

Summary
Transcription start points in bacteria are influenced by the nature of the RNA polymerase·promoter interaction. For Escherichia coli RNA polymerase holoenzyme containing σ70, it is presumed that specific sequence in one or more of the −10, extended −10 and −35 elements of the promoter guides the RNAP to select the cognate start point. Here, we investigated the promoter driving expression of the LEE1 operon in enteropathogenic Escherichia coli (EPEC) and found two promoters separated by 10 bp, LEE1 P1A (+1) and LEE1 P1B (+10) using various in vitro biochemical tools. A unique feature of P1B was the presence of multiple transcription starts from five neighboring As at the initial transcribed region. The multiple products did not arise from stuttering synthesis. Analytical software based on information theory was employed to determine promoter elements. The concentration of the NTP pool altered the preferred transcription start points, albeit the underlying mechanism is elusive. Under in vivo conditions, dominant P1B, but not P1A, was subject to regulation by IHF.
Introduction
Bacterial transcription initiates by binding of RNA polymerase (RNAP) to the promoter, leading to the formation of the closed complex (RPC). The RPC isomerizes to the open promoter complex (RPO), in which DNA immediately upstream of the transcription start point (tsp) is melted (Chamberlin, 1974, Neidhardt & Curtiss, 1996). After forming a stable initiation complex, RNAP often produces non-productive initiation products before entering elongation mode. The non-productive initiation includes abortive synthesis (Levin et al., 1987, Jacques & Susskind, 1990, Johnson, 1976) and stuttering synthesis (Han & Turnbough, 1998, Jacques & Susskind, 1990, Harley et al., 1990), the latter being ascribed to slippage of RNAP at short homopolymeric regions of the template. The tsp is largely determined by the distance from the −10 hexamer, with the 11th bp downstream from this site being optimal, although there is also a preference for purine (Lewis & Adhya, 2004).
Enteropathogenic Escherichia coli (EPEC) is the prototypic organism for pathogenic Gram-negative bacteria that cause attaching and effacing (A/E) intestinal lesions. The genes involved in the formation of A/E lesions are encoded within a chromosomal pathogenicity island named the locus of enterocyte effacement (LEE)(McDaniel et al., 1995). The LEE region contains five major operons, LEE1, LEE2, LEE3, LEE4, and LEE5 (Mellies et al., 1999, Elliott et al., 1998), whose expression is repressed by the global regulator H-NS (Dorman, 2004). Ler (LEE-encoded regulator) is a 15-kDa protein encoded by the first gene of the LEE1 operon and is a positive regulator required for the expression of most, if not all, of LEE genes that acts by counteracting H-NS mediated repression (Umanski et al., 2002, Haack et al., 2003, Mellies et al., 1999, Friedberg et al., 1999, Elliott et al., 2000, Bustamante et al., 2001). In addition, Ler acts as a specific autorepressor of LEE1 transcription (Berdichevsky et al., 2005). Consequently, environmental regulation of LEE expression occurs primarily at the level of Ler. The transcriptional regulation of LEE1 is very complex and involves various factors including QseA, FIS, IHF, H-NS, Hha, ClpXP, RcsCDB, Pch regulators, GrlA and GrlR, GrvA, EtrA and EivF [reviewed in (Mellies et al., 2007)].
The LEE1 operon in EPEC was first reported to have a single promoter located 175 bp upstream from the ler translational start site (Mellies et al., 1999). More recently, mutational analysis studies identified two different LEE1 promoters in EHEC serotype O157:H7 (Islam et al., 2011a, Islam et al., 2011b). Therefore, the aim of the present study was to undertake a detailed biochemical analysis of the LEE1 promoter in EPEC. The results showed that two promoters, separated by 10 bp, drive transcription of LEE1.
Results
Analysis of LEE1 P1-driven transcripts
In an attempt to elucidate the mechanism underlying LEE1 P1 regulation, we used an in vitro transcription assay using purified components. The DNA template was supercoiled plasmid DNA (pHJ12) into which LEE1 P1 DNA [−200~+80; numbering based on (Mellies et al., 1999)] that had been PCR amplified from EPEC strain E2348/69 was cloned between the EcoRI and PstI sites immediately upstream of the 54-bp Rho-independent transcription terminator of the rpoC gene of E. coli in pSA508 (Choy & Adhya, 1993, Squires et al., 1981) (Fig. 1A). α-32P UTP was included in the reaction to detect nascent RNA. The transcripts were analyzed on an 6% polyacrylamide DNA sequencing gel. As shown in Fig. 1B (lane 1), multiple transcripts were generated from the LEE1 P1 promoter with sizes varying between 125 to 135 nt in addition to the 105 nt rna1 transcript from the origin of plasmid replication (Choy & Adhya, 1993). To verify that these transcripts were of LEE1 P1, the plasmid DNA was linearized at the EcoRI, PstI, or BamHI site. The BamHI site was immediately downstream of the transcription terminator, and the EcoRI site was immediately upstream of the LEE1 P1 DNA. Transcription using the DNA linearized with either EcoRI or BamHI revealed virtually the same pattern of multiple transcripts (lane 2 and 3), although the band intensity was somewhat reduced. These results suggest that the bands did arise from the cloned LEE1 P1 promoter and not from the sequence outside of LEE1 P1 cloned in pSA508. No such transcripts were observed with the DNA linearized with PstI, which resulted in a break between the LEE1 P1 promoter and the terminator (lane 4). It was concluded that the multiple transcripts originated from LEE1 P1 and that they were generated independently of the supercoiling status of the DNA template.
Fig. 1.
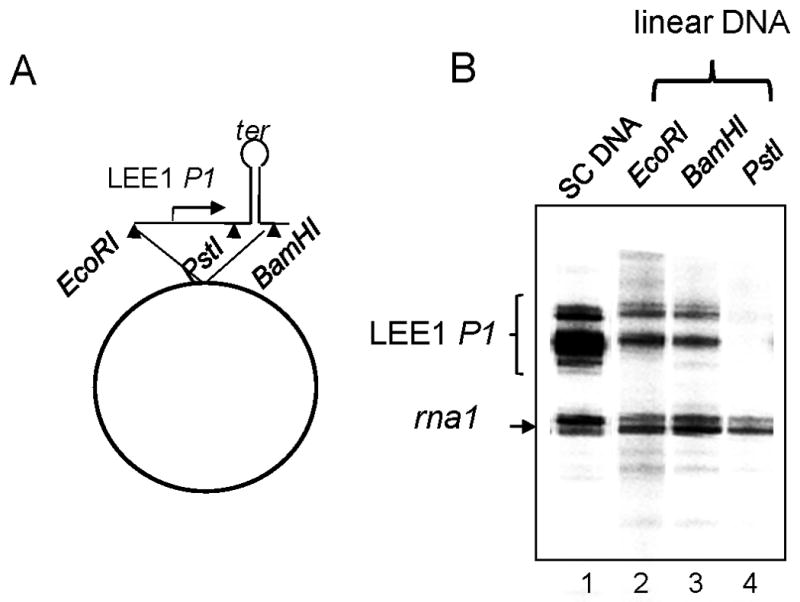
Identification of LEE1 P1 transcription initiation site. (A) Map of plasmid carrying LLE1 P1 promoter DNA from −200 to +80 in pSA508(Choy & Adhya, 1993), designated pHJ12. ter is a factor-independent transcription terminator. (B) Lanes 1 to 4 show in vitro transcription reaction with supercoiled plasmid pHJ101 (SC) and with the plasmid linearized with EcoR1, BamH1, and Pst1, respectively. The reaction products were analyzed by 8 M urea/6% polyacrylamide gel electrophoresis. The major transcripts from LEE1 P1 and rna1 are indicated.
The multiple transcripts could have arisen by inexact transcription termination at the terminator sequence of rpoC in pSA508. To test this, in vitro generated transcripts were analyzed by primer extension using a primer annealed to the sequence between +54~+74 of LEE1 transcript (Fig. 2A). The same pattern of multiple bands was detected, suggesting that inexact transcription termination was not the cause and that the multiple bands were due to the initiation of transcription at multiple sites. Since it is unusual to observe single promoter-driven transcripts with such heterogeneity, we suspected that the result may have been an in vitro artifact. To test this, RNA was extracted from E. coli strain MG1655 carrying the same pSA508:: LEE1 P1 and analyzed by primer extension using the same primer. The pattern of transcripts generated in vivo was essentially the same as in the in vitro assay, the multiple transcripts, suggesting that our original results were not an in vitro artifact. A previous study reported a similar pattern of multiple RNAs from LEE1 P1 by primer extension analysis of whole-cell RNA isolated from EPEC strain E2348/69; these RNAs were considered to arise from premature termination of reverse transcriptase (Mellies et al., 1999). Thus, it seems that multiple initiations are a characteristic feature of LEE1 P1. Careful examination of primer extension products suggested that these were from As neighboring the previously identified initiating nucleotide (G+1) (see below).
Fig. 2.

Analysis of the multiple transcripts from the LEE1 P1 promoter. (A) RNA generated from in vitro transcription reaction using pHJ12 as template (in vitro) and from E. coli (MG1655) carrying pHJ12 (in vivo) were determined by primer extension analysis using a primer annealing to +54~+74. (B) In vitro transcription reaction carried out with α32P UTP or γ32P ATP as indicated. (C) Transcription initiation points, shown as underlined As, near the previously assigned +1G (shown in bold).
In an attempt to identify the nucleotide at which transcription initiates from LEE1 P1, in vitro transcription was carried out in the presence of α-32P UTP or γ-32P ATP (Fig. 2B). The in vitro transcription assay was carried out with pSA508::LEE1 P1 (−200 to +20) so that shorter transcripts with distinct size were displayed on the 10% gel (panel B). Essentially, an identical transcript pattern was obtained with both α-32P UTP and γ-32P ATP. Since γ-32P ATP can be incorporated in the transcript only at the initiating nucleotide, this result indicated that all transcription initiated from As near the previously assigned +1G (Fig. 2C)(Friedberg et al., 1999). To further verify the tsp for the multiple transcripts, the RNAs were recovered by size fractionation on denaturing 10% polyacrylamide gels, eluted from the gel, ligated into 5′ adapters, amplified, cloned, and sequenced (Fig. S1) (Pfeffer et al., 2005). Subsequent sequencing confirmed that these transcripts started from the neighboring As.
Two promoters drive expression of ler
To identify the promoter element responsible for each transcript, the As near tsp were serially substituted and tested by in vitro transcription assay (Fig. 3). A careful examination of the transcripts from wild-type LEE1 P1 allowed us to reassign tsp to A+1CATTAGA+8A+9A+10A+11CA+13G, in which transcripts initiate from underlined As neighboring the previously assigned G+1 in bold (panel A). Substitution of A+3 with G reduced the transcripts initiated from the five As downstream of +1A, namely A+8A+9A+10A+11 and A+13. Substitution of A+1 reduced the transcript band intensity even further, while substitution of A−2 completely blocked transcript generation (lanes 2 to 4). The transcript from A+1 was unaffected by these substitutions. It was suggested that transcription governed by the −10 hexamer, T−3ACACA+3, has multiple initiation sites. The ranking of transcription initiation frequency, as determined by the density of each transcript, was A+9> A+10 >A+8= A+11 =A+13, where A+10 is the 7th nucleotide from the 3′ end of the −10 hexamer. Tentatively, A+10 was, therefore, assigned to +1 for LEE1 P1B. Substitution of A−11 or A−12 with G blocked transcription from A+1, suggesting that T−13AATGT−8 would constitute another −10 region for this transcript (lanes 5 and 6). In this case, the TGX sequence immediately upstream would confer the feature of an extended −10 hexamer. A weak transcript from A−2 was also removed, together with that from +1, suggesting that the transcription governed by the upstream −10 hexamer initiates from two As, A+1 and A−2. It was concluded that transcription of LEE1 is driven by two promoters: the upstream one was named P1A and the downstream one P1B. It should be noted that the P1B was dominant in vitro since ratio of P1A and P1B transcripts were approximately 1 to 3.8.
Fig. 3.

(A) In vitro transcription with wild-type LEE1 P1 (WT) and mutants carrying A to G substitutions close to the previously assigned +1G, as describe in the text. The assay was carried out with variant pSA508::LEE1 P1 (−200 to +20), and the reaction products were analyzed by 8 M urea/10% polyacrylamide gel electrophoresis. (B) tsp for P1A and P1B are indicated. The newly assigned +1A is underlined, and the putative −10 hexamers for P1A and P1B are shown in bold. −35 hexamer for P1A predicted by sequence walker (Supporting Information) is indicated.
Due to multiple transcription initiation sites, it was difficult to assign promoter elements, particularly for LEE1 P1B: T−3ACACA+3 deviates significantly from the −10 model used for conventional σ70 promoters. The promoter elements were determined using a program based on information theory, and the results were displayed using sequence walkers (Schneider & Stephens, 1990, Schneider, 1996, Shultzaberger et al., 2007) (Fig. S3). The analysis predicted at least four overlapping −10 elements within LEE1 P1 DNA (two of which were ruled out by genetic analysis; Fig. S2), in which the promoter elements for P1A, but not those for P1B, were included. This suggests that P1B is a nonconventional σ70 promoter (34), which might be responsible for the multiple tsp. Whereas, the −10 and −35 elements for P1A were predicted with optimal gap of 17 bp (Fig. 3, bottom) (http://alum.mit.edu/www/toms/papers/flexprom/). This promoter, with an unusual −35 (TTTTTT), could, however, be driven entirely from the extended −10 element.
RPO at the LEE1 P1 promoter was subsequently examined using KMnO4, which preferentially oxidizes thymidine residues within single-stranded DNA (Borowiec et al., 1987). LEE1 P1 DNA (−195 to +85) was PCR amplified using end labeled primers and analyzed following the addition of increasing amount of RNAP (Fig. 4). For the top strand, virtually all Ts between −20 and +5 were reactive upon the addition of RNAP (panel A). For the bottom strand, indicated Ts between −20 and +20 were reactive (panel B). Panel C shows a summary. To analyze strand melting following RNAP binding to P1A and P1B, mutants defective in either one of two promoters were included: A−2 to G substitution for P1A+/P1B−, and A−12 to G substitution for P1A−/P1B+. Different patterns were observed with the two mutant promoters. Since the base pairs involved in DNA wrapping around RNAP as well as those in transcription bubble are both reactive to KMnO4, exact position of melted region in RPO at the two promoters were only speculative (Borowiec et al., 1987, Shin et al., 2005). Nevertheless, it was evident that the two promoters use different mechanisms for RNAP binding and DNA unwinding following open complex formation by RNAP.
Fig. 4.

Structural changes induced by RNAP (0, 7 and 20 nM) binding to LEE1 P1 (−195 to +85) were probed by KMnO4 assay. Unpaired bases were identified for the top strand (A) and for the bottom strand (B). The first two lanes in each panel are DNA sequencing reactions for T and C. Arrows indicate unpaired bases in the presence of RNAP. Wild-type LEE1 P1 DNA and LEE1 P1A+/P1B− (A−2 to G) and LEE1 P1A−/P1B+ (A−12 to G) mutant DNA were analyzed. (C) Summary of the results from panels A and B. KMnO4-hypereactive bases induced by RNAP binding LEE1 P1 DNA are indicated with carets. The tsp for P1A and P1B are indicated, and the putative −10 hexamers are in bold.
NTP concentration-dependent changes of transcription start point (tsp)
Since all the initiating nucleotides were identified as As, we hypothesized that ATP concentration would alter the preferred initiation site, as in the case for abortive or stuttering RNA synthesis, both of which are [NTP]-dependent phenomena (Han & Turnbough, 1998, Qi & Turnbough, 1995). The in vitro transcription reaction was carried out in the presence of varying ATP concentrations, from 10 to 0.01 mM, with 0.1 mM being used in the standard reaction (Choy & Adhya, 1993) (Fig. 5). In the presence of 10 mM ATP, the transcription from P1B initiated predominantly from A+13, to a lesser extent from A+11, A+10, A+9, and only weakly from A+8. With decreasing ATP concentration, the preferred initiation nucleotide switched toward A+8. In the presence of 0.5 or 0.1 mM ATP, initiation was observed only weakly from A+11 and not from A+13, while a notable initiation from G+7 was clearly detected. Initiation from A+9 and A+10 remained constant. Subsequently, the pattern of transcription initiation induced by varying GTP or CTP concentrations was examined. Interestingly, changes in CTP concentration altered the initiation pattern from P1B, while changes in GTP concentration had no effect. In the presence of 1 or 0.5 mM CTP, the initiation from A+13 was as strong as that from A+9. As CTP concentration decreased below 0.1 mM, the initiation from both A+13 and A+11 decreased to an undetectable level, as was observed with decreasing ATP concentration. However, no initiation from the noble position was noted. The strong initiation from A+9, and the relatively weak initiation from A+8 and A+10, remained constant and were independent of [CTP]. It should be noted that C is at +12 and G is at +14 in the early transcribed region. Nevertheless, the P1B showed nucleotide concentration dependent change of the preferred tsp site. By contrast, no significant change in initiation from the P1A promoter or rna1 was noted. The effect of [UTP] was not analyzed since α-32P UTP in the presence of 0.01 mM cold UTP has been used for the detection of in vitro synthesized RNA (Choy & Adhya, 1993).
Fig. 5.

in vitro transcription assay with LEE1 P1 DNA in the presence of the indicated concentrations of rNTP. The concentrations of rNTPs used in routine in vitro transcription assays were 1 mM ATP, 0.1 mM GTP, 0.1 mM CTP, and 0.01 mM UTP were used.
In vivo activity of LEE1 P1A and B promoters
To determine the in vivo activity of each LEE1 P1 promoter, both wild type and the relevant mutant E. coli strains (MG1655) were transformed with pSA508 carrying the wild type or a mutant defective in either of the LEE1 P1 promoters. RNA was extracted from the bacteria during the exponential growth phase (A600≈1) and analyzed by primer extension (Fig. 6A). The wild type promoter construct generated transcripts from both P1A and P1B in wild type bacteria (lane 1). It is noteworthy that P1B transcripts were initiated mainly from G+7, A+8, A+9, and A+11. The P1A+/P1B− mutant construct generated a transcript that initiated only from A+1, but the intensity was somewhat increased, which may be due to unforeseen effects of P1B silencing (lane 4). P1A−/P1B+ constructgenerated identical transcripts from G+7 to A+11, as did the wild type promoter construct. These results confirmed that both P1A and P1B were active in vivo, although P1B was dominant. The ratio of P1A to P1B transcripts was approximately 1 to 49. In addition, the in vivo activity of each promoter was measured using the λ lysogen carrying wild-type or mutant LEE1 P1 promoters fused to promoter-less lacZYA (Fig. 6B). The same bacterial samples were prepared as for the primer extension analysis. Consistently, measurements of β-galactosidase activities revealed that P1B is more active than P1A but only about 6-fold, suggesting that the translation efficiencies of the transcripts from the two promoters are different
Fig. 6.

(A) LEE1 P1 activity measured in vivo. Both wild type and the indicated mutant E. coli (MG1655) strains were transformed with pSA508 carrying wild type (closed circles) or mutant LEE P1, LEE P1A+/P1B− (A−2 to G), or LEE P1A−/P1B+ (A−12 to G) as described in Fig. 2. Total RNA was extracted from the bacteria at A600=1.0 and the LEE1 P1 RNAs were analyzed by primer extension using the primer annealing to +54~+74. (B) LEE1 P1 activity measured in vivo using λ lysogens carrying wild-type or the mutant defective in either one of the promoters fused to lacZYA. Bacterial samples were taken as described in (A) for measurement of β-galactosidase activity (A420/min/ml/A600).
It has been reported that LEE1 P1 is activated by nucleoid-associated Fis and IHF on an EPEC strain background (Goldberg et al., 2001, Friedberg et al., 1999); therefore, relevant mutant strains, Ihf− (ihfA) and Fis− on an MG1655 background were used to test this (Fig. 6A, lanes 2 and 3). The LEE1 P1A and P1B transcripts generated in the Fis− mutant were virtually identical to those in the wild type; however, in the case of the IHF- mutant, the P1B transcript, but not P1A transcript, disappeared. This suggests that IHF activates the dominant promoter, LEE P1B, irrespective of the strain background, presumably by acting directly on the promoter as proposed by (Friedberg et al., 1999). In contrast, the regulatory effect of Fis may be strain specific.
Discussion
Transcription start points (tsp) are influenced by the RNAP-promoter interaction, which is regulated by specific sequences at the −10 and −35 elements, by the distance between the two elements, and by environmental factors such as nucleotide pools (Sorensen, 1993, Walker & Osuna, 2002, Liu & Turnbough, 1994, Jeong & Kang, 1994). In addition, the sequence context around the tsp strongly influences the selection of the initiating nucleotide. The results of the present study showed that the expression of LEE1-encoded genes is driven by two promoters, LEE1 P1A and LEE1 P1B. The most unusual feature associated with LEE1 P1B was multiple transcripts. Reiterative stuttering synthesis of transcripts is ascribed to the slippage of RNAP at short homopolymeric regions of template resulting in repetitive addition of the homopolymeric nucleotides to the 3′ end of the nascent transcript. The extent of reiterative transcription changes depending on the concentration of the hompolymeric nucleotide: reiterative transcription increases at galP2 (Jin, 1994) but decreases at carABp1 and pyrB1 (Liu & Turnbough, 1994, Han & Turnbough, 1998), as the concentration of homopolymeric nucleotide decreases. Instead, we observed a small shift of tsp with changing concentration of ATP and CTP. The physiological significance of the shift is unknown. Nevertheless, this suggested that stuttering synthesis did not contribute to the generation of multiple transcripts (Fig. 5). In RPO, the DNA duplex is disrupted over a stretch of 12–15 base pairs, around positions −12 and +2, which leads to the formation of a transcription bubble and assignment of the tsp accessible to the RNAP catalytic center (Kirkegaard et al., 1983, Murakami et al., 2002). In the case of LEE1 P1B, we suggest that the downstream end of the transcription bubble would lie within a stretch of As, which would result in variability in the position of the downstream end of the transcription bubble. At the same time, the flexibility of the DNA in the stretch of As would result in imprecise initiation of transcription from neighboring As, resulting in flexible transcription initiation from LEE1 P1B. Alternatively, the multiple transcripts could be due to RNAP backtracking in the stretch of As, although such backtracking has been demonstrated to only occur during transcription elongation (Nudler et al., 1997). This issue would be resolved by performing structural analyses of RPO at LEE1 P1B.
It should be noted that flexible transcription initiation events from positions near +1 are not uncommon, particularly with regard to eukaryotic RNA polymerase III-dependent transcription of tRNA genes. A study using a Tobacco in vitro transcription system showed that transcription of most tRNA genes in budding yeast and Arabidopsis initiate from multiple neighboring As (Yukawa et al., 2011). In E. coli, transcription from the fis promoter is initiated predominantly from +1C, but can also initiate from −2G, −1C, and +2T (Walker & Osuna, 2002). Interestingly, the preferred tsp for the fis promoter was found to be dependent on intracellular NTP concentration. Similar changes were observed for the LEE1 P1B tsp in vitro; in the presence of ATP at concentrations below 1 mM, transcription initiation shifted to G+7 (Fig. 5). Analysis of the transcripts generated in vivo showed that P1B transcription initiated at G+7, A+8, A+9, and A+11 with relatively equal intensity (Fig. 6); however, since the ATP pool during steady state bacterial growth (E. coli) is maintained at 2.1 ± 0.5 mM (Radchenko et al., 2010), this shift thus might also be a reflection of the physiochemical environment within the bacterial cytosol.
Analysis of the transcripts generated in vivo revealed that P1B was the dominant major promoter and was activated by IHF (Fig. 6). IHF activates transcription through direct protein-protein interactions between transcription activator proteins and the C-terminal domain of the α subunit of RNAP (Busby & Ebright, 1999). P1 P1A and P1B are separated by one helical turn (10 bp); therefore, the RNAP molecules bound to these sites should be in different geometric positions. It is unlikely that transcription activators make the same contacts with RNAPs on the two LEE1 P1 promoters; therefore only one of the two promoters should be activated at any one time. The data presented in the present study suggest that the major promoter, P1B, is subject to regulation by IHF, in addition to other environmental factors. Whereas, P1A might play a role in setting the basal level of expression only enough to autorepress LEE1 P1 promoter by Ler, encoded by the first gene of the LEE1 operon (Berdichevsky et al., 2005). This hypothesis could be confirmed by analyzing the two promoters in the EPEC strain background.
EPEC and EHEC, both of which cause disease by forming lesions that attach to and efface the intestinal wall, share genetic and phenotypic similarities; most notably the LEE pathogenicity island (Mellies et al., 2007). The promoter for the LEE1 operon plays a key role in regulating all LEE-encoded genes. LEE1 P1 in the two pathogenic strains of E. coli share virtually the same nucleotide sequence with few changes. EHEC carries a single-base-pair deletion in the stretch of Ts that comprise −35 of P1A, and C+12 is replaced with A (Porter 2005). Although these changes reduce the activity of the promoter compared with that in EPEC (Porter et al., 2005), regulation of LEE1 in these two strains is considered to be the same. The string of 6As (A+8 to A+13) identified in EHEC does not affect flexible transcription initiation from P1B in EPEC (Fig. S4). Both the tsp and the promoter elements of LEE1 in EPEC and EHEC have been reassigned several times. The −10 element and the tsp in EPEC were identified as TTTACAand G+7, respectively (Mellies et al., 1999). In the case of EHEC, the −10 element was originally identified as TACACA (Sperandio et al., 2002), but was later reassigned to a sequence two base pairs upstream; TTTACA (Porter et al., 2005, Russell et al., 2007, Sharp & Sperandio, 2007). Recently, Busby’s group identified TTGACA and TACACA as the functional −35 and −10 hexamer elements of LEE1 P1 based on mutational analysis (Islam et al., 2011a). In a subsequence report, the same group identified a cryptic promoter (Islam et al., 2011b). These are the same promoters identified in the present study, although the tsp are slightly different. The results of the present study showed that the confusion is mainly due to the presence of multiple tsp, which shift depending on the experimental conditions (in vivo or in vitro). Therefore, the different tsp reported in different experiments may all be correct. Nevertheless, it is now certain (based on genetic and biochemical analyses, including the measurement of RNA in vivo and in vitro) that transcription from LEE1 P1 is driven by two promoters, and that the downstream promoter, P1B, is the major one.
Experimental procedures
Strains and plasmids
The plasmids used for in vitro transcription assays were constructed by cloning LEE1 P1 DNA sequences between the EcoR1 and Pst1 sites of the transcriptional vector pSA508 (Choy & Adhya, 1993). LEE1 P1 DNA was obtained by PCR amplification from chromosomal DNA of EPEC strain E2348/69 (Levin et al., 1987). Mutants carrying altered LEE1 P1 sequences were generated by cloning the synthetic DNA oligomers. The primers used for PCR amplification of respective DNA are shown in Table 1. λ bacteriophages carrying various wild-type and mutant lerP::lacZYA were obtained by cloning PCR-amplified lerP fragments into the EcoR1 and BamH1 sites in pRS415 and subsequent homologous recombination with λRS45 (Simons et al., 1987). λ lysogens carrying recombinant prophages were prepared as described in (Simons et al., 1987). The bacterial strains constructed by P1 transduction and the relevant plasmids are listed in Table 2.
Table 1.
Primers used for PCR amplification of various LEE1 P1 DNA sequences
| Primer | Sequence | Product |
|---|---|---|
| −200 EcoRI | 5′-CGGAATTCCAGCTTGGTTTTTATTCTG-3′ | pHJ12, pHJ36 |
| +80 pstI | 5′-GGTTCTGCAGAGATAACGTTTATCTATC-3′ | pHJ12 |
| +20 pstI | 5′-GGTTCTGCAGATGTTATTATTCTCTGTTT-3′ | pHJ36 |
| A+3->G(R) | 5′-GGTTCTGCAGCTCTGTTTTCTAACGTGTAAAAATAGATTATC-3′ | pHJ56 |
| A+1->G(R) | 5′-GGTTCTGCAGCTCTGTTTTCTAATGCGTAAAAATAGATTATC-3′ | pHJ57 |
| A−2->G(R) | 5′-GGTTCTGCAGCTCTGTTTTCTAATGTGCAAAAATAGATTATC-3′ | pHJ58 |
| A−11->G(R) | 5′-GGTTCTGCAGCTCTGTTTTCTAATGTGTAAAAATAGACTATC-3′ | pHJ73 |
| A−12->G(R) | 5′-GGTTCTGCAGCTCTGTTTTCTAATGTGTAAAAATAGATCATC-3′ | pHJ74 |
Table 2.
Strains and plasmids used in this study
| Strains | Description | Reference |
|---|---|---|
| Escherichia coli | Wild type | |
| MG1655 | ||
| CH1018 | [argF-lac]Δ | (Kim et al., 2004) |
| HJ1098 | CH1018, Φ Lee1 P1 (−200~+80)::lacZYA | This work |
| HJ1103 | HJ1098, fis::cat | (Sircili et al., 2004) |
| HJ1104 | HJ1098, himA::tet | (Miller & Nash, 1981) |
| HJ1193 | CH1018, Φ Lee1 P1 (200~+80;A−2 substituted by G)::lacZYA | This work |
| KH1001 | CH1018, Φ Lee1 P1 (200~+80;A−12 substituted by G)::lacZYA | This work |
| Plasmids | ||
| pSA508 | Transcriptional vector, Ampr | (Choy & Adhya, 1993) |
| pHJ12 | pSA508 containing −200 to +80 of lEEP | This work |
| pHJ36 | pSA508 containing −200 to +20 of lEEP | This work |
| pHJ56 | pSA508 containing −200 to +20 of lEEP | This work |
| A+3 substituted by G | ||
| pHJ57 | pSA508 containing −200 to +20 of lEEP | This work |
| A+1 substituted by G | ||
| pHJ58 | pSA508 containing −200 to +20 of lEEP | This work |
| A−2 substituted by G | ||
| pHJ73 | pSA508 containing −200 to +20 of lEEP | This work |
| A−11 substituted by G | ||
| pHJ74 | pSA508 containing −200 to +20 of lEEP | This work |
| A−12 substituted by G |
β-galactosidase assay
The β-galactosidase assay was performed as described by Miller (Miller, 1972), using cells permeabilized with Koch’s lysis solution (Putnam & Koch, 1975). β-galactosidase–specific activity was expressed in Miller units (A420/min/A600).
Primer extension analysis
Primer extension analysis used the alkaline denaturation procedure described by (Rostoks et al., 2000)). The primer used annealed to +54~+74 of the LEE1 P1 RNA.
In vitro transcription assay
Transcription reactions were carried out as previously described (Choy & Adhya, 1993). Briefly, 2 nM DNA template, 1 mM ATP, 0.1 mM GTP, 0.1 mM CTP, 0.01 mM UTP and 10–20 μCi of [α-32P] UTP were preincubated in buffer (20 mM Tris-acetate, pH 7.8, 10 mM magnesium acetate, 100 mM potassium glutamate, 1 mM dithiothreitol) at 37°C for 5 min. Transcription was initiated by the addition of RNAP (20 nM) in a total volume of 20 μL and was terminated after 10 min at 37°C by the addition of an equal volume (20 μL) of RNA loading buffer (80% (v/v) deionized formamide, 1 × TBE (89 mM tris, 89 mM boric acid, 2 mM EDTA), 0.025% bromophenol blue, 0.025% xylene cyanole). The mixture was electrophoresed routinely in an 8 M urea/8% polyacrylamide sequencing gel (40 cm × 0.4 mm), for analysis. RNAP holoenzyme from the BL21 strain was purchased from Epicentre (Madison, WI, USA).
KMnO4 assay
LEE1 P1 promoter DNA (−200 to +80) was PCR amplified with the primers: CGGAATTCCAGCTTGGTTTTTATTCTG (27 mer) for the top strand and CGGGATCCGAGATAACGTTTATCTATC (27 mer) for the bottom strand. For analysis of the top strand, 32P-labeled top primer was used, and for analysis of the bottom strand, 32P-labeled bottom primer was used, respectively. KMnO4 reactions followed the protocol described by Rostoks et al.(Rostoks et al., 2000). The reaction conditions were the same as those for the in vitro transcription reactions except that nucleotides were omitted. Bases modified by KMnO4 were analyzed in the DNA sequencing gel.
Promoter analysis based on information theory
The LEE1 P1 region was scanned using an information theory based model for σ70 promoters developed previously (Shultzaberger et al., 2007) to identify -35 and -10 elements with appropriate spacing. Please see “Supporting Information” for further details.
Supplementary Material
Acknowledgments
This work was supported by the Korea Science and Engineering Foundation grant that was funded by MOST (No. 2007-04213). J.H.R. was supported by grant No. RTI05-01-01 from the Regional Technology Innovation Program of the Ministry of Knowledge Economy (MKE). T. S. was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research
Footnotes
Additional supporting information may be found in the online version of this article.
References
- Berdichevsky T, Friedberg D, Nadler C, Rokney A, Oppenheim A, Rosenshine I. Ler is a negative autoregulator of the LEE1 operon in enteropathogenic Escherichia coli. J Bacteriol. 2005;187:349–357. doi: 10.1128/JB.187.1.349-357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowiec JA, Zhang L, Sasse-Dwight S, Gralla JD. DNA supercoiling promotes formation of a bent repression loop in lac DNA. J Mol Biol. 1987;196:101–111. doi: 10.1016/0022-2836(87)90513-4. [DOI] [PubMed] [Google Scholar]
- Busby S, Ebright RH. Transcription activation by catabolite activator protein (CAP) J Mol Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- Bustamante VH, Santana FJ, Calva E, Puente JL. Transcriptional regulation of type III secretion genes in enteropathogenic Escherichia coli: Ler antagonizes H-NS-dependent repression. Mol Microbiol. 2001;39:664–678. doi: 10.1046/j.1365-2958.2001.02209.x. [DOI] [PubMed] [Google Scholar]
- Chamberlin MJ. The selectivity of transcription. Annu Rev Biochem. 1974;43:721–775. doi: 10.1146/annurev.bi.43.070174.003445. [DOI] [PubMed] [Google Scholar]
- Choy HE, Adhya S. RNA polymerase idling and clearance in gal promoters: use of supercoiled minicircle DNA template made in vivo. Proc Natl Acad Sci U S A. 1993;90:472–476. doi: 10.1073/pnas.90.2.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman CJ. H-NS: a universal regulator for a dynamic genome. Nat Rev Microbiol. 2004;2:391–400. doi: 10.1038/nrmicro883. [DOI] [PubMed] [Google Scholar]
- Elliott SJ, Sperandio V, Giron JA, Shin S, Mellies JL, Wainwright L, Hutcheson SW, McDaniel TK, Kaper JB. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect Immun. 2000;68:6115–6126. doi: 10.1128/iai.68.11.6115-6126.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott SJ, Wainwright LA, McDaniel TK, Jarvis KG, Deng YK, Lai LC, McNamara BP, Donnenberg MS, Kaper JB. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol Microbiol. 1998;28:1–4. doi: 10.1046/j.1365-2958.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- Friedberg D, Umanski T, Fang Y, Rosenshine I. Hierarchy in the expression of the locus of enterocyte effacement genes of enteropathogenic Escherichia coli. Mol Microbiol. 1999;34:941–952. doi: 10.1046/j.1365-2958.1999.01655.x. [DOI] [PubMed] [Google Scholar]
- Goldberg MD, Johnson M, Hinton JC, Williams PH. Role of the nucleoid-associated protein Fis in the regulation of virulence properties of enteropathogenic Escherichia coli. Mol Microbiol. 2001;41:549–559. doi: 10.1046/j.1365-2958.2001.02526.x. [DOI] [PubMed] [Google Scholar]
- Haack KR, Robinson CL, Miller KJ, Fowlkes JW, Mellies JL. Interaction of Ler at the LEE5 (tir) operon of enteropathogenic Escherichia coli. Infect Immun. 2003;71:384–392. doi: 10.1128/IAI.71.1.384-392.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Turnbough CL., Jr Regulation of carAB expression in Escherichia coli occurs in part through UTP-sensitive reiterative transcription. J Bacteriol. 1998;180:705–713. doi: 10.1128/jb.180.3.705-713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Lawrie J, Boyer HW, Hedgpeth J. Reiterative copying by E. coli RNA polymerase during transcription initiation of mutant pBR322 tet promoters. Nucleic Acids Res. 1990;18:547–552. doi: 10.1093/nar/18.3.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS, Bingle LE, Pallen MJ, Busby SJ. Organization of the LEE1 operon regulatory region of enterohaemorrhagic Escherichia coli O157:H7 and activation by GrlA. Mol Microbiol. 2011a;79:468–483. doi: 10.1111/j.1365-2958.2010.07460.x. [DOI] [PubMed] [Google Scholar]
- Islam MS, Pallen MJ, Busby SJ. A cryptic promoter in the LEE1 regulatory region of enterohaemorrhagic Escherichia coli: promoter specificity in AT-rich gene regulatory regions. Biochem J. 2011b;436:681–686. doi: 10.1042/BJ20110260. [DOI] [PubMed] [Google Scholar]
- Jacques JP, Susskind MM. Pseudo-templated transcription by Escherichia coli RNA polymerase at a mutant promoter. Genes Dev. 1990;4:1801–1810. doi: 10.1101/gad.4.10.1801. [DOI] [PubMed] [Google Scholar]
- Jeong W, Kang C. Start site selection at lacUV5 promoter affected by the sequence context around the initiation sites. Nucleic Acids Res. 1994;22:4667–4672. doi: 10.1093/nar/22.22.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin DJ. Slippage synthesis at the galP2 promoter of Escherichia coli and its regulation by UTP concentration and cAMP.cAMP receptor protein. J Biol Chem. 1994;269:17221–17227. [PubMed] [Google Scholar]
- Johnson DE, McClure WR. RNA Polymerase. 1976. [Google Scholar]
- Kim EY, Shin MS, Rhee JH, Choy HE. Factors influencing preferential utilization of RNA polymerase containing sigma-38 in stationary-phase gene expression in Escherichia coli. J Microbiol. 2004;42:103–110. [PubMed] [Google Scholar]
- Kirkegaard K, Buc H, Spassky A, Wang JC. Mapping of single-stranded regions in duplex DNA at the sequence level: single-strand-specific cytosine methylation in RNA polymerase-promoter complexes. Proc Natl Acad Sci U S A. 1983;80:2544–2548. doi: 10.1073/pnas.80.9.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JR, Krummel B, Chamberlin MJ. Isolation and properties of transcribing ternary complexes of Escherichia coli RNA polymerase positioned at a single template base. J Mol Biol. 1987;196:85–100. doi: 10.1016/0022-2836(87)90512-2. [DOI] [PubMed] [Google Scholar]
- Lewis DE, Adhya S. Axiom of determining transcription start points by RNA polymerase in Escherichia coli. Mol Microbiol. 2004;54:692–701. doi: 10.1111/j.1365-2958.2004.04318.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Turnbough CL., Jr Effects of transcriptional start site sequence and position on nucleotide-sensitive selection of alternative start sites at the pyrC promoter in Escherichia coli. J Bacteriol. 1994;176:2938–2945. doi: 10.1128/jb.176.10.2938-2945.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel TK, Jarvis KG, Donnenberg MS, Kaper JB. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A. 1995;92:1664–1668. doi: 10.1073/pnas.92.5.1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellies JL, Barron AM, Carmona AM. Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect Immun. 2007;75:4199–4210. doi: 10.1128/IAI.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellies JL, Elliott SJ, Sperandio V, Donnenberg MS, Kaper JB. The Per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler) Mol Microbiol. 1999;33:296–306. doi: 10.1046/j.1365-2958.1999.01473.x. [DOI] [PubMed] [Google Scholar]
- Miller HI, Nash HA. Direct role of the himA gene product in phage lambda integration. Nature. 1981;290:523–526. doi: 10.1038/290523a0. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in molecular genetics. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y: 1972. p. xvi.p. 466. [Google Scholar]
- Murakami KS, Masuda S, Campbell EA, Muzzin O, Darst SA. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- Neidhardt FC, Curtiss R. Escherichia coli and Salmonella: cellular and molecular biology. ASM Press; Washington, D.C: 1996. [Google Scholar]
- Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell. 1997;89:33–41. doi: 10.1016/s0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Lagos-Quintana M, Tuschl T. Cloning of small RNA molecules. Curr Protoc Mol Biol. 2005;Chapter 26(Unit 26–24) doi: 10.1002/0471142727.mb2604s72. [DOI] [PubMed] [Google Scholar]
- Porter ME, Mitchell P, Free A, Smith DG, Gally DL. The LEE1 promoters from both enteropathogenic and enterohemorrhagic Escherichia coli can be activated by PerC-like proteins from either organism. J Bacteriol. 2005;187:458–472. doi: 10.1128/JB.187.2.458-472.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam SL, Koch AL. Complications in the simplest cellular enzyme assay: lysis of Escherichia coli for the assay of beta-galactosidase. Anal Biochem. 1975;63:350–360. doi: 10.1016/0003-2697(75)90357-7. [DOI] [PubMed] [Google Scholar]
- Qi F, Turnbough CL., Jr Regulation of codBA operon expression in Escherichia coli by UTP-dependent reiterative transcription and UTP-sensitive transcriptional start site switching. J Mol Biol. 1995;254:552–565. doi: 10.1006/jmbi.1995.0638. [DOI] [PubMed] [Google Scholar]
- Radchenko MV, Thornton J, Merrick M. Control of AmtB-GlnK complex formation by intracellular levels of ATP, ADP, and 2-oxoglutarate. J Biol Chem. 2010;285:31037–31045. doi: 10.1074/jbc.M110.153908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostoks N, Park S, Choy HE. Reiterative transcription initiation from galP2 promoter of Escherichia coli. Biochim Biophys Acta. 2000;1491:185–195. doi: 10.1016/s0167-4781(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Russell RM, Sharp FC, Rasko DA, Sperandio V. QseA and GrlR/GrlA regulation of the locus of enterocyte effacement genes in enterohemorrhagic Escherichia coli. J Bacteriol. 2007;189:5387–5392. doi: 10.1128/JB.00553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD. Reading of DNA sequence logos: prediction of major groove binding by information theory. Methods Enzymol. 1996;274:445–455. doi: 10.1016/s0076-6879(96)74036-3. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FC, Sperandio V. QseA directly activates transcription of LEE1 in enterohemorrhagic Escherichia coli. Infect Immun. 2007;75:2432–2440. doi: 10.1128/IAI.02003-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Song M, Rhee JH, Hong Y, Kim YJ, Seok YJ, Ha KS, Jung SH, Choy HE. DNA looping-mediated repression by histone-like protein H-NS: specific requirement of Esigma70 as a cofactor for looping. Genes Dev. 2005;19:2388–2398. doi: 10.1101/gad.1316305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultzaberger RK, Chen Z, Lewis KA, Schneider TD. Anatomy of Escherichia coli sigma70 promoters. Nucleic Acids Res. 2007;35:771–788. doi: 10.1093/nar/gkl956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- Sircili MP, Walters M, Trabulsi LR, Sperandio V. Modulation of enteropathogenic Escherichia coli virulence by quorum sensing. Infect Immun. 2004;72:2329–2337. doi: 10.1128/IAI.72.4.2329-2337.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen L. Nobody gets off comfortably. Interview by Mette Fjordbo. Sygeplejersken. 1993;93:14–15. [PubMed] [Google Scholar]
- Sperandio V, Li CC, Kaper JB. Quorum-sensing Escherichia coli regulator A: a regulator of the LysR family involved in the regulation of the locus of enterocyte effacement pathogenicity island in enterohemorrhagic E. coli. Infect Immun. 2002;70:3085–3093. doi: 10.1128/IAI.70.6.3085-3093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires C, Krainer A, Barry G, Shen WF, Squires CL. Nucleotide sequence at the end of the gene for the RNA polymerase beta’ subunit (rpoC) Nucleic Acids Res. 1981;9:6827–6840. doi: 10.1093/nar/9.24.6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umanski T, Rosenshine I, Friedberg D. Thermoregulated expression of virulence genes in enteropathogenic Escherichia coli. Microbiology. 2002;148:2735–2744. doi: 10.1099/00221287-148-9-2735. [DOI] [PubMed] [Google Scholar]
- Walker KA, Osuna R. Factors affecting start site selection at the Escherichia coli fis promoter. J Bacteriol. 2002;184:4783–4791. doi: 10.1128/JB.184.17.4783-4791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yukawa Y, Dieci G, Alzapiedi M, Hiraga A, Hirai K, Yamamoto YY, Sugiura M. A common sequence motif involved in selection of transcription start sites of Arabidopsis and budding yeast tRNA genes. Genomics. 2011;97:166–172. doi: 10.1016/j.ygeno.2010.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.