zimring JC. Biphasic clearance of incompatible red blood cells through a novel mechanism requiring neither complement nor Fcgamma receptors in a murine model. doi: 10.1111/j.1537-2995.2012.03647.x. Transfusion. In press. [DOI] [PubMed] [Google Scholar]
- 84.Kumpel BM. Monoclonal anti-D development programme. Transplant Immunology. 2002;10(2-3):199–204. doi: 10.1016/s0966-3274(02)00066-7. [DOI] [PubMed] [Google Scholar]
- 85.Gorick BD, Hughes-Jones NC. Relative functional binding activity of IgG1 and IgG3 anti-D in IgG preparations. Vox Sanguinis. 1991;61(4):251–254. doi: 10.1111/j.1423-0410.1991.tb00955.x. [DOI] [PubMed] [Google Scholar]
- 86.Coopamah MD, Freedman J, Semple JW. Anti-D initially stimulates an Fc-dependent leukocyte oxidative burst and subsequently suppresses erythrophagocytosis via interleukin-1 receptor antagonist. Blood. 2003;102(8):2862–2867. doi: 10.1182/blood-2003-04-1029. [DOI] [PubMed] [Google Scholar]
- 87.Fudenberg H, Allen FH. Transfusion reactions in the absence of demonstrable incompatibility. The New England Journal of Medicine. 1957;256(25):1180–1184. doi: 10.1056/NEJM195706202562504. [DOI] [PubMed] [Google Scholar]
- 88.Greenbury CL, Moore DH, Nunn LA. The reaction with red cells of 7s rabbit antibody, its sub-units and their recombinants. Immunology. 1965;8:420–431. [PMC free article] [PubMed] [Google Scholar]
- 89.Economidou J, Hughes-Jones NC, Gardner B. Quantitative measurements concerning A and B antigen sites. Vox Sanguinis. 1967;12(5):321–328. doi: 10.1111/j.1423-0410.1967.tb03362.x. [DOI] [PubMed] [Google Scholar]
- 90.Rochna E, Hughes-Jones NC. The use of purified 125-I-labelled anti-gamma globulin in the quantitative determination of D-antigen sites. Bibliotheca Haematologica. 1965;23:490–491. doi: 10.1159/000384295. [DOI] [PubMed] [Google Scholar]
- 91.Rochna E, Hughes-Jones NC. The use of purified 125-I-labelled anti-gamma globulin in the determination of the number of D antigen sites on red cells of different phenotypes. Vox Sanguinis. 1965;10(6):675–686. doi: 10.1111/j.1423-0410.1965.tb05179.x. [DOI] [PubMed] [Google Scholar]
- 92.Hughes-Jones NC, Gardner B. The Kell system studied with radioactively-labelled anti-K. Vox Sanguinis. 1971;21(2):154–158. doi: 10.1111/j.1423-0410.1971.tb00572.x. [DOI] [PubMed] [Google Scholar]
- 93.Nicolas V, Van Kim CL, Gane P, et al. Rh-RhAG/ankyrin-R, a new interaction site between the membrane bilayer and the red cell skeleton, is impaired by Rhnull-associated mutation. Journal of Biological Chemistry. 2003;278(28):25526–25533. doi: 10.1074/jbc.M302816200. [DOI] [PubMed] [Google Scholar]
- 94.Lee S, Russo D, Redman C. Functional and structural aspects of the Kell blood group system. Transfusion Medicine Reviews. 2000;14(2):93–103. doi: 10.1016/s0887-7963(00)80001-2. [DOI] [PubMed] [Google Scholar]
- 95.Westhoff CM, Reid ME. Review: the Kell, Duffy, and Kidd blood group systems. Immunohematology. 2004;20(1):37–49. [PubMed] [Google Scholar]
- 96.Westhoff CM. The structure and function of the Rh antigen complex. Seminars in Hematology. 2007;44(1):42–50. doi: 10.1053/j.seminhematol.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gardas A, Kościelak J. A, B and H blood group specificities in glycoprotein and glycolipid fractions of human erythrocyte membrane. Absence of blood group active glycoproteins in the membranes of non-secretors. Vox Sanguinis. 1971;20(2):137–149. doi: 10.1111/j.1423-0410.1971.tb00546.x. [DOI] [PubMed] [Google Scholar]
- 98.Springer GF, Williamson P, Brandes WC. Blood group activity of gram-negative bacteria. The Journal of Experimental Medicine. 1961;113:1077–1093. doi: 10.1084/jem.113.6.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Springer GF, Horton RE. Blood group isoantibody stimulation in man by feeding blood group-active bacteria. Journal of Clinical Investigation. 1969;48(7):1280–1291. doi: 10.1172/JCI106094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Storry JR, Olsson ML. The ABO blood group system revisited: a review and update. Immunohematology. 2009;25(2):48–59. [PubMed] [Google Scholar]
- 101.Woodrow JC, Finn R, Krevans JR. Rapid clearance of Rh positive blood during experimental Rh immunisation. Vox Sanguinis. 1969;17(5):349–361. doi: 10.1111/j.1423-0410.1969.tb00407.x. [DOI] [PubMed] [Google Scholar]
- 102.Samson D, Mollison PL. Effect on primary Rh immunization of delayed administration of anti-Rh. Immunology. 1975;28(2):349–357. [PMC free article] [PubMed] [Google Scholar]
- 103.Devey ME, Voak D. A critical study of the IgG subclasses of Rh anti D antibodies formed in pregnancy and in immunized volunteers. Immunology. 1974;27(6):1073–1079. [PMC free article] [PubMed] [Google Scholar]
- 104.Wagener FADTG, Eggert A, Boerman OC, et al. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98(6):1802–1811. doi: 10.1182/blood.v98.6.1802. [DOI] [PubMed] [Google Scholar]
- 105.Simionatto CS, Cabal R, Jones RL, Galbraith RA. Thrombophlebitis and disturbed hemostasis following administration of intravenous hematin in normal volunteers. American Journal of Medicine. 1988;85(4):538–540. doi: 10.1016/s0002-9343(88)80092-5. [DOI] [PubMed] [Google Scholar]
- 106.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. Journal of the American Medical Association. 2005;293(13):1653–1662. doi: 10.1001/jama.293.13.1653. [DOI] [PubMed] [Google Scholar]
- 107.Baek JH, D 'Agnillo F, Vallelian F, et al. Hemoglobin-driven pathophysiology is an in vivo consequence of the red blood cell storage lesion that can be attenuated in guinea pigs by haptoglobin therapy. The Journal of Clinical Investigation. 2012;122:1444–1458. doi: 10.1172/JCI59770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Krevans JR, Jackson DP, Conley CL, Hartmann RC. The nature of the hemorrhagic disorder accompanying hemolytic transfusion reactions in man. Blood. 1957;12(9):834–843. [PubMed] [Google Scholar]
- 109.Josephson CD, Mullis NC, Van Demark C, Hillyer CD. Significant numbers of apheresis-derived group O platelet units have “high-titer” anti-A/A,B: implications for transfusion policy. Transfusion. 2004;44(6):805–808. doi: 10.1111/j.1537-2995.2004.03290.x. [DOI] [PubMed] [Google Scholar]
- 110.Brown DL, Lachmann PJ, Dacie JV. The in vivo behaviour of complement-coated red cells: studies in C6-deficient, C3-depleted and normal rabbits. Clinical and Experimental Immunology. 1970;7(3):401–421. [PMC free article] [PubMed] [Google Scholar]
- 111.Meryhew NL, Runquist OA. A kinetic analysis of immune-mediated clearance of erythrocytes. Journal of Immunology. 1981;126(6):2443–2449. [PubMed] [Google Scholar]
- 112.Ruddy S, Austen KF. C3b inactivator of man. II. Fragments produced by C3b inactivator cleavage of cell-bound or fluid phase C3b. Journal of Immunology. 1971;107(3):742–750. [PubMed] [Google Scholar]
- 113.Law SK, Fearon DT, Levine RP. Action of the C3b-inactivator on cell-bound C3b. Journal of Immunology. 1979;122(3):759–765. [PubMed] [Google Scholar]
- 114.Lachmann PJ, Pangburn MK, Oldroyd RG. Breakdown of C3 after complement activation. Identification of a new fragment, C3g, using monoclonal antibodies. Journal of Experimental Medicine. 1982;156(1):205–216. doi: 10.1084/jem.156.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Engelfriet CP, Von dem Borne AE, Beckers D, Reynierse E, Van Loghem JJ. Autoimmune haemolytic anaemias. V. studies on the resistance against complement haemolysis of the red cells of patients with chronic cold agglutinin disease. Clinical and Experimental Immunology. 1972;11(2):255–264. [PMC free article] [PubMed] [Google Scholar]
- 116.Jaffe CJ, Atkinson JP, Frank MM. The role of complement in the clearance of cold agglutinin sensitized erythrocytes in man. Journal of Clinical Investigation. 1976;58(4):942–949. doi: 10.1172/JCI108547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Akeroyd JH, O’Brien WA. Survival of group AB red cells in a group A recipient. Vox Sanguinis. 1958;3(5):330–335. doi: 10.1111/j.1423-0410.1958.tb04014.x. [DOI] [PubMed] [Google Scholar]
- 118.Jandl JH, Simmons RL. The agglutination and sensitization of red cells by metallic cations: interactions between multivalent metals and the red-cell membrane. British Journal of Haematology. 1957;3:19–38. doi: 10.1111/j.1365-2141.1957.tb05768.x. [DOI] [PubMed] [Google Scholar]
- 119.Qin X, Goldfine A, Krumrei N, et al. Glycation inactivation of the complement regulatory protein CD59: a possible role in the pathogenesis of the vascular complications of human diabetes. Diabetes. 2004;53(10):2653–2661. doi: 10.2337/diabetes.53.10.2653. [DOI] [PubMed] [Google Scholar]
- 120.Waitumbi JM, Donvito B, Kisserli A, Cohen JHM, Stoute JA. Age-related changes in red blood cell complement regulatory proteins and susceptibility to severe malaria. Journal of Infectious Diseases. 2004;190(6):1183–1191. doi: 10.1086/423140. [DOI] [PubMed] [Google Scholar]
- 121.Win N, Doughty H, Telfer P, Wild BJ, Pearson TC. Hyperhemolytic transfusion reaction in sickle cell disease. Transfusion. 2001;41(3):323–328. doi: 10.1046/j.1537-2995.2001.41030323.x. [DOI] [PubMed] [Google Scholar]
- 122.Salama A, Mueller-Eckhardt C. Binding of fluid phase C3b to nonsensitized bystander human red cells. A model for in vivo effects of complement activation on blood cells. Transfusion. 1985;25(6):528–534. doi: 10.1046/j.1537-2995.1985.25686071424.x. [DOI] [PubMed] [Google Scholar]
- 123.Wang RH, Phillips G, Jr., Medof ME, Mold C. Activation of the alternative complement pathway by exposure of phosphatidylethanolamine and phosphatidylserine on erythrocytes from sickle cell disease patients. Journal of Clinical Investigation. 1993;92(3):1326–1335. doi: 10.1172/JCI116706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Test ST, Woolworth VS. Defective regulation of complement by the sickle erythrocyte: evidence for a defect in control of membrane attack complex formation. Blood. 1994;83(3):842–852. [PubMed] [Google Scholar]
- 125.Reyes MA, Illoh OC. Hyperhemolytic transfusion reaction attributable to anti-Fy3 in a patient with sickle cell disease. Immunohematology. 2008;24(2):45–51. [PubMed] [Google Scholar]
- 126.Strait RT, Hicks W, Barasa N, et al. MHC class I-specific antibody binding to nonhematopoietic cells drives complement activation to induce transfusion-related acute lung injury in mice. The Journal of Experimental Medicine. 2011;208:2525–2544. doi: 10.1084/jem.20110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shaz BH, Stowell SR, Hillyer CD. Transfusion-related acute lung injury: from bedside to bench and back. Blood. 2011;117(5):1463–1471. doi: 10.1182/blood-2010-04-278135. [DOI] [PubMed] [Google Scholar]
- 128.Silliman CC, Dzieciatkowska M, Moore EE, et al. Proteomic analyses of human plasma: Venus versus Mars. Transfusion. 2012;52:417–424. doi: 10.1111/j.1537-2995.2011.03316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sahu A, Kay BK, Lambris JD. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. Journal of Immunology. 1996;157(2):884–891. [PubMed] [Google Scholar]
- 130.Inagi R, Miyata T, Hong K, Pramoonjago P, Maeda K, Inoue K. Decreased activity of complement-mediated immune complex clearance in hemodialysis patients. Clinical Immunology and Immunopathology. 1993;68(3):333–339. doi: 10.1006/clin.1993.1135. [DOI] [PubMed] [Google Scholar]
- 131.Miyata T, Fujita Y, Inagi R, Inoue I, Sugiyama S, Maeda K. Effectiveness of nafamostat mesilate on glomerulonephritis in immune-complex diseases. The Lancet. 1993;341(8856):p. 1353. doi: 10.1016/0140-6736(93)90869-i. [DOI] [PubMed] [Google Scholar]
- 132.Weisman HF, Bartow T, Leppo MK, et al. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249(4965):146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 133.Piddlesden SJ, Jiang S, Levin JL, Vincent A, Morgan BP. Soluble complement receptor 1 (sCR1) protects against experimental autoimmune myasthenia gravis. Journal of Neuroimmunology. 1996;71(1-2):173–177. doi: 10.1016/s0165-5728(96)00144-0. [DOI] [PubMed] [Google Scholar]
- 134.Pruitt SK, Baldwin WM, III, Marsh HC, Jr., Lin SS, Yeh CG, Bollinger RR. The effect of soluble complement receptor type 1 on hyperacute xenograft rejection. Transplantation. 1991;52(5):868–873. doi: 10.1097/00007890-199111000-00022. [DOI] [PubMed] [Google Scholar]
- 135.Roos A, Nauta AJ, Broers D, et al. Specific inhibition of the classical complement pathway by C1q-binding peptides. Journal of Immunology. 2001;167(12):7052–7059. doi: 10.4049/jimmunol.167.12.7052. [DOI] [PubMed] [Google Scholar]
- 136.Mqadmi A, Zheng X, Song J, Abramowitz S, Giclas P, Yazdanbakhsh K. Prevention of complement-mediated immune hemolysis by a small molecule compound. Biochemical and Biophysical Research Communications. 2004;325(4):1465–1471. doi: 10.1016/j.bbrc.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 137.Mqadmi A, Abdullah Y, Yazdanbakhsh K. Characterization of complement receptor 1 domains for prevention of complement-mediated red cell destruction. Transfusion. 2005;45(2):234–244. doi: 10.1111/j.1537-2995.2004.04163.x. [DOI] [PubMed] [Google Scholar]
- 138.Spitzer D, Unsinger J, Bessler M, Atkinson JP. ScFv-mediated in vivo targeting of DAF to erythrocytes inhibits lysis by complement. Molecular Immunology. 2004;40(13):911–919. doi: 10.1016/j.molimm.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 139.Basta M, Langlois PF, Marques M, Frank MM, Fries LF. High-dose intravenous immunoglobulin modifies complement-mediated in vivo clearance. Blood. 1989;74(1):326–333. [PubMed] [Google Scholar]
- 140.Basta M, Fries LF, Frank MM. High doses of intravenous Ig inhibit in vitro uptake of C4 fragments onto sensitized erythrocytes. Blood. 1991;77(2):376–380. [PubMed] [Google Scholar]
- 141.Hillmen P, Hall C, Marsh JCW, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. The New England Journal of Medicine. 2004;350(6):552–559. doi: 10.1056/NEJMoa031688. [DOI] [PubMed] [Google Scholar]
- 142.Moller G. Survival of H-2 incompatible mouse erythrocytes in untreated and isoimmune recipients. Immunology. 1965;8:360–374. [PMC free article] [PubMed] [Google Scholar]
- 143.Evans RS, Turner E, Bingham M. Chronic hemolytic anemia due to cold agglutinins: the mechanism of resistance of red cells to C’ hemolysis by cold agglutinins. Journal of Clinical Investigation. 1967;46(9):1461–1474. doi: 10.1172/JCI105638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Evans RS, Turner E, Bingham M, Woods R. Chronic hemolytic anemia due to cold agglutinins. II. The role of C’ in red cell destruction. Journal of Clinical Investigation. 1968;47(4):691–701. doi: 10.1172/JCI105764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mollison PL, Cutbush M. Use of isotope-labelled red cells to demonstrate incompatibility in vivo. The Lancet. 1955;265(6878):1290–1295. doi: 10.1016/s0140-6736(55)92057-2. [DOI] [PubMed] [Google Scholar]
- 146.Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51(3):503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 147.Mak TW, Penninger JM, Ohashi PS. Knockout mice: a paradigm shift in modern immunology. Nature Reviews Immunology. 2001;1(1):11–19. doi: 10.1038/3509551. [DOI] [PubMed] [Google Scholar]
- 148.Campbell-Lee SA, Liu J, Velliquette RW, et al. The production of red blood cell alloantibodies in mice transfused with blood from transgenic Fyb-expressing mice. Transfusion. 2006;46(10):1682–1688. doi: 10.1111/j.1537-2995.2006.00966.x. [DOI] [PubMed] [Google Scholar]
- 149.Desmarets M, Cadwell CM, Peterson KR, Neades R, Zimring JC. Minor histocompatibility antigens on transfused leukoreduced units of red blood cells induce bone marrow transplant rejection in a mouse model. Blood. 2009;114(11):2315–2322. doi: 10.1182/blood-2009-04-214387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Smith NH, Henry KL, Cadwell CM, et al. Generation of transgenic mice with antithetical KEL1 and KEL2 human blood group antigens on red blood cells. doi: 10.1111/j.1537-2995.2012.03641.x. Transfusion. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Smith NH, Hod EA, Spitalnik SL, Zimring JC, Hendrickson JE. Transfusion in the absence of inflammation induces antigen-specific tolerance to murine RBCs. Blood. 2012;119:1566–1569. doi: 10.1182/blood-2011-09-382655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. US Dept of Health and Human Services, Fatalities Reported to the FDA Following Blood Collection, 2011.