

Abstract
A reduction in low density lipoprotein (LDL) cholesterol or an increase in high density lipoprotein (HDL) cholesterol can reduce the risk of development of atherosclerosis through overlapping or independent mechanisms. However, the clinical outcome of combined therapy remains in debate. In this study, we first characterized effects of various constructs of helper-dependent adenoviral vector (HDAd) expressing apolipoprotein E3 or LDL receptor (LDLR) in vivo on plasma cholesterol levels. Using this information, we designed experiments and compared the effects of long-term (28 weeks) LDL cholesterol lowering or raising HDL cholesterol, or a combination of both on advanced atherosclerosis in Ldlr−/− mice, a mouse model of familial hypercholesterolemia. Our major findings are: (i) various factors influence in vivo functional activity, which appear to be context dependent; (ii) apolipoprotein AI (APOAI) gene transfer, which raises HDL cholesterol, retards progression of atherosclerosis but does not induce regression; (iii) LDLR or LDLR and APOAI combination gene therapy induces lesion regression; however, LDLR gene transfer accounts for the majority of the effects of combined gene therapy; (iv) LDLR gene therapy reduces interleukin-7, which is a master regulator of T-cell homeostasis, but APOAI gene therapy does not. These results indicate that LDL cholesterol lowering is effective and sufficient in protection against atherosclerosis and induction of regression of pre-existing atherosclerosis.
Keywords: Atherosclerosis regression, Apolipoprotein AI, Apolipoprotein E, Helper-dependent adenoviral vector, Familial hypercholesterolemia, Gene therapy, LDL receptor
Introduction
Despite availability of primary and secondary preventions, cardiovascular disease (CVD) is the leading cause of morbidity and mortality in industrial countries and this trend is extending to developing countries. Furthermore, an increasing incidence of obesity and diabetes in industrial countries may exacerbate the incidence of this disease [1]. Atherosclerosis is a chronic inflammatory disease and the major cause of CVD. This disease is characterized by the infiltration of immune cells and the presence of lipid-enriched macrophages within the arterial wall. Lipid lowering is a primary target for prevention of CVD associated with dyslipidemia [2] and despite its progressive nature, atherosclerosis is a reversible process [3]. Intensive statin (HMG-CoA reductase inhibitor) therapy has been reported not only to reduce LDL cholesterol (LDL-C) but also to induce regression of atherosclerosis in patients who require coronary angiography for a clinical indication [4]. Post-hoc analysis combining raw data from 4 prospective randomized trials in which 1455 patients were analyzed has also reported over 5% atheroma regression in patients with low LDLR-C and increased HDL-C [5]. However, statins exhibit pleiotropic effects. Therefore, it remains elusive whether the observed lesion regression is due to reduction of LDL-C, increase of HDL-C a combination of both, or pleiotropic effects. Nevertheless, there is considerable interest in the outcome of combined therapy that takes advantage of different pathways, which might have synergistic or additive effects on atherosclerosis, although combination therapy to lower LDL-C by statins and to raise HDL-C through inhibition of cholesteryl ester transfer from HDL to triglyceride-rich lipoproteins failed in clinical trials [6].
Helper-dependent adenoviral vector (HDAd) is a non integrating viral vector that has a large cloning capacity (~36 kb). HDAd devoid of all viral coding sequences contains only cis elements, inverted terminal repeat for replication and the packaging signal. All necessary proteins for packaging HDAd genome are supplied in trans using helper virus. Therefore, HDAd has an intrinsic safety profile compared to first or second generation Ad and allows sustained transgene expression in vivo [7]. Upon intravenous injection, HDAd mainly is taken up by the liver, which can result in relatively liver restricted expression especially when combined with use of a liver-specific promoter. The size and composition of HDAd influence vector stability and amplification during production and the expression of transgene following gene transfer [8,9]. Because of its large cloning capacity, various promoter expression cassettes and inclusion of DNA sequences that influence transcription and translation can be accommodated and compared.
In this study, we constructed various helper-dependent adenoviral vectors (HDAd) expressing human apolipoprotein E3 (apoE3) to study regulatory elements that might influence hepatic transgene expression in Apoe−/− mice, a mouse model of hypercholesterolemia, and examined the correlation between plasma cholesterol level and development of atherosclerosis. We then tested the effects of various HDAd constructs on plasma cholesterol levels through LDLR augmentation in Ldlr−/− mice. Finally, we tested whether lowering LDL-C and raising HDL-C have additive or synergistic effects on atherosclerosis. We induced atherosclerosis in Ldlr−/− mice by feeding a high cholesterol diet and treated with HDAd expressing LDLR to reduce LDL-C, or APOAI to raise HDL-C, or both vectors, and compared atherosclerosis in these mice. Despite the use of an expression cassette which lowered plasma cholesterol only moderately, LDLR gene therapy had profound effects on atherosclerosis and APOAI gene therapy did not provide additional benefits to that of LDLR gene therapy alone.
Methods
Construction of recombinant helper-dependent adenoviral vector (HDAd)
HDAd expressing human apoE3 or LDLR with various promoters were constructed using the C4HSU [10], pΔ21 [11] or pΔ28 backbone [12]. The construction of phosphoenolpyruvate carboxykinase (PEPCK) expression cassette has been described previously [12]. The Woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) [13] and the 1.7 kb PstI fragment containing APOE liver control region (LCR) [14] were added to this basic PEPCK expression cassette (Figure 1A). To construct the APOAI promoter expression cassette containing WPRE and LCR, the basic APOAI expression cassette [15] was used (Figure 1B). The human APOE promoter expression cassette was generated from a human APOE gene fragment. In brief, that fragment that contains 5′-flanking region to exon 2 was amplified by PCR using the following primers: 5′ –GAATTCTAGCCGTCGATTGGAGAAC– 3′ and 5′ – TGATCAGCGGCCGCCCGGGCCAATTGATCGATTGTCGAC – 3′ (artificial cloning sites are underlined). The exon 4 containing a polyadenylation signal was amplified using the following primers: 5′ - GTCGACCTCCGCGCAGCCTGCAGC - 3′ and 5′-GAATTCATACACGACACAGATGGAG – 3′. The 5.1 kb HindIII/AatII human APOE gene fragment, the 0.39 kb AatII/DraI fragment and the 0.43 kb DraI/SalI fragment derived from the PCR product containing the part of exon 2 were ligated to 1.9 kb SalI/BamHI fragment consisting of 0.2 kb SalI/EcoRI fragment that contains the part of exon 4 and the LCR into the HindIII/BamHI sites of the modified pLPBL-1 [12] (Figure 1C). To compare DNA stuffer, human APOE gene and LCR were cloned into both C4HSU [10] and pΔ21 [11] (Figure 1C and D). LDLR cDNA was cloned into the basic PEPCK and APOAI expression cassettes. In addition we cloned the adenovirus E4 promoter (AdE4) [10] in front of the right ITR to test the effect of this element on vector production and in vivo performance (Figure 2A). We also constructed HDAd in which the LDLR gene is expressed under the control of PEPCK promoter. To accommodate this large expression cassette, we generated pΔ0E4 which contains only cis elements and AdE4 (Figure 2B). HDAd expressing human apoAI that contains an 11 kb human APOAI gene has been described [16]. Empty vector (HDAd-0) which contained no expression cassette was used as a control [12]. Rescue and amplification of the HDAd were performed as described [17]. Helper virus contamination was analyzed by Southern blot analysis and by real time PCR, and was less than 0.1% in all vectors.
Figure 1.

Structure of helper-dependent adenoviral vectors expressing APOE3. A. Schematic presentation of helper-dependent adenoviral vectors (HDAd) containing phosphoenolpyruvate carboxykinase (PEPCK) promoter on C4HSU backbone. L-ITR and R-ITR indicate left and right adenovirus (Ad) inverted terminal repeat sequence, respectively; Ψ, Ad packaging signal; PEPCKpr, PEPCK promoter; AI intron, human apolipoprotein AI intron, hGH polyA, human growth hormone polyadenylation signal; WPRE, Woodchuck hepatitis virus post-transcriptional regulatory element; LCR, human APOE gene liver control region. B. HDAd containing human APOE gene promoter and liver specific enhancer, LCR. In HDAd-E-E3, the fragment containing exon 2–4 was removed and replaced with APOE3 cDNA. HDAd-gE3 contains APOE3 5′ flanking region as well as all exons. C. HDAd containing human APOAI promoter and 3′ flanking region. In HDAd-AI-E3, the APOAI gene corresponding to exon 2 to exon 4 was removed and replaced with APOE3 cDNA. D. HDAd containing human APOE3 gene and LCR on pΔ21 backbone. HPRT, intron region of human genomic HPRT; C346, cosmid C346 human genomic stuffer sequence.
Figure 2.

Structure of HDAd expressing low-density lipoprotein receptor (LDLR). A. HDAd vector containing LDLR cDNA on pΔ28 or pΔ21 backbone. HDAd-P-LDLR has the pΔ28 backbone and HDAd-AI-LDLR and HDAd-AI-LDLR-E4 have the pΔ21 backbone. AdE4, Ad E4 promoter. Other abbreviations are as described in Figure 1 legend. B. HDAd containing human LDLR gene. The intron 1 of LDLR gene was removed and replaced with cDNA.
Animals
All animal protocols were performed according to the Guidelines of the Institutional Animal Care and Usage Committee at Baylor College of Medicine. Female Apoe−/− mice (6–8 weeks of age, Stock No. 002052) and Ldlr−/− mice (Stock No. 002207) on a C57BL/6 background were purchased from The Jackson Laboratory. Apoe−/− mice were fed normal chow and Ldlr−/− mice were fed a diet containing 0.2% (w/w) cholesterol and 10% (v/w) coconut oil [12]. To compare various expression cassettes, mice were treated with intravenous (i.v.) injection of 5 × 1012 viral particles (v.p.)/kg body weight of HDAd vector, while for combined vector treatment mice were treated with a combined total of 1 × 1013 vp/kg of HDAd. We used 5 × 1012 vp/kg of HDAd-LDLR [17] + 5 × 1012 vp/kg HDAd-0 for HDAd-LDLR group, or 5 × 1012 vp/kg of HDAd-AI [16] + 5 × 1012 vp/kg HDAd-0 for HDAd-AI group and 5 × 1012 vp/kg of HDAd-LDLR + 5 × 1012 vp/kg of HDAd-AI for HDAd-LDLR + HDAd-AI group. The HDAd-0 group received 1 × 1013 vp/kg of HDAd-0. For combined vector treatment, Ldlr−/− mice were fed a diet containing 0.2% (w/w) cholesterol and 10% (v/w) coconut oil for 36 weeks to induce atherosclerosis. After measurement of plasma cholesterol, mice were separated into 5 groups. One group was euthanized for baseline analysis and the remaining groups were received HDAd vectors as described above. Blood was collected from the saphenous vein into EDTA-coated tubes at the specified times and plasma was immediately isolated by centrifugation and stored at −80°C until assay. Twenty eight weeks after vector treatment, mice were euthanized for analysis.
Immunohistochemistry, histology and quantification of atherosclerotic lesions
Aortic en face and cross sectional lesion areas were measured by quantitative morphometry using SigmaScanPro [12,16,18]. For immunohistochemistry, sequential sections (5 μm thick) of fresh-frozen OCT-embedded proximal aorta were stained with Mac3 (Santa Cruz, 1:100), goat anti-mouse IL-7 (Santa Cruz, 1:100) and rabbit anti-α actin (Abcam, 1:100). Goat anti-mouse or anti-rabbit antibodies or rabbit anti-goat antibodies (1:200, Vector Laboratories) were used as secondary antibodies. In brief, sections were incubated with primary antibodies at 4°C overnight. After blocking, they were incubated with the secondary antibody for 1 hour at room temperature. Immunoreactive proteins were detected by a Vextastain Elite ABC reagent and by a NovaRed substrate kit for peroxidase (Vector Laboratories). Oil-Red O and H&E staining were performed by standard techniques.
Other procedures
Lipid and FPLC (GE Healthcare) analyses were performed as previously described [12]. For FPLC analysis, pooled plasma was used. Human apoE3 levels were quantified by an ELISA using polyclonal goat anti-apoE antibody (Chemicon, 1:1000) and apoE3 (Calbiochem) as standard [18].
Data analysis
Data are presented as mean ± SD unless specified. Differences were determined by t-test for two group comparison or by one-way analysis of variance (ANOVA) for multiple group comparisons using SigmaStat (Systat Software, Inc.). Rank sum test was also used where necessary and data are presented as median, 25th and 75th percentile. Statistical significance was assigned at p<0.05.
Results
Multiple factors influence transgene expression
Hepatic transfer of APOE3 gene into Apoe−/− mice results in the lowering of plasma cholesterol which is associated with appearance of apoE3 protein in plasma. Using this system, we first compared various liver-specific expression cassettes. Female Apoe−/− mice fed a normal chow diet were treated at 8 weeks of age with intravenous (i.v.) injection of HDAd. Plasma cholesterol levels in the HDAd-P-E3 group was not significantly different from the PBS group 1 week after treatment, but were significantly reduced to 286 ± 67 mg/dL (p<0.001 vs. PBS) after 2 weeks and still significantly lower than those in the PBS group 32 weeks after treatment. The inclusion of WPRE and LCR incrementally lowered cholesterol. Plasma cholesterol levels in the HDAd-PW-E3 and the HDAd-PWL-E3 group were significantly reduced to 198 ± 55 mg/dL and 77 ± 26 mg/dL, respectively, 1 week after treatment (Figure 3A). The addition of WPRE and LCR to the APOAI expression cassette increased cholesterol lowering activity (Figure 3B). However, the incremental effect was not observed. There was no significant difference in cholesterol lowering among the expression cassettes, perhaps reflecting saturation of the ability of plasma APOE3 to restore normal cholesterol levels. In contrast to PEPCK and APOAI expression cassette, addition of WPRE to APOE expression cassette did not enhance but attenuated cholesterol lowering activity. Two weeks after treatment, plasma cholesterol levels in the HDAd-E-E3 group were 71 ± 8 mg/dL, whereas those in the HDAd-EW-E3 were 145± 24 mg/dL (p<0.001). Despite our experience with HDAd based on genomic fragments [11,19], HDAd containing APOE gene was not better in lowering cholesterol than those containing cDNA (HDAd-E-E3 vs. HDAd-gE3, Figure 2C). Therefore, we constructed HDAd-gE3 on the pΔ21 backbone (HDAd-gE3-D21). Cholesterol lowering by the HDAd on the pΔ21 backbone was more effective than that on the C4HSU backbone (Figure 2C and D). At 32 weeks, plasma cholesterol in the HDAd-gE3-D21 was significantly lower than that in the HDAd-gE3 group (84 ± 23 mg/dL vs. 145 ± 42 mg/dL, n=5/group, p<0.05).
Figure 3.

Plasma cholesterol levels. Six to eight week old Apoe−/− mice received i.v. injection of 5 × 1012 v.p./kg of HDAd expressing apoE3 and plasma cholesterol was measured over the following 32 weeks. A. PEPCK expression cassette. *p<0.05 vs. PBS, **p<0.001 vs. PBS, †p<0.01 vs. HDAd-P-E3, ‡p<0.05 vs. HDAd-P-E3 and HDAd-PW-E3. All time points in the HDAd-PW-E3 and HDAd-PWL-E3 groups were significantly different from the PBS group after vector treatment (p<0.001), but not indicated. B. APOAI expression cassette. p<0.001 vs. PBS, group, except in the HDAd-AI-E3 group 1 week after treatment. There was no statistical significance among treatment groups. C. APOE expression cassette. Plasma cholesterol levels in all treatment groups were significantly lower than those in the PBS group (p<0.001) except 1 week after treatment in the HDAd-E-E3 and HDAd-ghE3 groups. *p<0.05 vs. HDAd-EW-E3. **p<0.01 vs. HDAd-EW-E3 and HDAd-gE3. D. Plasma cholesterol levels in mice treated with HDAd-gE3 on pΔ21 backbone. *p<0.05 vs. HDAd-gE3, **p<0.01 vs. HDAd-gE3. n=5/group.
Plasma cholesterol level and apoE3 level are not linear in Apoe−/− mice
The reduction of plasma cholesterol levels in the Apoe−/− mice coincided with the appearance of apoE3 protein in plasma (Figure 4). Plasma apoE3 levels measured by ELISA were significantly higher in the treatment group than those in the PBS and the HDAd-0 groups (p<0.01, n=5/group). ApoE3 levels in the HDAd-PWL-E3 group were significantly higher than those in the HDAd-P-E3 group 1 week after treatment (14.7 ± 5.4 μg/dL vs. 5.5 ± 6.2, p<0.05, n=5); however, they were not different among treatment groups at other time points (Figure 4A). Similarly, one week after treatment apoE3 levels in the HDAd-AIW-E3 and HDAd-AIWL-E3 groups were significantly higher than those in the HDAd-AI-E3 group (p<0.01, p<0.05, respectively) and those in the HDAd-AIW-E3 group were higher than those in the HDAd-AI-E3 group 2 weeks after treatment (p<0.05). Plasma apoE3 levels among HDAd containing APOAI promoter were not different at other time points, though apoE3 levels in the HDAd-AI-E3 group were higher than those in the other groups after 8 weeks, which did not reach statistical significance (Figure 4B). ApoE3 produced by HDAd containing the APOE promoter was correlated with plasma cholesterol levels. The relationship between cholesterol and apoE3 was not linear. Although the HDAd-EW-E3 group had higher apoE3 levels than the HDAd-E-E3 group 1 week after treatment (7.8 ± 3.5 vs. 2.5 ± 2.2 μg/mL, p<0.05, n=5), apoE3 levels in the HDAd-E-E3 were higher than those in the HDAd-EW-E3 group after 2 weeks of treatment. HDAd-gE3 also showed apoE3 production similar to that of HDAd-EW-E3. The apoE3 level was higher at 1 week in the HDAd-gE3 group than in the HDAd-E-E3 groups, but plasma apoE3 level in the HDAd-gE3 group stayed lower than that in the HDAd-E-E3 group after 2 weeks of treatment (Figure 4C). HDAd containing human APOE3 genomic fragment and LCR on pΔ21 backbone produced significantly more apoE3 than that on the C4HSU backbone until 12 weeks after treatment, but such difference waned thereafter (Figure 4D).
Figure 4.

Human ApoE3 levels in Apoe−/− mice after treatment with HDAd. A. PEPCK expression cassette. *p<0.05 vs. HDAd-P-E3 (n=5/group). B. APOAI cassette. *p<0.05 vs. HDAd-AIW-E3 at 2 weeks, **p<0.01 vs. HDAd-AIW-E3 and HDAd-AIWL at 1 week. C. APOE expression cassette. *p<0.05 vs. HDAd-EW-E3 and HDAdgE3, **p<0.01 vs. HDAd-EW-E3 and HDAd-gE3, and †p<0.05 vs. HDAd-EW-E3. D. Plasma apoE3 levels in mice treated with HDAd-gE3-D21. *p<0.05 vs. HDAd-gE3, **p<0.01 vs. HDAd-gE3.
Progression of atherosclerosis was arrested by induction of hepatic APOE3
Thirty two weeks after treatment, mice were euthanized and atherosclerosis was measured by en face analysis. The degree of atherosclerosis correlated with plasma cholesterol. Progression of atherosclerosis was inhibited in all mice treated with HDAd expressing ApoE3 (Figure 5). The extent of atherosclerosis was significantly lower in mice treated with HDAd expressing apoE3 than that in the control groups. Progression of atherosclerosis was almost completely arrested in the experimental groups. Due to the limited number of animals used for analysis, the differences among HDAd with the same promoter (PEPCK, APOAI and APOE) were not statistically significant and the effects of additional elements were not observed (e.g., WPRE, LCR with PEPCK promoter). However, the lesion areas in mice treated with HDAd-gE3-D21 were significantly smaller than those in mice treated with HDAd-gE3 (p<0.01, n=5/group).
Figure 5.

En face quantification of aortic atherosclerotic lesions. 1. PBS (n=5); 2. HDAd-0 (n=4); 3. HDAd-P-E3 (n=3); 4. HDAd-PW-E3 (n=5); 5. HDAd-PWL-E3 (n=4); 6. HDAd-AI-E3 (n=2); 7. HDAd-AIW-E3 (n=5); 8. HDAd-AIWL-E3 (n=4); 9. HDAd-E-E3 (n=3); 10. HDAd-EW-E3 (n=4); 11. HDAd-gE3 (n=5), 12. HDAd-gE3 (n=5). HDAd-0 was on pΔ28 backbone and HDAd-gE3 was on pΔ21 backbone. All other HDAd vectors were on C4HSU backbone. *p<0.05 vs. PBS, **p<0.01 vs. HDAd-gE3. Mann-Whitney Rank Sum Test was used. Median, 25th percentile and 75th percentile values are indicated.
Multiple factors influence LDLR gene expression
ApoE is a secretory protein and less than 10% of the normal level of plasma apoE is sufficient to normalize plasma cholesterol [20,21], while LDLR is a membrane-associated protein and more than 50% of normal LDLR activity is required for normal function as shown in patients with heterozygous LDLR deficiency. To remove LDL- from circulation, levels of expression as well as extent of LDLR- expressing versus non-expressing hepatocytes may influence the cholesterol lowering activity. We generated HDAd using human LDLR cDNA under less potent PEPCK and potent APOAI promoters (Figure 2A) and injected them i.v. into 6–8 week old female Ldlr−/− mice fed a high cholesterol diet. Two weeks after vector injection, plasma cholesterol levels were reduced to 133 ± 54 mg/dL (HDAd-P-LDLR), 90 ± 12 mg/dL (HDAd-AI-LDLR) and 127 ± 40 mg/dL (HDAd-AI-LDLR-E4), respectively, which were significantly lower than that in the PBS control group (604 ± 160 mg/dL, p<0.001, n=5/group). After 12 weeks of treatment, plasma cholesterol in the HDAd-P-LDLR was no longer different from that in the PBS group, while that in the HDAd-AILDLR and in the HDAd-AI-LDLR-E4 groups was significantly lower than that in the PBS group even after 20 weeks of treatment (p<0.01, p<0.001 vs. PBS, respectively). However, perhaps due to limited sample size, plasma cholesterol levels among treatment groups were not significantly different. The 0.4 kb AdE4promoter has been reported to confer a growth advantage to HDAd and has been incorporated in C4HSU backbone [10]. Incorporation of this sequence into a HDAd on the pΔ21 backbone did not significantly affect either vector production (data not shown) or cholesterol lowering (Figure 6A).
Figure 6.

Plasma cholesterol levels in Ldlr−/− mice after hepatic LDLR gene transfer. 6–8 week old Ldlr−/− mice on high cholesterol diet were treated with a single injection of HDAd expressing LDLR and plasma cholesterol was measured at indicated time. All data are presented as mean ± SD. A. Effects of expression cassette and Ad E4 promoter. HDAd vectors are on pΔ21 or pΔ28 backbone. *p<0.01, **p<0.001 vs. PBS. n=5/group. At 12 weeks, plasma cholesterol levels in the HDAd-P-LDLR group were no longer different from those of the PBS group. B. Incorporation of LDLR gene structure into HDAd. n=3.
Variable factors may influence in vivo performance of HDAd. Transgene expression from genomic DNA has been reported to be significantly higher than that achieved using a cDNA expression cassette [9]. However, the human LDLR gene is larger than the packaging capacity of HDAd. In addition, the entirety of promoter elements necessary for hepatic expression has not been fully characterized. We modified the human LDLR gene to accommodate the HDAd vector size within the packaging limit and cloned into the PEPCK expression cassette containing WPRE (Figure 2B). Two weeks after vector injection, plasma cholesterol levels were reduced to 266 ± 74 mg/dL and further reduced to 183 mg/dL after 4 weeks, which was higher than that achieved by cDNA- based vectors (Figure 6A and B). However, cholesterol lowering persisted in the mice treated with HDAd-PW-hgLDLR-E4. Twenty weeks after treatment, plasma cholesterol levels were 186 ± 6 mg/dL, which was lower than that in mice treated with HDAd-AI-LDLR-E4 (299 ± 62 mg/dL). These results suggest that genomic DNA based HDAd does not influence levels, but the kinetics of expression.
Cholesterol lowering by LDLR accounts for most of the effects in combined treatment of Ldlr−/− mice
In our previous studies, we demonstrated that in the context of complete LDLR deficiency high level of LDLR expression induces host immune response against LDLR protein [17]. It is also possible that drastic cholesterol lowering by LDLR using a potent promoter masks the effects of raising HDL-C by APOAI gene transfer on atherosclerosis. Therefore, we used the least potent PEPCK promoter to express LDLR. For apoAI expression, we used HDAd-APOAI which contains human APOAI gene and has been extensively characterized for its effects on atherosclerosis [16]. After 36 weeks on high cholesterol diet to induce atherosclerosis beyond the advanced stage, plasma cholesterol was measured and mice were divided into 5 groups to assess similar mean plasma cholesterol levels. One group (baseline) was sacrificed to determine baseline atherosclerosis, and remaining groups were injected i.v. with total 1 × 1013 v.p./kg HDAd (Figure 7A). Two weeks after vector injection, plasma cholesterol of the HDAd-LDLR group dropped from 455 ± 64 mg/dL to 72 ± 17 mg/dL (n=7), then stayed at similar levels until 8 weeks (84 ± 22 mg/dL), but started to increase thereafter. At 28 weeks, plasma cholesterol in the HDAd-LDLR was significantly lower than that of the HDAd-0 group (300 ± 31 mg/dL vs. 415 mg/dL, p<0.001, Figure 7B). Plasma cholesterol of the HDAd-AI group was significantly higher than that of the control group 2 weeks after injection (462 ± 75 mg/dL vs. 353 ± 25 mg/dL, p<0.001), but after 4 weeks, there was no significant difference between the HDAd-AI and the HDAd-0 group. The group treated with both HDAd-LDLR and HDAd-AI exhibited significantly higher plasma cholesterol levels than the HDAd-LDLR group until 8 weeks, but the difference was not significant thereafter. We pooled plasma collected at 28 weeks and analyzed the lipoprotein profiles by FPLC. Compared with the HDAd-0 and HDAd-AI groups, the HDAd-LDLR and the HDAd-LDLR+HDAd-AI groups had remarkably reduced IDL/LDLR cholesterol and elevated HDL cholesterol (Figure 7C). The peak of HDL cholesterol in the HDAd-AI group shifted to the left, indicating a larger HDL particle.
Figure 7.

Comparison of single gene therapy and combined gene therapy on plasma cholesterol. A. Experimental design. 6–8 week old female Ldlr−/− mice were fed a diet containing 0.2% (w/w) cholesterol and 10% (v/w) coconut oil for 36 weeks to induce atherosclerosis. Mice were separated in 5 groups. One group (baseline) was sacrificed and other 4 groups received single i.v. injection of HDAd vector. A high cholesterol diet was maintained throughout experiment. n=13 (baseline), 13 (HDAd-0), 14 (HDAd-LDLR), 12 (HDAd-AI) and 15 (HDAd-LDLR + HDAd-AI). B. Plasma cholesterol levels. *p<0.01, **p<0.001 vs. HDAd-0. C. FPLC analysis. Plasma collected 28 weeks after vector treatment. 0.2 ml of pooled plasma was separated with FPLC and cholesterol was determined. VLDL: very low density lipoprotein; IDL/LDL: intermediate-density-lipoprotein/low density lipoprotein; HDL: high density lipoprotein.
The extent of atherosclerosis was analyzed by morphometry. Twenty eight weeks after treatment, the HDAd-0 group showed a progressive 2-fold increase in lesion area compared with the baseline (p<0.001), while the HDAd-AI group showed an arrest of atherosclerosis progression (58% inhibition vs. HDAd-0, p<0.05, Figure 8). In contrast, the lesion areas in the HDAd-LDL and the HDAd-LDL + HDAd-AI groups significantly decreased (54% and 57%, respectively, p<0.001) compared with the baseline value. There was no significant difference between the HDAd-LDLR and the HDAd-LDLR + HDAd-AI groups. Thus, LDLR gene therapy alone appears to be sufficient to induce lesion regression.
Figure 8.
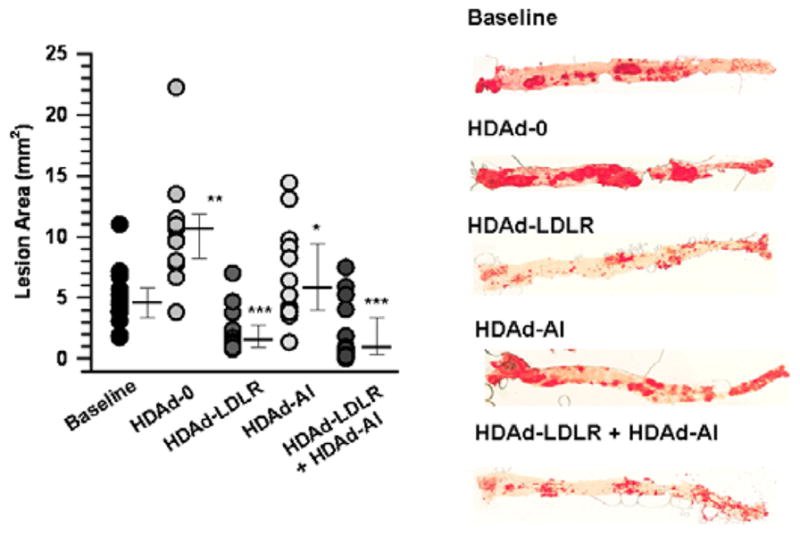
LDLR gene therapy induces regression of advanced atherosclerosis. A. En face quantification of aortic lesions. Atherosclerotic lesions were stained with Oil Red-O and quantified by morphometric analysis. Data were analyzed by Mann-Whitney Rank Sum Test. Median, 25th percentile and 75th percentile values are indicated. *p<0.05 (vs. HDAd-0), **p<0.001 (vs. baseline), ***p<0.001 (vs. baseline, HDAd-0, HDAd-AI). n=13 (baseline), 13 (HDAd-0), 14 (HDAd-LDLR), 12 (HDAd-AI), and 15 (HDAd-LDLR + HDAd-AI). Representative staining of aortas is shown in right panel.
IL-7 immunoreactivity in aortas is reduced in the HDAd- LDLR group but not in the HDAd-LDLR + HDAd-AI group
HDL has been proposed to serve an atheroprotective role through various mechanisms. Direct augmentation of apoAI indirectly increases HDL since apoAI is a major protein component of HDL [22]. To understand quantitative and qualitative differences between a single gene and combined gene treatment, we further characterized aortas by immunohistochemistry. Relative macrophages detected by Mac3 were significantly reduced in the HDAd-LDLR and the HDAd-LDLR + HDAd-AI groups (p<0.01 vs. baseline, n=5/group, Figure 9A and B), while relative α-actin positive areas were significantly increased in these groups (Figure 9C and D). IL-7 has been recently identified as an active participant in atherogenesis and downregulation of IL-7 is associated with lesion regression [18]. IL-7 positive areas were increased in the HDAd-0 group compared with the baseline, while they were significantly reduced in the HDAd-LDLR group (p<0.05 vs. baseline and HDAd-AI, p<0.01 vs. HDAd-0, Figure 9). Despite lesion regression in the HDAd-LDLR+HDAd-AI group, IL-7 positive areas were not reduced, but were similar to those of the HDAd-AI group.
Figure 9.

Immunological features in atherosclerotic lesions. A. Mac3 (macrophage) staining. *p<0.01 vs. baseline, HDAd-0 and HDAd-AI. Data are presented as mean ± SD. n=5/group. B. Representative Mac3 immunoreactivity in cross-section of aorta 28 weeks after treatment. Bar, 100 μm. C. Relative α-actin immuno positive areas. *p<0.01 vs. baseline and HDAd-0, and p<0.05 vs. HDAd-AI. **p<0.001 vs. baseline, HDAd-0 and HDAd-AI. n=4 (baseline), 3 (HDAd-0), 4 (HDAd-LDLR), 4 (HDAd-AI) and 4 (HDAd-LDLR+HDAd-AI). D. Representative α-actin immuno staining. Bar, 100 μm.
Discussion
In HDAd, the size and/or composition of the vector genome influences vector stability and amplification during production and the expression profile upon vector transfer into animals [9,23]. Using ApoE3 protein level as a reporter, we constructed a series of HDAd to test for the role of vector composition on in vivo performance. First, we constructed HDAd vector using the C4HSU backbone. With a PEPCK expression cassette, the addition of WPRE and LCR had an incremental reduction in plasma cholesterol levels and increase of plasma APOE3 levels in Apoe−/− mice (Figure 4A), while with an APOAI expression cassette additional elements did not change plasma cholesterol levels (Figure 4B). This may reflect the reaching of apoE3 levels that are saturating for cholesterol lowering due to potency of the APOAI promoter [20,21]. The enhancement of PEPCK driven transgene expression by WPRE has been previously reported in HDAd vector [24]. Plasma apoE3 levels, however, indicate that the addition of WPRE and LCR to the APOAI expression cassette did not significantly influence APOE3 expression but affected kinetics (Figure 5B). ApoE3 levels increased earlier (1 week after vector treatment) in the HDAd-AIW-E3 and HDAd-AI-AIWL-E3 groups than in the HDAd-AI-E3 group. In contrast, addition of WPRE to the APOE promoter cassette attenuated promoter activity, suggesting that WPRE may interfere with APOE transcriptional elements. Therefore, the benefit of additional elements appears to be dependent on the context of expression cassettes. We compared the effects of vector backbone (Figure 4C, D) and found differences in apoE3 expression. Transgene expression has been reported to be marginally affected by the location of the expression cassette [9], which supports the notion that the DNA stuffer influences expression level as well as duration of expression [8].
ApoE is a convenient reporter for optimization of expression cassettes in Apoe−/− mice since secretion of apoE reduces plasma cholesterol and protein can be measured by ELISA. Furthermore, Apoe−/− mice spontaneously develop atherosclerosis on normal chow. However, plasma cholesterol levels are not linearly correlated with plasma apoE3 levels, because less than 10% of normal plasma apoE levels are sufficient to maintain normal plasma cholesterol [20,21]. This may be why we did not observe significant effects of vector genome compositions on development of atherosclerosis. Alternatively, the sample size was not large enough to detect any difference, or normal cholesterol levels in mice may be below threshold for development of atherosclerosis and complete normalization of plasma cholesterol is not required to protect against development of atherosclerosis.
The critical parameters for optimal HDAd in vivo performance might be dependent on the functional property of proteins expressed. We examined effects of expression cassettes and vector composition on cholesterol lowering activity using LDLR in Ldlr−/− mice. The potency of promoter activity did not affect cholesterol levels in Ldlr−/− mice as much as anticipated from the results in Apoe−/− mice, suggesting that a strong promoter is not absolutely required for augmentation of LDLR in Ldlr−/− mice. Interestingly, cholesterol lowering by HDAd containing modified LDLR gene led to substantially prolonged LDLR expression compared to the HDAd containing cDNA, both of which were under PEPCK promoter (HDAd-P-LDLR vs. HDAd-PW-hgLDLR-E4, Figure 6). This is in agreement with the previous reports that expression of genes from genomic fragments or inclusion of genomic components improves expression [9,11,16].
Using this information, we designed the experiment to test whether raising HDL-C by increasing a major protein component of HDL, apoAI, in addition to lowering cholesterol provides additional benefits in treating atherosclerosis. Although fatty streaks and early atherosclerotic lesions have been reported to be regressed by direct infusion of HDL [25] or by short-term increase of apoAI by liver-directed apoAI gene transfer [26], instead, we and others have found that long-term hepatic APOAI gene transfer alone does not induce regression of more advanced lesions but instead attenuates lesion progression [16,27]. Furthermore, early fatty streaks spontaneously regress [28]. Therefore, the earlier findings are not relevant to human conditions. In agreement with previous studies [16,27,29], raising HDL-C by APOAI gene transfer alone did not induce regression of advanced atherosclerosis. Despite use of a less potent promoter, atherosclerosis regressed by treatment of HDAd expressing LDLR and additional expression of APOAI gene did not add significant effects (Figure 8). These results suggest that lowering LDL-C or total plasma cholesterol plays a major role in inducing regression of atherosclerosis and that reducing plasma cholesterol to threshold but not complete normalization of plasma cholesterol is sufficient to induce regression of advanced atherosclerosis, though the threshold plasma cholesterol level remains to be determined.
Potential mechanisms for lesion regression include reduction of atherogenic lipoproteins within the arterial wall, efflux of cholesterol and other lipids from plaques, emigration of foam cells, and influx of healthy macrophages to remove necrotic cell debris and other inflammatory components of the plaque [3]. Some mechanisms involving foam cell migration [30] and retention [31] have been proposed. CCR7, a mediator of dendritic cell migration, has been implicated in atherosclerosis regression [30]. However, a recent study has shown that macrophage emigration is not involved in regression induced by aggressive cholesterol lowering, but suppression of monocyte recruitment and a stable apoptosis accounts for loss of plaque macrophages [32]. It has also been proposed that activation of liver X receptor-α inhibits the progression of atherosclerosis and promotes regression of existing lesions by regulating macrophage target gene Arg1 through PU.1 and IRF8 [33]. We have reported that down-regulation of IL-7 in aortas is associated with regression of advanced lesions in Apoe−/− mice induced by long-term cholesterol lowering [18]. IL-7 recruits monocytes/macrohpages to atherosclerotic lesion through activation of endothelium. Therefore, the association of IL-7 downregulation with lesion regression is in agreement with the mechanisms proposed by Potteaux et al. [32]. In this study, we have shown reduced IL-7 immunoreactive protein in the HDAd-LDLR group compared with baseline, HDAd-0 and HDAd-AI. IL-7 immunoreactive areas were not reduced in the HDAd-AI group, which is consistent with our previous findings that downregulation of IL-7 is associated with cholesterol lowering and that oxidized LDL upregulated IL-7 in macrophages. IL-7 is a master regulator for T-cell homeostasis and plays an important role in inflammatory responses [34]. Despite lesion regression and cholesterol lowering in the HDAd-LDLR+AI group, IL-7 positive areas were not reduced. Although it is possible that the sample size was not large enough to detect subtle differences, it appears that increasing HDL by hepatic overexpression of APOAI gene counteracts the benefit of lowering plasma cholesterol. Another anti-atherogenic apolipoprotein well characterized is apoE, which mediates the uptake of chylomicron and VLDL remnants by serving as a ligand for LDLR. In spite of reports suggesting its anti-atherogenic role independent of LDLR’s function, coexpression of LDLR and APOE did not have an additive effect on atherosclerosis reduction [35].
One of mechanisms by which HDL is atheroprotective involves reverse cholesterol transport [22]. Reverse cholesterol transport is initiated by activation of ATP binding cassette transporter AI (ABCA1). A genetic disorder caused by deficiency of ABCA1 is Tangier Disease and results in unmeasurable HDL-C levels in homozygotes and half-normal HDL-C levels in heterozygotes. A modest risk of cardiovascular disease has been reported in patients with Tangier disease [36], however, in large population studies, low levels of HDL-C due to heterozygosity for ABCA1 mutations were not associated with an increased risk of ischemic heart disease [37]. The authors have suggested that low HDL cholesterol is associated with increased risk of IHD only in combination with a coexisting increase in triglycerides and atherogenic remnant lipoproteins [37]. Another approach to manipulate HDL-C is to inhibit cholesteryl ester transfer protein (CETP), though the initial study does not support the athero-protective role of CETP deficiency [38]. In the Investigation of Lipid Level Management to Understand its Impact in Atherosclerotic Events (ILLUMINATE), despite increasing HDL-C by 72% and decreasing LDL-C levels by 25%, the primary combined cardiovascular end point worsened [6]. One mechanism proposed for this unexpected result was off-target effects related to stimulation of aldosterone synthesis followed by increase of blood pressure [39]. It remains unsettled whether increase of HDL-C by inhibition of CETP is a therapeutic option and whether the concept of decreasing LDL-C with simultaneously raising HDL-C improves clinical outcome. At least two compounds, dalcetrapib [40] and ancetrapib [41] which apparently lack the off-target effects of torcetrapib, are under clinical trials, which will answer these questions. Another explanation for why coexpression of APOAI did not have an additive effect over cholesterol lowering is the dysfunctionality of the HDL produced by APOAI gene transfer [42]. It is possible that artificially raised HDL was not functional in reverse cholesterol transport.
There are limitations in attempting to extrapolate the present work in rodents to humans. There are substantial differences in lipoprotein metabolism between mice and humans. Mice have hepatic apoB editing activity and are deficient in CETP activity, which results in an intrinsic anti-atherogenic lipoprotein profile (higher HDL than LDL cholesterol levels). This cannot be achieved in humans by currently available treatments. Nevertheless, our results underscore the most effective approach to protect against atherosclerosis, which is to normalize LDL cholesterol levels.
Figure 10.

IL-7 immunoreactive areas are reduced in mice treated with HDAd-LDLR. A. Relative IL-7 immunoreactive area in aorta. *p<0.05 vs. baseline and HDAd-AI, and p<0.01 vs. HDAd-0. n=6 (baseline), 6 (HDAd-0), 4 (HDAd- LDLR), 3 (HDAd-AI), and 3 (HDAd-LDLR+HDAd-AI). Bar represents mean value. B. Representative staining of aortas. Bar, 100 μm.
Acknowledgments
This work was supported by NIH Grants (Grant numbers- HL73144, HL51586 and DK079638).
Abbreviations
- apoAI
Apolipoprotein AI
- CVD
Cardiovascular disease
- HDAd
Helper-dependent adenoviral vector
- LDL
Low density lipoprotein
- HDL
High density lipoprotein
- PEPCK
Phosphoenolpyruvate carboxykinase
- LCR
Liver control region
Footnotes
Conflict of Interest
None.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 2.Aikawa M, Libby P. Lipid lowering therapy in atherosclerosis. Seminars in Vascular Medicine. 2004;4:357–366. doi: 10.1055/s-2004-869592. [DOI] [PubMed] [Google Scholar]
- 3.Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med. 2008;5:91–102. doi: 10.1038/ncpcardio1086. [DOI] [PubMed] [Google Scholar]
- 4.Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–1565. doi: 10.1001/jama.295.13.jpc60002. [DOI] [PubMed] [Google Scholar]
- 5.Nicholls SJ, Tuzcu EM, Sipahi I, Grasso AW, Schoenhagen P, et al. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA. 2007;297:499–508. doi: 10.1001/jama.297.5.499. [DOI] [PubMed] [Google Scholar]
- 6.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 7.Brunetti-Pierri N, Ng P. Progress and prospects: gene therapy for genetic diseases with helper-dependent adenoviral vectors. Gene Ther. 2008;15:553–560. doi: 10.1038/gt.2008.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parks RJ, Bramson JL, Wan Y, Addison CL, Graham FL. Effects of stuffer DNA on transgene expression from helper-dependent adenovirus vectors. J Virol. 1999;73:8027–8034. doi: 10.1128/jvi.73.10.8027-8034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiedner G, Hertel S, Johnston M, Biermann V, Dries V, et al. Variables affecting in vivo performance of high-capacity adenovirus vectors. J Virol. 2002;76:1600–1609. doi: 10.1128/JVI.76.4.1600-1609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandig V, Youil R, Bett AJ, Franlin LL, Oshima M, et al. Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc Natl Acad Sci USA. 2000;97:1002–1007. doi: 10.1073/pnas.97.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim IH, Jozkowicz A, Piedra PA, Oka K, Chan L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc Natl Acad Sci USA. 2001;98:13282–13287. doi: 10.1073/pnas.241506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oka K, Pastore L, Kim IH, Merched A, Nomura S, et al. Long-term stable correction of low-density lipoprotein receptor-deficient mice with a helper-dependent adenoviral vector expressing the very low-density lipoprotein receptor. Circulation. 2001;103:1274–1281. doi: 10.1161/01.cir.103.9.1274. [DOI] [PubMed] [Google Scholar]
- 13.Donello JE, Loeb JE, Hope TJ. Woodchuck hepatitis virus contains a tripartite posttranscriptional regulatory element. J Virol. 1998;72:5085–5092. doi: 10.1128/jvi.72.6.5085-5092.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simonet WS, Bucay N, Lauer SJ, Taylor JM. A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J Biol Chem. 1993;268:8221–8229. [PubMed] [Google Scholar]
- 15.Koeberl DD, Sun B, Bird A, Chen YT, Oka K, et al. Efficacy of helper-dependent adenovirus vector-mediated gene therapy in murine glycogen storage disease type Ia. Mol Ther. 2007;15:1253–1258. doi: 10.1038/sj.mt.6300188. [DOI] [PubMed] [Google Scholar]
- 16.Belalcazar LM, Merched A, Carr B, Oka K, Chen KH, et al. Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation. 2003;107:2726–2732. doi: 10.1161/01.CIR.0000066913.69844.B2. [DOI] [PubMed] [Google Scholar]
- 17.Nomura S, Merched A, Nour E, Dieker C, Oka K, et al. Low-density lipoprotein receptor gene therapy using helper-dependent adenovirus produces long-term protection against atherosclerosis in a mouse model of familial hypercholesterolemia. Gene Ther. 2004;11:1540–1548. doi: 10.1038/sj.gt.3302310. [DOI] [PubMed] [Google Scholar]
- 18.Li R, Paul A, Ko KW, Sheldon M, Rich BE, et al. Interleukin-7 induces recruitment of monocytes/macrophages to endothelium. Eur Heart J. 2011 doi: 10.1093/eurheartj/ehr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oka K, Belalcazar LM, Dieker C, Nour EA, Nuno-Gonzalez P, et al. Sustained phenotypic correction in a mouse model of hypoalphalipoproteinemia with a helper-dependent adenovirus vector. Gene Ther. 2007;14:191–202. doi: 10.1038/sj.gt.3302819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hasty AH, Linton MF, Swift LL, Fazio S. Determination of the lower threshold of apolipoprotein E resulting in remnant lipoprotein clearance. J Lipid Res. 1999;40:1529–1538. [PubMed] [Google Scholar]
- 21.Thorngate FE, Rudel LL, Walzem RL, Williams DL. Low levels of extrahepatic nonmacrophage ApoE inhibit atherosclerosis without correcting hypercholesterolemia in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2000;20:1939–1945. doi: 10.1161/01.atv.20.8.1939. [DOI] [PubMed] [Google Scholar]
- 22.Degoma EM, Rader DJ. Novel HDL-directed pharmacotherapeutic strategies. Nat Rev Cardiol. 2011;8:266–277. doi: 10.1038/nrcardio.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muruve DA, Cotter MJ, Zaiss AK, White LR, Liu Q, et al. Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo. J Virol. 2004;78:5966–5972. doi: 10.1128/JVI.78.11.5966-5972.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mian A, McCormack WM, Jr, Mane V, Kleppe S, Ng P, et al. Long-term correction of ornithine transcarbamylase deficiency by WPRE-mediated overexpression using a helper-dependent adenovirus. Mol Ther. 2004;10:492–499. doi: 10.1016/j.ymthe.2004.05.036. [DOI] [PubMed] [Google Scholar]
- 25.Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–1241. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tangirala RK, Tsukamoto K, Chun SH, Usher D, Pure E, et al. Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation. 1999;100:1816–1822. doi: 10.1161/01.cir.100.17.1816. [DOI] [PubMed] [Google Scholar]
- 27.Van Craeyveld E, Gordts SC, Nefyodova E, Jacobs F, De Geest B. Regression and stabilization of advanced murine atherosclerotic lesions: a comparison of LDL lowering and HDL raising gene transfer strategies. J Mol Med (Berl) 2011;89:555–567. doi: 10.1007/s00109-011-0722-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 29.Rong JX, Li J, Reis ED, Choudhury RP, Dansky HM, et al. Elevating high-density lipoprotein cholesterol in apolipoprotein E-deficient mice remodels advanced atherosclerotic lesions by decreasing macrophage and increasing smooth muscle cell content. Circulation. 2001;104:2447–2452. doi: 10.1161/hc4501.098952. [DOI] [PubMed] [Google Scholar]
- 30.Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci USA. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2009;119:136–145. doi: 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pourcet B, Feig JE, Vengrenyuk Y, Hobbs AJ, Kepka-Lenhart D, et al. LXR{alpha} Regulates Macrophage Arginase 1 Through PU.1 and Interferon Regulatory Factor 8. Circ Res. 2011;109:492–501. doi: 10.1161/CIRCRESAHA.111.241810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kittipatarin C, Khaled AR. Interlinking interleukin-7. Cytokine. 2007;39:75–83. doi: 10.1016/j.cyto.2007.07.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawashiri M, Zhang Y, Usher D, Reilly M, Pure E, et al. Effects of coexpression of the LDL receptor and apoE on cholesterol metabolism and atherosclerosis in LDL receptor-deficient mice. J Lipid Res. 2001;42:943–950. [PubMed] [Google Scholar]
- 36.Serfaty-Lacrosniere C, Civeira F, Lanzberg A, Isaia P, Berg J, et al. Homozygous Tangier disease and cardiovascular disease. Atherosclerosis. 1994;107:85–98. doi: 10.1016/0021-9150(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 37.Frikke-Schmidt R, Nordestgaard BG, Stene MC, Sethi AA, Remaley AT, et al. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008;299:2524–2532. doi: 10.1001/jama.299.21.2524. [DOI] [PubMed] [Google Scholar]
- 38.Ishigami M, Yamashita S, Sakai N, Arai T, Hirano K, et al. Large and cholesteryl ester-rich high-density lipoproteins in cholesteryl ester transfer protein (CETP) deficiency can not protect macrophages from cholesterol accumulation induced by acetylated low-density lipoproteins. J Biochem. 1994;116:257–262. doi: 10.1093/oxfordjournals.jbchem.a124516. [DOI] [PubMed] [Google Scholar]
- 39.Forrest MJ, Bloomfield D, Briscoe RJ, Brown PN, Cumiskey AM, et al. Torcetrapib-induced blood pressure elevation is independent of CETP inhibition and is accompanied by increased circulating levels of aldosterone. Br J Pharmacol. 2008;154:1465–1473. doi: 10.1038/bjp.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz GG, Olsson AG, Ballantyne CM, Barter PJ, Holme IM, et al. Rationale and design of the dal-OUTCOMES trial: efficacy and safety of dalcetrapib in patients with recent acute coronary syndrome. Am Heart J. 2009;158:896–901. e3. doi: 10.1016/j.ahj.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 41.Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;363:2406–2415. doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- 42.Navab M, Anantharamaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, et al. Mechanisms of disease: proatherogenic HDL--an evolving field. Nat Clin Pract Endocrinol Metab. 2006;2:504–511. doi: 10.1038/ncpendmet0245. [DOI] [PubMed] [Google Scholar]