

Abstract
Noroviruses (NoVs) are the leading cause of outbreaks of sporadic acute gastroenteritis worldwide in humans of all ages. They are important cause of hospitalizations in children with a public health impact similar to that of Rotavirus. NoVs are RNA viruses of great genetic diversity and there is a continuous appearance of new strains. Five genogroups are recognized; GI and GII with their many genotypes and subtypes being the most important for human infection. However, the diagnosis of these two genotypes remains problematic, delaying diagnosis and treatment. 1, 2, 3
For RNA extraction from stool specimens the most commonly used method is the QIAmp Viral RNA commercial kit from Qiagen. This method combines the binding properties of a silica gel membrane, buffers that control RNases and provide optimum binding of the RNA to the column together with the speed of microspin. This method is simple, fast and reliable and is carried out in a few steps that are detailed in the description provided by the manufacturer.
Norovirus is second only to rotavirus as the most common cause of diarrhea. Norovirus diagnosis should be available in all studies on pathogenesis of diarrhea as well as in outbreaks or individual diarrhea cases. At present however norovirus diagnosis is restricted to only a few centers due to the lack of simple methods of diagnosis. This delays diagnosis and treatment 1, 2, 3. In addition, due to costs and regulated transportation of corrosive buffers within and between countries use of these manufactured kits poses logistical problems. As a result, in this protocol we describe an alternative, economic, in-house method which is based on the original Boom et al. method4 which uses the nucleic acid binding properties of silica particles together with the anti-nuclease properties of guanidinium thiocyanate.
For the detection and genogrouping (GI and GII) of NoVs isolates from stool specimens, several RT-PCR protocols utilizing different targets have been developed. The consensus is that an RT-PCR using TaqMan chemistry would be the best molecular technique for diagnosis, because it combines high sensitivity, specificity and reproducibility with high throughput and ease of use. Here we describe an assay targeting the open reading frame 1 (ORF1)-ORF2 junction region; the most conserved region of the NoV genome and hence most suitable for diagnosis. For further genetic analysis a conventional RT-PCR that targets the highly variable N-terminal-shell from the major protein of the capsid (Region C) using primers originally described by Kojima et al.5 is detailed. Sequencing of the PCR product from the conventional PCR enables the differentiation of genotypes belonging to the GI and GII genogroups.
Keywords: Virology, Issue 65, Medicine, Genetics, norovirus, gastroenteritis, RNA extraction, diarrhea, stool samples, PCR, RT-PCR, TaqMan, silica
Protocol
1. Stool Samples
Stool samples should be stored frozen to preserve the RNA. To make a 10% fecal suspension, take approximately 0.1 g of thawed stool sample and complete to 1 ml with PBS.
Aliquot in 200 μl to avoid repeated freezing and thawing. Store the aliquots at -70 °C.
Thaw and centrifuge aliquots at 4,000 g for 10 min before use in extraction.
2. Preparation of Silica Particles for Extraction with Guanidine & Silica
At RT, suspend 30 g of silicon dioxide and complete with dH2O up to 250 mL.
After 24 hrs of sedimentation, remove 215 mL of the supernatant by suction. Add dH2O up to 250 mL, and suspend the silica pellet by shaking.
After another 24 hrs, remove the supernatant by suction, and adjust the pH of the silica suspension to pH 2.0, using hydrochloric acid (HCl). Aliquot the silica suspension in glass bottles and autoclave.
3. Preparation of L6 Buffer for Extraction with Guanidine & Silica
At RT, prepare L6 buffer by dissolving 60 g of guanidinium thiocyanate (GuSCN) and 1.3 g of Triton X-100 in 50 mL of 0.1 M Tris hydrochloride, pH 6.4, and 11 mL of a 0.2 M EDTA, pH 8.0 solution.
4. Preparation of L2 Buffer for Extraction with Guanidine & Silica
At RT, prepare L2 buffer by dissolving 180 g of guanidinium thiocyanate (GuSCN) in 150 mL of 0.1 M Tris hydrochloride, pH 6.4.
5. Extraction Procedure
In each extraction a NoV positive stool sample, as positive control, and DEPC treated water, as negative control, should be included.
In a micro-centrifuge tube mix the following; 1 mL of buffer L6, 20 μL of silica particles, and add 200 μL of the 10% fecal extract suspension. Vortex briefly and leave at RT for 15 min.
Centrifuge the suspension at 6000 g for 10 s and wash the pellet with 1ml of buffer L2 twice, followed by two washes with 70% ethanol and one time with acetone. Dry the pellet in a dry heating block at 56 °C for 5 min.
Hydrate the RNA in the silica pellet by adding 50 μL of RNase-free dH20. Add 1 μL of RNasin, mix by inverting the tube and incubate at 56 °C for 15 min.
Centrifuge for 3 min at full speed, in a tabletop centrifuge, and collect 40 μL of the supernatant containing the RNA.
Quantify the extracted RNA samples using Nanodrop 2000 spectrophotometer (Thermo scientific). Then store at -70 °C until use, up to 6 months. (Note: A better way to preserve total RNA intact is converting it into cDNA).
6. Alternative Extraction Using the Commercial RNA QIAamp Viral RNA Kit
Full instructions are found in the booklet provided with the QIAGEN kit. Briefly the procedure is as follows:
Pipet 560 μL of Buffer AVL containing the carrier RNA into a 1.5 mL tube, and add 140 μL of the 10% stool suspension. Mix by pulse-vortexing for 15 sec. Incubate at RT for 10 min.
Add 560 μL of ethanol (96-100%) to the sample and mix by pulse-vortexing for 15 sec, then briefly centrifuge.
Apply 630 μL of this solution to a spin column placed in a 2 mL collection tube. Centrifuge for 1 min at 6,000 g; then place the spin column into a clean 2 mL tube.
Wash the column containing bound RNA with 500 μL of Buffer AW1 and centrifuge at 6,000 g for 1 min. Place column in a clean 2 mL collection tube.
Wash the column with 500 μL of Buffer AW2 and centrifuge at full speed (20,000 g or 14,000 rpm) for 3 min.
Place spin column in a clean 1.5 mL micro-centrifuge tube, add 40 μL DEPC-treated water and elute the RNA at full speed. Repeat once for a final 80 μL of eluted RNA.
Quantify the extracted RNA samples using Nanodrop 2000 spectrophotometer (Thermo scientific). Then store at -70 °C until use, up to 6 months. (Note: A better way to preserve total RNA intact is converting it into cDNA).
7. Detection of Norovirus in Extracted RNA by One-step Real Time RT-PCR with Specific Taqman Probes
Instrument StepOnePlus Real Time PCR Systems (Applied Biosystems).
Methodology
Wipe and Clean all work surfaces, pipettes, and centrifuges with RNase AWAY to remove any potential RNase contamination.
Pipet the NoV GI and GII screening master mixes according to Table 1. Primers and Taqman probes are listed in Table 2.
Aliquot 10 μL of appropriate master mix to each reaction tube.
Add 5 μL of undiluted unknown sample RNA, DEPC-treated water as negative control, or GI respectively GII positive control RNA, to the corresponding reaction wells. All samples should be run in duplicate. Positive controls and standards with concentration of 300 μg/μL and 500 μg/μL respectively were provided by National Calicivirus Laboratory Center for Disease Control and Prevention (CDC).
Centrifuge the reaction plate at 6,000 g for 10 sec.
In the experiment properties screen; define and select a type of experiment for the run. Make sure TaqMan reagents are displayed as the reagents type, and that Pre-PCR read and amplification are selected.
Run 15 μL reactions using the thermal profile in Table 3.
Analyze the results.
8. Genotyping of NoV GI and GII Using Conventional RT-PCR and Sequencing
(It's not mentioned in this video since it's a common and widely used method.)
Instrument. Applied Biosystems StepOne and StepOnePlus Real Time PCR Systems with the Quantitec template.
Methodology
Before genotyping, the genogroup (GI or GII) of the samples are determined by the screening RT-PCR described above. GI NoV RNA has to be amplified with the GI genotyping master mix and the GII NoV RNA with the GII genotyping master mix.
Pipet the NoV GI and GII genotyping master mixes according to Table 4. Primers GI SKF/SKR and G2 SKF/SKR are listed in Table 5.
Aliquot 45 μL of the appropriate master mix to 0.2 mL reaction tubes.
Add 5 μL of DEPC-treated water to each negative control tube, and 5 μL of pre-screened, NoV (GI and/or GII) positive RNA to the corresponding reaction tube.
Run 50 μL reactions using the thermal profile in Table 6.
Send PCR products containing at least 100 ng/μL of the amplified capsid region to Macrogen Company for purification and sequencing. (9700 Great Seneca Hwy. Rockville, MD 20850 and $10 per sample per sequencing).
9. Representative Results
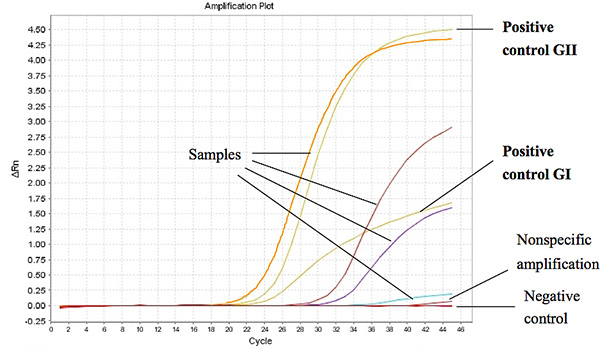 Figure 1 shows representative results from the Taqman One-Step RT-PCR when used to assay RNA extracted from stool samples from diarrheal children. The threshold cycle (Ct) for a positive sample was set at less or equal to Ct 37 for GI and Ct 39 for GII. The Ct-values for the positive controls were found to be less than 27 and 18 of the ORF1-ORF2 junction for GI and GII, respectively. Click here to view larger figure.
Figure 1 shows representative results from the Taqman One-Step RT-PCR when used to assay RNA extracted from stool samples from diarrheal children. The threshold cycle (Ct) for a positive sample was set at less or equal to Ct 37 for GI and Ct 39 for GII. The Ct-values for the positive controls were found to be less than 27 and 18 of the ORF1-ORF2 junction for GI and GII, respectively. Click here to view larger figure.
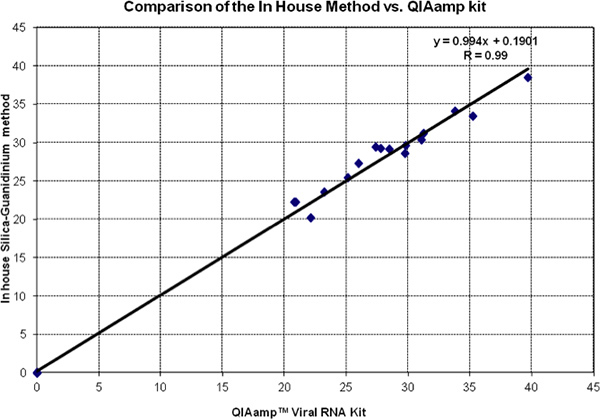 Figure 2 shows a head-to-head comparison of Ct values of in silica method guanidinium and QiAmp Viral RNA kit. Data of both tests fall very close to the equality line (slope = 1 and intercept = 0). This is confirmed by the Regression Analysis and the Correlation Coefficient. All negative controls were evaluated and found to be 0 by both methods.
Figure 2 shows a head-to-head comparison of Ct values of in silica method guanidinium and QiAmp Viral RNA kit. Data of both tests fall very close to the equality line (slope = 1 and intercept = 0). This is confirmed by the Regression Analysis and the Correlation Coefficient. All negative controls were evaluated and found to be 0 by both methods.
| Master Mix | Final Conc | Volume (μL) |
| H20 PCR | 2.875 | |
| Quantitect RT-PCR Master Mix | 1x | 6.250 |
| RT Mix | 1x | 0.125 |
| 50 μM Cog R primer | 1 μM | 0.250 |
| 50 μM Cog F primer | 1 μM | 0.250 |
| 10 μM Ring probe | 1 μM | 0.125/0.250* |
| 10.00 |
* 0.250 μL of each probe was used for GI testing, while 0.125 μL of each probe was used for GII testing.
Table 1. Qiagen Quantitect master mix for screening GI and GII (Trujillo et al., 2006).7
| Name | Geno- group | Use | Sequence (5' to 3') |
| Cog 1R | GI | Primer | CTT AGA CGC CAT CAT CAT TYA C |
| Cog 1F | GI | Primer | CGY TGG ATG CGN TTY CAT GA |
| Cog 2R | GII | Primer | TCG ACG CCA TCT TCA TTC ACA |
| Cog 2F | GII | Primer | CAR GAR BCN ATG TTY AGR TGG ATG AG |
| Ring 1A | GI | Probe | FAM-AGA TYG CGA TCY CCT GTC CA-BHQ-1 |
| Ring 1B | GI | Probe | FAM-AGA TCG CGG TCT CCT GTC CA-BHQ-1 |
| Ring2-TP | GII | Probe | FAM-TGG GAG GGC GAT CGC AAT CT-BHQ-1 |
Table 2. Primer and probe oligonucleotides used for real-time quantitative RT-PCR for genogroups I, II (Kageyama et al., 2003).8
| Step | Temp (°C) | Time (min) | |
| 1 | 50 | 30:00 | cDNA synthesis |
| 2 | 95 | 15:00 | HotStart Taq polymerase activation |
| 3 | 95 | 00:15 | |
| 60 | 01:00 | 45X cycles |
Table 3. Thermal profile for One-step Taqman Real time RT-PCR (Trujillo et al., 2006).7
| Master Mix | Final Conc | Volume (μL) |
| H20 PCR | 29.50 | |
| 5X Qiagen RT-PCR Buffer | 1x | 10.00 |
| 10 mM dNTP Mix | 0.4 mM | 2.00 |
| Enzyme Mix | 2.00 | |
| 40 U/μL RNAsin | 20 U/μL | 0.50 |
| 10 μM SKF | 0.1 μM | 0.50 |
| 10 μM SKR | 0.1 μM | 0.50 |
| 45.00 |
Table 4. Qiagen One-Step RT-PCR master mix for genotyping GI and GII.
| Name | Geno- group | Use | Sequence (5' to 3') |
| G1SKF | GI | Primer | 5'- CTG CCC GAA TTY GTA AAT GA - 3 |
| G1SKR | GI | Primer | 5'- CCA ACC CAR CCA TTR TAC A -'3 |
| G2SKF | GII | Primer | 5'- CNT GGG AGG GCG ATC GCA A - 3 |
| G2SKR | GII | Primer | 5'- CCR CCN GCA TRH CCR TTR TA CAT- 3 |
Table 5. Primers used for amplification of Region C of Capsid Region (Kojima et al., 2002).5
| Step | Temp (°C) | Time (min) | |
| 1 | 60 | 30:00 | cDNA synthesis |
| 2 | 96 | 15:00 | HotStart Taq polymerase activation |
| 3 | 94 | 00:30 | |
| 52 | 01:00 | 40X cycles | |
| 72 | 00:30 | ||
| 4 | 72 | 10:00 | Annealing |
Table 6. Thermal profile for Taqman One-Step RT-PCR.
Discussion
Using the economic in-house method for isolating nucleic acid from stool samples, we obtain equal results as with the commercial QIAmp Viral RNA kit from Qiagen, and together with the TaqMan RT-PCR developed in our laboratory we can detect a broad range of NoV genotypes belonging to the GI and GII genogroup. A recent publication of the region C protocol reported genotyping rates of 78%6. Since the diversity of the contemporary NoV strains has been increasing during previous years, the success rate will depend on the mismatches of primers in region C for the specimens being tested. The assay is useful for routine diagnostic as it eliminates post-amplification product processing, thus shortening the turn around time7. Apart from diagnosis, this protocol can also be used for clarifying the epidemiology of NoV infections, thus being useful for public health control of this disease.
When running the screening RT-PCR, negative control (NC) reactions for primer/probe sets should not exhibit fluorescence growth curves that cross the threshold line. If a false positive occurs with one or more of the primer/probe set's NC reactions, sample contamination may have occurred, and in all such cases, the whole run was invalidated and repeated with stricter adherence to the guidelines.
Cross reaction for the screening RT-PCR was tested using the GI and GII positive standards provided by CDC. No cross reaction was observed between GI primers and probes with GII RNA and vice versa. The reproducibility of the screening RT-PCR was high as Ct-values where similar for repeated runs with RNA from the positive controls. RNA from children stool samples that had been found positive for NoV and had Ct-values between 20 and 33 where subsequently amplified by the conventional RT-PCR and sent for genotyping. Five positive samples with Ct-values between 35 and 38 were not sent for sequencing as the viral load where found to be too low for subsequent amplification using the conventional RT-PCR.
Currently, the availability of norovirus diagnosis is limited because the methods are not able to be implemented in local laboratories with only basic equipment. This delays diagnosis and treatment in individual diarrhea cases or in outbreaks. The kit method is reliable and fast yet requires a significant amount of time and resources to transport materials into countries. The cost is three times that of our method. We have shown an auxiliary method using readily-available in-country reagents for the detection of NoV that is equally simple, fast, and reliable. This cost-effective method bypasses reliance on materials purchased abroad, the problems of international and intra-national regulations on its transportation which ultimately will lead to an accessible diagnosis and rapid treatment of one the most common viral causes of gastroenteritis in children.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank the National Calicivirus Laboratory Center for Disease Control and Prevention (CDC) for the kind gift of a standard and control positives for NoV, and Laboratories of the School of Public Health at Johns Hopkins for providing the reagents.
References
- Medici M. Molecular epidemiology of Norovirus infections in sporadic cases of viral gastroenteritis among children in Northern Italy. L. Medical Virology. 2006;78 doi: 10.1002/jmv.20723. [DOI] [PubMed] [Google Scholar]
- Vidal R. Novel recombinant Norovirus causing outbreaks of gastroenteritis in Santiago, Chile. J. Clinica Microbiology. 2006;4 doi: 10.1128/JCM.01890-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xavier M. Detection of caliciviruses associated with acute infantile gastroenteritis in Salvador, an urban center in Northeast Brazil. Braz. J. Med. Biol. Res. 2009;42 doi: 10.1590/s0100-879x2009000500007. [DOI] [PubMed] [Google Scholar]
- Boom R. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990;28:495–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S. Genogroup-specific PCR primers for detection of Norwalk-like viruses. J. Virol. Methods. 2002;100:107–114. doi: 10.1016/s0166-0934(01)00404-9. [DOI] [PubMed] [Google Scholar]
- Mattison K. Multicenter comparison of two norovirus ORF2-based genotyping protocols. J. Clin. Microbiol. 2009;47:3927–3932. doi: 10.1128/JCM.00497-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo AA. Use of TaqMan real-time reverse transcription-PCR for rapid detection, quantification, and typing of norovirus. J. Clin. Microbiol. 2006;44:1405–1412. doi: 10.1128/JCM.44.4.1405-1412.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama T. A broadly reactive and highly sensitive assay for Norwalk-like viruses on real-time quantitative RT-PCR. J. Clin. Microbiol. 2003;41:1548–1557. doi: 10.1128/JCM.41.4.1548-1557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]