

Abstract
The pulmonary circulation is a high flow and low pressure circuit, with an average resistance of 1 mmHg.min.L−1 in young adults, increasing to 2.5 mmHg.min.L−1 over 4–6 decades of life. Pulmonary vascular mechanics at exercise are best described by distensible models. Exercise does not appear to affect the time constant of the pulmonary circulation or the longitudinal distribution of resistances. Very high flows are associated with high capillary pressures, up to a 20–25 mmHg threshold associated with interstitial lung edema and altered ventilation/perfusion relationships. Pulmonary artery pressures of 40–50 mmHg, which can be achieved at maximal exercise, may correspond to the extreme of tolerable right ventricular afterload. Distension of capillaries that decrease resistance may be of adaptative value during exercise, but this is limited by hypoxemia from altered diffusion/perfusion relationships. Exercise in hypoxia is associated with higher pulmonary vascular pressures and lower maximal cardiac output, with increased likelihood of right ventricular function limitation and altered gas exchange by interstitial lung edema. Pharmacological interventions aimed at the reduction of pulmonary vascular tone have little effect on pulmonary vascular pressure-flow relationships in normoxia, but may decrease resistance in hypoxia, unloading the right ventricle and thereby improving exercise capacity. Exercise in patients with pulmonary hypertension is associated with sharp increases in pulmonary artery pressure and a right ventricular limitation of aerobic capacity. Exercise stress testing to determine multipoint pulmonary vascular pressures-flow relationships may uncover early stage pulmonary vascular disease.
1. Introduction
The pulmonary circulation is a low pressure, high flow circuit. Low pressure prevents fluid moving out of fluid from the pulmonary vessels into the interstitial space, and allows the right ventricle to operate at minimal energy cost. Exercise increases oxygen uptake (VO2) and carbon dioxide output (VCO2) up to some twenty fold above resting values, and this increase in gas exchange is matched by an increase in cardiac output up to some 6-fold. The 6-fold increase in cardiac output is needed for the increased convective transport demands of these gases to and from exercising muscles. Exercise may thus be a considerable stress on the pulmonary circulation, taking the entire cardiac output while keeping the lungs dry and the right ventricle compensated.
An additional stress is imposed upon the pulmonary circulation by decreased inspired PO2 at altitude. Hypoxia causes pulmonary arterioles and venules (to a lesser extent) to constrict, which may increase pulmonary capillary pressures (Pcp) to the point of excessive transudation, and pulmonary artery pressures (Ppa) to the point of excessive right ventricular afterload. Even though the maximum VO2 (VO2max) decreases in hypoxia, thereby decreasing the maximum pulmonary flow required for gas exchange, both Pcp and Ppa may reach the limits of compensation, resulting in lung edema or right heart failure, or both.
Since the essential stress of exercise imposed on the pulmonary circulation is an increase in cardiac output, with the additional burden of an increase in pulmonary vascular pressures in hypoxia, in this chapter we will first review concepts of pulmonary vascular pressure-flow relationships and right ventriculo-arterial coupling. We will then examine available data on invasive and non invasive measurements of pulmonary hemodynamics at exercise, in humans and in different animal species, in normoxia and in hypoxia. A final part of the chapter will be devoted to typical pathophysiological applications to patients with cardiopulmonary disease.
Only dynamic exercise will be considered here, as static exercise is associated with an increase in systemic vascular resistance, a limited increase in cardiac output or VO2, and eventual intrathoracic pressure changes (131), making it a more complex stress that is less relevant for the study of pulmonary vascular mechanics and right ventricular function. High levels of dynamic exercise may be generated with variable levels of resistive effort, but how this affects the pulmonary circulation is not exactly known. Therefore, in this chapter, exercise will be considered as a cause of increased cardiac output affecting pulmonary vascular pressures, and pulmonary hemodynamics at exercise will be explained using reference models of flow-related changes in pulmonary vascular mechanics.
2. Steady flow pulmonary hemodynamics
2.1. Pulmonary vascular resistance
How cardiac output affects the pulmonary circulation is usually understood in terms of “steady-flow” hemodynamics. This approach summarizes time-dependent pulmonary artery pressure and flow waves by their time-averaged or mean values, and defines pulmonary vascular pressure-flow relationships by a single point pressure difference-flow ratio calculated as a resistance. Accordingly, a single point resistance calculation serves to describe the flow-resistive properties of the pulmonary circulation, which is one aspect of pulmonary vascular function.
Thus pulmonary vascular resistance (PVR) is the difference between mean Ppa (Ppam) and mean left atrial pressure (Pla) divided by cardiac output Q:
This is most often implemented in clinical practice by measurements of Ppam, Pla and Q via fluid-filled thermodilution catheters. These catheters are balloon-tipped, allowing for an estimate of Pla by a wedged or a balloon occluded Ppa (Ppw or Ppao).
Sometimes a measurement of Pla or Ppao cannot be obtained, and a total PVR (TPVR) is calculated as
Since Pla is not negligible with respect to Ppa, TPVR is larger than PVR and this difference may be flow-dependent. Thus TPVR is not a correct characterization of the flow-resistive properties of the pulmonary circulation. On the other hand, TPVR may be a more realistic estimate of right ventricular afterload, as the right ventricle is exposed to Ppa and not to the difference between Ppa and Pla.
A resistance calculation derives from Ohm’s law first described for electric circuits. The resistance is determined by vessel and fluid properties. The physical law that governs laminar flows of Newtonian fluids through non-distensible, straight circular tubes was originally proposed by the French physician Poiseuille and later put in mathematical equation by the German physicist Hagen. Poiseuille showed experimentally that flow was inversely related to the fourth power of the internal radius, and confirmed previous demonstrations that arterial pressure remains high in arteries down to 2 mm in diameter while venous pressure is low in animals. The Hagen-Poiseuille law states that resistance R to flow of a single tube is equal to the product of the length l of the tube and viscosity η and a constant 8 divided by the product of π and the fourth power of the internal radius r. More generally R can be calculated as a pressure drop ΔP to flow (Q) ratio:
The fact that r in the equation is to the fourth power explains why R is exquisitely sensitive to small changes in tube caliber (a 10% change in radius results in almost 50% change in resistance). Accordingly, PVR is a good indicator of the state of constriction or dilatation of pulmonary resistive vessels and is useful for detecting changes in arteriolar vessel caliber due to changes in tone and/or structure.
The limits of normal resting pulmonary vascular pressures and flows as derived from invasive measurements reported by ourselves in resting supine young adult healthy volunteers are shown in Table 1 (134,146,161). In that study population, Q was lower in women, who are smaller than men, and thus PVR in women was higher. However, there were no gender differences in pulmonary hemodynamics after correction for body dimensions. More recently, Kovacs et al exhaustively reviewed the literature on invasive hemodynamic measurements in normal subjects (111). They retrieved 47 studies on a total of 1187 individuals, of whom 225 were identified as women and 717 as men. Resting supine pulmonary vascular pressures were in agreement with our own. In particular, no mean Ppa higher than 20 mmHg was found, and there were no gender differences in indexed PVR. However, cardiac output was somewhat higher in Kovacs’ group, averaging 7.3 vs 6.4 Lmin−1. This may be explained by the smaller proportion of female subjects (22 % in the report by Kovacs et al. vs. 40 % in our own studies) and possible uncertainties about quality of cardiac output measurements using the Fick method (57). It is obvious that the accuracy of the measurement of cardiac output is essential to resting or exercise PVR calculations.
Table 1.
Limits of normal of pulmonary blood flow and vascular pressures
| Variables | Mean | Limits of normal |
|---|---|---|
| Q L/min | 6.4 | 4.4 – 8.4 |
| Q, L/min.m2 | 3.8 | 2.5 – 4.5 |
| Heart rate, bpm | 67 | 4 – 100 |
| Ppa systolic, mmHg | 19 | 13 – 26 |
| Ppa diastolic, mmHg | 10 | 6 – 16 |
| Ppa, mean, mmHg | 13 | 7 – 19 |
| Ppao, mmHg | 9 | 5 – 13 |
| Pcp, mmHg | 10 | 8 – 12 |
| Pra, mmHg | 5 | 1 – 9 |
| PVR, dyne.s.cm−5 | 55 | 11 – 99 |
| Psa, mean, mmHg | 91 | 71 – 110 |
Legend: Q: cardiac output; Ppa: pulmonary artery pressure; Ppao: occluded Ppa; Pcp: pulmonary capillary pressure (measured by single occlusion); Pra: right atrial pressure: PVR: pulmonary vascular resistance; Psa: systemic arterial pressure; limits of normal: from mean − 2 SD to mean + 2 SD
Aging is associated with an increase in PVR. This is related to a slight increase in mean Ppa and a more important decrease in cardiac output leading to a doubling of PVR over a five decades life (11,68,69,88). According to the Hagen-Poiseuille law, assuming no changes in pulmonary vascular length or blood viscosity, a doubling of PVR should be associated with a 16% decrease in internal radius of small vessels. This type of change could be caused by remodeling or an increase in smooth muscle cell tone, both of which have been described to occur with age in arterioles of the systemic circulation (64,125).
Body position affects PVR through associated changes in systemic venous return. In the upright position, Pla, right atrial pressure (Pra) and cardiac output are lower than in the supine position. Because of pulmonary vascular de-recruitment, mean Ppa remains essentially the same. Accordingly, PVR is increased. This difference in upright vs. supine PVR is important to keep in mind when examining changes in PVR during exercise performed upright as compared to supine (11,57,68,69,111,178).
2.2. Pressure-flow relationships
A difficulty with interpreting PVR changes with exercise is the flow-dependency of this variable.
The inherent assumption in a PVR calculation is that the mean Ppa-Q relationship is constant and crosses the pressure axis at a value equal to Pla (such that when flow is zero, theoretically, Ppam = Pla). Then, PVR is constant, independent of the absolute pressure or flow (Fig 1, curve A) (155).
Figure 1.

Starling resistor model to explain the concept of closing pressure within a circulatory system. Flow (Q) is determined by the gradient between an inflow pressure, or mean pulmonary artery pressure (Ppa), and an outflow pressure which is either closing pressure (Pc) or left atrial pressure (Pla). When Pla > Pc, the (Ppa – Pla)/Q relationship crosses the origin (A curve) and PVR is constant. When Pc > Pla, the (Ppa - Pla)/Q relationship has a positive pressure intercept (B and C curves), and PVR decreases curvilinearly with increasing Q. The B and C curves are curvilinear a low flow representing recruitment. Also shown are possible misleading PVR calculations: PVR, the slope of (Ppa-Pla)/Q may remain unchanged in the presence of a vasoconstriction (from 1 to 2) or decrease (from 1 to 3) with no change in the functional state of the pulmonary circulation (unchanged pressure/flow line). Adapted from reference 155. Permission pending.
The relationship between (Ppam-Pla) and Q has been shown to be reasonably well described by a linear approximation over a limited “physiological” range of flows, with a zero extrapolated pressure intercept in well oxygenated lungs in supine resting intact animals including man (155,178,183). This suggests complete recruitment and minimal distension of the normal well oxygenated pulmonary circulation. However, hypoxia and a number of cardiac and respiratory diseases increase both the slope and the extrapolated intercepts of multipoint (Ppam – Pla) vs. Q plots (155).
While an increase in the slope of Ppam-Q is easily understood as being caused by a decreased cross-sectional area of pulmonary resistive vessels, the positive extrapolated pressure intercept has inspired various explanatory models.
2.3. The Starling resistor model of the pulmonary circulation
To explain the non-zero and positive extrapolated pressure intercept, Permutt et al. conceived a vascular “waterfall” or “Starling resistor” model made of parallel collapsible vessels with a distribution of non-zero and positive closing pressures (174). The waterfall analogy refers to the fact that the flow rate (Q) over a waterfall is independent of its height (the pressure difference between upstream and downstream). Instead, an “external” factor (in the case of a waterfall, fluid momentum) controls the flow rate. The Starling resistor itself was actually a device: a collapsible tube inside of a closed chamber that could be pressurized, thus providing an “external” control over the flow rate. Starling used this device in the circuit of his heart-lung preparation to control blood pressure (171). Permutt et al. postulated that in the pulmonary circulation, as flow decreased, arteries would be progressively de-recruited, accounting for a low flow Ppam-Q curve that is concave to the flow axis, and intercepts the pressure axis at the lowest closing pressure needed to be overcome to generate a flow. At higher flows, complete vessel recruitment and negligible distension account for a linear Ppam–Q curve with an extrapolated pressure intercept representing a weighted mean of closing pressures. In this model, the mean closing pressure is the effective outflow pressure of the pulmonary circulation. At higher flows, Pla is equal to the mean closing pressure. However, at lower flows, Pla is less than the mean closing pressure and becomes irrelevant to flow, analogous to the height of water below a waterfall. Instead, other factors analogous to the external pressure acting on collapsible tube in the Starling resistor determine the effective outflow pressure of the circuit. In that situation, a PVR calculation becomes misleading because of flow-dependency. That is, changes in flow then change PVR without changing the functional state of the pulmonary circulation (Fig 1, curve B). However, according to Permutt, when Pla in the PVR equation is replaced by the mean effective closing pressure, PVR reflects the functional state of the pulmonary circulation in all circumstances (174).
A characteristic typical of a vascular system made up of collapsible vessels is the functional dissociation between inflow, outflow pressures and flow rate when the closing pressure is higher than the (apparent) outflow pressure (151). For the pulmonary circulation, a useful experiment is to progressively increase Pla from near zero while keeping Q constant. As long as Pla is lower than the closing pressure, the increase in Pla does not affect Ppam. When Pla is higher than the closing pressure, any increase in Pla is transmitted to Ppa in a one-to-one ratio. Thus, the pressure at the inflection point of the Ppam vs. Pla relationship is the closing pressure and should be equal to the zero flow pressure intercept of the linear (Ppam – Pla) vs. Q relationship. This is indeed a feature of de-recruited upright lung zones described by West (232), and has been observed in pulmonary hypertension after acute oleic acid lung injury (121) (Fig 2). A pulmonary closing pressure may become higher than Pla because of either an increased alveolar pressure with or without increased tone, or because of a decreased Pla with respect to alveolar pressure in the presence of a low cardiac output.
Figure 2.

Mean pulmonary artery pressure (Ppa) as a function of cardiac output (Q) at constant left atrial pressure (Pla), left panel, and Ppa as a function of Pla at constant Q in an anesthetized dog before (stippled line) and after (full line) injection of oleic acid (OA) to produce an acute lung injury. Lung injury was associated with a shift of linear Ppa-Q relationships to higher pressures, with increased extrapolated pressure intercept (small stipple line). Pla was transmitted to Ppa in a close to 1/1 relationship before oleic acid, but only at a pressure equal to the extrapolated pressure intercept of Ppa-Q after oleic acid, which is compatible with an increased closing pressure becoming the effective downstream pressure of the pulmonary circulation. From reference 121.
However, experimental data have been accumulated that cannot be explained by the Starling resistor model of the pulmonary circulation In embolic pulmonary hypertension, multipoint (Ppam–Pla)-Q relationships were linear at physiological flow rates, but an inflection point in Ppam vs. Pla relationships could not always be identified (84). Furthermore, the slope of Ppam-Q was independent of embolism size, while one would expect larger size emboli to have a greater effect on incremental resistance (the slope of Ppam vs. Q) than on the extrapolated pressure intercepts of Ppam vs. Q (33). Moreover, in these experiments, inflection points of Ppam vs. Pla plots were not correlated to extrapolated pressure intercepts of Ppam vs. Q (144). It was also noted that in embolic pulmonary hypertension, the slope and pressure intercept of Ppam vs. Q was flow dependent, with lower incremental resistance and higher pressure intercepts in studies in which flow was increased by opening systemic arterio-venous fistulae rather than decreased by inflating an inferior vena cava balloon to decrease venous return (33,84,144).
Other concerns about the Starling resistor model of the pulmonary circulation are its inability to explain shifts of Ppa-Q relationships with increased extrapolated pressure intercepts in conditions of increased in hematocrit (105). The Hagen-Poiseuille equation and the Starling resistor model would indeed predict increased viscosity of the blood to increase the slope of (Ppam–Pla)-Q.
Finally, an implicit assumption in the use of Starling resistor model to date is that perfused pulmonary resistive vessels, or arterioles, can only be fully distended. This is in contradiction with experimental evidence (113).
Thus the Starling resistor model of the pulmonary circulation appears adequate to explain pressure-flow relationships in de-recruited lung regions in healthy states and possibly in certain pathological states such as lung edema, in situations of limited changes in distension caused pressure and/or cardiac output. However, it does not explain the effects of emboli or increased hematocrit on steady-flow pulmonary vascular pressure-flow relationships.
2.4. Distensible models of the pulmonary circulation
Zhuang et al. modeled the feline pulmonary circulation taking into account not only morphometry but also mechanics (i.e., distensibility) of the pulmonary vascular tree and rheological properties of each of its branched segments (242). The authors used the sheet flow theory of Fung et al for the capillary blood flow (61) and an analogous “fifth power law” for arteries and veins, and predicted Ppam vs. Q curves with a slight curvilinearity concave to the flow axis over physiological ranges of flow that was progressively enhanced at decreasing flow. This curvilinearity was generated via arterial, capillary and venous distensibility based on experimental data (195,196,238,239), with no need to invoke a closing pressure (242).
This distensible or compliant model has been shown to predict parallel shifts of Ppa-Q plots to higher pressures, induced by various interventions such as embolism, changes in lung volume, and hypoxia (21,141,144) and even a functional dissociation between Ppa and Pla at constant Q (21,144).
Marshall and Marshall (140) modified the model to account for hypoxic pulmonary vasoconstriction and were able to reasonably fit experimental data from dogs (141). Subsequently, Bshouty and Younes (21) added vasomotor tone and the effects of gravity, and confirmed that the nonlinearity in the Ppam–Q curve is primarily a function of the distensibility of the vascular bed. Similarly, Melot et al. (144) demonstrated that Ppam–Q curves altered by pulmonary embolism could be mimicked via a pressure-induced loss of distensibility coupled with arterial and capillary obstruction.
Experimentally, Nelin et al. showed in normoxic and hypoxic pigs that a sufficient number of Ppam-Q coordinates, between 5 and 10, enables visualization of a slight curvilinearity (166). The same observation was reported in isolated perfused mice lungs by Tuscherer et al. (219). As illustrated in Figure 3, constructed from their data, pulmonary embolization was associated with a shift of slightly curvilinear Ppam-Q plots to higher pressures. The best polynomial fit of these Ppam-Qm relationships may still be linear, but distensibility permits a proportionally less increased Ppam at the highest Q. One can also observe from the figure that pressure intercepts derived from the higher flow regions of the Ppam – Q curves are higher than those derived from the lower regions of the curves. Using the Starling resistor model, this would have been improperly interpreted as increased closing pressure with increased flow.
Figure 3.
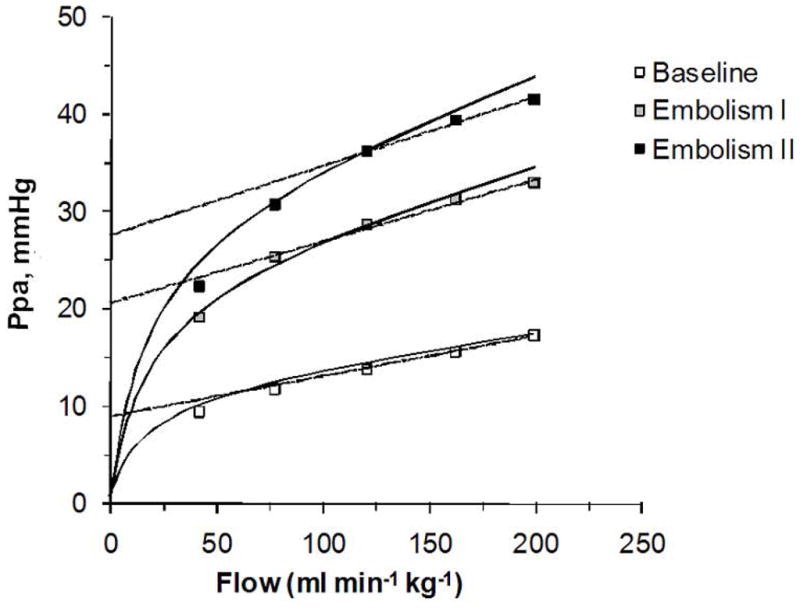
Pressure-flow relationships of an isolated perfused mouse lung before (empty sqares) and after (full squares) embolization. The shape of the multipoint pressure-flow relationship is increasingly curvilinear at decreasing flow. Extrapolated pressure intercept from best adjustments on “physiological” values for pressure and flow leads to spurious estimations of increased closing pressures. From reference 219
Linehan reasoned that previously reported compliant models were too complex, requiring a large number of parameters not identifiable from pressure and flow measurements alone (128). Accordingly, he developed a simpler distensibility model based on only two parameters, and showed its adequacy to describe Ppam-Q relationships at variable hematocrits in perfused dog lung lobes with the following equation:
where the hematocrit is captured in the total pulmonary vascular resistance at rest, R0 and the vessel distensibility α is assumed constant throughout the pulmonary vasculature. This model allows for prediction of Ppam at given levels of Pla, Q, hematocrit and distensibility. Nelin et al used it to obtain a very good fit of multipoint Ppam-Q curves from isolated pig lungs in normoxia and in hypoxia (166)
The Linehan equation can also be used to estimate vessel distensibnility α from given values of resistance, Ppam, Pla and Q. Reeves et al used pulmonary hemodynamic data obtained by right heart catheterization at rest and during exercise in normoxic healthy volunteers, and was able to recalculate a value of α averaging 2 %/mmHg (182). This value was remarkably identical to the 2 %/mmHg distensibility measured on isolated vessels from a variety of mammalian species (113). Reeves et al also showed that α is a the lower limit of the range of estimated values in exercising horses, and tends to decrease with aging and with chronic but not acute hypoxic exposure (182). Similar values of α were calculated by Argiento et al from echocardiographic measurements of pulmonary vascular pressures and flows in normal volunteers (4).
2.5. Left atrial pressure, pulmonary artery wedge and capillary pressures
Whatever the reference model, experimental data clearly demonstrate that Pla is transmitted upstream, in a close to 1:1 manner, in the fully recruited pulmonary circulation. It is then obvious that changes in Pla will affect Pcp as well. Pulmonary capillary pressure must be less than Ppa and greater than Pla, which is typically estimated by wedged or occluded Ppa (Ppw or Ppao). Reasonably accurate estimates of pulmonary capillary pressure can be obtained from the analysis of Ppa decay curves after arterial occlusion (29). Normal resting human values have been reported by Maggiorini et al (134) (Table 1). Based on measured distribution of resistances in perfused normal lungs, with 60 % arterial resistance and 40 % capillary + venous resistance, Gaar et al showed (62) that Pcp can be approximated by the equation:
However, when normal lungs are perfused with very high flows, up to 7–10 times normal, like during high levels of exercise, the distribution of resistances is altered, such that upstream resistance decreases and the Gaar equation cannot be used (20). The arterial occlusion method to estimate Pcp at high flows has been validated using the isofiltration technique (20). To our knowledge, estimates of Pcp by single arterial occlusion have not been reported in normal exercising humans.
3. Pulsatile flow pulmonary hemodynamics
3.1. Pulmonary vascular impedance
The study of the pulmonary circulation as a steady flow system is a simplification, since pulmonary arterial pulse pressure, or the difference between systolic and diastolic Ppa is on the order of 40 to 50 % of mean pressure, and instantaneous flow varies from a maximum at mid-systole to around zero in diastole. Accordingly, an improved evaluation of pulmonary arterial function should theoretically be offered by the relationship between pulsatile pressure and pulsatile flow. This relationship can be quantified by the calculation of pulmonary vascular impedance (PVZ).
Pulmonary vascular impedance is derived from a spectral analysis of the pulmonary arterial pressure and flow waves (235). This analysis is possible because the pulmonary circulation behaves nearly linearly, i.e., a purely sinusoidal flow oscillation produces a purely sinusoidal pressure oscillation of the same frequency. The sinusoidal pressure and flow waves can be related by the ratio of their amplitudes (modulus) and the difference in their phases (phase angle). To do so, both pressure and flow waves are decomposed into a series of sine waves with frequencies 1, 2, 3, etc times the heart rate, each with its amplitude and phase angle. Pulmonary vascular impedance modulus is then the ratio of amplitudes of the pressure and flow sine waves and PVZ angle is the phase difference between pressure and flow sine waves. The PVZ modulus and phase angle as a function of frequency are often represented graphically. Typical PVZ spectra at rest and at exercise in a dog are illustrated in figure 4 (48).
Figure 4.

Pulmonary vascular impedance spectra in a dog, at rest and running. Running was associated with a decrease in 0 Hz impedance, but an increase in the ratio of pressure and flow moduli (in dyne.s.cm−5) at all frequencies, and a decrease in low frequency phase angle, suggestive of decreased proximal compliance of the pulmonary arterial tree. Drawn after reference 48
Pulmonary arterial impedance at zero Hz, Z0, (the ratio of mean pressure to mean flow, Ppa/Q) corresponds to TPVR. This parameter is mainly determined by the small resistance vessels as well as Pla. As frequency increases, the impedance is affected by more proximal elements of the arterial tree (235). The modulus of the impedance decreases from Z0 rapidly to a first minimum at 2–3 times the heart rate and then oscillates about a constant value. The impedance phase increases from a negative value at low frequencies, indicating that flow leads pressure, to zero at higher frequencies. The precipitous fall in modulus and the negative phase of the impedance are a measure of the total arterial compliance. At high frequencies the rather constant modulus and nearly zero phase angle are a measure of the proximal arterial compliance.
The impedance modulus at high frequencies, when impedance phase is nearly zero and therefore wave reflections can be ignored, is the characteristic impedance ZC. It is typically measured as the average modulus at higher frequencies (usually 10–20 times the heart rate). It can also be measured as the slope of the early systolic pulmonary artery pressure-flow relationship in the time domain (41).
The oscillations of the impedance modulus about its mean value result from distinct wave reflections. Increased magnitude of these oscillations implies increased reflections. A shift of the first minimum of modulus to higher frequencies indicates an increased wave velocity or a decreased distance to the dominant reflection site.
Characteristic impedance is dependent on the ratio of inertia to compliance of the pulmonary circulation, and can be approximated by the equation:
Where ρ is the density of blood, r the mean interrnal radius, ρ/πr4 the inertance and Δπr2/ΔP the compliance of the pulmonary arterial tree.
The human PVZ spectrum has the same pattern as reported in canine studies, but with lower Z0 and ZC (48,154). Resting Z0 and ZC were 482 and 147 dyne.s.cm−5, respectively in dogs, and 73 and 22 dyne.s.cm−5 in humans (48,154). These differences can be explained by differences in body size, and thus also in cardiac output and oxygen uptake, while vascular pressures are essentially the same (235).
Exercise in normal dogs has been shown by Elkins and Milnor to decrease PVR but to shift the PVZ spectrum to higher pressure and flow moduli at all frequencies with an increase in ZC a shift of the first minimum of the ratio of pressure and flow moduli to higher frequencies and more negative phase angle (Fig 4). The authors explained the increase in ZC by a decreased area compliance of the proximal pulmonary arterial tree because of increased distending pressure related to increased flow, with possibly a contribution of exercise-associated sympathetic nervous system activation. Sympathetic nervous system activation may indeed increase ZC without significantly changing PVR (170). However, Elkins and Milnor also mentioned the possibility that an increase in proximal arterial dimensions with exercise was prevented by the electromagnetic flow probe placed around the main pulmonary artery, which caused a spurious increase in ZC (48).
In the dog experiments reported by Elkins and Milnor, the ratio of oscillatory to total hydraulic power decreased from 0.4 at rest to 0.33 at exercise. This is counterintuitive in the presence of stiffer proximal pulmonary arteries, but was explained by an improved matching of impedances because of increased cross-sectional area of peripheral resistive vessels and subsequently decreased wave reflection.
Slife et al. investigated the effects of exercise on pulmonary arterial compliance (Ca), ZC and PVR in 8 normal patients referred to the catheterization laboratory for the evaluation of a chest pain syndrome, but in whom no cardiovascular abnormality could be found (193). The hemodynamic measurements were performed using multisensor micromanometer technology, and Ca, ZC and PVR either calculated or modeled with a three-element Windkessel model. Direct calculation versus modeling of PVR, ZC and Ca yielded some differences, but the changes associated with exercise were the same. Exercise was associated with a 50 % decrease in PVR and a 30 % increase in Ca (P = 0.06) while ZC did not change (Fig 5). The apparent discrepancy between changes in ZC and Ca is explained by the sensitivity of ZC to proximal stiffness and dimensions, while Ca integrates the distensibility of the entire pulmonary circulation. Thus unchanged ZC would be explained by the balanced effects of proximal stiffening and increased cross-sectional area.
Figure 5.
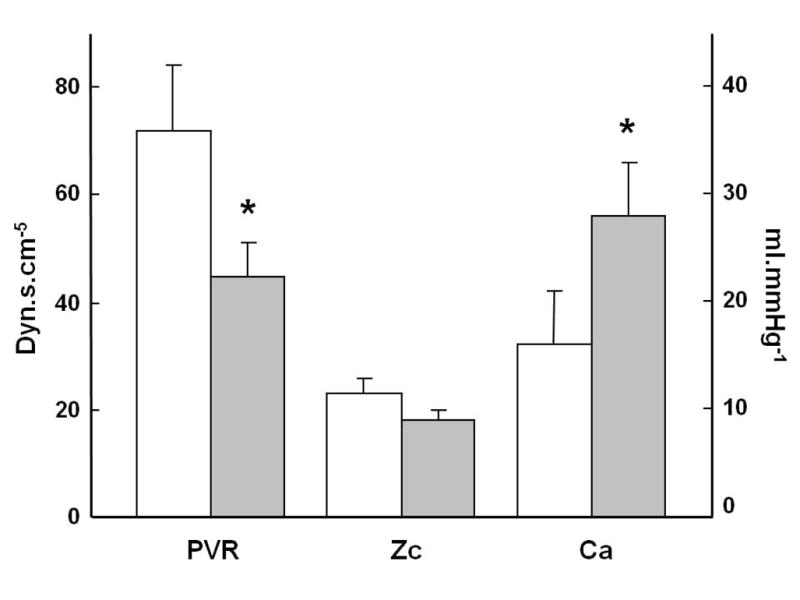
Effects of exercise (shaded columns) on pulmonary vascular resistance (PVR), characteristic impedance (Zc) and arterial compliance (Ca) in healthy human subjects. Exercise decreased PVR and increased Ca, while there was no significant change in Zc. Ddrawn from data in reference 193.
3.2. The time constant of the pulmonary circulation
Impedance calculations have not gained clinical acceptance because of perceived mathematical complexity and difficulty interpreting data in the frequency domain.
Accordingly, Lankhaar et al modeled PVZ as being determined by a dynamic interaction between PVR, Ca and Zc (118,119). These authors showed PVR and Ca are related in a hyperbolic fashion and that their product, or the time constant (τ=RC) of the pulmonary circulation, is remarkably constant at approximately 0.7 seconds from normal to variable severities and etiologies of pulmonary hypertension. Saouti et al. subsequently showed that the constancy of τ holds for individual lungs (189). These authors also reported on hemodynamic and magnetic resonance imaging measurements indicating that proximal arterial compliance given by the sum of main right and left pulmonary arterial compliance is only 19 % of total arterial compliance Ca (189). The time constant RC recalculated from the data of Slife et al (193) averaged 0.86 s at rest and 0.8 s at exercise (P NS), which suggests that the product of resistance and compliance also remains constant at exercise.
The hyperbolic relationship between compliance and resistance of the pulmonary circulation demonstrates that mild increases in PVR may markedly increase right ventricular (RV) afterload because of associated decreases in Ca. This analysis was used by Bonderman et al to explain persistent dyspnea at exercise in patients after successful pulmonary endarterectomy and no or mild residual increase in PVR (12). In that study, exercise increased PVR with little or no decrease in Ca in the patients, as opposed to healthy controls in which exercise caused a mild decrease in PVR and a marked increase in Ca. Furthermore, in these patients, Ca rather than PVR showed up as an independent predictor of exercise capacity.
The stability of the time constant of the pulmonary circulation explains the reported tight correlation between systolic, diastolic and mean Ppa in normal subjects and animals during a variety of activities, and in patients with pulmonary hypertension of all possible etiologies (27,212). Accordingly, Ppam can be calculated from systolic Ppa (Ppas) using a simple formula: (27)
This notion is of practical relevance as non-invasive evaluations of the pulmonary circulation in clinical practice often rely on the measurement of a maximum velocity of tricuspid regurgitation (TRV) to calculate a Ppas using the simplified form of the Bernoulli equation and a measurement or estimate of Pra (240):
4. Right ventricular function
Because pulmonary artery pressures and flow are linearly related in the physiological range, and very high Q and thus also Ppa can be reached by trained endurance athletes (178), the question has been raised as to whether intense endurance exercise might exhaust RV adaptation to increased afterload (116), and be a cause of arrhythmogenic remodeling (117). It is therefore necessary to take a closer look at the coupling of RV to afterload, and what is known about it at exercise.
4.1. Right ventricular hydraulic load
Increased Ppa at exercise requires increased RV hydraulic power to sustain adequate forward flow. Hydraulic power is made of two components: the energy per unit time (power) to produce steady flow, which is the product of Ppam and Q, and the power to produce the pulsatile component of Ppa and Q. The latter can be calculated from the difference between the product of the integrations of instantaneous Ppa and Q waves and the product of Ppam and Q. Since mean flow determines oxygen transport, mean power may be considered useful whereas oscillatory power is “wasted”. As a consequence, the ratio of oscillatory to mean power, or oscillatory to total (mean plus oscillatory) power, should preferably be small (150).
Because of the proportional relationship between systolic, diastolic and mean Ppa, one would expect the ratio of oscillatory to total RV power to remain constant. This was indeed recently reported by Saouti et al. (190). These authors measured pulmonary artery pressures with right heart catheterization and pulmonary arterial flows by magnetic resonance imaging in 35 patients with idiopathic pulmonary arterial hypertension and in 14 patients without pulmonary hypertension. The range of Ppa was wide, from normal to markedly increased. Total power increased with severity of pulmonary hypertension, but the ratio of oscillatory to total power remained reasonably constant, at 23 %. Accordingly, the authors proposed that the total power of the RV should be equal to 1.3 times mean power in all circumstances. They acknowledged previous experimental and clinical studies showing a decreased oscillatory power fraction in pulmonary hypertension, part of which could be of methodological origin. Since the proportionality of pulmonary artery pressures appears to be maintained at exercise (212) the calculation of total RV power as 1.3 times mean power is probably transposable to exercise, but this will require confirmation by further studies.
4.2. Right ventriculo-arterial coupling
The calculation of RV power includes a measurement of cardiac output, and thus depends not only on the impedance of the pulmonary circulation, but also on ventricular function.
It has been suggested that power transfer and ventricular efficiency are near maximal in normal subjects. A simplified approach to test this has been given by Sunagawa et al. (209). These authors proposed a graphical analysis based on the right ventricular pressure-volume diagram (187,206).
The pressure-volume diagram allows for the determination of maximal ventricular elastance (Emax), which is the best possible load-independent measurement of contractility, and of arterial elastance Ea ≈ PVR/T (with T the R-R interval), as a measurement of afterload as it is “seen” by the ventricle. The ratio of Emax to Ea is a measure of the coupling of the ventricle and the arterial load. The optimal matching of heart and load, i.e. where power transfer and cardiac efficiency are close to maximal, is found when Emax/Ea is about 1.5 in humans. An isolated increase in Ea (increased vascular resistance), or decrease in Emax (decreased cardiac contractility), decreases Emax/Ea indicating uncoupling of the ventricle from its arterial system, i.e lower ventricular-vascular coupling efficiency. Assuming no change in the vasculature, a decrease in Emax/Ea is necessarily accompanied by a decrease in stroke volume. On the other hand, an isolated increase in preload associated with an increase in stroke volume leads to unaltered ventriculo-arterial coupling.
Calculating the Emax to Ea ratio requires measurement of at least two end-systolic pressures and volumes, since the end-systolic pressure-volume relation does not go through the origin. A common approach is to reduce RV diastolic filling and measure a series of pressure-volume loops (187,206), but bedside manipulations of venous return are too invasive to be ethically acceptable. In addition, when applied to intact beings, changes in venous return are associated with reflex sympathetic nervous system activation, which affects the ventricular as well as vascular function. These problems can be overcome by a single beat approach, which was developed by Sunagawa for the left ventricle (210), and later shown to be applicable to the RV (17). The single beat method is based on the calculation of a maximal RV pressure Pmax from a non linear extrapolation of the early and late systolic portions of a RV pressure curve, and a synchronized relative change in RV volume from integrated pulmonary arterial flow. The extrapolated Pmax has been validated against direct measurements of pressures generated by a non ejecting RV beat in an experimental experimental intact animal preparation (17).
The single beat method applied to intact animal preparations has shown that Emax/Ea for the RV is decreased by propranolol, increased by dobutamine, and maintained in the presence of increased Ea due to acute hypoxic pulmonary vasoconstriction. In fact, Emax increases in proportion to increased Ea in hypoxia, even in the presence of adrenergic blockade, which is compatible with the concept of homeometric adaptation of RV contractility (17).
The notion that RV functional adaptation to afterload is essentially systolic, through an increase in contractility, with secondary diastolic changes and increased RV dimensions (187) has been confirmed by a study which applied the single beat mathod to derive Emax and Ea from magnetic imaging and invasive hemodynamic measurements in patients with severe pulmonary hypertension (114).
An alternative and maybe simpler approach was developed by Elzinga and Westerhof in 1978 (49). The authors described RV pump function curves by plotting mean RV pressure as a function of stroke volume (SV). The RV pump function curve is built from measurements of mean RV pressure and stroke volume, a calculated maximum RV pressure at zero stroke volume, and a parabolic extrapolation to a zero pressure stroke volume. In this representation, an increase in preload shifts the curve to greater stroke volumes with no shape change, while an increased contractility leads to a higher Pmax with no change in SVmax.
This analysis has been used to explain more severe RV failure in the face of lower Ppam in pulmonary hypertension associated with systemic sclerosis, probably due to myocardial involvement by the disease process (169).
As present, there have been no reports on RV pump function curves or RV-arterial coupling analysis at exercise. This area is thus still open to investigation. It is hoped that developments in echocardiographic and magnetic resonance imaging techniques will be implemented for a better understanding of RV function at exercise, and be able to address the unsolved issue of validation against above mentioned gold standard invasive measurements.
5. Pulmonary hemodynamics at exercise in healthy humans
5.1. Pulmonary artery pressures: invasive measurements
The first catheterizations of the right heart in normal humans at exercise were reported in the late 1940’s. The results showed a small increase, no change and sometimes a decrease in Ppam as cardiac output was increased with moderate exercise (87,185). In 1950, Cournand et al. observed a sharp increase in Ppam when cardiac output was increased to three and a half times the resting value (30). A similar sharp rise in Ppam at flows above 350 % of normal was reported a few years later in isolated perfused lungs (120). However, subsequent studies repeatedly reported a linear increase in Ppam as a function of flow in isolated perfused lungs (59,236) as well as in intact human beings studied using the unilateral balloon occlusion technique (to double the flow in the contralateral lung) and/or exercise (18,44). There has been a recent report of a “take-off” pattern of Ppam-Q relationships in normal subjects at exercise (216) reminiscent of the pioneering work of Cournand. The reasons why a small minority of studies report such an inflection point in Ppam-Q relationships are not entirely clear. In an editorial about pulmonary hemodynamics at exercise published in 1969, Fowler concluded that Ppam-Q relationships are generally best described by a linear approximation until the highest physiologically possible flows, and that previously reported take-off patterns were probably explained by a deterioration of the experimental preparation in isolated perfused lung studies, and either methodological problems or diastolic dysfunction with an increase in Pla in intact human studies (58).
It has to be emphasized that these considerations are relevant to studies at progressively increased cardiac output during incremental exercise stress tests. When the degree of exercise is constant, Ppam tends to return to baseline after approximately 5–10 min and during the following hour, in both the recumbent (188,194) and the sitting upright (43) positions, while cardiac output is maintained. This is explained by the initial sympathetic nervous system activation related to some degree of anxiety or excitement as a cause of blood displacement from the legs, with associated changes in Pla.
The rate of recovery of Ppam after an exercise test, is rapid, with values back to resting baseline or even below within the first minutes of post-exercise rest (194).
In 1989, Reeves, Dempsey and Grover reviewed the published data on invasive pulmonary hemodynamic measurements at progressively increased levels of bicycle exercise normal subjects (178). They compiled 196 measurements in the supine posture in 91 subjects (37,69,77,88,221) and 104 measurements in the upright posture in 24 subjects (11,73,181, and unpublished data provided by Wagner and Moon). Only healthy subjects with normal Ppam were included in the analysis, and the quality of reported measurements was checked. The authors referred the measurements to limits of normal of Ppam previously established by Reeves and Groves in a total of 106 subjects 6–83 years old, which showed an average Ppam of 14 mmHg with a SD of 3 mmHg, defining a normal range of 9–20 mmHg (179). These limits of normal have been repeatedly confirmed (111,134,146,161).
This analysis established that supine exercise is associated with only a slight decrease in PVR, in keeping with the notion that the normal pulmonary circulation is maximally dilated and relaxed at rest. In the upright position, the initial PVR was found to be higher, which can be explained by a de-recruitment caused by a lower cardiac output (via decreased venous return). The initially higher PVR accounts for more marked decrease in PVR at exercise reported in upright subjects. However, the upright and supine PVR-workload relationships converge at moderate levels of exercise, in keeping with the notion that Ppam-Q relationships are identical in fully recruited supine or erect lungs (Fig 6).
Figure 6.
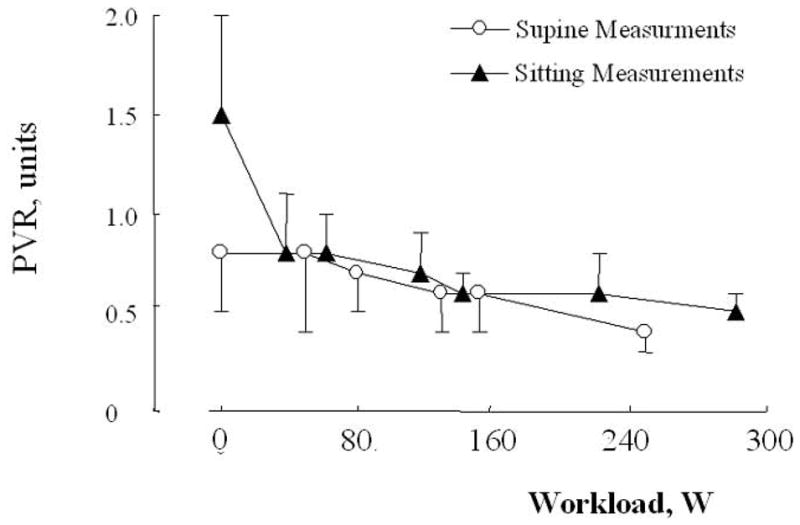
Effects of increasing levels of exercise in the supine and in the sitting position on mean ± SD (vertical bars) pulmonary vascular resistance (PVR, Wood units or mmHg.L−1) in healthy volunteers. Initial PVR was higher in the sitting position, but otherwise PVR decreased slightly with increasing levels of workload, and this was similar in the supine and in the sitting positions. Redrawn after reference 179.
Reeves et al were able to find measurements at rest and at least two levels of exercise in 63 subjects (including 21 women), so that they could calculate linear regressions relating Ppam to Q in each of them. On average, each liter per minute of increase in cardiac output was accompanied by 1 mmHg increase in Ppam in young adult men and women exercising in the supine position (Table 2). The same average increase in Ppam per liter per minute of flow was found in 21 subjects exercising in the upright position Aging to 60–80 years was found to be associated with a more than doubling of the slope of Ppam-Q relationships (Table 2) such that:
Table 2.
Slopes of pulmonary vascular pressure-flow relationships in normal subjects
| Supine measurements | Upright measurements | |||
|---|---|---|---|---|
| Normal young men and women | ||||
| Variables | n | Slope | n | Slope |
| Q, Ppam | 63 | 1.00 | 21 | 1.01 |
| SD | 0.91 | 0.43 | ||
| Q, Ppw | 53 | 0.30 | 16 | 0.84 |
| SD | 0.35 | 0.39 | ||
| Normal old men | ||||
| Q, Ppam | 14 | 2.54 | ||
| SD | 0.77 | |||
| Q, Ppw | 1.93 | |||
| SD | 0.94 | |||
Legend: Q: cardiac output; Ppam: mean pulmonary artery pressure; Ppw: wedged Ppa
Young adults were aged 20–30 yearsd, old men were aged 60–80 years
Souce: Reference 178
However, there was a large individual variation, as shown by SDs of 0.94 in the supine exercise group, and 0.43 in the upright exercise group. This wide variability in individual responses of Ppam to exercise was noted in earlier studies as well (58).
5.2. Pulmonary artery pressures: non invasive measurements
Repeating invasive hemodynamic studies in normal subjects is met with increasing ethical reticence, so that not many additional data are likely to be reported to address remaining unsolved issues regarding pulmonary vascular function at exercise. On the other hand, non invasive methods have improved along with technological advances allowing for increasingly valid estimations of Ppam, Q, Pla and indices of RV function. The most commonly used approaches for the evaluation of the pulmonary circulation rely on the estimation of systolic Ppa from the maximum velocity of tricuspid regurgitation (241) and recalculation of a Ppam based on the constant relationship between systolic and mean pulmonary artery pressures (27,212). Cardiac output can be estimated from the Doppler aortic flow (28) and Pla estimated from the ratio of transmitral Doppler and mitral annulus tissue Doppler ratio of early diastolic E and E′ waves (163).
Grunig et al used systolic Ppa estimated from the maximum velocity of tricuspid regurgitation to show excessive responses to exercise in healthy relatives of patients with idiopathic pulmonary arterial hypertension as compared to controls (75). In 32 % of healthy relatives of pulmonary hypertensive patients, the maximum velocity of tricuspid regurgitation exceeded 3.1 m/s (or systolic Ppa > 40 mmHg) as compared to 10 % of the healthy controls. The cut-off value of 3.1 m/s (or 40 mmHg) corresponded to values in excess of 90 % quartile of controls. Bossone et al. observed markedly increased maximum velocities of tricuspid regurgitation in exercising athletes (13), who often reach calculated systolic Ppa well in excess of 40 mmHg at exercise, as would be expected from previously reported increases in cardiac output at high levels of exercise in invasive studies (162,178). However, these studies did not report on estimations of cardiac output and Pla, so that pulmonary vascular pressure-flow relationships to evaluate pulmonary vascular function could not be determined.
Acknowledging the methodological limitations of echocardiographic pulmonary vascular pressures and flow measurements at rest and even more so at exercise, Argiento et al investigated the shape of multipoint Ppam-Q relationships at exercise in 25 healthy volunteers (4). Mean Ppa increased from 14 ± 3 to 30 ± 7 mmHg (mean ± SD) at the highest achieved levels of exercise, at which workload was 170 ± 51 W, cardiac output was 18 ± 4 L.min−1, close to predicted maximal values.
As illustrated in Fig 7, Ppam decreased to 19 ± 4 mmHg after 5 min recovery, and had returned to baseline values after 20 min. The recovery of Q was fast as well, though somewhat slower, with a persistent 15–20 % elevation above baseline after 20 min. Rapid recovery, but at different rates, of Ppam and Q after exercise testing limits the validity of post-exercise stress measurements to evaluate the functional state of the pulmonary circulation (31,156).
Figure 7.
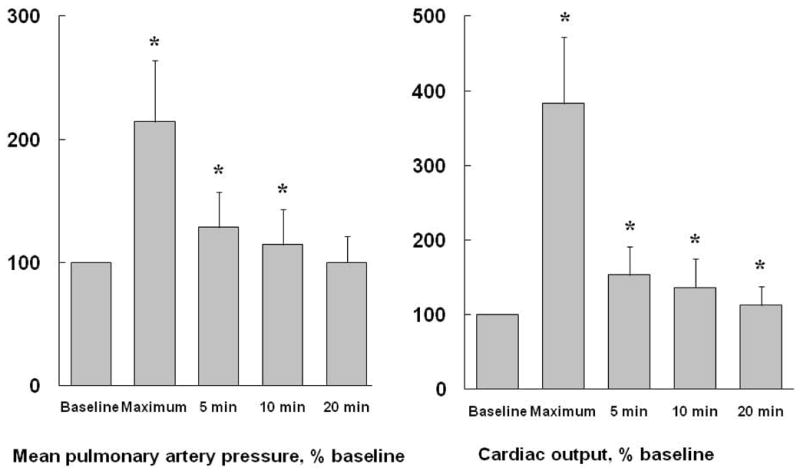
Rapid recovery of mean pulmonary artery pressure (mPpa) and cardiac output (Q) after maximal exercise. Values are reported as mean ± SD (vertical bars). *: P < 0.05 compared to resting baseline. After 20 min, Q is still higher than resting baseline. Drawn from reference 4, permission pending.
Argiento et al measured 6 to 12 Ppam-Q coordinates at increasing levels of exercise. These pressure-flow relationships could be fitted using the distensibility model equations reported by Linehan et al. (128). This allowed for the calculation of a distensibility coefficient α of 1.7 ± 1.8 %/mmHg, which is remarkably similar to those previously calculated based on invasive measurements in humans (182). On the other hand, checking for the best fit of these multipoint Ppam-Q plots, by testing for increasing order polynomials, it could be shown that the data were best described by a linear approximation. In addition, there was a marked inter-individual variability in the slopes of Ppam-Q plots, with maximal achievable Ppam around 40 mmHg. Such a variability is actually a feature of pulmonary hemodynamic responses to exercise. However, when the authors applied a normalization procedure to correct for intra-individual variablility (176), they found a slope of Ppam vs. Q of 1.37 ± 0.65 mmHg.min.L−1, indicating a normal range from 0.03 to 2.67 mmHg.min.L−1.
Further studies are obviously needed for a better definition of the limits of normal pulmonary hemodynamics at exercise.
5.3. Right and left atrial pressure
It has long been acknowledged that Ppw increases with exercise. Bevegaard et al found Ppw up to 25 mmHg in exercising athletes (11), while Granath et al frequently observed Ppw over 30 mmHg in elderly normal subjects (68,69). This increase in Ppw has been found to be strongly correlated to Pra, even though Pra rises less than Ppw. This is illustrated in Fig 9, which presents data from young healthy volunteers who participated in hypobaric chamber experiments on the effects of simulated altitudes up the summit of Mount Everest (180,181).
Figure 9.
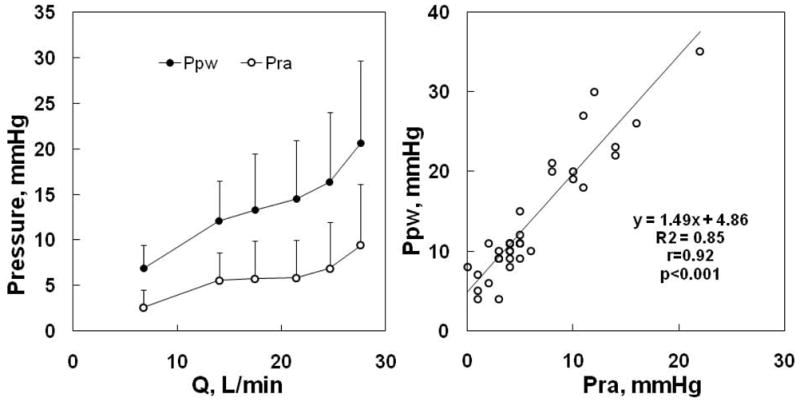
Mean ± SD (vertical bars) values of mean pulmonary artery wedge pressure (Ppw) and right atrial pressure (Pra) as a function of cardiac output (Q) during progressive exercise in normal volunteers, left panel, and Ppw vs Pra of the same subjects, right panel. Both Ppw and Pra increase with Q, but the gradient between the pressures tended to increase. The right panel shows that Ppw is correlated to Pra, but is higher than Pra by 5 mmHg at rest, and this increases with a slope of 1.45 mmHg increase in Ppw for every mmHg increase of Pra at exercise. From references 180 and 181.
In their analysis of the pulmonary vascular pressure-flow relationships from at least 3 points during exercise, Reeves et al found a linear increase in Ppw with cardiac output. In 53 young adult men and women, the slope of Ppw vs. Q relationships was 0.30 ± 0.35 mmHg.min.L−1, increasing to 1.93 ± 0.94 mmHg.min.L−1 in older subjects (Table 2) (178).
In a further analysis pooling all available Ppam and Ppw, Reeves and Taylor found a high correlation between these measurements during either supine or upright exercise, with slopes very close to one (183), suggesting a one-for-one mmHg upstream transmission of Pla (Fig 10).
Figure 10.

Mean ± SD (vertical bars) values of mean pulmonary artery pressure (Ppa) and wedged Ppa (Ppw) as a function of cardiac output (Q) during progressive exercise in normal volunteers, left panel, and Ppa vs Prw of the same subjects, right panel. Both Ppa and Ppw increase with Q, but the gradient between the pressures tends to remain unchanged. The left panel shows that Ppa is correlated to Prw, but is higher than Ppw by 9 mmHg at rest, and this increases with a slope of 1.1 mmHg increase in Ppa for every mmHg increase of Ppw at exercise. From references 180 and 181
Eighty % of the variability of Ppam at exercise in these studies was explained by Ppw (183).
A close to one for one upstream transmission of Ppw to Ppa corresponds to the prediction of Starling resistor as well as distensibility models of the pulmonary circulation (144).
Reeves and Taylor discussed the possible causes of increased Ppw at exercise, and whether Ppw in exercising conditions might be an accurate and precise estimate of Pla. They discarded intrathoracic pressure changes and swings at exercise as a cause of increased Ppw or increased gradient between Ppw and Pla, arguing that mean intrathoracic pressure averaged over several respiratory cycles would be near to zero and unchanged from the resting state. Spurious overestimation of Pla by Ppw was excluded on the basis of a previous report of right and left heart catheterization studies in 10 normal male subjects investigated for chest pain syndromes (217). Iin that study, a high correlation between Ppw and left ventricular end-diastolic pressure was found, confirming pioneer studies in the 1950’s, even though the agreement did not seem optimal, with most left ventricular end-diastolic pressures on the left of the line of identity. In a more recent retrospective study on over 11000 patients undergoing simultaneous right and left heart catheterization, Halpern and Taichman found that Ppw was correlated to left ventricular end-diastolic pressure, but rather loosely, with Bland-Altman limits of agreement from −15 to + 10 mmHg (79). Most recently, Tonelli et al studied 37 patients undergoing a right heart catheterization for suspected pulmonary hypertension, and showed that Ppw can be falsely elevated by balloon inflation volume, with a mean difference of 4 mmHg for full and half inflation (217). Thus while there is little doubt that increased Ppw reflects increased Pla at exercise, there is uncertainty as to how closely the measurements are related.
As for the cause of increased Pla at exercise, this may be explained by the left ventricle using the Starling mechanism to matching its flow output to peripheral demand (183). Left ventricular (LV) diastolic compliance decreases with increasing diastolic volume, which reaches a maximum at mild to moderate levels of exercise (208). This is due to intrinsic mechanical properties of the LV, with contribution of competition for space with the RV within the relatively non-distensible pericardium and possible sympathetic nervous system activation (180). Pericardial constraint may play an important role in decreasing diastolic LV compliance with increased venous return. This mechanism has been used to explain limited increases in stroke volume and end-diastolic volume but one-to-one transmission of increases in Pla to Pra with exercise (102). Elite endurance athletes have parallel increases of Pla and Pra at high levels of exercise, whereas less well trained or sedentary subjects have smaller increases in Pra than Ppw with exercise (201), which suggests that pericardial constraint is acting in the elite athletes at very high levels of exercise. Further confirmation of this theory is provided by studies with athletic animals in which removal of the the pericardium resulted in increased peak end-diastolic volume, stroke volume and maximal oxygen uptake (80,202).
Given the curvilinear relationship between diastolic ventricular pressure and volumes, it is curious that available data rather suggest a linear relationship between Ppw and cardiac output during exercise (183). A possible explanation for this apparent paradox is in the limited number of individual Ppw-Q points usually reported for each exercising subject. Stickland et al. addressed the issue in an invasive study in 8 healthy subjects of variable fitness, of whom 3 were considered to present with a low aerobic exercise capacity, as defined by a maximal oxygen uptake of less than 55 ml/kg/min, and 5 were considered to present with a high aerobic exercise capacity defined by a maximal oxygen uptake higher than 55 ml/kg/min (201). The authors hypothesized that while stroke volume normally reaches a plateau during submaximal exercise (5), this would not occur in endurance-trained athletes, due to improved left ventricular diastolic compliance (66,126). As illustrated in Fig 11, the fittest subjects indeed presented with a delayed increase in Ppw. It is interesting to note that neither Ppw nor Pra increased above normal before oxygen uptake values of approximately 30 ml/kg/min, corresponding to cardiac outputs in the range of 20 L.min−1. Above this level of exercise, both Ppw and Pra increased rapidly in the less fit subjects, but was very much delayed in the fittest subjects, in whom as expected stroke volume continued to increase. The observation of late increase in Ppw and Pra at exercise levels corresponding to cardiac outputs higher than 20–25 L.min−1 was confirmed by Stickland et al in another study exploring the effects of lower body positive pressure on pulmonary gas exchange (200).
Figure 11.
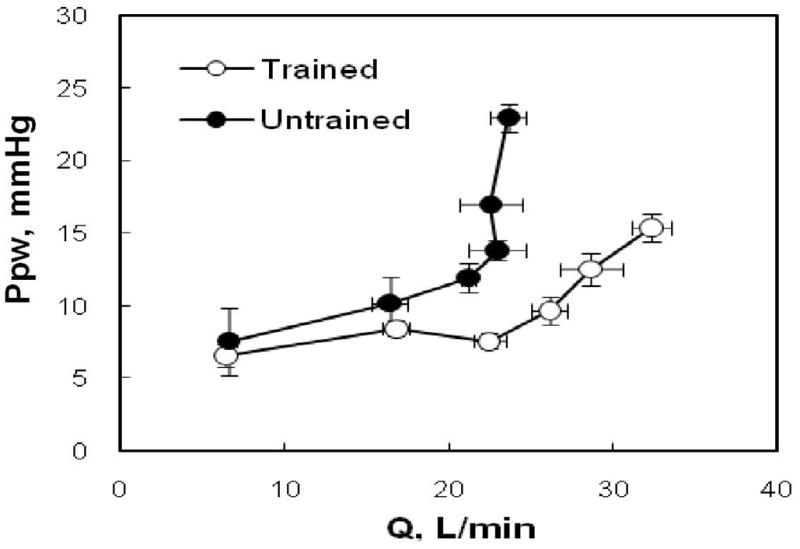
Individual mean ± SE values of pulmonary artery wedge pressure (Ppw) as a function of cardiac output (Q) in subjects with a low (untrained) vs a high exercise capacity (trained). The increase in Ppw was delayed until higher Q in the fittest subjects, suggesting improved ventricular compliance. From reference 201.
Argiento et al calculated Pla from the mitral E/E′ ratio in normal exercising individuals and could not find a significant change over a range of cardiac outputs from 10 to 26 L/min, mean 18 L.min−1 (4). Although the measurement has been validated at exercise (214), the authors wondered if this negative result might have been artifactual, or related to the fact that most subjects did not exercise at levels high enough to be associated with a significant increase in Pla.
Why the increase in Pla as a consequence of a pericardial constraint or altered left ventricular diastolic function would vary considerably from one subject to another remains unclear. A possible role of diastolic ventricular interaction may be invoked in relation to variable increases in Ppam at given levels of flow and exercise capacity, leading to variable increases in RV diastolic volumes and competition for space with the LV within the non-distensible pericardium.
6. Mechanisms of decreased PVR at exercise
The question has been asked whether there is a humoral or neural contribution to decreased PVR at exercise. The pulmonary endothelium releases vasoconstrictors, such as endothelin-1, and vasodilators such as prostacyclin and nitric oxide (NO) that control pulmonary vascular structure and function (54). In systemic arteries, the ability of increased shear stress to stimulate endothelial NO synthase production to release NO and cause vasodilation is well recognized. In the pulmonary circulation, there is some evidence of this phenomenon in ferret lungs, in hypoxia, not in normoxia (25). In patients with pulmonary arterial hypertension, who often have excessive pulmonary artery smooth muscle cell tone, therapies based on the administration of endothelin receptor antagonists, prostacyclin analogues, or inhibitors of phosphodiesterase-5 to potentiate NO-cyclic guanosine monophosphate signaling have shown efficacy in controlling progression of vascular remodeling (143).
The administration of L-arginine analogues to inhibit NO synthase has been reported to increase PVR in resting sheep (110), horses (136), pigs (42,149) and healthy normal volunteers (197). The administration of the phosphodiesterase-5 inhibitor sildenafil in resting normal volunteers has been reported to be either without effect (76) or to decrease PVR (53). Sildenafil shifted Ppam-Q relationships to lower pressures at exercise (53), but, the exercise-induced decrease in PVR was similar in subjects treated with or without L-arginine analogues or sildenafil (42,53,110,136,149).
The administration of the dual endothelin A and B receptor antagonist tezosentan did not affect PVR at rest but decreased PVR at exercise in swine, while the selective endothelin A receptor antagonist EMD122946 had no effect, which suggested that endothelin B receptor mediates increased pulmonary vascular tone in the porcine species (148). However, neither the dual endothelin A and B receptor blocker bosentan nor the selective endothelin A receptor blocker sitaxsentan affected PVR at rest or at exercise in normoxic volunteers, suggesting no participation of endothelin-1 to resting or exercise pulmonary vascular tone in healthy humans (51,158).
The interaction of endothelin and angiotensin II signaling pathways was considered in studies on pulmonary and systemic hemodynamics at exercise in male and female swine (32). The angiotensin II type I receptor blocker irbesartan decreased systemic vascular resistance at rest in both male and female swine, but paradoxically increased pulmonary vascular tone, in female swine only. The dual endothelin A/B blocker tezosentan decreased pulmonary vascular tone at exercise, and this effect was enhanced by irbesartan in female swine only. These results indicate that angiotensin II limits the vasoconstrictor influence of endothelin 1 in female but not in male swine for reasons that are poorly understood but may be related to cross-talk between estrogen and endothelin signaling.
The possible role of prostacyclin in decreased PVR with exercise has also been investigated in experimental animals. Cyclooxygenase inhibition with meclofenamate in dogs (127) and in sheep (167) increased PVR at rest, but PVR fell by the same amount in treated and untreated animals at exercise. The cyclooxygenase inhibitor indomethacin did not affect resting or exercise PVR in swine (149)
Altogether, these experimental animal and human results indicate that low resting pulmonary vascular tone at rest and at exercise is maintained by a tonic release of prostacyclin and NO, with possibly a gender-related participation of angiotensin II to limit endothelin-related increase in pulmonary vascular tone in the porcine species. However, these mechanisms are not clearly involved in exercise-induced decreases in PVR. Endothelin signaling does not appear to participate in the control of pulmonary vascular tone at rest or at exercise in normoxic humans.
Since exercise activates the sympathetic nervous system, the question has also been raised whether this could play a role in observed changes in PVR. Increased sympathetic nervous system tone has consistently been reported to stiffen the large pulmonary arteries and thereby to increase ZC (16,41,159,170). However, the associated changes in PVR are dependent on the level of activation and the balance of α and β receptor stimulation principally in the distal arteries of the lung. A low level of sympathetic nervous system tone may decrease PVR because of a predominance of β receptor-mediated vasodilation, whereas a high level of sympathetic nervous system tone increases PVR because of a predominance of α receptor-mediated vasoconstriction (16,170). In exercising sheep (107) and swine (203) β-adrenergic blockade with propranolol increased resting PVR and prevented the decrease in PVR associated with exercise. Alpha-adrenergic blockade with phentolamine tended to enhance the fall in PVR, but combined α- and β-adrenergic blockade did not affect PVR. It is thus possible that β-adrenergic stimulation contributes to low PVR both at baseline and at exercise, but it is unclear if this mechanism contributes to the exercise-induced decrease in PVR.
Parasympathetic nervous system activation has been reported to decrease pulmonary vascular tone in cats (165) but this result was not confirmed in dogs (122). In swine, blocking the muscarinic receptors with atropine decreased PVR at rest but less so at exercise, in keeping with decreased parasympathetic nervous system activity at exercise (204).
In a review on the control of pulmonary vascular tone during exercise in healthy subjects and those pulmonary hypertension, Merkus et al. considered the effects of endothelium-derived mediators, neuro-humoral activation, the renin-angiotensin system, natriuretic peptides, serotonin, adenosine, cyclic neucleotides, reactive oxygen species and potassium channel activation, and concluded that even if some of these interventions slightly decrease PVR at rest or at exercise, their role in exercise-induced decrease in pulmonary vascular tone remains uncertain (147).
The PVR of healthy lungs is already very low, leaving little room for further decrease, and multipoint pulmonary vascular pressure-flow relationships at exercise closely conform to model predictions which do not take into account possible smooth muscle cell-mediated changes in PVR. Reeves et al. calculated a mean difference between measured Ppam and predicted Ppam (by the Linehan distensibility model) of 0.008 ± 1.656 mmHg on 267 measurements from healthy humans at rest and at exercise (180). As illustrated in Fig 12, a regression line through these data was calculated as y = 0.059 x − 1.4 (r2 0.078, P < 0.05). Therefore, the most likely explanation for decreased PVR or slopes of multipoint Ppam-Q plots less than the unit with exercise is vascular distension.
Figure 12.
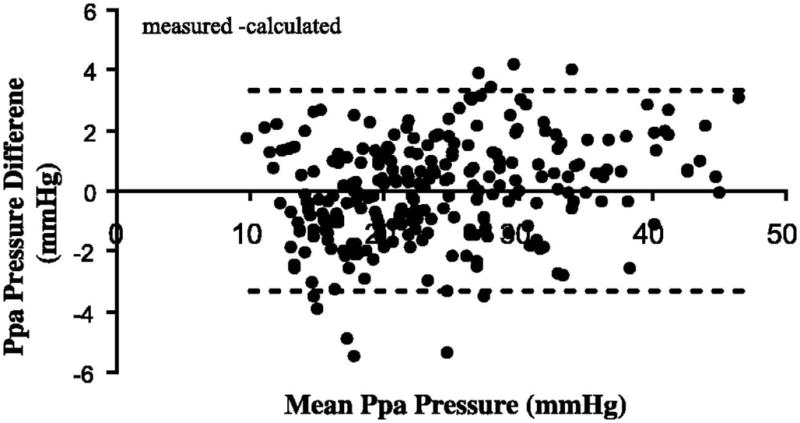
Predicted minus measured mean pulmonary artery pressure (Ppa) as a function of mean Ppa in healthy exercising volunteers. The stippled line shows 2 SD, which is equal to 1.7 mmHg. This presentation is suggestive of a good agreement. The prediction of Ppa was obtained using the distensibility model of Linehan. From reference 182.
7. Animal data
It is possible to train large animals for hemodynamic measurements at exercise. The obvious advantages to studying experimental animals instead of human volunteers are easier access to invasive manipulations of the cardio-respiratory system and sampling of biological material. However, studies of the pulmonary circulation of different animal species for the purpose of comparative physiology is difficult because of different levels of metabolism and body size, which complicate the analysis of pulmonary vascular pressure-flow relationships, sequellae of general anesthesia and thoracotomy, and insertion of pulmonary flow probes that can alter proximal pulmonary arterial compliance and which requires pericardiactomy that delays the increase in Pla at exercise (183). There also may be differences in pulmonary vascular structure and function with a tendency for animal species without collateral ventilation to present with higher PVR and pulmonary vascular reactivity (229). Despite these limitations, some relevant studies are reviewed here.
Measurements in sheep suggest a delayed increase in Pla with exercise as compared to humans, but otherwise similar increases in Ppam as a function of changes in flow (107,168). This delayed increase in Pla has been ascribed to the removal of the pericardium for the insertion of pulmonary flow probes (183).
The porcine species has been reported to have higher resting PVR and ZC compared to dogs and goats once adjusted for body dimensions (229). Pigs have no collateral ventilation, so that increased pulmonary vascular reactivity is needed to preserve ventilation/perfusion (VA/Q) relationships. Submaximal exercise in miniswine (average weight 26 kg) increasing oxygen uptake by a factor of 2.6 increased cardiac output by 10 L.m−1 and increased Ppam by an average of 18 mmHg, corresponding to an average increase in slope (i.e., TPVR) of 1.8 mmHg.min.L−1 (95). In 2–3 months-old swine (average weight 22 kg), exercise to a 3–4 fold increase in oxygen uptake increased cardiac output by 4 L.min−1 and increased Ppam by 17 mmHg, corresponding to an average increase in slope of 4.3 mmHg.min.L−1 (32). In these experiments, cardiac output was determined either by an aortic flow probe (32) or by the Fick method (95), avoiding mechanical constraints by measurement devices on the main pulmonary artery or confounding effects of pericardiectomy. With correction for body dimensions by a factor of 3, , these slopes are in the range of those of exercising humans.
Exercise in horses (430 to 559 kg) up to approximately 60 % of their maximal oxygen uptake increased Ppam from 28 to 62 mmHg and cardiac output from 40 to 310 L.min−1 (90). This allows for the calculation of a slope of 0.3 mmHg.min.L−1, which is similar to humans if we assume a correction for body dimensions by a factor of 1/7. However, because of huge cardiac outputs achieved with exercise by horses, Ppam are also very high, around 60 mmHg at moderately severe exercise, up to 80–100 mmHg at maximal levels of exercise (183). Exercising horses also present with very high Ppw, up to 40–60 mmHg, which was shown to be associated with proportional increases in Pcp (137). Typical increases in Ppam and Ppw in exercising horses are illustrated in Fig 13. These pressure-flow relationships likely present extremes of right ventricular afterload and capillary pressure, and consequently extreme stresses on cardiac and lung function. It is remarkable that measured equine Ppam-Q relationships agree perfectly with the predictions of the Linehan distensibility model, with an α value of approximately 1 %/mmHg (182).
Figure 13.

Mean values for pulmonary artery pressure (Ppa) and pulmonary artery wedge pressure (Ppw) as a function of cardiac output (Q) in exercising horses. Exercise in horses is associated with marked increases in pulmonary vascular pressures. From reference 183.
8. Gas exchange at exercise
Exercise stresses the gas exchange function of the lungs because of the huge increase, up to factors of twenty, of oxygen uptake (VO2) and carbon dioxide production (VCO2) in the presence of markedly decreased oxygen content of mixed venous blood and shortened transit time of red blood cells. Two main adjustments take place: a three-fold increase in capillary blood volume, allowing to compensate for the decreased red blood cell transit time, and an increase in mean VA/Q relationship by a factor of three to increase alveolar PO2 (PAO2) and decrease alveolar PCO2 (PACO2) (34). These adaptations explain why pulmonary gas exchange is generally well preserved up to high levels of exercise, as assessed by maintained arterial PO2 (PaO2) and PaCO2, and by chemoreflex decrease in PaCO2 above the ventilatory threshold.
In 1984, Dempsey et al. reported on exercise-induced hypoxemia in highly trained athletes, suggesting that extreme levels of exercise may reach the limits of lung function adaptation (35). In that study, the lowest PaO2 were observed in those athletes with lesser decrease in PaCO2, which is an indication of lung mechanical constraints accounting for exercise-induced hypoxemia associated with abnormally well maintained capnia.
However, moderate exercise may already affect gas exchange as assessed by an increased gradient between alveolar and arterial PO2 (A-aPO2) which widens until maximal exercise capacity. Studies in exercising healthy subjects using the multiple gas elimination technique (MIGET) have demonstrated that this is attributable in part to VA/Q inhomogeneity and, at the highest levels of exercise, to a diffusion limitation (63,67,81,162,226). Both VA/Q imbalance and diffusion limitation may be related to the state of the pulmonary circulation at exercise.
8.1. VA/Q relationships
Exercise-induced VA/Q inhomogeneities have been demonstrated by the MIGET. This technique is based on the use of inert gases with different solubility to calculate the distributions of VA and Q to 50 compartments of a mathematical lung model with VA/Q increased from zero (shunt) to the infinite (dead space) passing through an ideal VA/Q around one. The difficulty of this representation is that changes in distributions may be due either to changes in VA or Q for any given compartment (145). For example, an apparent increase in flow distribution to a lung unit with a VA/Q of 0.1 may as well be caused by a ten times less important decrease in ventilation. Thus changes in MIGET VA/Q distributions can always be explained by changes in the distribution of perfusion and/or ventilation but one cannot discern the precise mechanism.
Gravity normally imposes a vertical gradient of pulmonary perfusion, which causes of physiologic inhomogeneity of VA/Q (232). Increased Ppam at exercise counteracts the effects of gravity. Accordingly, radioactive tracer studies of VA and Q distributions at exercise have shown more homogeneous VA/Q because of a redistribution of Q to the upper parts of the lung (19,82).
However, MIGET studies have demonstrated a deterioration in VA/Q distributions in proportion to level of exercise and increased Ppam in normal exercising humans (63,67,81,162,226), independent of level of exercise or inspired PO2 (PIO2) (63,162,226,228). The most likely explanation is that increased pulmonary vascular pressures are associated with an increased capillary filtration, causing interstitial edema. This alters the distribution of VA, because of narrowing of airways and decrease in alveolar compliance, but may also alter the distribution of Q because of compression of blood vessels. Left atrial pressure and Ppw increase at exercise in proportion to Ppam, and it is easy to calculate that at some point, corresponding to a Pcp of 20–25 mmHg (78), Starling forces governing pulmonary capillary filtration become disturbed such that excess fluid leaves the capillaries, overcoming the capacities of the lung lymphatic drainage. In a recent discussion about the mechanisms of altered gas exchange at exercise, Hopkins argued in favor of a role of interstitial edema based on a series of reports showing a typical VA/Q deteriorations, persistent of VA/Q mismatch after recovery, worsening of VA/Q with exercise duration or severity of hypoxia, and improved hypoxic VA/Q with oxygen breathing (89). The notion that exercise-induced deterioration in VA/Q matching is caused by the development of interstitial edema is supported by experimental animal data. Lung lymphatic flow increases rapidly after the onset of exercise in sheep, in proportion to increased cardiac output and microvascular pressures (168). There is histologic evidence of interstitial lung edema in heavily exercised pigs (191). However, VA/Q matching is better preserved during exercise in horses (9,99,227) than in pigs (92,95) while in both species a diffusion limitation does not seem to explain increased A-aPO2 gradients, which remains poorly understood.
Refined measurements of the spatial distribution of pulmonary perfusion in healthy volunteers have more recently been made possible by the advent of functional MRI arterial spin labeling (93). This method has allowed the demonstration of a perfusion heterogeneity correlated to MIGET indices of VA/Q mismatch in normal subjects exercising for 45 min at 70 % of their VO2max (22). The only possible explanation for these findings is exercise-induced interstitial lung edema, even though the method could not detect a change in mean lung density.
8.2. Diffusion
Exercise increases lung diffusing capacity (Fig 14). Studies using the low O2 breathing method have demonstrated that exercise increases both the membrane and the capillary volume components of carbon monoxide diffusing capacity (DLCO), in proportion to increased cardiac output (104,184). Assuming a maximum expansion of capillary blood volume by a factor of 2.5 compensating for decreased red blood cell transit time, Dempsey calculated that for an athlete exercising at a high VO2 and cardiac output with a PAO2 of 100 mmHg and a mixed venous PO2 down to 20 mmHg, the mean red blood transit time would be shorter than the minimal transit time needed (0.6 s) to allow for complete end-capillary equilibrium (34). It is curious that DLCO does not become asymptotic to some limiting value as cardiac output increases up to maximal exercise, as illustrated in Fig 14. It is possible that increased Pla, Pwp and Pcp at high levels exercise serve to maximally increase capillary blood volume, to improve gas exchange, but that this is nevertheless counteracted by excessive red blood cell velocity, and eventual appearance of interstitial lung edema.
Figure 14.
Relation of pulmonary carbon monoxide diffusion capacity (DLCO) to pulmonary blood flow (Q) at increasing levels of exercise. DLCO increases in proportion of Q, even in Olympic cyclists with very high Q. From references 104 and 183.
As pulmonary venous pressure and flow increase with exercise, capillaries may undergo a “stress failure” for Pcp’s > 40 mmHg (234). Stress-failure of the pulmonary capillaries occurs in race horses presenting which present with severe hypoxemia and hemorrhagic lung edema after high levels of exercise (233). However, alveolar edema as a cause of pulmonary shunting and increased A-aPO2 has not been identified by MIGET studies in exercising humans (63,67,81,162,226).
Dempsey and Wagner analyzed relative contributions and mechanisms of inadequate ventilatory response, VA/Q inequality and diffusion limitation to decreased arterial oxygenation and increased A-aPO2 at exercise (36). They noted that exercise-induced hypoxemia depends on exercise intensity independent of age, gender and body position, and that the greatest inefficiencies in gas exchange occur in the animal species such as the dog or the horse, which have the highest body size-corrected VO2max. While mechanical constraints on ventilation emerged as the predominant cause of hypoxemia, increased A-aPO2 at maximum exercise was exclusively explained by approximately equal contributions of VA/Q inequality and diffusion limitation, without identifiable pulmonary or cardiac shunting.
8.3. Pulmonary shunting of agitated contrast
In 2004, Eldridge, Stickland and colleagues reported on the occurrence of exercise-induced shunting demonstrated by agitated saline contrast echocardiography in subjects with otherwise no evidence of intrapulmonary or intracardiac shunt at rest (46,199). In these studies, exercise-induced pulmonary shunting of bubbles was correlated to cardiac output, Ppam and A-aPO2 at exercise, suggesting an impact on gas exchange (198). Agitated saline or gelatine contrast echocardiography is standard practice for the detection of cardiac shunts. The bubbles are of similar size, 10–35 mm, and do not normally traverse the pulmonary circulation. In the case of a cardiac right-to-left shunt, the appearance of contrast in the left heart chambers is immediate. In the case of pulmonary shunt, the appearance of contrast in the left heart chambers is delayed by 3–5 beats (Fig 15). However, there has been some controversy about the location and dimension of pulmonary vessels through which the bubbles travel, and the relationship of this phenomenon to gas exchange (94,129). Pulmonary shunting of agitated contrast could be explained by pressure-induced pulmonary capillary distension, causing a diffusion/perfusion imbalance with or without a true shunt measured by pure oxygen breathing or the MIGET, like seen in early stage hepato-pulmonary syndrome (157). In exercising athletes, intrapulmonary shunting measured by the retention of the less soluble gases in MIGET studies has always be found to be less than 1 % of cardiac output, averaging 0.2 % in normoxia and 0.1 % in hypoxia, which cannot explain more than 7 % of the total A-aPO2 in normoxia and much less, < 1 % in hypoxia (94). It is not always possible to find a correlation between presence or graded severity of agitated saline contrast shunting and A-aPO2 in either exercising athletes (116) or in patients with pulmonary vascular dilatations (145).
Figure 15.

Echocardiographic apical 4 chamber views at rest (left panel) and at exercise (middle and right panels) after the injection of agitated contrast, showing appearance in the left heart chambers after 4–5 beats at moderate exercise, and more so at intense exercise. Courtesy of A La Gerche.
9. Right ventricular function at exercise
Traditional thinking about the determinants of exercise capacity focused on left ventricular function (186), even in conditions of hypoxic exposure and associated increase in pulmonary artery pressures that increase RV afterload (181,182,205). However, heavy exercise is associated with doubling or tripling of Ppam, which imposes a marked increase in afterload added on to the stress of venous return-induced increase in preload on the normally thin-walled RV. This may explain why prolonged intense exercise results in altered indices of cardiac fatigue which disproportionately affect the RV (39,117,153,165). Right ventricular mass and dimensions have been shown by large scale MRI studies to increase in relation to the level of physical activity, independent of measures of left ventricular structure and function (1). It has been hypothesized that RV remodeling associated with a chronic high level of training in athletes might be a cause of cardiac rhythm disturbances (86,115)
9.1. Acute effects of exercise on right ventricular function
Inspection of individual pulmonary artery pressure-flow relationships at exercise suggests that subjects able to achieve higher maximum cardiac output and oxygen uptake also present shallower slopes, so that maximum levels of pulmonary artery pressures are in the range of 35–40 mmHg Ppam (4,116). Echocardiographic measurements have been reported suggesting that pulmonary shunting of agitated contrast would be associated with enhanced pulmonary vascular reserve and RV function at exercise (116). As an example, when cardiac output increased from 5 to 15 L.min−1 there was a 28 % reduction in PVR in the subjects with the greatest transpulmonary passage of agitated contrast, compared with a 4 % decrease in PVR in subjects with minimal shunting of contrast. By tissue Doppler examination, subjects with the highest transit of agitated contrast had also higher RV maximal systolic myocardial velocity and isovolumic acceleration, while indices of LV function were not different. Furthermore, the exercise-induced increase in circulating B-type natriuretic peptide was lower in the subjects with the highest transit of agitated contrast, suggesting less RV distension. All of these measurements are in keeping with the idea of a better coupling of RV function to afterload in subjects with the highest transit of agitated saline. However, the concept of exercise-induced pulmonary vasodilatation allowing for improved maximal RV output but potentially worse pulmonary gas exchange remains debated (130). Decreased PVR allowing possibly for a higher RV flow output at exercise may also be explained by a higher left ventricular compliance (201).
Taking a step further, La Gerche et al investigated end-sysolic wall stress of the right and left ventricles using MRI and echocardiography in endurance athletes and controls at exercise (115). Endurance athletes showed greater RV than LV remodeling, and peak exercise RV wall stress was greater in endurance athletes than in non athletic controls. These results were explained by greater RV enlargement and wall thickening corresponding to more loading in endurance athletes. In that study, end-systolic wall stress was calculated as the product of peak systolic pressure times internal radius, divided by wall thickness. Maximal wall stress is a good definition of afterload, as an alternative to arterial elastance (or end systolic pressure divided by stroke volume). However, this measure of maximal wall stress relies on peak systolic pressure, which is dependent on the mechanical properties of the arterial system to which the ventricle is connected.
The ratio of peak systolic pressure to end-systolic volume would appear to be a more acceptable surrogate of end-systolic elastance to measure contractility, as previously reported in normal subjects exercising in hypoxia (205), and a ratio of Ppam (instead of end-systolic) divided by stroke volume a reasonable surrogate to arterial elastance. Still the relationships between compliance and resistance are not identical in systemic and pulmonary circulations, so that approximations become inadequate for comparisons (188,235).
Thus, in spite of previous application to patients with pulmonary hypertension, MRI and echocardiographic estimates of RV wall stress based on systolic Ppa and end-systolic volumes and wall thickness that have shown good reproductibility (177), the measurement is composite and probably requires more validation against gold standards. However, the report by La Gerche and his colleagues is consistent with the emerging notion that athletes do not develop a balanced hypertrophy (193), but that RV changes are predominant (218).
9.2. Pharmacological interventions
There is little pulmonary vascular tone at exercise, so pharmacological attempts at decreasing PVR would not be expected to affect RV afterload or exercise capacity. The effects of drugs effective in the treatment of PAH have been investigated in normal volunteers exercising in normoxia, in acute normobaric hypoxia or in chronic hypobaric hypoxia. In normoxic conditions, sildenafil, bosentan or sitaxsentan did not affect maximum workload or VO2max (51,53,65,96,158). It must be emphasized that the RV at high levels of exercise is stressed by combined increases in preload and afterload, and that pulmonary vasodilators also tend to increase systemic venous return, which could counteract any slight decrease in afterload obtained through a decrease in PVR.
10. Exercise in hypoxia
10.1. Hypoxic pulmonary vasoconstriction and remodeling
Hypoxic exposure induces pulmonary vasoconstriction followed by arteriolar remodeling. The hypoxic pressor response is characterized by marked inter-individual and inter-species variability, as it is intense in pig, horse and cow, moderate in rodents and humans and very low in dog, guinea pig, yak and llama (71). Chronic hypoxia induces pulmonary hypertension, in proportion to initial vasoconstriction. Initial hypoxic vasoconstriction is induced by inhibition of voltage-gated potassium channels localized in smooth muscle cells, which explains a quasi-immediate response, and there is secondary modulation by endothelium-derived mediators (230). Hypoxic vasoconstriction strengthens during the first 1–2 hours of hypoxic exposure in humans (74), but has been found to remain stable during one hour in anesthetized intact experimental animals (159).
The time-course of hypoxia-induced remodeling is less well known. In humans, chronic hypoxic exposure induces a rapid 5-minute increase in PVR followed by a slower further increase, leading to a stabilization after 2 hours (74) and a maximal response after approximately 24 hours (73,134). After six hours of hypoxic exposure, re-oxygenation immediately decreases PVR, without however a complete return to normal (38). The reversibility of increased PVR with re-oxygenation is largely lost after 24 to 48 hours exposure to hypoxia (73,134). These observations indicate that hypoxic exposure is associated with pulmonary vascular remodeling occurring within the first 24 hours.
The first description of human hypoxic pulmonary vasoconstriction was reported by Motley et al in 1947 in 5 conscious normal subjects who consented to a right heart catheterization. These subjects were acutely challenged with a fraction of inspired O2 (FIO2) of 0.1, which increased Ppam from 13 to 23 mmHg (152). The authors somehow selected strong « responders » to hypoxia. In later studies on a total of 39 normal subjects challenged with a FIO2 of 0.125 (PaO2, 40 mmHg), hypoxia-induced increases in Ppam ranged from 0 to 20 mmHg (146,161,162). Hypoxia did not increase PVR in 8 of these subjects, which is a 20 % proportion of “non responders” that is corroborated by other studies on the acute effects of normobaric hypoxia in normal volunteers (73).
A difficulty in studying the effects of hypoxia on the pulmonary circulation in conscious intact animals is the interference with the hypoxic ventilatory response. This decreases PACO2, increasing PAO2 at any given PIO2. An acute normobaric hypoxic exposure with a fraction of FIO2 progressively dimished to 0.12 does not much affect ventilation (40), so that PaCO2 remains essentially unchanged or only slightly decreased (134,146,161) With prolonged hypoxic exposure, there is a progressive increase in ventilation allowing for adaptation with improved PAO2 and PaO2. This takes a few days at a given level of hypoxia, and is an essential feature of the adaptation to altitude (123). It has been shown that markedly increased ventilation and hypocapnia are indispensable to the adaptation to extreme altitudes (231). However, the ventilatory response to hypoxia decreases over years, so that high altitude dwellers present with levels of ventilation at rest which are identical or only slightly increased compared to measurements in usual lowlanders at sea level (123). Further loss of ventilatory adaptation is a feature of chronic mountain sickness, or “Monge’s disease” characterized by severe hypoxemia and polycythemia (172,231). Since the main determinant of hypoxic vasoconstriction is PAO2 (139), studies on the pulmonary circulation at high altitudes must always take the ventilatory adaptation into account. The same PaO2 around 40 mmHg at rest is seen in high-altitude natives living at Morococha (Peru), at 4,540 m (172), and in lowlanders acclimatized in a few weeks at 7,000 m (73).
Another adjustment to take into account is in the stimulus-response curve for hypoxic pulmonary vasoconstriction. The relationship between FIO2 or PAO2 and hypoxic vasoconstriction expressed as a change in Ppam at a given flow or as an amount of flow diversion at a given Ppam has been generally found in experimental animal preparations to be either sigmoid-shape (7) or linear (70), with a continued constriction as long as FIO2 or PAO2 decreased. However, in isolated pig lungs at a constant flow, in which particular attention was paid to reach a steady state before each measurement, the Ppa/PAO2 curve was shown to be biphasic, with a maximum at a PAO2 between 30 and 60 mmHg, and a down-sloping portion called “hypoxic pulmonary vasodilation” at lower PAO2 (211). In dogs, a species with a pulmonary vasoreactivity to hypoxia comparable to that of man, the relationship between Ppam, or PVR, and FIO2 progressively decreased from 1 to 0.06, has been shown to be biphasic as well, with a downsloping at a FIO2 lower than of 0.1, corresponding to a PaO2 of 36–38 mmHg (15).
Evidence of hypoxic pulmonary vasodilation in man was obtained during Operation Everest II. In these studies, normal subjects decompressed in a hypobaric chamber for 40 days to an atmospheric pressure equivalent to the summit of Mount Everest, presented an average Ppam of 34 mmHg at rest and of 54 mmHg at exercise at a barometric pressure (Pb) of 282 mmHg (7,620 m, resting PaO2 37 mmHg), that decreased to 33 and 48 mmHg respectively at a Pb of 240 Torr (8,840 m, resting PaO2 30 mmHg) (73). Because of a relative hypoventilation compared to acclimatized lowlanders, high altitude natives present a resting PaO2 of 40 mmHgr at relatively lower altitudes, around 4,500 m.
In part related to the physiological inhomogeneity of VA/Q distributions and associated variations in regional PAO2 and PACO2, hypoxic vasoconstriction is inhomogeneous. This has been demonstrated using the fluorescent microsphere technique in swine (92) and functional MRI in humans (91). In the latter study, hypoxia-induced perfusion heterogeneity was more marked in subjects with a previous history of high altitude pulmonary edema (HAPE) (91).
Hypoxic pulmonary vasoconstriction is particularly intense in few percent of normal human subjects (75). Because hypoxia does not affect the longitudinal distribution of resistances (134), these subjects present with increased Pcp and, accordingly, are predisposed to HAPE (74,134). Lowlanders with a brisk hypoxic vasoconstriction traveling to high altitudes may also be exposed to the risk of right heart failure. This complication of high altitude exposure has been reported in high altitude cattle (85), infants of Chinese Han brought to high altitudes in Tibet (208), Indian soldiers transported to high altitude borders with China (3) and occasional Europeans travelling to the South American altiplano (98).
10.2. Hypoxic pulmonary hypertension
Because of hypoxic pulmonary vasoconstriction and subsequent remodeling, Ppam is increased at altitude, acutely as well as chronically. Maggiorini et al. catheterized subjects with or without a previous history of HAPE in normoxia, in acute normobaric hypoxia, and after 1–2 days at the altitude of 4559 m. In subjects without previous HAPE, Ppam increased from 14 mmHg in normoxia to 22 mmHg in normobaric hypoxia (PaO2 38 mmHg) and to 26 mmHg in hypobaric hypoxia (PaO2 45 mmHg). In subjects with previous HAPE, Ppam increased from 16 mmHg in normoxia to 27 mmHg in normobaric hypoxia, and to 38 mmHg in hypobaric hypoxia. Subjects with a previous HAPE had a higher PVR than controls in hypoxia and in normoxia, and PVR increased in hypoxia from acute to more chronic conditions (134). Similar observations have been reported using non invasive echocardiographic measurements (53).
Subjects with a previous HAPE have repeatedly been reported to present with an enhanced hypoxic pulmonary vasoconstriction, measured either invasively (47,101,108,134,222) or noninvasively (56,74,237). It is interesting that in these studies, normoxic PVR at rest as well as at exercise has been repeatedly shown to be increased in HAPE-susceptibles compared to controls (47,108,134,237), which points at a constitutional abnormality somehow linked to an increased pulmonary vasoreactivity.
Chronic hypoxic exposure in high altitude dwellers is characterized by a variable increase in Ppam. Penaloza and Arias-Stella plotted Ppam reported in high altitude studies as a function of arterial O2 saturation (SaO2) and found a hyperbolic relationship (172) ( Fig 16). However, when they plotted Ppam as a function of altitude of residence, they found that there were disproportionally high Ppam in Leadville, and low Ppam in Lhasa (Fig 16)
Figure 16.

Curvilinear relationships between mean pulmonary artery pressure (PPA) and arterial oxygen saturation (SaO2) in healthy highlanders (empty symbols) and highlanders with chronic mountain sickness (full symbols), left palel, and between mean pulmonary artery pressure (PPA) and altitude of locations (right panel). Extremes are observed in Lhasa and in Leadville. Hypoxia is a major determinant of Ppa, but there is ethnic variability. From reference 172. Permission pending.
The reasons why relatively higher Ppam were recorded in Leadville (223) as compared to Lhasa (72) may be related to genetic predisposition. Centuries of living at high altitudes in the Himalayas has probably allowed for selection of fitter individuals with lower hypoxic pulmonary pressore response allowing for better survival and reproduction.. No such selection pressure has acted on human residents of Leadville, where the population is transient and similar to the rest of the US. Selection pressure may have acted on cattle in the North American high altitude pastures; in the first few years after introduction of cattle to these sites, Brisket’s disease was more common than it is today.
Echocardiographic studies indicate that South American highlanders tend to present with a lower Ppam compared to acclimatized lowlanders at the same cardiac output (97,135,204). This is not apparent in invasive hemodynamic studies (6,173), but comparisons are difficult because of subject selection biases and maybe also technical limitations of echocardiographic estimates of pressures and flows.
The question has been asked whether pulmonary hypertension and right heart failure is characteristic of chronic mountain sickness, or “Monge’s disease” defined by symptomatic polycythemia, sevee hypoxemia and hemoglobin levels higher than 21 g/dl in men and 19 g/dl in women (124). Penaloza and Arias-Stella showed that patients with Monge’s disease present with higher Ppam and lower arterial SaO2 extrapolated on the same hyperbolic relationship as in healthy highlanders, suggesting identical pathogenesis of pulmonary hypertension (Fig 16).
There are very fews studies looking into the effects of hypoxic exposure on pulsatile pulmonary hemodynamics, and all of these are on experimental animals. Acute hypoxia has been reported to increase ZC in pigs and in goats, but not in dogs (55,132,229). Chronic hypoxia decreased ZC in mice (220), in spite of demonstrated in vitro stiffening of the proximal pulmonary arteries (109). Associated changes in pulmonary arterial compliance are not exactly known, and even less is known about the effects of exercise.
Studies using the arterial occlusion technique in dogs (55,133) and minipigs (133) have shown that hypoxia-induced increase in PVR does not affect the longitudinal distribution of resistances. This has been confirmed in humans with or without a previous HAPE, in acute normobaric hypoxia as well as after 1–2 days of acclimatization to high altitude (134). Thus in resting human subjects in hypoxia, Pcp increases in proportion to Ppam in a manner is predicted by the Gaar equation. There are no reported Pcp estimates using the occlusion technique in human volunteers at exercise. As the arterial resistance decreases more than the capillary-venous resistance in lungs perfused at very high flows (20), it is possible that Pcp increases proportionally more than Ppam at exercise. However, this is hypothetical in the absence of reported measurements.
10.3. The pulmonary circulation during hypoxic exercise
Exercise in acute normobaric or hypobaric hypoxia, or in chronic hypobaric hypoxia has been reported to shift Ppam-Q relationships to higher pressures (6,52,53,56,73). Invasive measurements performed on lowlanders progressively decompressed in a hypobaric chamber during the Operation Everest II show progressively increased slopes of Ppam-Q relationships with increasing altitudes (Fig 17) (73). In these experiments, there was a decrease in Ppw compared to sea level exercise, excluding upstream transmission of increased left ventricular filling pressure as a mechanism of increased slope.
Figure 17.

Mean pulmonary artery pressure (Ppa) as a function of cardiac output (Q) at exercise in healthy subjects at sea level, barometric pressure (Pb) 760 mmHg, and after acclimatization at two levels of simulated altitudes. Baseline Ppa and the slope of Ppa-Q increase with altitude. From reference 73.
Exercise in acute hypoxic conditions in lowlanders with a previous HAPE has been shown to be associated with markedly increased Ppam and PVR as compared to controls (47,108).
Exercise in subjects born and living permanently at high altitiudes is associated with particularly steep Ppam-Q relationships. This is illustrated in Fig 18 constructed with right heart catheterization and non invasive echo-Doppler data from high altitude inhabitants investigated at high altitude versus lowlanders at sea level (4,6,11,13,73,173,204,223). Echo-Doppler studies were generally associated with lower cardiac outputs, at rest and at exercise, which is explained by differences in exercise protocols, the difficulty in obtaining good quality signals at high levels of exercise, and maybe a systematic underestimation. On the other hand, with the exception of Tibetans, highlanders have much steeper slopes of their Ppam-Q relationships, in spite of only moderate pulmonary hypertension at rest. The highest baseline Ppam values at rest and the steepest slopes of Ppam-Q were recorded by Penaloza and Sime in patients with Monge’s disease, with Ppam of 37 mmHg at rest climbing to 82 mmHg at mild exercise (173). Vogel et al reported resting Ppam of 25 mmHg increasing to 54 mmHg at moderate exercise with workloads ranging from 25 to 100 W in subjects living in Leadville, at the altitude of 3000 m (223). Banchero et al reported resting Ppam of 29 mmHg increasing to 60 mmHg at workloads around 80 W in subjects born and living in Morococha, at the altitude of 4540 m (6) In contrast, Groves et al measured a Ppam of 15 mmHg increasing to 35 mmHg at workloads ranging from 60 to 180 W in subjects living in Lhasa, at 3658 m (72), indicating slopes of Ppam-Q relationships at the upper limit of normoxic normal. In the only non invasive exercise study reported to date in highlanders, Stuber et al reported on echocardiographic measurements at rest and at a 50 W workload in subjects born and living in La Paz, at altitudes of 3600–4000 m (204). The results showed a Ppam of 20 mmHg at rest and 29 mmHg (slope of Ppam-Q: 3 mmHg.L−1.min−1) at mild exercise in highlanders without Monge’s disease, and 23 mmHg increasing to 39 mmHg (slope of Ppam-Q 5: mmHg/L/min−1) at mild exercise in highlanders with Monge’s disease.
Figure 18.
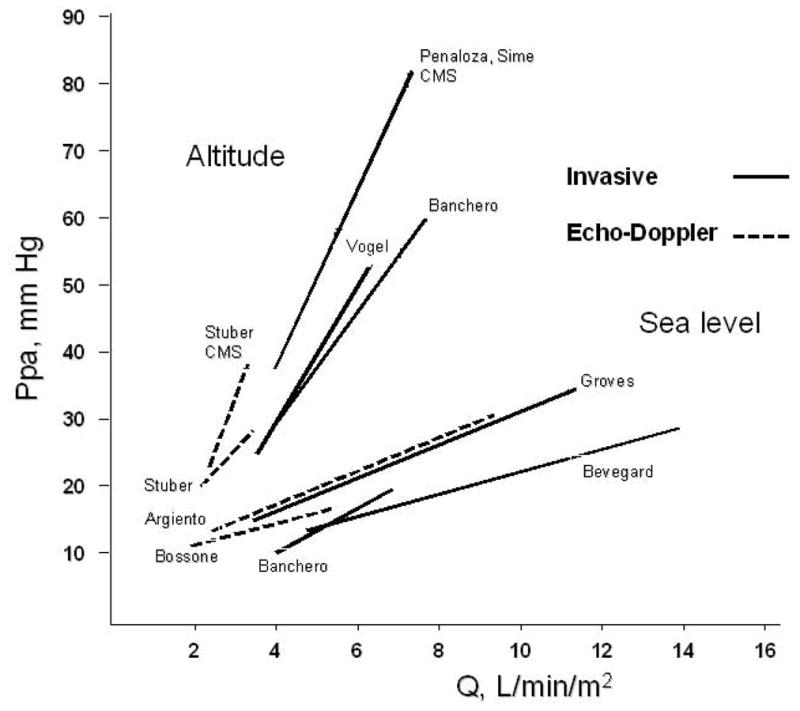
Averaged mean pulmonary artery pressures (Ppa) versus indexed cardiac output (Q) plots measured invasively (cardiac cath, full lines) or noninvasively (echo-Doppler, stippled lines) in highlanders exercising at high altitude and in lowlanders exercising at sea leve. From references 1,4,6,11,13,73,173,204,223, with indications of names of first authors in the figure. With the exception of Tibetans in lhasa, highlanders present with higher resting Ppa and increased slopes of Ppa-Q. Slopes of Ppa-Q are particularly steep on patients with chronic mountain sickness (CMS). Courtesy of Dante Penaloza.
Reeves et al. used the individual data of the Operation Everest II measurements to recalculate the effects of chronic hypoxic exposure in normal humans on TPVR and the pulmonary distensibility coefficient α (28). The results showed an increase in TPVR from 2.3 ± 0.2 to 3.9 ± 0.2 Wood units and a decrease in α from 0.02 ± 0.002 to 0.008 ± 0.001 mmHg−1. The same calculations on the individual data of acute hypobaric exposure experiments reported by Wagner et al in another group of healthy volunteers (226) did not show ant difference in α. However, in that study, PVR remained unchanged in hypoxia as well, so that the unchanged distensibility could be simply related to absent hypoxic pulmonary vasoconstriction. Further studies on larger groups of unselected subjects are needed to evaluate the effects of acute hypoxia on pulmonary vascular distensibility. There are no individual data reported that would enable distensibility to be calculated in highlanders with or without Monge’s disease.
10.4. Pulmonary gas exchange during hypoxic exercise
Exercise in hypoxia is associated with a marked deterioration in VA/Q relationships. Wagner et al. reported on MIGET studies during Operation Everest II which showed a deterioration in VA/Q matching that increased with severity of hypoxic exposure and exercise (226). The dispersion of blood flow as an index of VA/Q inhomogeneity was correlated to Ppam, not to cardiac output, Ppw or ventilation. Alveolar-end-capillary diffusion limitation of O2 was observed at a VO2 > 3 L.min−1 at sea level, of > 1–1.2 L.min−1 at a barometric pressure of 428 mmHg, and approximately of 1 L.min−1 at the highest simulated altitudes. The recovered VA/Q patterns and relationship to Ppam were compatible with alveolar interstitial edema as the primary cause of VA/Q inequality.
Wagner et al. further investigated pulmonary gas exchange with the MIGET at the altitude of 5260 m in Bolivian highlanders usually living at the altitude of La Paz (altitude 3600–4000 m) versus acclimatized lowlanders who were relatively well matched for body weight and age (225). Maximum workload, cardiac output and VO2 were the same, but PaO2 was better preserved in highlanders in spite of decreased ventilatory response to exercise, and this was explained by a greater pulmonary diffusing capacity. There were no pulmonary hemodynamic measurements reported, so that the possible impact of Ppam on increased A-aPO2 was not assessed.
Because both hypoxia and exercise are associated with an increase in Ppam, a critical Pcp as a cause of excessive capillary filtration would be more easily reached in conditions of acute hypoxic exposure. On the other hand, exercise capacity is decreased in hypoxia, so that the flow-dependent increase in Ppam is limited as well. Still there is a significant risk of over-perfused pulmonary capillaries, due to high Pcp and inhomogenous hypoxic vasoconstriction, so that a patchy lung edema typical of HAPE may develop in a proportion of susceptible subjects (8). Heavy exercise may be a cause of increased red blood cells and protein in broncho-alveolar lavage fluid, and this is markedly increased when exercise is performed in hypoxia (45). Steeper Ppam-Q relationships in subjects with a previous HAPE could be a cause of marked increase in Pcp, further increased in overperfused lung regions, leading to VA/Q mismatch and eventual pulmonary capillary stress-failure (175)
10.5. The right ventricle as a limiting factor of hypoxic exercise capacity
Decreased maximum exercise capacity at high altitude is explained by a decrease in arterial oxygen content and a limitation in maximal cardiac output (60). With acclimatization, the arterial oxygen content may be restored to sea level values because of an increased hemoglobin concentration, but maximal cardiac output remains depressed (23). The decrease in maximal cardiac output at altitude has been tentatively explained by the combined effects of decreased blood volume, hypocapnia, increased viscosity of the blood, autonomic nervous system changes, or depressed myocardial function, but has also been modeled by an altered coupling of convectional and diffusional oxygen transport systems (224).
An additional factor might be a limitation in RV flow output secondary to hypoxic pulmonary hypertension. An improvement in maximal workload and VO2max together with a decrease in Ppam has indeed been reported after the intake of sildenafil, bosentan or sitaxsentan in hypoxic healthy volunteers (51,53,65,96,158). A decreased Ppam with an improved VO2max has been observed in subjects susceptible to HAPE taking dexamethasone before altitude exposure (56). However, improved exercise capacity could not unequivocally be ascribed to associated inhibition of hypoxic pulmonary vasoconstriction when the pharmacological intervention was also associated with an improved arterial oxygenation, as occurs with sildenafil (53,65,96) or dexamethasone (56). Improved arterial oxygenation is not observed with the selective or non selective endothelin receptor blockers sitaxsentan and bosentan, which decreased the hypoxic pressure response by approximately 50 % in either acute normobaric hypoxia or more chronic hypobaric hypoxia (51,158). Significant inverse correlations between systolic Ppa (echocardiography) and VO2max have been observed (Fig 19), which is compatible with the notion that increased Ppam at high altitudes limits exercise capacity by reducing maximal RV flow output (158). However, further studies are needed to confirm this hypothesis.
Figure 19.

Correlation between hypoxia-induced increase in mean pulmonary artery pressure (Ppam) and decrease maximal oxygen uptake (VO2max) in healthy volunteers in either acute normobaric or more chronic hypobaric hypoxic conditions. From reference 158. Permission pending.
Considering the coupling of RV function to the pulmonary circulation, it is generally assumed that physiological levels of oxygen deprivation, up to an altitude equivalent to the summit of Mount Everest, do not negatively affect cardiac function. Echocardiography was used in 8 volunteers progressively decompressed in 40 days to the simulated altitude of Mount Everest (Operation Everest II), for measurements of LV volumes and evaluation of systolic function by peak systolic (brachial artery sphygmomanometry) versus end-systolic volume (echocardiography) relationships. This is a surrogate for completely invasive end-systolic elastance measurements as a gold standard of load-independent contractility. The results showed a decrease in the volumes of both the LV and the RV, compatible with hypovolemia, but a normal ejection fraction that increased adequately with exercise. The ratio of peak systolic pressure to end-systolic volume at rest and at exercise increased at all altitudes, indicating enhanced contractility, up to the simulated altitude of approximately 8400 m (205). Thus contractility appeared to be well preserved in healthy subjects at high altitudes, indicating excellent tolerance of the normal myocardium to extreme environmental oxygen deprivation.
However, the relative change in afterload conditions of the RV in hypoxia make that cardiac chamber potentially more vulnerable to increased PVR. Accordingly, additional resting echocardiographic measurements were reported in a similar series of experiments in 8 healthy volunteers at simulated altitudes of 5000 m to 8000 m (Operation Everest III) (14). The measurements confirmed previously reported mild pulmonary hypertension, preserved LV contractility, and decreased preload of both ventricles, but showed an abnormal LV filling pattern with decreased early filling and greater contribution of atrial contraction, without elevation of end-diastolic pressure. The authors thought that these LV diastolic changes could be explained by the combined effects of tachycardia and reduced preload, with possibly also effects of ventricular interdependence or hypoxia. Similar results were reported in 41 healthy volunteers who ascended in 24 hours to the altitude of 4559 m in the Italian Alps (2).
A complete evaluation right and left ventricular function by Doppler echocardiography with tissue Doppler imaging was reported in 25 healthy volunteers acutely breathing a fraction of inspired oxygen of 0.12 (99). These results suggested that short-term hypoxic exposure is associated with a preserved RV systolic function, either sympathetically-mediated or via homeometric adaptation to mild pulmonary hypertension. Also, there were early RV diastolic changes that probably reflected an increased afterload. Changes in diastolic filling patterns of both ventricles and increased LV contractility would be explained by acute hypoxia-induced activation of the sympathetic nervous system.
These measurements were most recently repeated in 15 healthy high altitude inhabitants of the Bolivian altiplano, at approximately 4000 m, as compared to age and body surface area -matched acclimatized lowlanders (97). Acute exposure to high altitude in lowlanders caused an increase in Ppam to 20–25 mmHg, with mildly altered LV and RV diastolic function, and maintained RV systolic function. This profile was essentially unchanged after acclimatization and ascending to 4850 m, except for a higher Ppam. The highlanders presented with relatively lower Ppam and higher SaO2, but more pronounced alteration in indices of diastolic function of both ventricles, a decreased LV ejection fraction, and decreased indices of RV systolic function. The estimated LV filling pressure was somewhat lower in the highlanders. Thus the cardiac adaptation of highlanders appeared qualitatively similar, with slight but significant deterioration of indices of both systolic and diastolic function, in spite of less marked pulmonary hypertension and better oxygenation. The authors explained these results by combined effects of a lesser degree of sympathetic nervous system activation, relative hypovolemia and may be some negative inotropic effects of a long lasting hypoxic exposure.
Echocardiographic measurements have also been reported in a study on high altitude Peruvian dwellers with a diagnosis of Monge’s disease (135). The patients presented with trans-tricuspid gradients at an average of 34 mmHg, which is in the range reported in acclimatized lowlanders, and a RV dilatation with more alteration in indices of function than reported in either acclimatized lowlanders or healthy high altitude inhabitants. The reasons for more altered RV function in patients with Monge’s disease are not entirely clear, but it is thought that repetitive bouts of pulmonary hypertension at exercise could play a role.
Recent progress in portable Doppler echocardiography has allowed improved evaluation of RV function in lowlanders with a reactive pulmonary circulation and exposed to high altitude. It is possible that a proportion of these subjects presents with a clinically silent high altitude acute right heart failure, characterized by a severe pulmonary hypertension, dilatation of right heart chambers with a septal shift, dilatation of the inferior vena cava with loss of inspiratory collapse, indicating increased right atrial pressure, and altered RV function with “post systolic shortening” (98). Post systolic shortening reflects ischemia and asynchronic contraction the RV, much like recently reported by MRI studies in severe pulmonary arterial hypertension (138).
There is thus indirect evidence that the RV during hypoxic exercise might be overloaded and functionally limited and thereby contribute to decreased exercise capacity. Chronic RV overload and/or hypoxia migt be a cause of impaired RV function.
11. Exercise with cardiac or lung disease
Increased baseline PVR in patients with pulmonary hypertension implies that any increase in cardiac output associated with physical activity may be the cause of an excessive increase in Ppam and RV afterload. The dyspnea-fatigue symptomatology that is characteristic of pulmonary hypertension is essentially explained by the inability of the RV to adapt to increased afterload (26). The slope of Ppam-Q with exercise in pulmonary hypertension reflects the severity of pulmonary vascular remodeling and increased tone, but is also affected by associated changes in arterial and mixed venous blood gases, filling pressures of the left heart, sometimes alveolar pressure, and sympathetic nervous system activation (71). Accordingly, adding exercise to resting hemodynamic measurements in patients with pulmonary hypertension may help to understand changes in functional state and effects of pharmacological interventions. Exercise stress testing may be particularly useful for the detection of early or latent pulmonary vascular disease.
11.1 Exercise in patients with pulmonary hypertension
Pulmonary vascular pressure-flow relationships have been reported in patients with a variety of lung and cardiac diseases with flow increased by unilateral balloon occlusion and/or exercise, sometimes with inotropic drug interventions (50,83,103,106,160,241). A summary of this data is represented in Fig 20.
Figure 20.

Two point mean pulmonary artery pressure (Ppa) cardiac output (Q) plots in normal subjects (N), or patients with mitral stenosis (MS), acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), left ventricular failure (LVF) and pulmonary arterial hypertension (PAH). All Ppa-Q plots are shifted upward to higher pressure with an increased extrapolated pressure intercept (stippled lines). Left atrial pressure was equal to the extrapolated pressure intercept in LVF. Contructed with mean data reported in references 50,83,103,106,160,241.
In these studies, Ppam-Q relationships were assumed to be best described by linear approximations, and extrapolated pressure intercepts higher than Ppw or Pla were explained by an increased pulmonary vascular closing pressure (142). Such an interpretation refers to the Starling resistor model of the pulmonary circulation, while, as already mentioned, distensible models might be more realistic. A common feature of Ppa-Q relationships in patients with pulmonary hypertension is that, like in normal subjects, PVR decreases with increased flow (142,155).
Janicki et al. investigated the pressure-flow responses of the pulmonary circulation in patients with left heart failure without and with pulmonary hypertension (defined by a Ppam higher than 19 mmHg), and in patients with various pulmonary vascular diseases (103). The authors were able to record 3 to 11 Ppam-Q coordinates, while patients were exercising with workload increased every 2 minutes up to submaximal or maximal levels. In patients with no pulmonary hypertension, the slope of Ppam-Q was 3 mmHg.min.L−1. In patients with pulmonary hypertension secondary to heart failure the slope increased to 5.1 mmHg.min.L−1, reaching 7.2 mmHg.min.L−1 in patients with pulmonary arterial hypertension (PAH). The changes in extrapolated pressure intercept of Ppam-Q with exercise were variable, with often values below the resting Ppw, and even negative in a few patients. This is physically impossible in any model of the pulmonary circulation, and has to be explained by exercise-induced pulmonary vasoconstriction and/or increased left atrial pressures at the highest levels of exercise.
Kafi et al. compared the effects of exercise to those of an infusion of a low dose of dobutamine assumed to be without effect on pulmonary vascular tone in patients with idiopathic PAH (106). Slopes and extrapolated pressure intercepts of Ppam-Q plots were respectively higher and lower with exercise (Fig 21) which was explained by exercise-induced vasoconstriction related to decreased pH and mixed venous blood PO2 and may be also to sympathetic nervous system activation.
Figure 21.
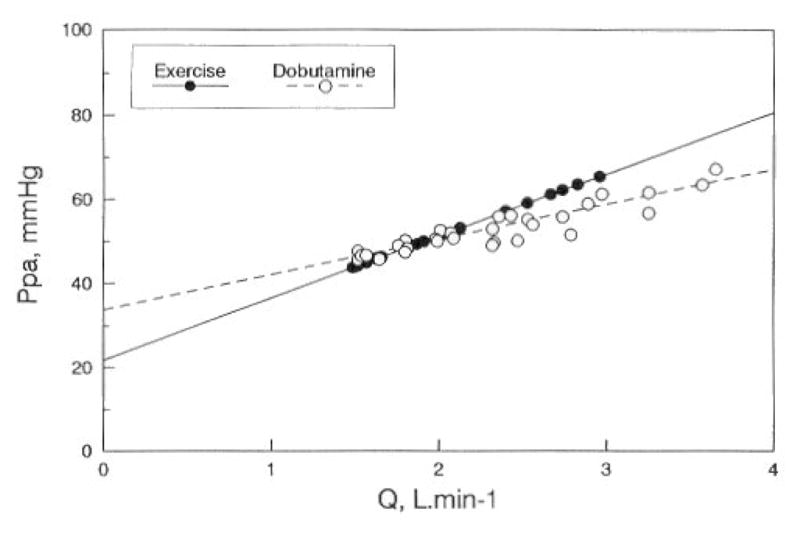
Plots of mean pulmonary artery pressure (Ppa) as a function cardiac output (Q) in patients with idiopathic pulmonary arterial pressure, with Q increased at exercise or by an infusion of low-dose dobutamine. The slope of Ppa-Q was higher with exercise. From reference 106. Permission pending.
Castelain et al. investigated the effects of intravenous epoprostenol in patients with idiopathic PAH (24). Six weeks of treatment with epoprostenol increased exercise capacity as assessed by a 6-minute walk distance, but resting PVR was unchanged. Hemodynamic measurements at exercise showed a decreased slope of Ppam-Q relationships, explaining improved exercise capacity by a decreased RV afterload. The average slope of Ppam-Q decreased from 12 to 7 mmHg.L.min−1.m2, which was highly significant. It may be noted that, because of a dramatic decrease in PVR with exercise, the extrapolated intercepts increased from 18 to 23 mmHg, underscoring the difficulties of the functional interpretation of these extrapolations
One could argue that exercise measurements of Ppam-Q relationships do not allow for valid estimations of RV afterload, because the steady-flow approach neglects the effects of of systolic Ppa or peak RV pressure. However, PVR has been reported to be inversely correlated to pulmonary arterial compliance, with unchanged time constant of the pulmonary circulation, in various types of pulmonary hypertension and therapies (118,119) suggesting limited variability in responses of the functional state of the pulmonary circulation to disease processes. On the other hand, PVR divided by heart rate may be considered as a reasonable approximation of pulmonary arterial elastance (187). Therefore exercise-induced changes in PVR or slopes of multipoint Ppam-Q may provide clinically acceptable approximations of RV afterload.
Exercise in patients with pulmonary hypertension is associated with an increase in Ppw. In the study by Janicki et al, Ppw increased by 14–16 mmHg in patients with heart failure, and by 10 mmHg in patients with pulmonary vascular diseases (103). In heart failure, Ppam closely followed Ppw, while Ppam was less tightly correlated in pulmonary vascular disease. The authors noted that Ppw also increases at exercise in normal subjects, with reported increments up to 5 mmHg in young adults, but to 10–15 mmHg in elderly normal men, highlighting that the limits of normal of Ppw-Q relationships are not exactly known. Nevertheless, tight relationships between Ppam and Ppw are characteristic of left heart failure. After cardiac transplantation in patients with severe left heart failure, pulmonary hypertension, and very high Ppw, the postoperative decrease in Ppw was followed by a proportional decrease in Ppam (160)
In summary, exercise hemodynamic studies in patients with pulmonary hypertension allow for an improved understanding of functional state, exercise capacity and effects of therapeutic interventions. Exercise stress tests provide less information about pulmonary vascular function, as the slope and probably also shape of Ppam-Q relationships are affected by exercise-induced pulmonary vasoconstriction and changes in left atrial pressure. Studies of pulmonary vascular pressure-flow characteristics for different types of pulmonary hypertension would ideally rely on interventions which cause purely passive increases of flow. Whether this might be achieved by low-dose dobutamine, as suggested by previous experimental animal (17) and clinical (106) studies requires further confirmation.
11.2 Exercise stress tests for the detection of latent pulmonary vascular disease
While efficacious targeted therapies have been developed for PAH, disappointment remains about persistently high failure rates and mortality (143). Accordingly, the idea has emerged that earlier administration of these drugs might be more effective, and that exercise stress hemodynamics might detect latent or early pulmonary vascular disease. Invasive right heart catheterization studies at exercise have been reported in high risk patients (112,216), while non invasive stress echocardiographic approaches are being developed for the purpose of larger scale application in lower risk patients (13,31,75,156).
Diagnosis of early pulmonary vascular disease requires an unequivocal definition of limits of normal. At the latest expert consensus conference on pulmonary hypertension, held in Dana Point in 2008, pulmonary hypertension was defined by a Ppam above than 25 mmHg at rest (143). This is higher than the upper limit of normal (111,133,146,161,179). Thus more work is needed to define the range of normal values for Ppam, that have no impact on exercise capacity or survival.
Tolle et al. catheterized patients referred for unexplained dyspnea and suspicion of pulmonary hypertension. The results allowed the authors to correlate exercise-induced pulmonary hypertension, defined by a Ppam higher than 30 mmHg, to dyspnea-fatigue symptoms, supporting the notion of exercise-induced pulmonary hypertension as a disease entity (216). Kovacs et al. catheterized patients with systemic sclerosis, and showed that Ppam higher than 30 mmHg and PVR at the upper limits of normal are associated with decreased exercise capacity (112). Tolle and colleagues identified patterns of Ppam-VO2 (or Q) relationships associated with exercise-induced pulmonary hypertension (216). They found a “take-off” pattern to be characteristic of normal subjects, while patients more often presented with “plateau” patterns. “Take-off” patterns of Ppam-VO2 relationships might reflect increased Ppw at higher levels of exercise, or a progressive increase of the resistive component of exercise, affecting the intrathoracic pressure regimen, with increased pressures caused by active expirations or Valsalva phenomena. Reasons for “plateau” patterns of these relationships are not entirely clear, but might relate to expected slightly curvilinear shape of Ppam-Q curves together with a limitation in the incease of stroke volume. However, Kovacs et al found exercise-induced increase in Ppam to be linearly related to cardiac output (112).
The validation of stress echocardiography for the diagnosis of early pulmonary hypertension is on-going. It becomes clearer that pulmonary vascular pressures have to be related to pulmonary blood flow rather than to workload, and that internal controls are needed to avoid errors on single signal reading (156). It has already been possible to show that patients with systemic sclerosis and no abnormal increase in Ppam at rest may present with excessive increase in the slope of Ppam-Q, a decrease in maximal workload, but similar maximal Ppam compared to controls (100). These results underscore the need for adequate functional evaluation of the pulmonary circulation by pressure-flow relationships.
12. Conclusions and recommendations for further research
Dynamic exercise increases pulmonary blood flow and vascular pressures. The relationships between pulmonary blood flow, pulmonary arterial pressures and pulmonary artery wedge pressure have been characterized in experimental animal preparations and mathematically modeled. The most realistic description of pulmonary vascular pressure-flow relationships is provided by simple distensible models relating pressure, resistance, distensibility and flow. Proportional changes in pulmonary vascular resistance and compliance have been shown, allowing for prediction of a stable time constant of the pulmonary circulation in all circumstances. The understanding of proportional changes in pulmonary artery pressures and pulmonary capillary pressures has been achieved on the basis of measurements of longitudinal distributions of resistances. Valid approaches for the definition of right ventricular afterload have been developed.
However, several important aspects of the relationships between the pulmonary circulation and gas exchange or RV function remain incompletely understood. Areas still open to urgent investigation are listed below:
Improved validation against invasive gold standards of non invasive approaches for the study of pulmonary hemodynamics and right ventricular function.
Definition of the limits of age and gender-related normal of averaged slopes of multipoint Ppam-Q relationships at exercise in normoxic and in hypoxic conditions.
Definition of the limits of normal of derived distensibility in acute and in chronic hypoxia
Characterization of multipoint pulmonary vascular pressure-flow relationships in chronic mountain sickness and cardiac and pulmonary vascular diseases associated with pulmonary hypertension.
Definition of the time constant of the pulmonary circulation at exercise in relation to the time constant at rest.
Definition of expected increase in pulmonary capillary pressure with increased pulmonary artery pressures at high levels of exercise, and relation to gas exchange and exercise-induced lung edema
Measurements of right ventricular function at exercise, and relation to steady or pulsatile pulmonary hemodynamic measurements.
Effects of resistive versus dynamic exercise
Effects of environmental conditions such as immersion or extreme changes in temperature.
Figure 8.
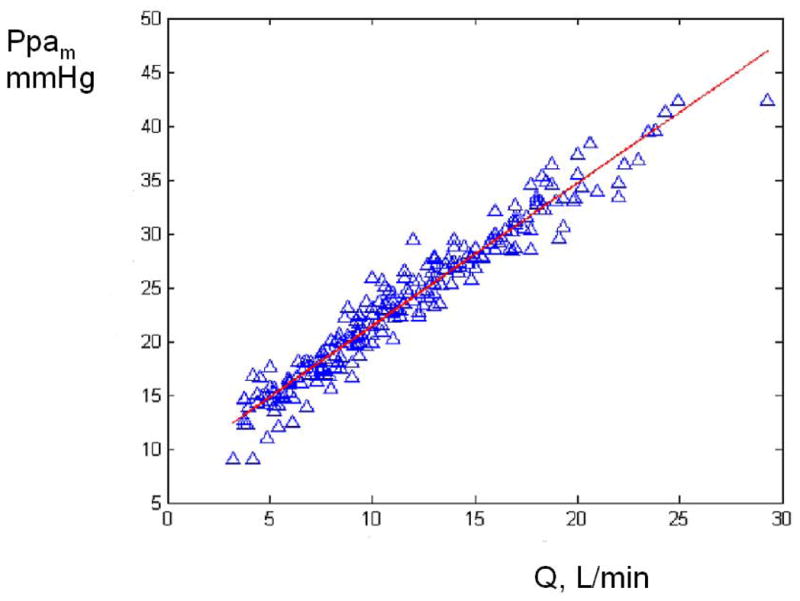
Linear relationship between mean pulmonary artery pressure (Ppam) and cardiac output (Q) in 25 healthy subjects during progressively severe exercise until maximum tolerated. The slope of Ppam-Q was 1.37 mmHg/L.min−1. From reference 4. Permission pending.
Acknowledgments
Constructive and stimulating interactions with N Westerhof contributed greatly to the sections about pulmonary vascular mechanics and right ventricular function.
Extensive discussions and exchange of information with D Penaloza contributed greatly to the sections about the pulmonary circulation in hypoxia.
We also wish to thank Paola Argiento, Diana Bonderman, Andre La Gerche, Daphne Merkus and Massimiliano Mulè for valuable contributions in the preparation of this manuscript.
The technical assistance of Pascale Jespers and Amira Khoulied was greatly appreciated.
Financial support from the Belgium-Luxembourg Fulbright Scholar Program (NCC) and National Institutes of Health grants 1R01HL086939 (NCC) are gratefully acknowledged.
Contributor Information
R NAEIJE, Email: rnaeije@ulb.ac.be, Department of Physiology, Erasme Campus of the Free University of Brussels, CP 604, 808, Lennik road, B-1070 Brussels, BELGIUM, Tel +32 2 5553322, Fax +32 2 5554124.
N CHESLER, Email: chesler@engr.wisc.edu, University of Wisconsin at Madison, 2146 Engineering Centers Building, 1550 Engineering drive, Madison, Wisconsin 53706-1609, USA, Tel +1 608 265 8920, Fax +1 608 265 9239.
References
- 1.Aaron CP, Tandri H, Barr RG, Johnson WC, Bagiella E, Chahal H, Jain A, Kizer JR, Bertoni AG, Lima JA, Bluemke DA, Kawut SM. Physical activity and right ventricular structure and function. The MESA-right ventricle study. Am J Respir Crit Care Med. 2010 doi: 10.1164/rccm.201003-0469OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allemann Y, Rotter M, Hutter D, Lipp E, Sartori C, Scherrer U, Seiler C. Impact of acute hypoxic pulmonary hypertension on LV diastolic function in healthy mountaineers at high altitude. Am J Physiol Heart Circ Physiol. 2004;286:H856–862. doi: 10.1152/ajpheart.00518.2003. [DOI] [PubMed] [Google Scholar]
- 3.Anand I, Malhotra R, Chandrashekhar Y, Bali HK, Chauhan SS, Jindal SK, Bhandari RK, Wahi PL. Adult subacute mountain sickness-a syndrome of congestive heart failure in man at very high altitude. Lancet. 1990;335:561–5. doi: 10.1016/0140-6736(90)90348-9. [DOI] [PubMed] [Google Scholar]
- 4.Argiento P, Chesler N, Mulè M, D’Alto M, Bossone E, Unger P, Naeije R. Exercise stress echocardiography for the study of the pulmonary circulation. Eur Respir J. 2010;35:1273–8. doi: 10.1183/09031936.00076009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Astrand PO, Cuddy TE, Saltin B, Tenberg J. Cardiac output during submaximal and maximal work. J Appl Physiol. 1964;19:268–274. doi: 10.1152/jappl.1964.19.2.268. [DOI] [PubMed] [Google Scholar]
- 6.Banchero N, Sime F, Penaloza D, Cruz J, Gamboa R, Marticorena E. Pulmonary pressure, cardiac output, and arterial oxygen saturation during exercise at high altitude and at sea level. Circulation. 1966;33:249–262. doi: 10.1161/01.cir.33.2.249. [DOI] [PubMed] [Google Scholar]
- 7.Barer GR, Howard P, Shaw JW. Stimulus-response curves for the pulmonary vascular bed to hypoxia and hypercapnia. J Physiol. 1970;21:139–155. doi: 10.1113/jphysiol.1970.sp009271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bärtsch P, Mairbäurl H, Maggiorini M, Swenson E. Physiologic aspects of high-altitude pulmonary edema. J Appl Physiol. 2005;98:1101–1110. doi: 10.1152/japplphysiol.01167.2004. [DOI] [PubMed] [Google Scholar]
- 9.Bernard SL, Glenny RW, Erickson HH, Fedde MR, Polissar N, Basaraba RJ, Hlastala MP. Minimal redistribution of blood flow with exercise in race horses. J Appl Physiol. 1996;81:1062–1070. doi: 10.1152/jappl.1996.81.3.1062. [DOI] [PubMed] [Google Scholar]
- 10.Bevegaard S, Holmgren A, Jonsson B. The effect of body position on the circulation at rest and during exercise, with special reference to the influence of stroke volume. Acta Physiol Scand. 1960;49:279–298. doi: 10.1111/j.1748-1716.1960.tb01953.x. [DOI] [PubMed] [Google Scholar]
- 11.Bevegaard S, Holmgren A, Jonsson B. Circulatory studies in well trained athletes at rest and during heavy exercise, with special reference to stroke volume and the influence of body position. Acta Physiol Scand. 1963;57:26–50. doi: 10.1111/j.1748-1716.1963.tb02572.x. [DOI] [PubMed] [Google Scholar]
- 12.Bonderman D, Martischnig AM, Vonbank K, Nikfardjam M, Meyer B, Heinz G, Naeije R, Lang IM. Reduced exercise capacity after successful pulmonary endarterectomy is due to decreased pulmonary arterial compliance. Chest. 2011;139:122–127. doi: 10.1378/chest.10-0348. [DOI] [PubMed] [Google Scholar]
- 13.Bossone E, Rubenfire M, Bach DS, Ricciardi M, Armstrong WF. Range of tricuspid regurgitation velocity at rest and during exercise in normal adult males: implications for the diagnosis of pulmonary hypertension. J Am Coll Cardiol. 1999;33:1662–1666. doi: 10.1016/s0735-1097(99)00055-8. [DOI] [PubMed] [Google Scholar]
- 14.Boussuges A, Molenat F, Burnet H, Cauchy E, Gardette B, Sainty JM, Jammes Y, Richalet JP. Operation Everest III (Comex ’97): Modifications of cardiac function secondary to altitude-induced hypoxia. Am J Respir Crit Care Med. 2000;161:264–270. doi: 10.1164/ajrccm.161.1.9902096. [DOI] [PubMed] [Google Scholar]
- 15.Brimioulle S, Lejeune P, Vachiéry JL, Delcroix M, Hallemans R, Leeman M, Naeije R. The stimulus-response curve of hypoxic pulmonary vasoconstriction in intact dogs: effects of ASA. J Appl Physiol. 1994;77:476–480. doi: 10.1152/jappl.1994.77.1.476. [DOI] [PubMed] [Google Scholar]
- 16.Brimioulle S, Maggiorini M, Stephanazzi J, Vermeulen F, Lejeune P, Naeije R. Effects of low flow on pulmonary vascular pressure-flow curves and pulmonary vascular impedance. Cardiovasc Res. 1999;42:183–192. doi: 10.1016/s0008-6363(98)00301-0. [DOI] [PubMed] [Google Scholar]
- 17.Brimioulle S, Wauthy P, Ewalenko P, Rondelet B, Vermeulen F, Kerbaul F, Naeije R. Single-beat estimation of right ventricular end-systolic pressure-volume relationship. Am J Physiol Heart Circ Physiol. 2003;284:H1625–1630. doi: 10.1152/ajpheart.01023.2002. [DOI] [PubMed] [Google Scholar]
- 18.Brofman BL, Charms BL, Kohn PM, Elder J, Newman B, Rizika M. Unilateral pulmonary occlusion in man. Control studies. J Thoracic Surg. 1957;34:206–227. [PubMed] [Google Scholar]
- 19.Bryan AC, Bentibaglio LG, Beerel F, MacLeish H, Zidulka A, Bates DV. Factors affecting regional distribution of ventilation and perfusion in the lung. J Appl Physiol. 1964;19:395–402. doi: 10.1152/jappl.1964.19.3.395. [DOI] [PubMed] [Google Scholar]
- 20.Bshouty Z, Younes M. Arterial occlusion versus isofiltration pulmonary capillary pressures during very high flow. J Appl Physiol. 1987;62:1174–1178. doi: 10.1152/jappl.1987.62.3.1174. [DOI] [PubMed] [Google Scholar]
- 21.Bshouty Z, Younes M. Distensibility and pressure-flow relationship of the pulmonary circulation. II. Multibranched model. J Appl Physiol. 1990;68:1514–1527. doi: 10.1152/jappl.1990.68.4.1514. [DOI] [PubMed] [Google Scholar]
- 22.Burnham KJ, Arai TJ, Dubowitz DJ, Henderson AC, Holverda S, Buxton RB, Prisk GK, Hopkins SR. Pulmonary perfusion heterogeneity in increased in sustained heavy exercise in humans. J Appl Physiol. 2009;107:1559–1568. doi: 10.1152/japplphysiol.00491.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calbet JA, Boushel R, Radegran G, Sondergaard H, Wagner PD, Saltin B. Why is VO2 max after altitude acclimatization still reduced despite normalization of arterial O2 content? Am J Physiol Regul Integr Comp Physiol. 2003;284:R304–316. doi: 10.1152/ajpregu.00156.2002. [DOI] [PubMed] [Google Scholar]
- 24.Castelain V, Chemla D, Humbert M, Sitbon O, Simonneau G, Lecarpentier Y, Hervé P. Pulmonary artery pressure-flow relations after prostacyclin in primary pulmonary hypertension. Am J Respir Crit Care Med. 2002;165:338–340. doi: 10.1164/ajrccm.165.3.2106033. [DOI] [PubMed] [Google Scholar]
- 25.Chammas JH, Rickaby DA, Guarin M, Linehan JH, Hanger CC, Dawson CA. Flow-induced vasodilation in the ferret lung. J Appl Physiol. 1997;83:495–502. doi: 10.1152/jappl.1997.83.2.495. [DOI] [PubMed] [Google Scholar]
- 26.Champion HC, Michelakis ED, Hassoun PM. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation. 2009;120:992–1007. doi: 10.1161/CIRCULATIONAHA.106.674028. [DOI] [PubMed] [Google Scholar]
- 27.Chemla D, Castelain V, Humbert M, Hébert JL, Simonneau G, Lecarpentier Y, Hervé P. New formula for predicting mean pulmonary artery pressure using systolic pulmonary artery pressure. Chest. 2004;126:1313–1317. doi: 10.1378/chest.126.4.1313. [DOI] [PubMed] [Google Scholar]
- 28.Christie J, Sheldahl LM, Tristani FE, Sagar KB, Ptacin MJ, Wann S. Determination of stroke volume and cardiac output during exercise: comparison of two-dimensional and Doppler echocardiography, Fick oximetry, and thermodilution. Circulation. 1987;76:539–547. doi: 10.1161/01.cir.76.3.539. [DOI] [PubMed] [Google Scholar]
- 29.Cope DK, Grimbert F, Downey JM, Taylor AE. Pulmonary capillary pressure: A review. Crit Care Med. 1992;20:1043–1056. doi: 10.1097/00003246-199207000-00024. [DOI] [PubMed] [Google Scholar]
- 30.Cournand A, Riley RL, Himmelstein A, Austrial R. Pulmonary circulation and alveolar ventilation-perfusion relationships after pneumonectomy. J Thoracic Surg. 1950;19:80–116. [PubMed] [Google Scholar]
- 31.D’Alto M, Ghio S, D’Andrea A, Pazzano AS, Argiento P, Camporotondo R, Allocca F, Scelsi L, Cuomo G, Caporali R, Cavagna L, Valentini G, Calabrò R. Inappropriate exercise-induced increase inpulmonary artery pressure in patients with systemic sclerosis. Heart. 2011;97:112–117. doi: 10.1136/hrt.2010.203471. [DOI] [PubMed] [Google Scholar]
- 32.de Beer VJ, de Graaff HJD, Hoekstra M, Duncker DJ, Merkus D. Integrated control of pulmonary vascular tone by endothelin and angiotensin II in exercising swine depends on gender. Am J Physiol Heart Circ Physiol. 2010;298:H1976–H1985. doi: 10.1152/ajpheart.00459.2009. [DOI] [PubMed] [Google Scholar]
- 33.Delcroix M, Mélot C, Vachiery JL, Lejeune P, Leeman M, Vanderhoeft P, Naeije R. Effects of embolic size on hemodynamics and gas exchange in canine embolic pulmonary hypertension. J Appl Physiol. 1990;69:2254–2261. doi: 10.1152/jappl.1990.69.6.2254. [DOI] [PubMed] [Google Scholar]
- 34.Dempsey JA. Is the lung build for exercise? Med Sci Sports Exerc. 1986;18:143–155. [PubMed] [Google Scholar]
- 35.Dempsey JA, Hanson P, Henderson K. Exercise-induced alveolar hypoxemia in healthy human subjects at sea level. J Physiol (Lond) 1984;355:161–175. doi: 10.1113/jphysiol.1984.sp015412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dempsey JA, Wagner PD. Exercise-induced arterial hypoxemiua. J Appl Physiol. 1999;87:1997–2006. doi: 10.1152/jappl.1999.87.6.1997. [DOI] [PubMed] [Google Scholar]
- 37.Dexter L, Whittenberger JL, Haynes FW, Goodale WT, Gorlin R, Sawyer CG. Effects of exercise on circulatory dynamics of normal individuals. J Appl Physiol. 1951;3:439–453. doi: 10.1152/jappl.1951.3.8.439. [DOI] [PubMed] [Google Scholar]
- 38.Dorrington KL, Clar C, Young JD, Jonas M, Tansley JG, Robbins PA. Time course of the human pulmonary vascular response to 8 hours of isocapnic hypoxia. Am J Physiol. 1997;273:H1126–1134. doi: 10.1152/ajpheart.1997.273.3.H1126. [DOI] [PubMed] [Google Scholar]
- 39.Douglas PS, O(Toole ML, Hiller WD, Reichek N. Different effects of prolonged exercise on the right and the left ventricles. J Am Coll Cardiol. 1990;15:64–69. doi: 10.1016/0735-1097(90)90176-p. [DOI] [PubMed] [Google Scholar]
- 40.Dripps RD, Comroe JH., Jr The effect of the inhalation of high and low oxygen concentrations on respiration, pulse rate, ballistocardiogram and arterial oxygen saturation (oximeter) of normal individuals. Am J Physiol. 1947;149:277–283. doi: 10.1152/ajplegacy.1947.149.2.277. [DOI] [PubMed] [Google Scholar]
- 41.Dujardin JP, Stone DN, Forcino CD, Paul LT, Pieper HP. Effects of blood volume changes on characteristic impedance of the pulmonary artery. Am J Physiol. 242 doi: 10.1152/ajpheart.1982.242.2.H197. [DOI] [PubMed] [Google Scholar]; Heart Circ Physiol. 1982;(11):H197–H202. [Google Scholar]
- 42.Duncker DJ, Stubenitsky R, Tonino PA, Verdouw PD. Nitric oxide contributes to the regulation of vasomotor tone but does not modulate O2 consumption in exercising swine. Cardiovasc Res. 2000;47:738–748. doi: 10.1016/s0008-6363(00)00143-7. [DOI] [PubMed] [Google Scholar]
- 43.Ekelund LG, Holmgren A. Circulatory and respiratory adaptation during long-term, non-steady state exercise in the sitting position. Acta Physiol Scand. 1964;62:240–255. doi: 10.1111/j.1748-1716.1964.tb03971.x. [DOI] [PubMed] [Google Scholar]
- 44.Ekelund LG, Holmgren A, Ovenfors CO. Heart volume during prolonged exercise in the supine and sitting position. Acta Physiol Scand. 1967;70:88–98. doi: 10.1111/j.1748-1716.1967.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 45.Eldridge MW, Braun RK, Yoneda KY, Walby WF. Effects of altitude and exercise on pulmonary capillary integrity: evidence for subclinical high-altitude pulmonary edema. J Appl Physiol. 2006;100:972–980. doi: 10.1152/japplphysiol.01048.2005. [DOI] [PubMed] [Google Scholar]
- 46.Eldridge MW, Dempsey JA, Haverkamp HC, Lovering AT, Hokanson JS. Exercise-induced intrapulmonary arteriovenous shunting in healthy humans. J Appl Physiol. 2004;97:797–805. doi: 10.1152/japplphysiol.00137.2004. [DOI] [PubMed] [Google Scholar]
- 47.Eldridge MW, Podolsky A, Richardson RS, Johnson DH, Knight DR, Johnson EC, Hopkins SR, Michimata H, Grassi B, Feiner J, Kurdak SS, Bickler PE, Wagner PD, Severinghaus JW. Pulmonary hemodynamic response to exercise in subjects with prior high-altitude pulmonary edema. J Appl Physiol. 1996;81:911–921. doi: 10.1152/jappl.1996.81.2.911. [DOI] [PubMed] [Google Scholar]
- 48.Elkins RC, Milnor WR. Pulmonary vascular response to exercise in the dog. Circ Res. 1971;29:591–599. doi: 10.1161/01.res.29.6.591. [DOI] [PubMed] [Google Scholar]
- 49.Elzinga G, Westerhof N. The effect of an increase in inotropic state and end-diastolic volume on the pumping ability of the feline left heart. Circ Res. 1978;42:620–628. doi: 10.1161/01.res.42.5.620. [DOI] [PubMed] [Google Scholar]
- 50.Even P, Duroux P, Ruff F, Caubarrere I, de Vernejoul P, Brouet G. The pressure/flow relationship of the pulmonary circulation in normal man and in chronic obstructive pulmonary diseases. Effects of muscular exercise. Scand J Respir Dis. 1971;77(suppl):72–76. [PubMed] [Google Scholar]
- 51.Faoro V, Boldingh S, Moreels M, Martinez S, Lamotte M, Unger P, Brimioulle S, Huez S, Naeije R. Bosentan decreases pulmonary vascular resistance and improves exercise capacity in acute hypoxia. Chest. 2009;135:1215–1222. doi: 10.1378/chest.08-2222. [DOI] [PubMed] [Google Scholar]
- 52.Faoro V, Huez S, Giltaire S, Pavelescu A, van Osta A, Moraine JJ, Guenard H, Martinot JB, Naeije R. Effects of acetazolamide on aerobic exercise capacity and pulmonary hemodynamics at high altitudes. J Appl Physiol. 2007;103:1161–1165. doi: 10.1152/japplphysiol.00180.2007. [DOI] [PubMed] [Google Scholar]
- 53.Faoro V, Lamotte M, Deboeck G, Pavelescu A, Huez S, Guenard H, Martinot JB, Naeije R. Effects of sildenafil on exercise capacity in hypoxic normal subjects. High Alt Med Biol. 2007;8:155–163. doi: 10.1089/ham.2007.1058. [DOI] [PubMed] [Google Scholar]
- 54.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- 55.Fesler P, Pagnamenta A, Rondelet B, Kerbaul F, Naeije R. Effects of sildenafil on hypoxic pulmonary vascular function in dogs. J Appl Physiol. 2006;101:1085–1090. doi: 10.1152/japplphysiol.00332.2006. [DOI] [PubMed] [Google Scholar]
- 56.Fischler M, Maggiorini M, Dorschner L, Debrunner J, Bernheim A, Kiencke S, Mairbäurl H, Bloch KE, Naeije R, Brunner-La Rocca HP. Dexamethasone but not tadalafil improves exercise capacity in adults prone to high altitude pulmonary edema. Am J Respir Crit Care Med. 2009;180:346–352. doi: 10.1164/rccm.200808-1348OC. [DOI] [PubMed] [Google Scholar]
- 57.Fishman AP, Mc Clement J, Himmelstein A, Cournand A. Effects of acute anoxia on the circulation and respiration in patients with chronic obstructive pulmonary disease studied during the “steady state”. J Clin Invest. 1952;31:770–781. doi: 10.1172/JCI102662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fowler NO. The normal pulmonary arterial pressure-flow relationships during exercise. Am J Med. 1969;47:1–6. doi: 10.1016/0002-9343(69)90235-6. [DOI] [PubMed] [Google Scholar]
- 59.Fowler NO, Holmes JC. Pulmonary arterial pressure at high pulmonary flow. J Clin Invest. 1965;44:2040–2050. doi: 10.1172/JCI105311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fulco CS, Rock PB, Cymerman A. Maximal and submaximal exercise performance at altitude. Aviat Space Environ Med. 1998;69:793–801. [PubMed] [Google Scholar]
- 61.Fung YC, Sobin SS. Pulmonary alveolar blood flow. Circ Res. 1972;30:470–490. doi: 10.1161/01.res.30.4.470. [DOI] [PubMed] [Google Scholar]
- 62.Gaar KA, Jr, Taylor AE, Owens LJ, Guyton AC. Pulmonary capillary pressure and filtration coefficient in the isolated perfused lung. Am J Physiol. 1967;213:910–914. doi: 10.1152/ajplegacy.1967.213.4.910. [DOI] [PubMed] [Google Scholar]
- 63.Gale GE, Torre-Bueno JR, Moon RE, Saltzman HA, Wagner PD. Ventilation-perfusion inequality in normal humans during exercise at sea level and simulated altitude. J Appl Physiol. 1985;58:978–988. doi: 10.1152/jappl.1985.58.3.978. [DOI] [PubMed] [Google Scholar]
- 64.Geary GG, Buchholz JN. Effects of aging on cerebrovascular tone and [Ca2+]i. J Appl Physiol. 2003;95:1746–1754. doi: 10.1152/japplphysiol.00275.2003. [DOI] [PubMed] [Google Scholar]
- 65.Ghofrani HA, Reichenberger F, Kohstall MG, Mrosek EH, Seeger T, Olschewski H, Seeger W, Grimminger F. Sildenafil increased exercise capacity during hypoxia at low altitudes and at mount Everest base camp: a randomized, double-blind, placebo-controlled crossover trial. Ann Intern Med. 2004;141:169–177. doi: 10.7326/0003-4819-141-3-200408030-00005. [DOI] [PubMed] [Google Scholar]
- 66.Gledhill N, Cox D, Jamnik R. Endurance athletes stroke volume does not plateau: major advantage is diastolic function. Med Sci Sports Exerc. 1994;26:1116–1124. [PubMed] [Google Scholar]
- 67.Gledhill N, Froese AB, Buick FJ, Bryan AC. VA/Q inhomogeneity and AaDO2 in man during exercise: effect of SF6 breathing. J Appl Physiol. 1978;45:512–515. doi: 10.1152/jappl.1978.45.4.512. [DOI] [PubMed] [Google Scholar]
- 68.Granath A, Jonsson B, Strandell T. Circulation in healthy old men, studied by right heart catheterization at rest and during exercise in supine and sitting position. Acta Med Scand. 1964;176:425–446. doi: 10.1111/j.0954-6820.1964.tb00949.x. [DOI] [PubMed] [Google Scholar]
- 69.Granath A, Strandell T. Relationships between cardiac output, stroke volume, and intracardiac pressures at rest and during exercise in supine position and some anthropometric data in healthy old men. Acta Med Scand. 1964;176:447–466. doi: 10.1111/j.0954-6820.1964.tb00950.x. [DOI] [PubMed] [Google Scholar]
- 70.Grant BJ, Davies EE, Jones HA, Hughes JM. Local regulation of pulmonary blood flow and ventilation-perfusion ratios in the coatimundi. J Appl Physiol. 1976;40:216–228. doi: 10.1152/jappl.1976.40.2.216. [DOI] [PubMed] [Google Scholar]
- 71.Grover RF, Wagner WW, McMurtry IF, Reeves JT. Handbook of Physiology. The Cardiovascular System. Peripheral Circulation and Organ Blood Flow. part 1. III. Bethesda MD: Am Physiol Soc sect 2; 1983. Pulmonary Circulation; pp. 103–36. Chap 4. [Google Scholar]
- 72.Groves BM, Droma T, Sutton JR, McCullough RG, McCullough RE, Zhuang J, Rapmund G, Sun S, Janes C, Moore LG. Minimal hypoxic pulmonary hypertension in normal Tibetans at 3,658 m. J Appl Physiol. 1993;74:312–318. doi: 10.1152/jappl.1993.74.1.312. [DOI] [PubMed] [Google Scholar]
- 73.Groves BM, Reeves JT, Sutton JR, Wagner PD, Cymerman A, Malconian MK, Rock PB, Young PM, Houston CS. Operation Everest II: elevated high altitude pulmonary resistance unresponsive to oxygen. J Appl Physiol. 1987;63:521–530. doi: 10.1152/jappl.1987.63.2.521. [DOI] [PubMed] [Google Scholar]
- 74.Grünig E, Mereles D, Hildebrandt W, Swenson ER, Kübler W, Kuecherer H, Bärtsch P. Stress Doppler echocardiography for identification of susceptibility to high altitude pulmonary edema. J Am Coll Cardiol. 2000;35:980–987. doi: 10.1016/s0735-1097(99)00633-6. [DOI] [PubMed] [Google Scholar]
- 75.Grünig E, Weissmann S, Ehlken N, Fijalkowska A, Fischer C, Fourme T, Galié N, Ghofrani A, Harrison RE, Huez S, Humbert M, Janssen B, Kober J, Koehler R, Machado RD, Mereles D, Naeije R, Olschewski H, Provencher S, Reichenberger F, Retailleau K, Rocchi G, Simonneau G, Torbicki A, Trembath R, Seeger W. Stress-Doppler-echocardiography in relatives of patients with idiopathic and familial pulmonary arterial hypertension: results of a multicenter European analysis of pulmonary artery pressure response to exercise and hypoxia. Circulation. 2009;119:1747–57. doi: 10.1161/CIRCULATIONAHA.108.800938. [DOI] [PubMed] [Google Scholar]
- 76.Guazzi M, Tumminello G, Di Marco F, Fiorentini C, Guazzi MD. The Effects of Phosphodiesterase-5 Inhibition with sildenafil on pulmonary hemodynamics and diffusion capacity, exercise ventilatory efficiency, and oxygen uptake kinetics in chronic heart failure. J Am Coll Cardiol. 2004;44:2339–48. doi: 10.1016/j.jacc.2004.09.041. [DOI] [PubMed] [Google Scholar]
- 77.Gurtner HP, Walser P, Fassler B. Normal values for pulmonary hemodynamics at rest and during exercise in man. Progr Resp Res. 1975;9:295–315. [Google Scholar]
- 78.Guyton AC, Lindsay AE. Effects of elevated left atrial pressures and decreased plasma protein in the development of lung edema. Circ Res. 1959;7:649–655. doi: 10.1161/01.res.7.4.649. [DOI] [PubMed] [Google Scholar]
- 79.Halpern SD, Taichman DB. Misclassification of pulmonary hypertension due to reliance on pulmonary capillary wedge pressure rather than left ventricular end-diastolic pressure. Chest. 2009;136:37–43. doi: 10.1378/chest.08-2784. [DOI] [PubMed] [Google Scholar]
- 80.Hammond HK, White FC, Bhargava V, Shabetai R. Heart size and maximal cardiac output are limited by the pericardium. Am J Physiol Heart Circ Physiol. 1992;263:H1675–H1681. doi: 10.1152/ajpheart.1992.263.6.H1675. [DOI] [PubMed] [Google Scholar]
- 81.Hammond MD, Gale GE, Kapitan KS, Reis A, Wagner PD. Pulmonary gas exchange in humans during exercise at sea level. J Appl Physiol. 1986;60:1590–1598. doi: 10.1152/jappl.1986.60.5.1590. [DOI] [PubMed] [Google Scholar]
- 82.Harf A, Pratt T, Hughes JMB. Regional distribution of VA/Q in man at rest and with exercise measured with krypton-81m. J Appl Physiol. 1978;44:115–123. doi: 10.1152/jappl.1978.44.1.115. [DOI] [PubMed] [Google Scholar]
- 83.Harris P, Segel N, Bishop JM. The relation between pressure and flow in the pulmonary circulation of normal subjects and in patients with chronic bronchitis and mitral stenosis. Cardiovasc Res. 1968;2:73–84. doi: 10.1093/cvr/2.1.73. [DOI] [PubMed] [Google Scholar]
- 84.Hasinoff L, Ducas J, Schick U, Prewitt RM. Pulmonary vascular pressure-flow characteristics in canine pulmonary embolism. J Appl Physiol. 1990;68:462–467. doi: 10.1152/jappl.1990.68.2.462. [DOI] [PubMed] [Google Scholar]
- 85.Hecht HH, Kuida H, Lange RL, Thorne JL, Brown AM. Brisket disease. II. Clinical features and hemodynamic observations in altitude-dependent right heart failure of cattle. Am J Med. 1962;32:171–183. doi: 10.1016/0002-9343(62)90288-7. [DOI] [PubMed] [Google Scholar]
- 86.Heidbuchel H, Hoogsteen J, Fagard R, Vanhees L, Ector H, Willems R, Van Lierde J. High prevalence of right ventricular involvement in endurance athletes with ventricular arrythmias. Role of an electrophysiological study in risk stratification. Eur Heart J. 2003;24:1473–1480. doi: 10.1016/s0195-668x(03)00282-3. [DOI] [PubMed] [Google Scholar]
- 87.Hickman JB, Cargill WH. Effect of exercise on cardiac output and pulmonary artery pressure in normal persons and in patients with cardiovascular disease and pulmonary emphysema. J Clin Invest. 1948;27:10–23. doi: 10.1172/JCI101912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Holmgren A, Jonsson B, Sjostrand T. Circulatory data in normal subjects at rest and during exercise in the recumbent position, with special reference to the stroke volume at different working intensities. Acta Physiol Scand. 1960;49:343–363. doi: 10.1111/j.1748-1716.1960.tb01957.x. [DOI] [PubMed] [Google Scholar]
- 89.Hopkins SR. Last word on Point Counterpoint: pulmonary edema does/does not occur in human athletes performing sea level exercise. J Appl Physiol. 2010;109:1281. doi: 10.1152/japplphysiol.01353.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hopkins SR, Bayly WM, Slocombe RF, Wagner H, Wagner PD. Effect of prolonged heavy exercise on exercise gas exchange in horses. J Appl Physiol. 1998;84:1723–1730. doi: 10.1152/jappl.1998.84.5.1723. [DOI] [PubMed] [Google Scholar]
- 91.Hopkins SR, Garg J, Bolar DS, Balouch J, Levin DL. Pulmonary blood flow heterogeneity during hypoxia and high altitude pulmonary edema. Am J Respir Crit Care Med. 2005;171:83–87. doi: 10.1164/rccm.200406-707OC. [DOI] [PubMed] [Google Scholar]
- 92.Hopkins SR, Kleinsasser A, Bernard S, Loeckinger A, Faber E, Neradilek B, Polissar NL, Hlastala MP. Hypoxia has a greater effect than exercise on the distribution of pulmonary blood flow in swine. J Appl Physiol. 2007;103:2112–1254. doi: 10.1152/japplphysiol.00306.2007. [DOI] [PubMed] [Google Scholar]
- 93.Hopkins SR, Levin DL, Emami K, Kadlecek S, Yu J, Ishii M, Rizi RR. Advances in Magnetic Resonance Imaging on lung physiology. J Appl Physiol. 2007;102:1244–1254. doi: 10.1152/japplphysiol.00738.2006. [DOI] [PubMed] [Google Scholar]
- 94.Hopkins SR, Olfert IM, Wagner PD. Point: Exercise-induced intrapulmonary shunting is imaginary. J Appl Physiol. 2009;107:993–994. doi: 10.1152/japplphysiol.91489.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hopkins SR, Stary CM, Falor E, Wagner H, Wagner PD, McKirnan MD. Pulmonary gas exchange during exercise in pigs. J Appl Physiol. 1999;86:93–100. doi: 10.1152/jappl.1999.86.1.93. [DOI] [PubMed] [Google Scholar]
- 96.Hsu AR, Barnholt KE, Grundmann NK, Lin JH, McCullum SW, Friedlander AL. Sildenafil improves cardiac output and exercise performance during acute hypoxia, but not normoxia. J Appl Physiol. 2006;100:2031–2040. doi: 10.1152/japplphysiol.00806.2005. [DOI] [PubMed] [Google Scholar]
- 97.Huez S, Faoro V, Guénard H, Martinot JB, Naeije R. Echocardiographic and tissue Doppler imaging of cardiac adaptation to high altitude in native highlanders versus acclimatized lowlanders. Am J Cardiol. 2009;103 :1605–1609. doi: 10.1016/j.amjcard.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 98.Huez S, Faoro V, Vachiery JL, Unger P, Martinot JB, Naeije R. Images in cardiovascular medicine. High-altitude-induced right-heart failure. Circulation. 2007;6(115):e308–9. doi: 10.1161/CIRCULATIONAHA.106.650994. [DOI] [PubMed] [Google Scholar]
- 99.Huez S, Retailleau K, Unger P, Pavelescu A, Vachiery JL, Derumeaux G, Naeije R. Right and left ventricular adaptation to hypoxia: a tissue Doppler imaging study. Am J Physiol Heart Circ Physiol. 2005;289:H1391–H1398. doi: 10.1152/ajpheart.00332.2005. [DOI] [PubMed] [Google Scholar]
- 100.Huez S, Roufosse F, Vachièry JL, Pavelescu A, Derumeaux G, Wautrecht JC, Cogan E, Naeije R. Isolated right ventricular dysfunction in systemic sclerosis: latent pulmonary hypertension? Eur Respir J. 2007;30:928–936. doi: 10.1183/09031936.00025607. [DOI] [PubMed] [Google Scholar]
- 101.Hultgren HN, Grover RF, Hartly LH. Abnormal circulatory responses to high altitude in subjects with a previous history of high-altitude pulmonary edema. Circulation. 44:759–770. doi: 10.1161/01.cir.44.5.759. [DOI] [PubMed] [Google Scholar]
- 102.Janicki JS. Influence of the pericardium and ventricular interdependence on left ventricular diastolic and systolic function in patients with heart failure. Circulation. 1990;81:III15–III20. [PubMed] [Google Scholar]
- 103.Janicki JS, Weber KT, Likoff MJ, Fishman AP. The pressure-flow response of the pulmonary circulation in patients with heart failure and pulmonary vascular disease. Circulation. 1985;72:1270–1278. doi: 10.1161/01.cir.72.6.1270. [DOI] [PubMed] [Google Scholar]
- 104.Johnson RL, Spicer WS, Bishop JM, Forster RE. Pulmonary capillary blood volume, flow and diffusing capacity during exercise. J Appl Physiol. 1960;15:893–902. doi: 10.1152/jappl.1960.15.5.893. [DOI] [PubMed] [Google Scholar]
- 105.Julien M, Hakim TS, Vahi R, Chang HK. Effect of hematocrit on vascular profile in dog lungs. J Appl Pjysiol. 1985;58:743–748. doi: 10.1152/jappl.1985.58.3.743. [DOI] [PubMed] [Google Scholar]
- 106.Kafi SA, Mélot C, Vachiéry JL, Brimioulle S, Naeije R. Partitioning of pulmonary vascular resistance in primary pulmonary hypertension. J Am Coll Cardiol. 1998;31:1372–1376. doi: 10.1016/s0735-1097(98)00091-6. [DOI] [PubMed] [Google Scholar]
- 107.Kane DW, Tesauro T, Newman JH. Adrenergic modulation of the pulmonary circulation during strenuous exercise in the sheep. Am Rev Respir Dis. 1993;147:1233–1238. doi: 10.1164/ajrccm/147.5.1233. [DOI] [PubMed] [Google Scholar]
- 108.Kawashima A, Kubo K, Kobayashi T, Sekiguchi M. Hemodynamic response to acute hypoxia, hypobaria, and exercise in subjects susceptible to high-altitude pulmonary edema. J Appl Physiol. 1982–1989;67 doi: 10.1152/jappl.1989.67.5.1982. [DOI] [PubMed] [Google Scholar]
- 109.Kobs RW, Muvarak NE, Eickhoff JC, Chesler NC. Linked mechanical and biological aspects of remodelling in mouse pulmonary arteries with hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2005;288:H1209–H2017. doi: 10.1152/ajpheart.01129.2003. [DOI] [PubMed] [Google Scholar]
- 110.Koizumi T, Gupta R, Banerjee M, Newman JH. Changes in pulmonary vascular tone during exercise. Effects of nitric oxide (NO) synthase inhibition, L-arginine infusion and NO inhalation. J Clin Invest. 1994;94:2285–2282. doi: 10.1172/JCI117590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kovacs G, Berghold A, Scheid S, Olschewski H. Pulmonary artery pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34:888–894. doi: 10.1183/09031936.00145608. [DOI] [PubMed] [Google Scholar]
- 112.Kovacs G, Maier R, Aberer E, Brodmann M, Scheidl S, Tröster N, Hesse C, Salmhofer W, Graninger W, Gruenig E, Rubin LJ, Olschewski H. Borderline pulmonary arterial pressure is associated with decreased exercise capacity in scleroderma. Am J Respir Crit Care Med. 2009;180:881–886. doi: 10.1164/rccm.200904-0563OC. [DOI] [PubMed] [Google Scholar]
- 113.Krenz GS, Dawson CA. Flow and pressure distributions in vascular networks consisting of distensible vessels. Am J Physiol Heart Circ Physiol. 2003;284:H2192–H2203. doi: 10.1152/ajpheart.00762.2002. [DOI] [PubMed] [Google Scholar]
- 114.Kuehne T, Yilmaz S, Steendijk P, Moore P, Groenink M, Saaed M, Weber O, Higgins CB, Ewert P, Fleck E, Nagel E, Schulze-Neick I, Lange P. Magnetic resonance imaging analysis of right ventricular pressure-volume loops: in vivo validation and clinical application in patients with pulmonary hypertension. Circulation. 2004;110:2010–2016. doi: 10.1161/01.CIR.0000143138.02493.DD. [DOI] [PubMed] [Google Scholar]
- 115.La Gerche A, Heidbuchel H, Burns AT, Mooney DJ, Taylor AJ, Pfluger HB, Inder WJ, Macisaac AI, Prior DL. Disproportionate exercise load and remodeling of the athlete’s right ventricle. Med Sci Sports Exerc. 2010 doi: 10.1249/MSS.0b013e31820607a3. [DOI] [PubMed] [Google Scholar]
- 116.La Gerche A, MacIsaac AL, Burns AT, Mooney DJ, Inder WJ, Voigt JU, Heidbüchel H, Prior DL. Pulmonary transit of agitated contrast is associated with enhanced pulmonary vascular reserve and right ventricular function at exercise. J Appl Physiol. 2010;109:1307–1317. doi: 10.1152/japplphysiol.00457.2010. [DOI] [PubMed] [Google Scholar]
- 117.La Gerche A, Robberecht C, Kuiperi C, Nuyens D, Willems R, de Ravel T, Matthijs G, Heidbüchel H. Lower than expected desmosomal gene mutation prevalence in endurance athletes with complex ventricular arrhythmias of right ventricular origin. Heart. 2010;96:1268–1274. doi: 10.1136/hrt.2009.189621. [DOI] [PubMed] [Google Scholar]
- 118.Lankhaar JW, Westerhof N, Faes TJ, Gan CT, Marques KM, Boonstra A, van den Berg FG, Postmus PE, Vonk-Noordegraaf A. Pulmonary vascular resistance and compliance stay inversely related during treatment of pulmonary hypertension. Eur Heart J. 2008;29:1688–1695. doi: 10.1093/eurheartj/ehn103. [DOI] [PubMed] [Google Scholar]
- 119.Lankhaar JW, Westerhof N, Faes TJ, Marques KM, Marcus JT, Postmus PE, Vonk-Noordegraaf A. Quantification of right ventricular afterload in patients with and without pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2006;291:H1731–1737. doi: 10.1152/ajpheart.00336.2006. [DOI] [PubMed] [Google Scholar]
- 120.Lategola MT. Pressure-flow relationships in the dog lung during acute subtotal vascular occlusion. Am J Physiol. 1958;192:613–619. doi: 10.1152/ajplegacy.1958.192.3.613. [DOI] [PubMed] [Google Scholar]
- 121.Leeman M, Lejeune P, Closset J, Vachiéry JL, Mélot C, Naeije R. Nature of pulmonary hypertension in canine oleic acid pulmonary edema. J Appl Physiol. 1990;69:293–298. doi: 10.1152/jappl.1990.69.1.293. [DOI] [PubMed] [Google Scholar]
- 122.Lejeune P, Vachiéry JL, Leeman M, Brimioulle S, Hallemans R, Mélot C, Naeije R. Absence of parasympathetic control of pulmonary vascular pressure/flow plots in hyperoxic and hypoxic dogs. Respir Physiol. 1989;78:123–133. doi: 10.1016/0034-5687(89)90046-7. [DOI] [PubMed] [Google Scholar]
- 123.Lenfant C, Sullivan KN. Adaptation to high altitude. N Engl J Med. 1971;284:1298–309. doi: 10.1056/NEJM197106102842305. [DOI] [PubMed] [Google Scholar]
- 124.León-Velarde F, Maggiorini M, Reeves JT, Aldashev A, Asmus I, Bernardi L, Ge RL, Hackett P, Kobayashi T, Moore LG, Penaloza D, Richalet JP, Roach R, Wu T, Vargas E, Zubieta-Castillo G, Zubieta-Calleja G. Consensus statement on chronic and subacute high altitude diseases. High Alt Med Biol. 2005;6:147–157. doi: 10.1089/ham.2005.6.147. [DOI] [PubMed] [Google Scholar]
- 125.Leung H, Wang JJ, Rochtchina E, Tan AG, Wong TY, Klein R, Hubbard LD, Mitchell P. Relationships between Age, Blood Pressure, and Retinal Vessel Diameters in an Older Population. Invest Ophthalmol Vis Sci. 2003;44:2900–2904. doi: 10.1167/iovs.02-1114. [DOI] [PubMed] [Google Scholar]
- 126.Levine BD, Lane LD, Buckey JC, Friedman BD, Blomqvist CG. Left ventricular pressure-volume and Frank-Starling relation in endurance athletes. Implications for orthostatic tolerance and exercise performance. Circulation. 1991;84:1016–1023. doi: 10.1161/01.cir.84.3.1016. [DOI] [PubMed] [Google Scholar]
- 127.Lindenfeld J, Reeves JT, Horwitz LD. Low exercise pulmonary resistance is not dependent on vasodilator prostaglandins. J Appl Physiol. 1983;55:558–561. doi: 10.1152/jappl.1983.55.2.558. [DOI] [PubMed] [Google Scholar]
- 128.Linehan JH, Haworth ST, Nelin LD, Krenz GS, Dawson CA. A simple distensible model for interpreting pulmonary vascular pressure-flow curves. J Appl Physiol. 1992;73:987–994. doi: 10.1152/jappl.1992.73.3.987. [DOI] [PubMed] [Google Scholar]
- 129.Lovering AT, Eldridge MW, Stickland MK. Counterpoint: Exercise-induced intrapulmonary shunting is real. J Appl Physiol. 2009;107:994–998. doi: 10.1152/japplphysiol.91489.2008a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lovering AT, Stickland MK. Not hearing is believing: novel insight into cardiopulmonary function using agitated contrast and ultrasound. J Appl Physiol. 2010;109:1290–1291. doi: 10.1152/japplphysiol.01083.2010. [DOI] [PubMed] [Google Scholar]
- 131.MacDougall JD, McKelvie RS, Moroz DE, Sale DG, McCartney N, Buick F. Factors affecting blood pressure during heavy weight lifting and static contractions. J Appl Physiol. 1992;73:1590–1597. doi: 10.1152/jappl.1992.73.4.1590. [DOI] [PubMed] [Google Scholar]
- 132.Maggiorini M, Brimioulle S, De Canniere D, Delcroix M, Wauthy P, Naeije R. Pulmonary vascular impedance responses to hypoxia in dogs and in minipigs : effects of inhaled nitric oxide. J Appl Physiol. 1995;79:1156–1162. doi: 10.1152/jappl.1995.79.4.1156. [DOI] [PubMed] [Google Scholar]
- 133.Maggiorini M, Mélot C, Gilbert E, Vermeulen F, Naeije R. Nature of pulmonary vascular resistance in dogs and minipigs. Respir Physiol. 1998;111:213–222. doi: 10.1016/s0034-5687(97)00120-5. [DOI] [PubMed] [Google Scholar]
- 134.Maggiorini M, Mélot C, Pierre S, Pfeiffer F, Greve I, Sartori C, Lepori M, Hauser M, Scherrer U, Naeije R. High altitude pulmonary edema is initially caused by an increased capillary pressure. Circulation. 2001;103:2078–2083. doi: 10.1161/01.cir.103.16.2078. [DOI] [PubMed] [Google Scholar]
- 135.Maignan M, Privat C, Leon-Velarde F, Richalet JP, Pham I. Pulmonary pressure and cardiac function in chronic mountain sickness patients. Chest. 2008;135:499–504. doi: 10.1378/chest.08-1094. [DOI] [PubMed] [Google Scholar]
- 136.Manohar M, Goetz TE. L-NAME does not affect exercise-induced pulmonary hypertension in thoroughbred horses. J Appl Physiol. 1998;84:1902–1908. doi: 10.1152/jappl.1998.84.6.1902. [DOI] [PubMed] [Google Scholar]
- 137.Manohar M, Hutchens E, Corney E. Frusemide attenuates the exercise-induced rise in pulmonary capillary blood pressure in horses. Equine Vet J. 1994;26:51–54. doi: 10.1111/j.2042-3306.1994.tb04331.x. [DOI] [PubMed] [Google Scholar]
- 138.Marcus JT, Gan CT, Zwanenburg JJ, Boonstra A, Allaart CP, Götte MJ, Vonk-Noordegraaf A. Interventricular mechanical asynchrony in pulmonary arterial hypertension: left-to-right delay in peak shortening is related to right ventricular overload and left ventricular underfilling. J Am Coll Cardiol. 2008;51:750–757. doi: 10.1016/j.jacc.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 139.Marshall BE, Marshall C. Site and sensitivity for stimulation of hypoxic pulmonary vasoconstriction. J Appl Physiol. 1983;55:711–716. doi: 10.1152/jappl.1983.55.3.711. [DOI] [PubMed] [Google Scholar]
- 140.Marshall BE, Marshall C. A model for hypoxic constriction of the pulmonary circulation. J Appl Physiol. 1988;64:68–77. doi: 10.1152/jappl.1988.64.1.68. [DOI] [PubMed] [Google Scholar]
- 141.Marshall BE, Marshall C, Benumof J, Saidman LJ. Hypoxic pulmonary vasoconstriction in dogs: effects of lung segment size and oxygen tension. J Appl Physiol. 1981;51:1543–1551. doi: 10.1152/jappl.1981.51.6.1543. [DOI] [PubMed] [Google Scholar]
- 142.McGregor M, Sniderman A. On pulmonary vascular resistance: the need for more precise definition. Am J Cardiol. 1985;55:217–221. doi: 10.1016/0002-9149(85)90332-7. [DOI] [PubMed] [Google Scholar]
- 143.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, Mathier MA, McGoon MD, Park MH, Rosenson RS, Rubin LJ, Tapson VF, Varga J, Harrington RA, Anderson JL, Bates ER, Bridges CR, Eisenberg MJ, Ferrari VA, Grines CL, Hlatky MA, Jacobs AK, Kaul S, Lichtenberg RC, Lindner JR, Moliterno DJ, Mukherjee D, Pohost GM, Rosenson RS, Schofield RS, Shubrooks SJ, Stein JH, Tracy CM, Weitz HH, Wesley DJ ACCF/AHA. ACCF/AHA 2009 Expert Consensus Document on Pulmonary Hypertension. A Report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association. Circulation. 2009;119:2250–2294. doi: 10.1161/CIRCULATIONAHA.109.192230. [DOI] [PubMed] [Google Scholar]
- 144.Mélot C, Delcroix M, Lejeune P, Leeman M, Naeije R. Starling resistor versus viscoelastic models for embolic pulmonary hypertension. Am J Physiol. 267 doi: 10.1152/ajpheart.1995.268.2.H817. [DOI] [PubMed] [Google Scholar]; Heart Circ Physiol. 1995;(36):H817–H827. [Google Scholar]
- 145.Mélot C, Naeije R. Pulmonary vascular diseases. Compr Physiol. 2011;1:593–619. doi: 10.1002/cphy.c090014. [DOI] [PubMed] [Google Scholar]
- 146.Mélot C, Naeije R, Hallemans R, Lejeune P, Mols P. Hypoxic pulmonary vasoconstriction and pulmonary gas exchange in normal man. Respir Physiol. 1987;68 :11–27. doi: 10.1016/0034-5687(87)90073-9. [DOI] [PubMed] [Google Scholar]
- 147.Merkus D, de Beer VJ, Houweling B, Duncker DJ. Control of pulmonary vascular tone during exercise in health and pulmonary hypertension. Pharmacol Ther. 2008;119 :242–263. doi: 10.1016/j.pharmthera.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 148.Merkus D, Houweling B, Mirza A, Boomsma F, van den Meiracker AH, Duncker DJ. Contribution of endotheline and its receptors to the regulation of vascular tone during exercise is different in the systemic, coronary and pulmonary circulation. Cardiovasc Res. 2003;59:745–754. doi: 10.1016/s0008-6363(03)00479-6. [DOI] [PubMed] [Google Scholar]
- 149.Merkus D, Houweling B, Zarbanoui A, Duncker DJ. Interaction between prostanoids and nitric oxide in regulation of systemic, pulmonary and coronary vascular tone in exercising swine. Am J Physiol. 2004;286:H1114–H1123. doi: 10.1152/ajpheart.00477.2003. [DOI] [PubMed] [Google Scholar]
- 150.Milnor WR, Bergel DH, Bargainer JD. Hydraulic power associated with pulmonary blood flow and its relation to heart rate. Circ Res. 1966;19:467–480. doi: 10.1161/01.res.19.3.467. [DOI] [PubMed] [Google Scholar]
- 151.Mitzner W, Chang HK. Hemodynamics of the pulmonary circulation. In: Chang HK, Paiva M, editors. Respiratory Physiology: An Analytical Approach. New York: Dekker; 1989. pp. 561–631. The Pulmonary Circulation in Health and Disease. [Google Scholar]
- 152.Motley HL, Cournand A, Werkö L, Himmelstein A, Dresdale D. The influence of short periods of induced acute anoxia upon pulmonary artery pressures in man. Am J Physiol. 1947;150:315–320. doi: 10.1152/ajplegacy.1947.150.2.315. [DOI] [PubMed] [Google Scholar]
- 153.Moussavi N, Czarnecki A, Kumar K, Fallah-Rad N, Lytwyn M, Han SY, Francis A, Walker JR, Kirkpatrick ID, Neilan TG, Sharma S, Jassal DS. Relation of biomarkers and cardiac magnetic resonance imaging after marathon running. Am J Cardiol. 2009;103:1467–1472. doi: 10.1016/j.amjcard.2009.01.294. [DOI] [PubMed] [Google Scholar]
- 154.Murgo JP, Westerhof N. Input impedance of the pulmonary arterial system in normal man. Effects of respiration and comparison to systemic impedance. Circ Res. 1984;54:666–673. doi: 10.1161/01.res.54.6.666. [DOI] [PubMed] [Google Scholar]
- 155.Naeije R. Pulmonary vascular resistance: a meaningless variable? Intens Care Med. 2003;29:526–529. doi: 10.1007/s00134-003-1693-3. [DOI] [PubMed] [Google Scholar]
- 156.Naeije R. In defence of exercise stress tests for the diagnosis of pulmonary hypertension. Heart. 2011;97:94–95. doi: 10.1136/hrt.2010.212126. [DOI] [PubMed] [Google Scholar]
- 157.Naeije R, Faoro V. Pathophysiological insight into bubbles. J Appl Physiol. 2010;107:999–1000. doi: 10.1152/japplphysiol.00660.2009. [DOI] [PubMed] [Google Scholar]
- 158.Naeije R, Huez S, Lamotte M, Retailleau K, Neupane S, Abramowicz D, Faoro V. Pulmonary artery pressure limits exercise capacity at high altitudes. Eur Respir J. 2010;36:1049–1055. doi: 10.1183/09031936.00024410. [DOI] [PubMed] [Google Scholar]
- 159.Naeije R, Lejeune P, Leeman M, Melot C, Closset J. Pulmonary vascular responses to surgical chemodenervation and chemical sympathectomy in dogs. J Appl Physiol. 1989;66:42–50. doi: 10.1152/jappl.1989.66.1.42. [DOI] [PubMed] [Google Scholar]
- 160.Naeije R, Lipski A, Abramowicz M, Lejeune P, Mélot C, Antoine M, De Smet JM, Leclerc JL, Primo G. Nature of pulmonary hypertension in congestive heart failure. Effects of cardiac transplantation. Am J Respir Crit Care Med. 1994;147:881–887. doi: 10.1164/ajrccm.149.4.8143050. [DOI] [PubMed] [Google Scholar]
- 161.Naeije R, Mélot C, Mols P, Hallemans R. Effects of vasodilators on hypoxic pulmonary vasoconstriction in normal man. Chest. 1982;82 :404–410. doi: 10.1378/chest.82.4.404. [DOI] [PubMed] [Google Scholar]
- 162.Naeije R, Mélot C, Niset G, Delcroix M, Wagner PD. Improved arterial oxygenation by a pharmacological increase in chemosensitivity during hypoxic exercise in normal subjects. J Appl Physiol. 1993;74:1666–1671. doi: 10.1152/jappl.1993.74.4.1666. [DOI] [PubMed] [Google Scholar]
- 163.Nagueh S, Middleton K, Kopelen H, Zoghbi W, Quinones M. Doppler tissue imaging: a noninvasive technique for evaluation of left ventricular relaxation and estimation of filling pressures. J Am Coll Cardiol. 1997;30:1527–33. doi: 10.1016/s0735-1097(97)00344-6. [DOI] [PubMed] [Google Scholar]
- 164.Nandiwada PA, Hyman AL, Kadowitz PJ. Pulmonary vasodilator responses to vagal stimulation and acetylcholine in the cat. Circ Res. 1983;53:86–95. doi: 10.1161/01.res.53.1.86. [DOI] [PubMed] [Google Scholar]
- 165.Neilan TG, Januzzi JL, Lee-Lewandowski E, Ton-Nu TT, Yoerger DM, Jassal DS, Lewandrowski KB, Siegel AJ, Marshall JE, Douglas PS, Lawlor D, Picard MH, Wood MJ. Myocardial injury and ventricular dysfunction related to training levels among nonelite participants to the Boston marathon. Circulation. 2006;114:2325–2333. doi: 10.1161/CIRCULATIONAHA.106.647461. [DOI] [PubMed] [Google Scholar]
- 166.Nelin LD, Krenz GS, Rickaby DA, Linehan JH, Dawson CA. A distensible vessel model applied to hypoxic pulmonary vasoconstriction in the neonatal pig. J Appl Physiol. 1992;73:987–994. doi: 10.1152/jappl.1993.74.5.2049. [DOI] [PubMed] [Google Scholar]
- 167.Newman JH, Butka BJ, Brigham KL. Thromboxane A2 and prostacyclin do not modulate pulmonary hemodynamics during exercise in sheep. J Appl Physiol. 1986;61:1706–1711. doi: 10.1152/jappl.1986.61.5.1706. [DOI] [PubMed] [Google Scholar]
- 168.Newman JH, Cochran CP, Roselli RJ, Parker RE, King LS. Pressure and flow changes in the pulmonary circulation of exercising sheep: evidence for elevated microvascular pressure. Am Rev Respir Dis. 1993;147:921–926. doi: 10.1164/ajrccm/147.4.921. [DOI] [PubMed] [Google Scholar]
- 169.Overbeek MJ, Lankhaar JW, Westerhof N, Voskuyl AE, Boonstra A, Bronzwaer JG, Marques KM, Smit EF, Dijkmans BA, Vonk-Noordegraaf A. Right ventricular contractility in systemic sclerosis-associated and idiopathic pulmonary arterial hypertension. Eur Respir J. 2008;31:1160–1166. doi: 10.1183/09031936.00135407. [DOI] [PubMed] [Google Scholar]
- 170.Pace JB. Sympathetic control of pulmonary vascular impedance in anesthetized dogs. Circ Res. 1971;29 :555–567. doi: 10.1161/01.res.29.5.555. [DOI] [PubMed] [Google Scholar]
- 171.Patterson SW, Piper H, Starling EH. The regulation of the heart beat. J Physiol (London) 1914;48:465–513. doi: 10.1113/jphysiol.1914.sp001676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Penazola D, Arias-Stella J. The heart and pulmonary circulation at high altitudes. Healthy highlanders and chronic mountain sickness. Circulation. 2007;115:1132–1146. doi: 10.1161/CIRCULATIONAHA.106.624544. [DOI] [PubMed] [Google Scholar]
- 173.Penaloza D, Sime F. Chronic cor pulmonale due to loss of acclimatization (chronic mountain sickness) Am J Med. 1971;50:728–743. doi: 10.1016/0002-9343(71)90181-1. [DOI] [PubMed] [Google Scholar]
- 174.Permutt S, Bromberger-Barnea B, Bane HN. Alveolar pressure, pulmonary venous pressure and the vascular waterfall. Med Thorac. 1962;19:239–260. doi: 10.1159/000192224. [DOI] [PubMed] [Google Scholar]
- 175.Podolsky A, Eldridge MW, Richardson RS, Knight DR, Johnson EC, Hopkins SR, Johnson DH, Michimata H, Grassi B, Feiner J, Kurdak SS, Bickler PE, Severinghaus JW, Wagner PD. Exercise-induced VA/Q inequality in subjects with prior high-altitude pulmonary edema. J Appl Physiol. 1996;81:922–932. doi: 10.1152/jappl.1996.81.2.922. [DOI] [PubMed] [Google Scholar]
- 176.Poon CS. Analysis of linear and mildly nonlinear relationships using pooled subject data. J Appl Physiol. 1988;64:854–858. doi: 10.1152/jappl.1988.64.2.854. [DOI] [PubMed] [Google Scholar]
- 177.Quaife RA, Chen MY, Lynch D, Badesch DB, Groves RM, Wolfel E, Robertson AD, Bristow MR, Voelkel NF. Importance of right ventricular end-systolic regional wall stress in idiopathic pulmonary hypertension: a new method for estimation of right ventricular wall stress. Eur J Med Res. 2006;11:214–220. [PubMed] [Google Scholar]
- 178.Reeves JT, Dempsey JA, Grover RF. Pulmonary circulation during exercise. In: Weir EK, Reeves JT, editors. Pulmonary Vascular Physiology and Physiopathology. chap 4. New York: Marcel Dekker; 1989. pp. 107–133. [Google Scholar]
- 179.Reeves JT, Groves BM. Approach to the patient with pulmonary hypertension. In: Weir EK, Reeves JT, editors. Pulmonary Hypertension. Futura; Mt Kisco, NY: pp. 1–44. [Google Scholar]
- 180.Reeves JT, Groves BM, Cymerman A, Sutton JR, Wagner PD, Turkevich D, Houston CS. Operation Everest II: cardiac filling pressures during cycle exercise at sea level. Respir Physiol. 1990;80:147–154. doi: 10.1016/0034-5687(90)90078-d. [DOI] [PubMed] [Google Scholar]
- 181.Reeves JT, Groves BM, Sutton JR, Wagner PD, Cymerman A, Malconian MK, Rock PB, Young PM, Houston CS. Operation Everest II: preservation of cardiac function at extreme altitude. J Appl Physiol. 1987;63:531–539. doi: 10.1152/jappl.1987.63.2.531. [DOI] [PubMed] [Google Scholar]
- 182.Reeves JT, Linehan JH, Stenmark KR. Distensibility of the normal human lung circulation during exercise. Am J Physiol Lung Cell Mol Physiol. 2005;288:L419–25. doi: 10.1152/ajplung.00162.2004. [DOI] [PubMed] [Google Scholar]
- 183.Reeves JT, Taylor AE. Pulmonary hemodynamics and fluid exchange in the lungs at exercise. Handbook of Physiology. 1999;Section 12:585–613. [Google Scholar]
- 184.Riley RL, Shepard RH, Cohn JE, Carroll DG, Armstrong BW. Maximal diffusing capacity of the lungs. J Appl Physiol. 1954;10:573–587. doi: 10.1152/jappl.1954.6.10.573. [DOI] [PubMed] [Google Scholar]
- 185.Riley RL, Himmelstein A, Motley HL, Weiner H, Cournand A. Studies of the pulmonary circulation at rest and during exercise in normal individuals and in patients with chronic pulmonary disease. Am J Physiol. 1948;152:372–382. doi: 10.1152/ajplegacy.1948.152.2.372. [DOI] [PubMed] [Google Scholar]
- 186.Rowland T. Echocardiography and circulatory response to progressive endurance exercise. Sports Med. 2008;38:541–551. doi: 10.2165/00007256-200838070-00002. [DOI] [PubMed] [Google Scholar]
- 187.Sagawa K, Maughan L, Suga H, Sunagawa K. Cardiac contraction and the pressure-volume relationship. Oxford University Press; New York: 1988. [Google Scholar]
- 188.Sancetta SM, Rakita L. Response of pulmonary artery pressure and total pulmonary resistance of untrained, convalescent man to prolonged mild steady state exercise. J Clin Invest. 1957;36:1138–1149. doi: 10.1172/JCI103510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Saouti N, Westerhof N, Helderman F, Marcus JT, Stergiopulos N, Westerhof BE, Boonstra A, Postmus PE, Vonk-Noordegraaf A. RC time constant of single lung equals that of both lungs together: a study in chronic thromboembolic pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2009;297:H2154–160. doi: 10.1152/ajpheart.00694.2009. [DOI] [PubMed] [Google Scholar]
- 190.Saouti N, Westerhof N, Helderman F, Marcius JT, Boonstra A, Postmus PE, Vonk Noordegraaf A. Right ventricular oscillatory power is a constant fraction of total power irrespective of pulmonary artery pressure. Am J Respir Crit Care Med. 2010;182:1315–1320. doi: 10.1164/rccm.200910-1643OC. [DOI] [PubMed] [Google Scholar]
- 191.Schaffartzik WD, Polle DC, Derion T, Tsukimoto K, Mathieu CO, Wagner PD. Pulmonary interstitial edema in the pig after heavy exercise. J Appl Physiol. 1992;72:1657–1667. doi: 10.1152/jappl.1993.75.6.2535. [DOI] [PubMed] [Google Scholar]
- 192.Scharhag J, Schneider G, Urhausen A, Rochette V, Kramann B, Kindermann W. Athlete’s heart: right and left ventricular mass and function in male endurance athletes and untrained individuals determined by magnetic resonance imaging. J Am Coll Cardiol. 2002;40:1856–1863. doi: 10.1016/s0735-1097(02)02478-6. [DOI] [PubMed] [Google Scholar]
- 193.Slife DM, Latham RD, Sipkema P, Westerhof N. Pulmonary arterial compliance at res and at exercise in normal humans. Am J Physiol. 258 doi: 10.1152/ajpheart.1990.258.6.H1823. [DOI] [PubMed] [Google Scholar]; Heart Circ Physiol. 1990;(27):H1823–H1828. [Google Scholar]
- 194.Slonim NB, Ravin A, Balchum OJ, Dressler RH. The effect of mild exercise in the supine position on the pulmonary arterial pressure of five normal human subjects. J Clin Invest. 1954;33:1022–1030. doi: 10.1172/JCI102969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sobin SS, Fung YC, Tremer HM, Rosenquist TH. Elasticity of the pulmonary alveolar microvascular sheet in the cat. Circ Res. 1972;30:440–450. doi: 10.1161/01.res.30.4.440. [DOI] [PubMed] [Google Scholar]
- 196.Sobin SS, Lindal RG, Fung YC, Tremer HM. Elasticity of the smallest noncapillary pulmonary blood vessels in the cat. Microvasc Res. 1978;15:57–68. doi: 10.1016/0026-2862(78)90005-5. [DOI] [PubMed] [Google Scholar]
- 197.Stamler JS, Loh E, Reddy MA, Creager MA. Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation. 1994;89:2035–40. doi: 10.1161/01.cir.89.5.2035. [DOI] [PubMed] [Google Scholar]
- 198.Stickland MK, Lovering AT. Exercise-induced intrapulmonary arteriovenous shunting and pulmonary gas exchange. Exerc Sport Sci Rev. 2006;34:99–106. doi: 10.1249/00003677-200607000-00003. [DOI] [PubMed] [Google Scholar]
- 199.Stickland MK, Welsh RC, Haykowsky MJ, Petersen SR, Anderson WD, Taylor DA, Bouffard M, Jones RL. Intrapulmonary shunt and pulmonary gas exchange during exercise in humans. J Physiol. 2004;561:321–329. doi: 10.1113/jphysiol.2004.069302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Stickland MK, Welsh RC, Haykowsky MJ, Peterson SR, Anderson WD, Taylor DA, Bouffart M, Jones RL. Effect of acute increases in pulmonary vascular pressures on exercise pulmonary gas exchange. J Appl Physiol. 2006;100:1910–1917. doi: 10.1152/japplphysiol.01484.2005. [DOI] [PubMed] [Google Scholar]
- 201.Stickland MK, Welsh RC, Petersen SR, Tyberg JV, Anderson WD, Jones RL, Taylor DA, Bouffard M, Haykowsky MJl. Does fitness level modulate the cardiovascular hemodynamic response to exercise? J Appl Physiol. 2006;100:1895–901. doi: 10.1152/japplphysiol.01485.2005. [DOI] [PubMed] [Google Scholar]
- 202.Stray-Gunderson J, Musch TI, Haidot GC, Swain DP, Ordway GA, Mitchell JH. The effects of pericardiectomy on maximal oxygen consumption and maximal cardiac output in untrained dogs. Circ Res. 1986;58:523–530. doi: 10.1161/01.res.58.4.523. [DOI] [PubMed] [Google Scholar]
- 203.Stubenetsky R, Verdouw PD, Duncker DJ. Autonomic control of cardiovascular performance and whole body O2 delivery and utilization in swine during treadmill exercise. Cardiovasc Res. 1999;39:459–474. doi: 10.1016/s0008-6363(98)00102-3. [DOI] [PubMed] [Google Scholar]
- 204.Stuber T, Sartori C, Schwab M, Jayet PY, Rimoldi SF, Garcin S, Thalmann S, Spielvogel H, Salmòn CS, Villena M, Scherrer U, Allemann Y. Exaggerated pulmonary hypertension during mild exercise in chronic mountain sickness. Chest. 2010;137:288–292. doi: 10.1378/chest.09-1355. [DOI] [PubMed] [Google Scholar]
- 205.Suarez J, Alexander JK, Houston CS. Enhanced left ventricular systolic performance at high altitude during Operation Everest II. Am J Cardiol. 1987;60:137–142. doi: 10.1016/0002-9149(87)91000-9. [DOI] [PubMed] [Google Scholar]
- 206.Suga H, Sagawa K, Shoukas AA. Load independence of the instantaneous pressure-volume ratio of the canine left ventricle and the effect of epinephrine and heart rate on the ratio. Circ Res. 1973;32:314–322. doi: 10.1161/01.res.32.3.314. [DOI] [PubMed] [Google Scholar]
- 207.Sui G, Liu Y, Cheng X, Anand IS, Harris E, Harris P, Heath D. Subacute infantile mountain sickness. J Pathol. 1988;155 :161–170. doi: 10.1002/path.1711550213. [DOI] [PubMed] [Google Scholar]
- 208.Sullivan MJ, Cobb FR, Higginbotham MB. Stroke volume increases by similar mechanisms during upright exercise in men and women. Am J Cardiol. 1991;67:1405–1412. doi: 10.1016/0002-9149(91)90472-w. [DOI] [PubMed] [Google Scholar]
- 209.Sunagawa K, Maughan WL, Sagawa K. Optimal arterial resistance for the maximal stroke work studied in the isolated canine left ventricle. Circ Res. 1985;56:586–595. doi: 10.1161/01.res.56.4.586. [DOI] [PubMed] [Google Scholar]
- 210.Sunagawa K, Yamada A, Senda Y, Kikuchi Y, Nakamura M, Shibahara T. Estimation of the hydromotive source pressure from ejecting beats of the left ventricle. IEEE Trans Biomed Eng. 1980;57:299–305. doi: 10.1109/TBME.1980.326737. [DOI] [PubMed] [Google Scholar]
- 211.Sylvester JT, Harabin AL, Peake MD, Frank RS. Vasodilator and constrictor responses to hypoxia in isolated pig lungs. J Appl Physiol. 1980;49:820–825. doi: 10.1152/jappl.1980.49.5.820. [DOI] [PubMed] [Google Scholar]
- 212.Syyed R, Reeves JT, Welsh D, Raeside D, Johnson MK, Peacock AJ. The relationship between the components of pulmonary artery pressure remains constant under all conditions in both health and disease. Chest. 2008;133:633–639. doi: 10.1378/chest.07-1367. [DOI] [PubMed] [Google Scholar]
- 213.Tabima DM, Chesler NC. The effects of vasoactivity and hypoxic pulmonary hypertension on extralobar pulmonary artery biomechanics. J Biomech. 2010;43:1864–1869. doi: 10.1016/j.jbiomech.2010.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Talreja DR, Nishimura RA, Oh JK. Estimation of left ventricular filling pressure with exercise by Doppler echocardiography in patients with normal systolic function: a simultaneous echocardiographic-cardiac catheterization study. J Am Soc Echocardiogr. 2007;20:477–79. doi: 10.1016/j.echo.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 215.Thadani U, Parker JO. Hemodynamics at rest and during supine and sitting exercise in normal subjects. Am J Cardiol. 1978;41:52–59. doi: 10.1016/0002-9149(78)90131-5. [DOI] [PubMed] [Google Scholar]
- 216.Tolle JJ, Waxman AB, Van Horn TL, Pappagianopoulos PP, Systrom DM. Exercise-induced pulmonary arterial hypertension. Circulation. 2008;118:2183–2189. doi: 10.1161/CIRCULATIONAHA.108.787101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tonelli AR, Mubarak KK, Li N, Carrie R, Alnuaimat H. Effect of balloon inflation volume on pulmonary artery occlusion pressure in patients with and without pulmonary hypertension. Chest. 2011;139:115–121. doi: 10.1378/chest.10-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Trivax JE, Franklin BA, Goldstein JA, Chinnaiyan KM, Gallagher MJ, de Jong AT, Colar JM, Haines DE, McCullough PA. Acute effects of marathon running. J Appl Physiol. 2010;108:1148–1153. doi: 10.1152/japplphysiol.01151.2009. [DOI] [PubMed] [Google Scholar]
- 219.Tuchscherer HA, Webster EB, Chesler NC. Pulmonary vascular resistance and impedance in isolated mouse lungs: effects of pulmonary emboli. Ann Biomed Eng. 2006;34:660–668. doi: 10.1007/s10439-005-9050-z. [DOI] [PubMed] [Google Scholar]
- 220.Vanderpool RR, Naeije R, Chesler NC. Impedance in isolated mouse lungs for the determination of site of action of vasoactive agents and disease. Ann Biomed Eng. 2010;38:1854–1861. doi: 10.1007/s10439-010-9960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Varnauskas E, Korsgren M, Hoak P, Prerovsky I. Studies in hypertensive cardiovascular disease. Scand J Clin Lab Invest suppl. 1965;86:100–117. [Google Scholar]
- 222.Viswanathan R, Jain SK, Subramanian S, Subramanian TAV, Dua GL, Giri J. Pulmonary edema of high altitude. II. Clinical, aerohemodynamic and biochemical studies in a group with history of pulmonary edema of high altitude. Am Rev Respir Dis. 1969;100:334–349. doi: 10.1164/arrd.1969.100.3.334. [DOI] [PubMed] [Google Scholar]
- 223.Vogel JHK, Weaver WF, Rose RL, Blount SG, Grover RF. Pulmonary hypertension on exertion in normal man living at 10150 feet (Leadville, Colorado) Med Thorac. 1962;19:269–285. doi: 10.1159/000192254. [DOI] [PubMed] [Google Scholar]
- 224.Wagner PD. Reduced maximal cardiac output at altitude. Mechanisms and significance. Respir Physiol. 2000;120:1–11. doi: 10.1016/s0034-5687(99)00101-2. [DOI] [PubMed] [Google Scholar]
- 225.Wagner PD, Araoz M, Boushel R, Calbet JA, Jessen B, Rådegran G, Spielvogel H, Søndegaard H, Wagner H, Saltin B. Pulmonary gas exchange and acid-base state at 5,260 m in high-altitude Bolivians and acclimatized lowlanders. J Appl Physiol. 2002;92:1393–1400. doi: 10.1152/japplphysiol.00093.2001. [DOI] [PubMed] [Google Scholar]
- 226.Wagner PD, Gale GE, Moon RE, Torre-Bueno J, Stolpe BW, Saltzman HA. Pulmonary gas exchange in humans at sea level and simulated altitude. J Appl Physiol. 1986;61:260–270. doi: 10.1152/jappl.1986.61.1.260. [DOI] [PubMed] [Google Scholar]
- 227.Wagner PD, Gillespie JR, Landgren GL, Fedde MR, Jones BW, de Bowes RM, Peischel RL, Erickson HH. Mechanisms of exercise-induced hypoxemia in horses. J Appl Physiol. 1989;66:1227–1233. doi: 10.1152/jappl.1989.66.3.1227. [DOI] [PubMed] [Google Scholar]
- 228.Wagner PD, Sutton JR, Reeves JT, Cymerman A, Groves BM, Malconian MK. Operation Everest II : pulmonary gas echange during a simulated ascent of Mt Everest. J Appl Physiol. 1987;63:2348–2359. doi: 10.1152/jappl.1987.63.6.2348. [DOI] [PubMed] [Google Scholar]
- 229.Wauthy P, Pagnamenta A, Vassali F, Brimioulle S, Naeije R. Right ventricular adaptation to pulmonary hypertension. An interspecies comparison. Am J Physiol Heart Circ Physiol. 2004;286:H1441–H1447. doi: 10.1152/ajpheart.00640.2003. [DOI] [PubMed] [Google Scholar]
- 230.Weir EK, Archer SL. The mechanism of acute hypoxic pulmonary vasoconstriction: the tale of two channels. FASEB J. 1995;9:183–189. doi: 10.1096/fasebj.9.2.7781921. [DOI] [PubMed] [Google Scholar]
- 231.West JB. The physiological basis of high altitude diseases. Ann Intern Med. 2004;141:789–800. doi: 10.7326/0003-4819-141-10-200411160-00010. [DOI] [PubMed] [Google Scholar]
- 232.West JB, Dollery CT, Naimark A. Distribution of blood flow in isolated lung: relation to vascular and alveolar pressures. J Appl Physiol. 1964;19:713–724. doi: 10.1152/jappl.1964.19.4.713. [DOI] [PubMed] [Google Scholar]
- 233.West JB, Mathieu-Costello O, Jones JH, Birks EK, Logemann RB, Pascoe JR, Tyler WS. Stress failure of pulmonary capillaries in race horses with exerecise-induced pulmonary hemorrhage. J Appl Physiol. 1993;75:1097–1109. doi: 10.1152/jappl.1993.75.3.1097. [DOI] [PubMed] [Google Scholar]
- 234.West JB, Tsukimoto K, Mathieu-Costello O, Prediletto R. Stress failure in pulmonary capillaries. J Appl Physiol. 1991;70:1731–1742. doi: 10.1152/jappl.1991.70.4.1731. [DOI] [PubMed] [Google Scholar]
- 235.Westerhof N, Stergiopulos N, Noble MIM. Snapshots of hemodynamics an aid for clinical research and graduate education. 2. New York, NY: Springer; 2010. [Google Scholar]
- 236.Williams MH., Jr Relationships between pulmonary artery pressure and blood flow in the dog lung. Am J Physiol. 1954;179:243–245. doi: 10.1152/ajplegacy.1954.179.2.243. [DOI] [PubMed] [Google Scholar]
- 237.Yagi H, Yamada H, Kobayashi T, Sekiguchi M. Doppler assessment of pulmonary hypertension induced by hypoxic breathing in subjects susceptible to high altitude pulmonary edema. Am Rev Respir Dis. 1990;142:796–801. doi: 10.1164/ajrccm/142.4.796. [DOI] [PubMed] [Google Scholar]
- 238.Yen RT, Fung YC. Model experiments on apparent blood viscosity and hematocrit in pulmonary alveoli. J Appl Physiol. 1973;35:510–517. doi: 10.1152/jappl.1973.35.4.510. [DOI] [PubMed] [Google Scholar]
- 239.Yen RT, Fung YC, Bingham N. Elasticity of small pulmonary arteries in the cat. J Biomech Eng. 1980;102:170–177. doi: 10.1115/1.3138218. [DOI] [PubMed] [Google Scholar]
- 240.Yock P, Popp R. Noninvasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation. Circulation. 1984;70:657–662. doi: 10.1161/01.cir.70.4.657. [DOI] [PubMed] [Google Scholar]
- 241.Zapol WM, Snider MT. Pulmonary hypertension in severe acute respiratory failure. N Engl J Med. 1977;296:476–480. doi: 10.1056/NEJM197703032960903. [DOI] [PubMed] [Google Scholar]
- 242.Zhuang FY, Fung YC, Yen RT. Analysis of blood flow in cat’s lung with detailed anatomical and elasticity data. J Appl Physiol. 1983;55:1341–1348. doi: 10.1152/jappl.1983.55.4.1341. [DOI] [PubMed] [Google Scholar]