

Abstract
Urea-based methionyl-tRNA synthetase inhibitors were designed, synthesized and evaluated for their potential towards treating human African trypanosomiasis (HAT). With the aid of a homology model and a structure-activity-relationship approach, low nM inhibitors were discovered that show high selectivity towards the parasite enzyme over the closest human homolog. These compounds inhibit parasite growth with EC50 values as low as 0.15 μM while having low toxicity to mammalian cells. Two compounds (2 and 26) showed excellent membrane permeation in the MDR1-MDCKII model, and encouraging oral pharmacokinetic properties in mice. Compound 2 was confirmed to enter the CNS in mice. Compound 26 had modest suppressive activity against T. brucei rhodesiense in the mouse model, suggesting that more potent analogs or compounds with higher exposures need to be developed. The urea-based inhibitors are thus a promising starting point for further optimization towards the discovery of orally available and CNS active drugs to treat HAT.
Introduction
Approximately 60 million people in sub-Saharan Africa are at risk for human African trypanosomiasis (HAT) caused by Trypanosoma brucei. HAT caused by T. brucei gambiense progresses slowly from early stage to late stage disease, whereas disease caused by T. brucei rhodesiense progresses rapidly.1, 2 In late stage HAT, the central nervous system (CNS) becomes infected, and the untreated disease is uniformly fatal. Depending on the stage of the disease and the subspecies of the causative agent, HAT is treated either with suramin, pentamidine, melarsoprol, eflornithine, or a combination of nifurtimox and eflornithine.1, 2 These currently used drugs are either highly toxic and/or need to be administered by injection. Thus, there is an urgent need to develop new therapeutics that are effective, safe, affordable, orally administered, and easily stored in tropical conditions (http://www.dndi.org/diseases/hat/target-product-profile.html).
Methionyl-tRNA synthetase (MetRS), as one of the aminoacyl-tRNA synthetases (aaRS), plays an essential role in the core biological process of translating nucleotide-encoded gene sequences into proteins. The enzymatic reaction of aaRS generally consists of the following steps: the recognition of a specific amino acid and ATP, the formation of an aminoacyl-adenylate, the recognition of a specific tRNA, and the transfer of the aminoacyl group to the 3′-end of the tRNA.3 We recently showed by RNAi knockdown that the single MetRS of T. brucei is essential for parasite survival.4 Moreover, we synthesized a series of potent aminoquinolone-based inhibitors of parasite MetRS that inhibited parasite growth in culture, further demonstrating that MetRS is an attractive protein drug target for T. brucei.4 However, aminoquinolone-based MetRS inhibitors were known from antibacterial work to have poor bioavailability.5, 6 In our own hands, this series of compounds indeed exhibited poor permeability in the MDR1-MDCKII model that predicts CNS penetration,7-9 making it unlikely that they would cross the blood brain barrier for treating late stage HAT. These pharmacokinetic (PK) liabilities prompted us to explore other scaffolds to block T. brucei MetRS in our investigations towards anti-HAT therapeutics. In this paper, we report that using a urea moiety to replace the aminoquinolone group resulted in selective MetRS inhibitors that show good potency in parasite growth inhibition assays and promising improvements in bioavailability.
Results and Discussion
Design of urea-based inhibitors
The starting point for the work in this paper is the predicted binding mode of aminoquinolone-based compound 1 in a homology model of T. brucei MetRS that we reported earlier.4 We were able to create a high quality model because of the disclosure in a conference poster by the Replidyne company of a co-crystal structure of a related aminoquinolone-based inhibitor bound to Clostridium difficile MetRS.10 Compound 1 was successfully docked into the model, filling two binding pockets. The benzyl fragment occupies the mostly hydrophobic methionine substrate pocket and one of the meta-chlorine atoms resides essentially in the same position as the sulfur atom of methionine. The aminoquinolinone ring occupies an adjacent pocket, only created upon inhibitor binding, and forms hydrogen bonds through its NHs with the carboxylate of Asp 287 (Figure 1).
Figure 1. Docking study of aminoquinolone 1 and the design of urea and guanidine analogs using a T. brucei MetRS homology model4.

A) Docked pose of 1 with the two NHs of the aminoquinolone forming hydrogen bonds with Asp287; b) design of urea 2 and guanidine 3; c) overlaid poses of 1 (carbons in green) and 2 (carbons in yellow) after docking.
The Replidyne data and our docking study indicated the importance of a planar NH-X-NH in the aminoquinolone ring system for forming hydrogen bonds with the carboxylate of Asp287. This aspartate residue is strictly conserved in all MetRS enzymes based on a BLAST search that involved 250 sequence alignments, and is responsible for substrate binding by forming a salt bridge to the α-amino group of methionine.11 Thus the aminoquinolone targets an enzyme active site amino acid residue that is unlikely to mutate, which is advantageous for drug discovery. However, the very same aminoquinolone moiety was suspected to be the potential cause of the inhibitor’s poor bioavailability.5, 6 Therefore, we decided to move away from aminoquinolones but to keep a planar NH-X-NH moiety in our next generation of inhibitors. Conceptually dissecting the hetero ring system of the aminoquinolone led to a urea 2 or a guanidine 3 (Figure 1b). Literature search revealed that GlaxoSmithKline (GSK) has previously reported only one urea-based MetRS inhibitor for bacterial targets with moderate cellular activity.6 In addition, Ibis Therapeutics reported a series of similar urea-based compounds for anti-bacterial chemotherapy with moderate activities although the compounds’ target of action was not identified in their publication.12 Therefore, urea or guanidine-based inhibitors against T. brucei MetRS warrant further systematic investigation using structure-based approaches. Molecular modeling demonstrated that the preferred binding conformations of urea or guanidine analogs superimposed nicely onto the aminoquinolone-based inhibitor (Figure 1c for urea 2), and maintained the key hydrogen bond interactions with Asp287 as described before. As a result, compounds 2 and 3 were chosen as the initial testing candidates for synthesis, following Scheme 1.
Scheme 1. Synthetic routes.

(a) Ar-CHO, AcOH, NaCNBH3, MeOH, rt; (b) 2-acetyldimedone, DIPEA, CH2Cl2, MeOH rt; (c) di-tert-butyl dicarbonate, K2CO3, MeCN, water, rt; (d) hydrazine, THF, microwave (65 °C, with 200 W maximum power input); (e) 1-isothiocyanatobenzene, CH2Cl2, rt; (f) HgCl2, 7 M NH3 in MeOH; (g) TFA/CH2Cl2 (1:4 v/v), rt; or H2O/TIPS/TFA (3:3:94, v/v), rt; (h) Ar-NCO, CH2Cl2, rt; (i) Ar-NH2, CDI, DMF, rt; or (j) Ar-NH2, p-nitrophenyl chloroformate, DIPEA, THF, rt.
For the aminoquinolone-based inhibitors we previously reported an excellent correlation between T. brucei growth inhibition (EC50) and T. brucei MetRS thermal shift (ΔTm),4 a proxy for binding affinity where a two-degree shift usually indicates significant binding.13-15 By the same thermal shift assay 2 and 3 exhibited Tm=7.8±0.3 °C and 4.5±0.5 °C, respectively, indicating that the urea-based 2 binds to the MetRS enzyme more tightly than the guanidine-based 3. Hence, we selected the urea-based 2 as the template in our exploration for inhibitors with good permeability properties.
Affinity, activity, and selectivity of urea-based inhibitors
In order to explore the structure-activity-relationship (SAR), first the benzyl ring of 2 was modified by adding a hydrophobic substituent (−Cl, −OMe, and –OEt, see Scheme 1 and Table 1) to fill the mainly hydrophobic pocket. These compounds were synthesized using the second route in Scheme 1 due to the need to vary the benzyl group on the left part of the molecule, which was attached last during synthesis. All three substitution positions (ortho-, meta-, and para-) exhibited a preference for the smaller substituents (−Cl, −OMe), with the meta- position exhibiting the highest Tm values among all other tested mono- and non-substituted compounds 6 – 15.
Table 1. Binding of urea compounds to T. brucei MetRS by a thermal shift assay.
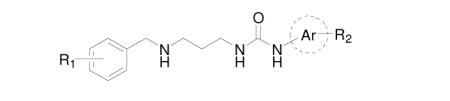
| Compound | Ri | Aromatic ring |
R2 |
T. brucei MetRS ΔTm (°C)a |
|---|---|---|---|---|
| 2 | 3,5-diCl | phenyl | H | 7.8±0.3 |
| 4 | 3-Cl,5-OMe | phenyl | H | 7.7±0.1 |
| 5 | 2,3,5-TriCl | phenyl | H | 6.4±0.3 |
| 6 | 3-Cl | phenyl | H | 5.5±0.1 |
| 7 | 3-OMe | phenyl | H | 4.7±0.1 |
| 8 | 3-OEt | phenyl | H | 2.7±i.3 |
| 9 | 4-Cl | phenyl | H | 2.5±0.i |
| 10 | 2-Cl | phenyl | H | 1.9±0.2 |
| 11 | 4-OMe | phenyl | H | 1.1±0.3 |
| 12 | 2-OEt | phenyl | H | 1.1±0.3 |
| 13 | H | phenyl | H | 1.3±0.1 |
| 14 | 2-OMe | phenyl | H | 1.1±0.1 |
| 15 | 4-OEt | phenyl | H | 0.5±0.2 |
|
| ||||
| 16 | 3,5-diCl | Phenyl | 2-OH | 8.4±0.3 |
| 17 | 3,5-diCl | Phenyl | 3-OH | 7.7±0.i |
| 18 | 3,5-diCl | Phenyl | 3-F | 7.7±0.i |
| 19 | 3,5-diCl | phenyl | 2-F | 7.3±0.i |
| 20 | 3,5-diCl | Phenyl | 4-F | 6.3±0.5 |
| 21 | 3,5-diCl | Phenyl | 4-Cl | 5.7±0.2 |
| 22 | 3,5-diCl | Phenyl | 3-Cl | 5.2±0.3 |
| 23 | 3,5-diCl | Phenyl | 4-OH | 5.8±0.3 |
| 24 | 3,5-diCl | Phenyl | 2-NH2 | 4.7±0.2 |
| 25 | 3,5-diCl | phenyl | 3-NH2 | 4.3±0.2 |
|
| ||||
| 26 | 3,5-diCl | 3-thiophene | H | 8.3±0.9 |
| 27 | 3-Cl,5-OMe | 3-thiophene | H | 7.9±0.i |
| 28 | 3,5-diCl | 2-thiophene | H | 7.6±0.3 |
| 29 | 3,5-diCl | 3-pyridine | H | 7.1±0.1 |
| 30 | 3,5-diCl | 2-pyridine | H | 4.5±0.2 |
| 31 | 3,5-diCl | 4-pyridine | H | 3.1±0.1 |
ΔTm values are the average of three independent runs.
Subsequently, the SAR around the aryl urea moiety was examined while keeping 3,5-dichlorobenzyl moiety on the other side of the molecule fixed (Scheme 1 and Table 1). Compounds were prepared using the first route shown in Scheme 1 so that the common part containing the 3,5-dichlorobenzyl moiety was synthesized first. As the aromatic ring of the aryl-urea is predicted to reside in a pocket that is created upon inhibitor binding, the pocket was anticipated to be somewhat flexible. Because the pocket consists of both hydrophilic and hydrophobic residues, a variety of substituents (−OH, −NH2, −F, and −Cl) as well as hetero-aromatic rings (thiophene and pyridine) were tested. From the Tm values it can be seen that a hydroxyl group in meta- and ortho- positions is tolerated but that an amino group binds less favorably. Fluorine is preferred over chlorine. Substituted thiophene compounds 26 – 28 bind as well as the phenyl-based 2. Among pyridine substituted compounds 29 – 31, only the 3-pyridine 29 was nearly as active as 2.
Choosing a minimum Tm threshold of 7.5 °C we selected a subset of compounds for confirmation of their inhibitory activity in an aminoacylation enzyme inhibition assay implemented for the T. brucei MetRS enzyme (Table 2). In this assay the incorporation of 3H-methionine into aminoacylated tRNA is measured. The IC50’s ranged from 19 to 110 nM, thereby confirming the value of the Tm screen to find effective inhibitors, and 27 was the most potent.
Table 2. Enzymatic and cellular Inhibitory activity of urea compounds.
| Enzyme Assays |
Cell Assays |
|||||
|---|---|---|---|---|---|---|
| Selectivity Index |
T. brucei brucei
|
Mammalian cell lines |
||||
| Compound |
T. brucei MetRS IC50 (nM) |
Human Mito MetRS IC50 (nM) |
Human IC50 / T. brucei IC50 |
EC50 (nM) | CRL-8155 IC50 (nM) |
Hep G2 EC50 (nM) |
| 27 | 19 | 6591 | 347 | 150 | 22,000 | 22,000 |
| 17 | 29 | 5430 | 187 | 170 | 12,000 | 12,000 |
| 16 | 39 | 3720 | 95 | 210 | >20,000 | >20,000 |
| 26 | 28 | 4349 | 155 | 220 | 12,000 | 13,000 |
| 4 | 45 | 8348 | 186 | 330 | 20,000 | 20,000 |
| 2 | 57 | >10,000 | >175 | 415 | 12,000 | 20,000 |
| 28 | 64 | 3056 | 48 | 350 | 9,500 | 14,500 |
| 18 | 110 | 4246 | 39 | 757 | 43,200 | 17,900 |
Enzyme inhibition assay was measured in aminoacylation assays.
Selectivity Index: human mitochondrial MetRS IC50 over T. brucei MetRS IC50.
We also examined these potent T.brucei MetRS inhibitors for selectivity. In the active site the sequence identity between T. brucei and human mitochondrial MetRS is 79%. With the human cytosolic enzyme the identity in the active site is only 38%. Earlier, Replidyne scientists found that aminoquinolone inhibitors were 1,000-fold better inhibitors of bacterial MetRS than the human mitochondrial enzyme and that they were virtually inactive against the human cytosolic enzyme.16 Therefore, we tested our urea-based inhibitors only against human mitochondrial MetRS. As shown in Table 2, the tested compounds exhibited IC50’s ranging from 4,349 to >10,000 nM, corresponding to a selectivity index of 39 or better.
The best urea-based inhibitors were tested in a T. brucei brucei growth inhibition assay (Table 2). The EC50’s ranged from 150 nM to 757 nM, and 27 was most potent. An excellent linear correlation (R2 = 0.95) between T. brucei EC50 and T. brucei MetRS IC50 values was observed for the tested compounds, strongly supporting that these compounds inhibit T. brucei growth by targeting the MetRS enzyme (Figure 2).
Figure 2. Correlation between enzyme (IC50) and parasite growth (EC50) inhibition for compounds listed in Table 2.
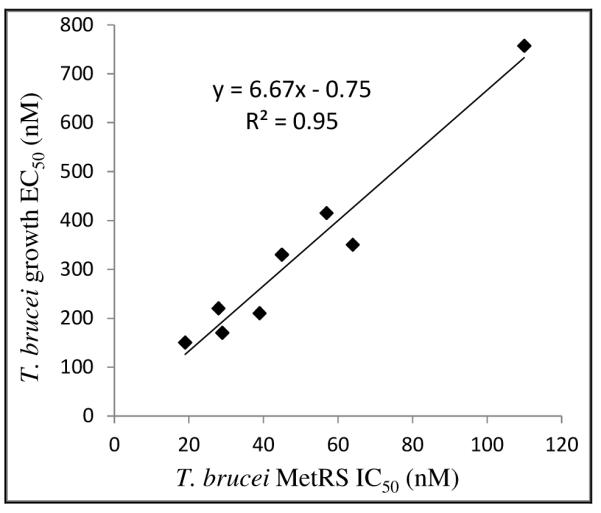
Correlation coefficient R2 is 0.95 (p < 0.0001).
To further establish that the MetRS inhibitors were acting “on target” in T. brucei, we performed experiments to demonstrate in vivo inhibition of protein synthesis. T. brucei cultures were incubated for 4 hours (approximately 1 doubling time) with 3H-histidine while in the presence of various test compounds. Newly synthesized protein was collected by precipitation with trichloracetic acid and quantified by scintillation counting. Both the aminoquinolone MetRS inhibitor 1 and the urea compound 26 resulted in a dose dependent decrease of incorporation of 3H-amino acid into proteins similar to the protein synthesis inhibitor, cycloheximide (Figure 3). Two control compounds were also tested that act by mechanisms not directly related to protein synthesis: difluoromethylornithine (DFMO) that inhibits ornithine decarboxylase and subsequently polyamine biosynthesis, and, a protein farnesyltransferase inhibitor (PFT inh) with anti-T. brucei activity.17 Incubating T. brucei with these two compounds did not result in a dose dependent decrease in protein synthesis (Figure 3). These results provide evidence that the MetRS inhibitors are blocking protein synthesis in cultured T. brucei as is expected by their mechanism of interfering with the normal processing of tRNA.
Figure 3. Protein synthesis inhibition in T. brucei cells.
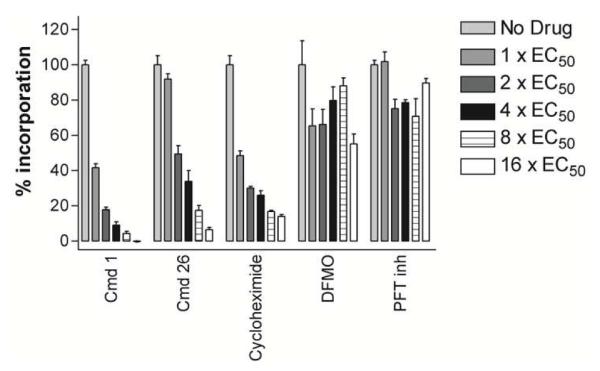
Mid-log bloodstream form T. brucei were cultured with 3H-histidine and various compounds at the indicated concentrations relating to their 48hr-EC50. After 4 hrs, the cultures were harvested, proteins precipitated with trichloroacetic acid, and radiation quantified by scintillation counting.
The compounds were also tested for host cell toxicity using a human lymphoblast cell line (CRL-8155) and a hepatocellular carcinoma cell line (Hep G2) (Table 2). These compounds showed toxicities to mammalian cell lines with EC50’s ranging from 9.5 to 43 μM, which translates into a selectivity index between 27 and 147. Compound 27, which was the most potent T. brucei growth inhibitor with an EC50 of 150 nM, showed the highest selectivity index of 147.
Binding mode to the target MetRS
Recently, we have obtained crystal structures of T. brucei MetRS in complex with six of the inhibitors discussed here. The binding of inhibitors to T. brucei MetRS resulted in large degree of structural changes in the protein and will be the subject of another manuscript (Koh et al., manuscript under preparation). However, the binding mode of the inhibitors to the active site is in strong agreement with the model generated because the Replidyne poster had allowed us to incorporate conformational changes in the homology model. For example, superposition (Cα-trace based) of the T. brucei MetRS:compound 2 crystal structure with the docking model of the same inhibitor revealed minimum differences in the binding site (Figure 4). Comparing the two, the differences in atomic positions of the inhibitor are all less than 1Å. The chlorine substituted benzyl ring binds to the methionine pocket while the urea linked benzene forms a planar moiety that fits into the auxiliary pocket, as predicted by the model. The hydrophobic parts of Tyr250 and Tyr472-Val473 stack against the urea linked benzene, validating the importance of planarity in this part of the inhibitors. All other major interactions are otherwise similar. The hydrogen bonds between the urea NHs and Asp287, as well as the positioning of a meta-chlorine in the same site of the methionine sulfur atom are all predicted correctly by the model. Therefore, the validity of using the docking model to guide the design of new inhibitors is confirmed by the crystal structure.
Figure 4. Superposition of T. brucei MetRS: 2 complex (protein in pink) with the docking model of the same inhibitor (protein in cyan).
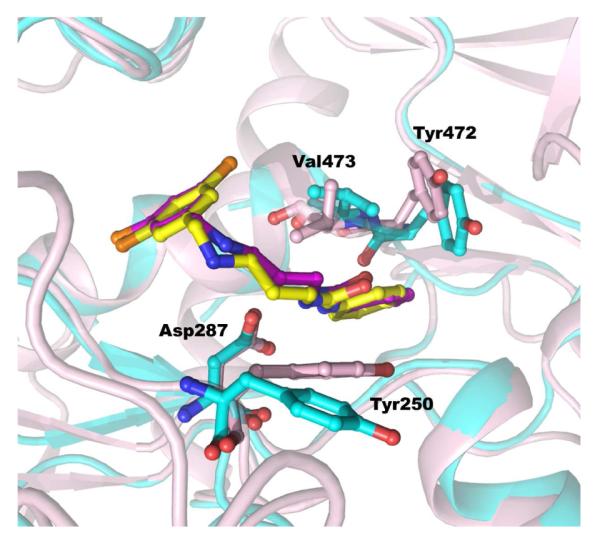
The binding mode of 2 in the crystal structure (carbons in magenta) at the active site is in good agreement with that of the model (carbons in yellow).
Membrane and brain permeability
After we achieved high affinity binding to the target, potent activity against T. brucei cultures, and selectivity versus human model cells, it was particularly important to analyze the potential permeability of compounds to the CNS in order to develop drugs for treating late stage HAT. Hence, compounds 2, 4, 17, 26, and 27 were tested for permeability across a monolayer of MDR1-MDCKII cells that mimic the blood brain barrier (Table 3).7-9 Consistent with previous reports of poor oral absorption of the aminoquinolone compounds,6 compound 1 was observed to have poor permeability through the MDR1-MDCKII monolayer (Table 3). Compounds that enter the central nervous system are typically associated with an apparent permeability Papp >150 nm/s.8 This liability makes the aminoquinolone scaffold a poor candidate for HAT development since it is unlikely to be orally absorbed or pass through the blood-brain barrier. Two compounds in the urea series, 2 and 26, exhibited a Papp >300 nm/s in the presence of P-gp inhibitor 34 (N-[4-[2-(6,7-dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)ethyl]phenyl]-5-methoxy-9-oxo-10H-acridine-4-carboxamide, GF120918),18 indicating excellent intrinsic permeability of the compounds. Permeation of compounds 4, 17, and 27 were relatively low and were limited by P-gp efflux. Compound 26 exhibited a moderate Papp value of 172±55 nm/s measured in the absence of 34 compared to Papp value of 317±82 nm/s in the presence of 34, with an absorptive quotient (AQ)9 value of 0.42±0.28, suggesting that compound 26 is a moderate substrate for P-gp. The permeability of compound 2 in the absence of 34 remained very high (Papp, 356±83 nm/s), with an AQ value of 0.21±0.21, indicating that P-gp efflux did not significantly limit the permeation of this compound.
Table 3. Cell permeability of urea compounds in the MDR1-MDCKII model.
| MDR1-MDCKII Papp (nm/s) |
|||
|---|---|---|---|
| Compound | − 34a | +34a | AQb |
| 1 | 29±8 | 52±20 | 0.36±0.33 |
| 2 | 356±83 | 455±62 | 0.21±0.21 |
| 26 | 172±55 | 317±82 | 0.42±0.28 |
| 27 | 97±21 | 191±3 | 0.53±0.12 |
| 4 | 73±9 | 150±31 | 0.49±0.16 |
| 17 | 29±3 | 83±31 | 0.62±0.13 |
Compound 34 is a P-gp pump inhibitor (see main text).
AQ: absorptive quotient.9
To corroborate the in vitro permeability experiments, two compounds were tested for brain permeability in mice by a single dose of 5 mg/Kg. As predicted from the in vitro model above, compound 1 demonstrated nearly undetectable brain levels at 60 minutes after IP injection. In contrast, compound 2 was actually concentrated in the brain (1.80 ± 0.09 μM) compared to plasma (0.12 ± 0.02 μM) (Figure 5). The mechanism of concentration remains to be characterized, but this feature is likely to be advantageous for treating second stage trypanosomiasis.
Figure 5.
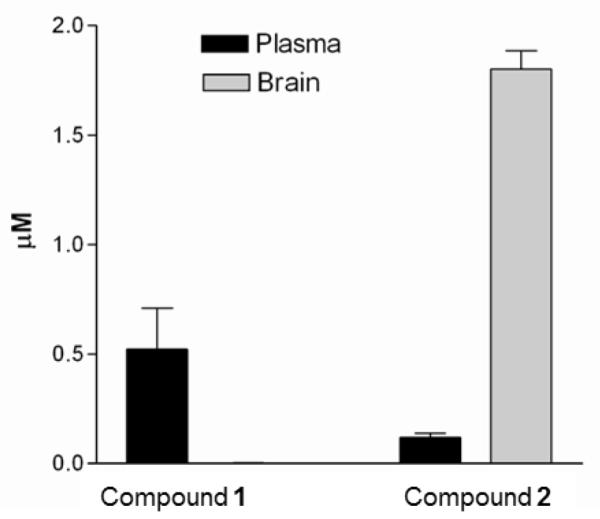
Concentrations of compounds 1 and 2 sixty minutes after IP injection in mice (n=3 per compound). The concentrations of 1 in the brain were at or below the limit of detection <0.003 μM.
Oral bioavailability and in vivo efficacy of urea-based inhibitors
To further analyze the PK properties of the urea series, oral mouse PK experiments were performed with compounds 2, 26, and 27 (Figure 6). Compound 26 exhibited the best Cmax at 3.7±0.01 μM and a half-life of 63±15 min with a moderate AUC level of 464±69 μM-min as an early lead compound. The overall mouse PK data suggested that the metabolic stability of compounds 2, 26, and 27 is reasonably good but may need further improvement by identifying and chemically modifying metabolically vulnerable fragments to further advance these aryl-urea moieties in the future. Regardless, the demonstrated oral bioavailability (Figure 6) and brain permeability (Figure 5) for the urea series of compounds are dramatic improvements of pharmacological properties over the aminoquinolone compounds.6
Figure 6. Pharmacokinetics characteristics of compounds 2, 26, and 27 in a mouse model.

Mice (n = 3 per group) were orally administered compounds by a single dose of 50 mg /kg.
Next, we selected compound 26 to test in the murine model of acute T. brucei infection (Figure 7). This model uses the highly pathogenic T. brucei rhodesiense subspecies that causes rapidly rising parasitemia and death within about 4-5 days of infection. The mice were given 26 or vehicle starting 1 day after infection. At 48, 72, and 96 h post-infection, parasitemia was significantly reduced in the 26-treated group compared to the vehicle-treated group by 94%, 80%, and 63%, respectively (Figure 7). Unfortunately, parasitemia continued to rise in the 26 group and the animals needed to be euthanized when they became moribund at 120 h post-infection. No toxicity attributable to 26 was observed. Although there was a significant suppressive effect on parasitemia and an extension of survival for 1 day, the modest in vivo activity is easily explained by considering the pharmacodynamics of the experiment. First, when tested 26 in vitro against the rhodesiense STIB900 strain, it has an EC50 of 0.42 μM. Based on the pharmacokinetic studies (Figure 4), the plasma concentrations of 26 are above the IC50 for less than 6 hours of the twelve hour dosing interval, and this is not considering the possibility that plasma protein binding may further reduce free compound levels in blood. In previous work, we found that T. brucei needed to be maintained with MetRS inhibitor at 8-times the EC50 for 72 hours to lead to irreversible killing in vitro.4 Based on these considerations, it is actually satisfying to see a significant in vivo suppression of parasitemia with 26. It is evident that more potent compounds (e.g. EC50 of at least 0.05 μM or less) with good pharmacokinetic profiles will be necessary to produce complete clearance of parasites in mice. With the aid of the recent crystal structure of urea compounds bound to the T. brucei MetRS, we hope to make progress in designing and synthesizing new compounds with the needed level of potency. For example, future plans to improve the potency of the urea inhibitors may involve making more extensive interactions with the methionyl substrate pocket as it appears that the current inhibitors do not fill up the entire volume of the pocket as confirmed by the crystal structure. Studies to address potential metabolic liabilities are also in progress, which may provide information on metabolism “hot-spots” that may be modified using structure-based design.
Figure 7. Suppression of T. brucei parasitemia in mice with 26.
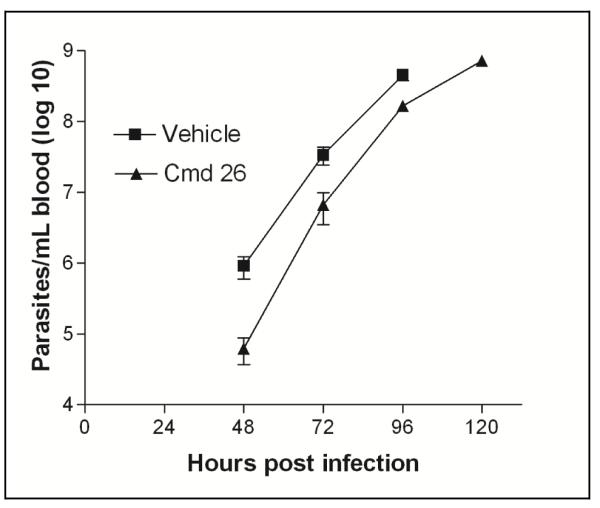
Animals (n=5 per group) were infected with T. brucei rhodesiense at time 0, and treatment was initiated at 24 h post-infection. Mice received 26 or vehicle every 12 hours for 4 days by oral administration. Mice were sacrificed when they became moribund (vehicle-treated mice were all sacrificed on day 4 and 26-treated mice were sacrificed on day 5). Differences in parasitemia were significant at 48, 72, and 96 hours (p = 0.030, p = 0.027, p = 0.008, respectively). The Y-axis is plotted on a log10 scale.
Conclusions
In summary, we used the T brucei MetRS homology model to guide rational modifications of the aminoquinolone scaffold and arrived at the new urea-based compounds that have improved PK properties. The homology model proved to be a high quality representation according to the X-ray structure of compound 2 bound to T. brucei MetRS. We were able to achieve EC50 values of 150 nM with a 147-fold selectivity over mammalian cell lines (compound 27). Protein labeling studies provide evidence that the MetRS inhibitors block protein synthesis in T. brucei cells. Importantly, the new urea compounds (2 and 26) have high cell permeability according to MDR1-MDCKII cell assays and compound 2 demonstrated high levels of brain penetration in mice. Additionally, the urea compounds were observed to give good plasma levels in mice after oral dosing. Compound 26 led to significant suppression of T. brucei rhodesiense in mice, but was not curative probably due to its modest potency of ~0.4 μM against this strain. Through structure-based drug design, we hope to develop more potent compounds with the needed pharmacological properties to advance the urea series of MetRS inhibitors through pre-clinical development.
Experimental Methods
Chemistry
All starting materials were purchased from various chemical vendors and used without further purification unless noted. Silica was EMD 1.07734 Silica Gel 60 0.063-0.200 MM, 70-230 Mesh ASTM and TLC plates were Silica Gel IB-F. 1H and 13C NMR spectra were recorded on a Bruker AV-300 or AV-500. Chemical shifts (δ) are reported in parts per million (ppm) relative to internal CD3OD (δ 3.31 1H NMR, δ 49.2 13C NMR) or CDCl3(δ 7.24 1H NMR), the coupling constants (J) are given in Hz. The abbreviations used are as follows: s, singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet; p, pentet. ESI-MS were recorded on an Agilent 1100 LC/MSD Trap mass spectrometer. The purity of all final compounds 2 – 31 (>98%) was confirmed by reverse phase HPLC analysis monitored at 230 nm.
1-(3-(3,5-Dichlorobenzylamino)propyl)-3-phenylurea (2)
To a solution of tert-butyl 3-aminopropylcarbamate (36 L, 0.21 mmol) in anhydrous CH2Cl2 (1 mL) at 0 °C was added 1-isocyanatobenzene (25 L, 0.21 mmol). After the mixture was stirred overnight at room temperature, the solvent was removed in vacuo. Silica gel flash chromatography (30 – 60% EtOAt, hexane) yielded tert-butyl 3-(3-phenylureido)propylcarbamate (70 mg, quantitative) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.13 – 7.39 (m, 5H), 7.01 – 7.09 (m, 1H), 5.73 – 5.81 (m, 1H), 5.02 – 5.10 (m, 1H), 3.23 – 3.31 (m, 2H), 3.15 – 3.21 (m, 2H), 1.56 – 1.66 (m, 2H), 1.46 (s, 9H). ESI-MS m/z 294.1 (M + H)+. To a solution of TFA:CH2Cl2 (1:4) was added tert-butyl 3-(3-phenylureido)propylcarbamate (20 mg, 0.068 mmol). After the mixture was stirred for 3 h at room temperature, the solvent was removed in vacuo. The residues were re-dissolved in MeOH (10 mL). Sodium methoxide (4 mg, 0.07 mmol) was added to the mixture, and stirred for 15 min. Acetic acid (250 L) and 3,5-dichlorobenzaldehyde (12 mg, 0.068 mmol) were added to the mixture, and stirred for 30 min, and then NaCNBH3 (13 mg, 0.2 mmol) was added. After the mixture was stirred overnight at room temperature, the solvent was removed in vacuo. Silica gel flash chromatography (5% (10% NH4OH, 90% MeOH), CH2Cl2) yieled 2 (13 mg, 54%) as a white solid (mp 96 – 97 °C). 1H NMR (500 MHz, MeOD) δ 7.32 – 7.45 (m, 5H), 7.26 (t, J = 7.9 Hz, 2H), 7.00 (t, J = 7.3 Hz, 1H), 3.76 (s, 2H), 3.27 (t, J = 6.7 Hz, 2H), 2.66 (t, J = 7.1 Hz, 2H), 1.66 – 1.83 (m, 2H), 13C NMR (126 MHz, MeOD) δ 157.08, 143.58, 139.55, 134.67, 128.39, 126.75, 126.64, 122.04, 118.89, 52.01, 45.80, 37.18, 29.61. ESI-MS m/z 353.3 (M + H)+.
N1-(3,5-Dichlorobenzyl)propane-1,3-diamine (32)
3,5-dichlorobenzaldehyde (100 mg, 0.57 mmol) was added to a solution of propyldiamine (500 L, 6 mmol) and AcOH (3 mL) in MeOH (10 mL). After the mixture was stirred for 30 min, NaCNBH3 (50 mg, 0.79 mmol) was added. The mixture was stirred overnight and the solvent was removed in vacuo. The residues were washed with EtOAc and 1 M NaOH (aq.). The organic layer was dried over MgSO4, and concentrated in vacuo, yielding the desired product 32 (120 mg, 90 %) as a colorless oil. ESI-MS m/z 234.1 (M + H)+. 1H NMR (300 MHz, CDCl3) δ 7.25 – 7.15 (m, 3H), 3.74 (s, 2H), 2.80 (t, J = 6.9 Hz, 2H), 2.68 (t, J = 6.9 Hz, 2H), 1.66 (p, J = 7.5 Hz, 2H).
tert-Butyl 3,5-dichlorobenzyl(3-(1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl-amino)propyl)carbamate (33)
2-acetyldimedone (Dde-OH) (70 mg, 0.39 mmol) was added to a solution of 32 (90 mg, 0.39 mmol) and DIPEA (0.13 mL, 0.77 mmol) in MeOH (0.5 mL) and CH2Cl2 (10 mL). After the mixture was stirred overnight at room temperature, the solvent was removed in vacuo. Silica gel flash chromatography (4% MeOH, CH2Cl2) afforded the desired intermediate. Di-tert-butyl dicarbonate (72 mg, 0.33 mmol) was added to a solution of the intermediate (130 mg, 0.33 mmol) and K2CO3 (45 mg, 0.33 mmol) in 10% water and 90% MeCN (15 mL). After the reaction mixture was stirred overnight at room temperature, the solvent was removed in vacuo. The residues were washed with EtOAt and brine. The organic layer was dried over anhydrous MgSO4, filtered, and then concentrated in vacuo. Silica gel flash chromatography (10 – 50% EtOAt, hexane) afforded the corresponding product 33 (100 mg) as a colorless oil. 1H NMR (500 MHz, MeOD) δ 7.34 (s, 1H), 7.24 (s, 2H), 4.44 (s, 2H), 3.46 – 3.53 (m, 2H), 3.34 – 3.44 (m, 2H), 2.53 (s, 3H), 2.37 (s, 4H), 1.88 – 1.96 (m, 2H), 1.47 (s, 9H), 1.03 (s, 6H). ESI-MS m/z 498.1 (M + H)+.
1-[3-(3,5-Dichloro-benzylamino)-propyl]-3-thiophen-3-yl-urea (26)
Anhydrous hydrazine (6.7 L, 0.20 mmol) was added to a solution of 33 (35 mg, 0.071 mmol) in anhydrous THF (1 mL). The mixture was irradiated with microwave (a CEM Discover system, 200 Watt maximum input power) to maintain at 65 °C for 30 min. The solvent was removed in vacuo and the residue was re-dissolved in anhydrous CH2Cl2 (0.5 mL). To the mixture at 0 °C was added 3-isocyanatothiophene (9 mg, 0.071 mmol) and the mixture was stirred overnight at room temperature. The solvent was removed in vacuo and the residues were purified by silica gel flash chromatography (EtOAt, hexane). The purified intermediate was added to a solution of 3% H2O, 3% TIPS, 94% TFA (1 mL). The solvent was removed in vacuo and silica gel flash chromatography (8% (10% NH4OH, 90% MeOH), CH2Cl2) yielded the desired product 26 (20 mg, 79%) as a white solid (mp 103 – 104 °C). 1H NMR (500 MHz, MeOD) δ 7.31 – 7.40 (m, 3H), 7.27 (dd, J = 3.2, 5.2 Hz, 1H), 7.14 (dd, J = 1.4, 3.2 Hz, 1H), 6.94 (dd, J = 1.4, 5.2 Hz, 1H), 3.77 (s, 2H), 3.27 (t, J = 6.6 Hz, 2H), 2.66 (t, J = 7.1 Hz, 2H), 1.70 – 1.79 (m, 2H). 13C NMR (126 MHz, MeOD) δ 156.96, 143.17, 137.28, 134.69, 126.81, 126.75, 123.69, 121.06, 106.40, 51.91, 45.73, 37.22, 29.49. ESI-MS m/z 359.2 (M + H)+.
1-[3-(3-Chloro-5-methoxy-benzylamino)-propyl]-3-thiophen-3-yl-urea (27)
3-isocyanatothiophene (36 mg, 0.29 mol) was added to a solution of tert-butyl 3-aminopropylcarbamate (50 mg, 0.29 mmol) in anhydrous CH2Cl2 at 0 °C. After the reaction was stirred overnight at room temperature, the solvent was removed in vacuo. Silica gel flash chromatography (30 – 60% EtOAt, hexane) afforded tert-butyl 3-(3-(thiophen-3-yl)ureido)propylcarbamate (60 mg, 70%). To a solution 3% H2O, 3% TIPS, 94% TFA (1 mL) was added tert-butyl 3-(3-(thiophen-3-yl)ureido)propylcarbamate (28 mg, 0.094 mol). After the mixture was stirred for 1 h at room temperature, the solvent was removed in vacuo. Residues were re-dissolved in MeOH (10 mL). Sodium methoxide (5 mg, 0.094 mol) was added to the mixture and stirred for 15 min. Acetic acid (250 L) and 3-chloro-5-methoxybenzaldehyde (16 mg, 0.094 mol) were added to the mixture, stirred for 30 min, and then NaCNBH3 (9 mg, 0.14 mmol) was added to the mixture. After the mixture was stirred overnight, the solvent was removed in vacuo. Silica gel flash chromatography (9% (10% NH4OH, 90% MeOH), CH2Cl2) yielded the desired product 27 (15 mg, 45%) as a colorless oil. 1H NMR (500 MHz, MeOD) δ 7.28 (ddd, J = 1.8, 3.2, 5.1 Hz, 1H), 7.16 (dd, J = 1.4, 3.2 Hz, 1H), 7.03 (q, J = 1.6 Hz, 1H), 6.97 (dd, J = 1.4, 5.1 Hz, 1H), 6.94 (q, J = 1.8 Hz, 1H), 6.91 (q, J = 2.0 Hz, 1H), 3.85 (s, 2H), 3.82 (s, 3H), 3.29 (t, J = 6.6 Hz, 2H), 2.78 (t, J = 7.1 Hz, 2H), 1.81 (p, J = 5.7, 6.6 Hz, 2H). 13C NMR (126 MHz, MeOD) δ 160.78, 157.14, 140.34, 137.23, 134.65, 123.72, 121.07, 120.64, 113.19, 112.75, 106.47, 54.70, 52.00, 45.46, 36.93, 28.92. ESI-MS m/z 354.9 (M + H)+.
Protein expression and purification
Full-length T. brucei MetRS was expressed and purified as previously described4 and used throughout the study except for crystallization experiments. Human mitochondrial MetRS was expressed as reported19 with plasmid provided by Dr. Spremulli.
Protein crystallization and structure determination
Detailed descriptions of protein crystallization and structure determination will be reported in a separate manuscript (Koh et al., manuscript under preparation). The structure of the complex of T. brucei MetRS and 2 was deposited to the Protein Data Bank (PDB ID code: 3TUN)
Thermal shift assay
The thermal shift assay was performed as previously described4, 15 with the modifications that samples contained the MetRS enzyme (0.4 mg/ml for T. brucei enzyme), 100 M of inhibitor, and 5% DMSO. The assays were repeated three times independently.
T. brucei methionyl-tRNA synthetase aminoacylation assay
Enzyme activity was quantified by the attachment of [3H]methionine to tRNA in the presence of the T. brucei MetRS enzyme. Reactions were performed in 96-well filter plates with Durapore® membranes (MSHVN4B10; Millipore) in volumes of 75 μl. The reaction was performed with 25 mM HEPES (pH 7.9), 10 mM MgCl2, 50 mM KCl, 0.2 mM spermine, 0.1 mg/mL bovine serum albumin, 2.5 mM dithiothreitol, 1% DMSO, and 1 U/mL pyrophosphatase (I1643; Sigma). Recombinant enzyme (10 nM) and compound inhibitors (starting concentration varied depending on potency and included 12 serial two dilutions) were mixed with the buffer and preincubated for 15 min. To start the reaction, 400 μg/mL bulk Escherichia coli tRNA (R4251; Sigma), 0.1 mM ATP, and 250 nM [3H]methionine (80 Ci/mmol) were added. The plate was incubated without shaking at room temperature for 120 min. The reactions were stopped by the addition of 100 μL cold 10% trichloroacetic acid. The reaction components were separated from tRNA by filtration through a vacuum manifold and washed three times with cold 10% trichloroacetic acid. The filter plates were dried overnight, scintillation fluid was added, and the counts on the plates were determined in a scintillation plate counter. Samples were run in quadruplicate and percent inhibition was calculated using two different controls (no enzyme and no test compound) with the following formula:
Where Mnd is the average no drug control, Dtd is each test drug value, and Mne is the average no enzyme control. IC50 values were then calculated by non-linear regression (sigmoidal dose response) in Prism 3.0.
Human mitochondrial methionyl-tRNA synthetase aminoacylation assay
Assays were performed as described above except for the following changes. Enzyme activity was quantified by the attachment of [3H]methionine to tRNA in the presence of the human mitochondrial MetRS enzyme. The reaction was performed with 50 mM Tris-HCl (pH 8.0), 2.5 mM MgCl2, 2.5 mM KCl, 0.2 mM spermine, 0.2 mg/mL bovine serum albumin, 2.5 mM dithiothreitol, and 1 U/mL pyrophosphatase (I1643; Sigma). Recombinant enzyme (20 nM) and compound inhibitors (starting concentration varied depending on potency and included 12 serial two dilutions) were mixed with the buffer and preincubated for 15 min. To start the reaction, 200 μg/mL bulk Escherichia coli tRNA (R4251; Sigma), 2.5 mM ATP, and 250 nM [3H]methionine (80 Ci/mmol) were added. The plate was incubated without shaking at 37°C for 120 min.
T. brucei growth inhibition assay
T. brucei brucei (bloodstream form strain 427 from K. Stuart, Seattle BioMed, Seattle, WA) were cultured in HMI-9 medium containing 10% fetal bovine serum, penicillin, and streptomycin at 37°C with 5% CO2 and T. brucei rhodesiense (bloodstream form STIB900 from Simon Croft, London School of Hygiene and Tropical Medicine, UK) were grown in HMI-18 containing 20% fetal bovine serum, penicillin, and streptomycin.20 Drug sensitivity of the T. brucei strain was determined in 96-well microtiter plates in triplicate with an initial inoculum of 5 × 104 trypomastigotes per well. Compound stock solutions were prepared in DMSO at 20 mM and added in serial dilutions for a final volume of 200 μl/well. Parasite growth was quantified at 48 h by the addition of AlamarBlue (Alamar Biosciences, Sacramento, CA).21 Pentamidine isethionate (Sigma-Aldrich,) was included in each assay as a positive control. Standard errors within assays were consistently less than 15%.
Protein synthesis inhibition assay
Mid-log phase T. brucei cells (5 × 105) were added to wells of a 96-well plate in a 100 ul volume of HMI-9 media. Compounds at concentrations ranging from 1 to 16 times the 48 hour EC50 were added to the plated cells in triplicate, and allowed to incubate for 15 minutes, with a no drug control included for each plate. [3H]-L-Histidine (1 mCi/ml, 47.9 Ci/mmol) (Moravek Biochemicals, Brea, CA) in a volume of 20 ul of PBS was added to each well and incubated with the parasites for 4 hours. Cycloheximide and difluoromethylornithine were purchased from Sigma-Aldrich; the PFT inhibitor corresponds to compound 4g in a previous publication.17 The experiment was stopped by adding 100 ul of 20% trichloroacetic acid and placed at 4°C for 15 minutes. Plates were harvested onto a PerkinElmer Unifilter®-96 GF/B filter using an Inotech Biosystems cell harvester and washed twice with 5% trichloroacetic acid. The filters were dried overnight in a vacuum oven then finished by raising the temperature to 40°C for 3 hours. Scintillation fluid was added, and the filters were counted using a PerkinElmer MicroBeta2 scintillation counter. These methods were adapted from Rock et al.22
Mammalian cell growth inhibition assays
The human lymphocytic cell line CRL-1855 (American Type Culture Collection) were grown in RPMI medium (Lonza, Walkersville, MD) with 10% fetal bovine serum at 37°C with 5% CO2. The human hepatocellular cell line HepG2 (American Type Culture Collection) were grown in DMEM/F-12 medium (Lonza, Walkersville, MD) with 10% fetal calf serum. Cells (5 × 103/well) were added to 96-well plates and incubated with serial dilutions of compounds for 48 h. At that time, cell viability was quantified by addition of AlamarBlue, and plates were incubated for an additional 4 h at 5% CO2, 37°C. Absorbance readings (OD570-600) were used to calculate viability referenced against cells grown with no inhibitors.
MDR1-MDCKII assay
Compounds and the controls, Amprenavir (Moravek Biochemicals) and Propranolol (Sigma), were tested in triplicate at a final concentration of 3 μM with and without 2 μM 34 (Elacridar, Toronto Research Chemicals Inc.). Amprenavir gives moderate-high permeability without 34 (typically Papp >200), but is reversed by 34 (AQ >0.5), whereas Propranolol gives high permeability without reversal with 34 (typically Papp >300 and AQ <0.5). MDR1-MDCKII cells were cultured at 37°C in 5% CO2 in cell culture media containing 500 mL Dulbecco’s Modification of Eagle’s Medium with GlutaMAX (GIBCO) supplemented with 50 mL Fetal Bovine Serum (GIBCO) and 5 mL Penicillin-Streptomycin (Sigma). 3.3 × 105 MDR1-MDCKII cells were plated in the hanging insert (apical chamber) of the 12-well Transwell® plates (Corning-Costar) in 0.5 mL of cell culture media. An additional 1.5 mL of cell culture media was added to the bottom (basolateral) chamber. The assay was conducted three days later at 37°C in 5% CO2 in transport media containing 500 mL Hank’s Balanced Salt Solution (Sigma) supplemented with 12.5 mL 1 M glucose (Sigma) and 12.5 mL 1 M HEPES buffer (Sigma).
On the day of the assay, the cell culture media was replaced with transport media with and without 2 μM 34 and pre-incubated for approximately 30 minutes. Transepithelial Electrical Resistance (TEER) values were then recorded using the Millicell®-ERS (Millipore). The transport media was then aspirated and compounds and controls were added to the inserts at a final concentration of 3 μM in 0.4 mL of transport media with and without 2 μM 34. Plates were incubated with shaking (~60 rpm) for 60 minutes. Immediately following incubation two 100 μL aliquots were taken from both chambers and the amount of drug was measured by mass spectroscopy. Lucifer Yellow (Aldrich), a fluorescence compound used to test membrane integrity during the assay, was then added to the inserts at a final concentration of 250 μM and incubated for 60 min with shaking (~60 rpm). After this final incubation, 100 uL aliquots were transferred to a black 96-well plate (BD Falcon) and florescence was measured for each membrane and TEER values were again recorded. Mass spectroscopy analysis was performed on samples with Lucifer Yellow Papp(A-B) values <20 nm/s and TEER values >200 ohm/cm2. The Papp (A-B) or Apparent Permeability Coefficient for each compound was calculated and reported in nm/s.
Where dQ is the amount of compound that has crossed the membrane, dt is the incubation time of the assay, A is the area of the cell monolayer, and Co is the initial concentration in the donor well.
The absorptive quotient (AQ) previously described by Troutman and Thakker9 identifies compounds that may be P-gp pump substrates by quantifying the functional activity of the P-gp pump during absorptive transport. An AQ value ≥0.5 is associated with high affinity for the P-gp pump.
Measurement of brain permeability in mice
All studies described in this paper using mice were done under an approved IACUC protocol at University of Washington. Six to 8 week-old Swiss Webster female mice (CFW®, Charles River, Wilmington, MA) were administered compound 1 or 2 by IP injection with a dose of 5 mg/Kg. The compounds were dissolved in a solution of 5% DMSO, 3% EtOH, 7% Tween 80 in saline. Each compound was administered to three mice and at 1 hour they were sacrificed by cervical dislocation, blood was collected in heparinized capillary tubes, and brains removed and frozen at −80C. Plasma was separated from whole blood and frozen for later analysis. Ten microliters of plasma were extracted with 30 μL of acetonitrile containing an extraction standard. Thawed brain tissue was homogenized in PBS containing an extraction standard using a Pyrex Tissue Grinder, then extracted with 4 mL of acetonitrile. Blank mouse brains spiked with known amount of compound were used to standardize the assay. The samples were centrifuged and the supernatants dried under vacuum. Samples were dissolved again in acetonitrile and compounds concentrations were quantified on a Waters Quattro Micro in MS/MS mode attached to an Agilent HPLC system as described.23
Pharmacokinetic studies in mice
Pharmacokinetic studies of compounds 2, 26 and 27 were performed by orally administering to mice 50 mg/kg of compounds dissolved in 5% DMSO, 3% EtOH, 7% Tween 80 in saline solution. Three mice were used per group. 40 ul of blood was collected at timepoints of 0.5, 1, 2, 3,4 and 6 hours post injection via tail snips. The plasma samples were extracted with acetonitrile and analyzed as described above.23
Efficacy studies in mice
Five female 6-8 week Swiss Webster (CFW®) mice (Charles River, Wilmington, MA) were used per group. At time 0, each mouse was infected by IP injection with 1×104 bloodstream forms of T. rhodesiense STIB900 (gift of Simon Croft, London School of Hygiene and Tropical Medicine, UK) in 0.2 ml. Treatment with compounds was initiated at 24 h post-infection. Compound 26 was mixed in vehicle of 5% DMSO, 3% EtOH, 7% Tween 80 in saline solution and administered at a dose of 50 mg/kg every 12 h for 4 days by oral gavage (0.2 mL per mouse). A second group received vehicle alone every 12 h for 4 days (0.2 mL) by oral gavage. Parasitemia was monitored daily by microscopic examination of tail blood on wet mounts. Statistical analysis on individual days was performed by unpaired t-tests.
Supplementary Material
Acknowledgement
Support for this research was provided by the National Institutes of Health grants AI067921 and AI084004. We are grateful to Colin McMartin and Regine Bohaceck for providing the FLO/QXP software; Robert Jacobs and Cindy Rewerts from Scynexis for advice and assistance in MDR1-MDCKII assay; Zhongsheng Zhang and Baoshun Zhang for technical assistance; Sharon Creason for performing mammalian cell cytotoxicity assays; Matthew Hulverson for assisting with pharmacokinetic experiments; Linda Spremulli for providing expression plasmid of human MetRS; and Wesley Van Voorhis, Alberto Napuli, and the protein production unit of Medical Structural Genomics of Pathogenic Protozoa program for protein expression.
Abbreviations Used
- HAT
human African trypanosomiasis
- MetRS
methionyl-tRNA synthetase
- aaRS
aminoacyl-tRNA synthetase
- MDCK
Madin-Darby Canine Kidney
- BLAST
Basic Local Alignment Search Tool
- AQ
absorptive quotient
Footnotes
Supporting Information Available. Additional synthesis and compound characterization details. This material is available free of charge via the Internet at http://pubs.acs.org.
PDB ID Code: 3TUN
References
- 1.Rodgers J. Human African trypanosomiasis, chemotherapy and CNS disease. J. Neuroimmunol. 2009;211:16–22. doi: 10.1016/j.jneuroim.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- 3.Sheppard K, Yuan J, Hohn MJ, Jester B, Devine KM, Soll D. From one amino acid to another: tRNA-dependent amino acid biosynthesis. Nucl. Acids Res. 2008;36:1813–1825. doi: 10.1093/nar/gkn015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shibata S, Gillespie JR, Kelley AM, Napuli AJ, Zhang Z, Kovzun KV, Pefley RM, Lam J, Zucker FH, Van Voorhis WC, Merritt EA, Hol WG, Verlinde CL, Fan E, Buckner FS. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei Infection in Mice. Antimicrob. Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jarvest RL, Berge JM, Berry V, Boyd HF, Brown MJ, Elder JS, Forrest AK, Fosberry AP, Gentry DR, Hibbs MJ, Jaworski DD, O’Hanlon PJ, Pope AJ, Rittenhouse S, Sheppard RJ, Slater-Radosti C, Worby A. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against gram-positive pathogens. J. Med. Chem. 2002;45:1959–1962. doi: 10.1021/jm025502x. [DOI] [PubMed] [Google Scholar]
- 6.Jarvest RL, Armstrong SA, Berge JM, Brown P, Elder JS, Brown MJ, Copley RCB, Forrest AK, Hamprecht DW, O’Hanlon PJ, Mitchell DJ, Rittenhouse S, Witty DR. Definition of the heterocyclic pharmacophore of bacterial methionyl tRNA synthetase inhibitors: potent antibacterially active non-quinolone analogues. Bioorg. Med. Chem. Lett. 2004;14:3937–3941. doi: 10.1016/j.bmcl.2004.05.070. [DOI] [PubMed] [Google Scholar]
- 7.Wang Q, Rager JD, Weinstein K, Kardos PS, Dobson GL, Li J, Hidalgo IJ. Evaluation of the MDR-MDCK cell line as a permeability screen for the blood-brain barrier. Int. J. Pharm. 2005;288:349–359. doi: 10.1016/j.ijpharm.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Mahar Doan KM, Humphreys JE, Webster LO, Wring SA, Shampine LJ, Serabjit-Singh CJ, Adkison KK, Polli JW. Passive Permeability and P-Glycoprotein-Mediated Efflux Differentiate Central Nervous System (CNS) and Non-CNS Marketed Drugs. J. Pharmacol. Exp. Ther. 2002;303:1029–1037. doi: 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- 9.Troutman MD, Thakker DR. Novel experimental parameters to quantify the modulation of absorptive and secretory transport of compounds by P-glycoprotein in cell culture models of intestinal epithelium. Pharm. Res. 2003;20:1210–1224. doi: 10.1023/a:1025001131513. [DOI] [PubMed] [Google Scholar]
- 10.Evans R, Green L, Sun X, Guiles J, Lorimer D, Burgin A, Janjic N, Jarvis T, Davies D. Co-crystal structure of REP3123 bound to Clostridium difficile methionyl tRNA synthetase, poster F1-2114; Abstr. 47 th Int. Conf. Antimicrob. Agents Chemother; American Society for Microbiology: Washington, DC. 2007. [Google Scholar]
- 11.Larson ET, Kim JE, Zucker FH, Kelley A, Mueller N, Napuli AJ, Verlinde CLMJ, Fan E, Buckner FS, Van Voorhis WC, Merritt EA, Hol WGJ. Structure of Leishmania major methionyl-tRNA synthetase in complex with intermediate products methionyladenylate and pyrophosphate. Biochimie. 2011;93:570–582. doi: 10.1016/j.biochi.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seth PP, Ranken R, Robinson DE, Osgood SA, Risen LA, Rodgers EL, Migawa MT, Jefferson EA, Swayze EE. Aryl urea analogs with broad-spectrum antibacterial activity. Bioorg. Med. Chem. Lett. 2004;14:5569–5572. doi: 10.1016/j.bmcl.2004.08.059. [DOI] [PubMed] [Google Scholar]
- 13.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J. Biomol. Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 14.Lo MC, Aulabaugh A, Jin GX, Cowling R, Bard J, Malamas M, Ellestad G. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal. Biochem. 2004;332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 15.Crowther GJ, Napuli AJ, Thomas AP, Chung DJ, Kovzun KV, Leibly DJ, Castaneda LJ, Bhandari J, Damman CJ, Hui R, Hol WG, Buckner FS, Verlinde CL, Zhang Z, Fan E, van Voorhis WC. Buffer optimization of thermal melt assays of Plasmodium proteins for detection of small-molecule ligands. J. Biomol. Screen. 2009;14:700–707. doi: 10.1177/1087057109335749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green LS, Bullard JM, Ribble W, Dean F, Ayers DF, Ochsner UA, Janjic N, Jarvis TC. Inhibition of Methionyl-tRNA Synthetase by REP8839 and Effects of Resistance Mutations on Enzyme Activity. Antimicrob. Agents Chemother. 2009;53:86–94. doi: 10.1128/AAC.00275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nallan L, Bauer KD, Bendale P, Rivas K, Yokoyama K, Horney CP, Pendyala PR, Floyd D, Lombardo LJ, Williams DK, Hamilton A, Sebti S, Windsor WT, Weber PC, Buckner FS, Chakrabarti D, Gelb MH, Van Voorhis WC. Protein farnesyltransferase inhibitors exhibit potent antimalarial activity. J. Med. Chem. 2005;48:3704–3713. doi: 10.1021/jm0491039. [DOI] [PubMed] [Google Scholar]
- 18.Polli JW, Wring SA, Humphreys JE, Huang LY, Morgan JB, Webster LO, Serabjit-Singh CS. Rational use of in vitro P-glycoprotein assays in drug discovery. J. Pharmacol. Exp. Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- 19.Spencer AC, Heck A, Takeuchi N, Watanabe K, Spremulli LL. Characterization of the human mitochondrial methionyl-tRNA synthetase. Biochemistry. 2004;43:9743–9754. doi: 10.1021/bi049639w. [DOI] [PubMed] [Google Scholar]
- 20.Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
- 21.Raz B, Iten M, Grether-Buhler Y, Kaminsky R, Brun R. The Alamar Blue assay to determine drug sensitivity of African trypanosomes (T.b. rhodesiense and T.b. gambiense) in vitro. Acta Trop. 1997;68:139–147. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 22.Rock FL, Mao W, Yaremchuk A, Tukalo M, Crepin T, Zhou H, Zhang YK, Hernandez V, Akama T, Baker SJ, Plattner JJ, Shapiro L, Martinis SA, Benkovic SJ, Cusack S, Alley MR. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- 23.Van Voorhis WC, Rivas KL, Bendale P, Nallan L, Horney C, Barrett LK, Bauer KD, Smart BP, Ankala S, Hucke O, Verlinde CL, Chakrabarti D, Strickland C, Yokoyama K, Buckner FS, Hamilton AD, Williams DK, Lombardo LJ, Floyd D, Gelb MH. Efficacy, pharmacokinetics, and metabolism of tetrahydroquinoline inhibitors of Plasmodium falciparum protein farnesyltransferase. Antimicrob. Agents Chemother. 2007;51:3659–3671. doi: 10.1128/AAC.00246-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.