

Abstract

Synthesis and preliminary biological evaluation of a 35-member library of bistramide A stereoisomers are reported. All eight stereoisomers of the C1-C13 tetrahydropyran fragment of the molecule were prepared utilizing crotylsilane reagents 9 and 10 in our [4+2]-annulation methodology. In addition, the four isomers of the C14-C18 γ-amino acid unit were accessed via a Lewis acid mediated crotylation reaction using both enantiomers of organosilane 11. The spiroketal subunit of bistramide A was modified at the C39-alcohol to give another point of stereochemical diversification. The fragments were coupled using standard peptide coupling protocol to provide 35 stereoisomers of the natural product. These stereochemical analogs were screened for their effects on cellular actin and cytotoxicity against cancer cell lines (UO-31 renal and SF-295 CNS). The results of these assays identified one analog, 1.21, with enhanced potency relative to the natural product, bistramide A.
Introduction
Actin is a cytoskeletal protein that plays a crucial role in maintenance of cell shape and other crucial processes including motility, cytokinesis, phagocytosis and intracellular transport. These functions are important for normal cell viability as well as for the aberrant processes vital to cancer cell growth, invasion, and metastasis. In order to function properly, actin is actively switched in a dynamic equilibrium between the G-actin and F-actin, both targets of natural products.1 The complex nature of actin regulation is under active investigation, and cell signaling pathways that include the small GTPases Rho, Rac and Cdc42 have been found to play coordinated, important roles in the assembly and disassembly of actin-rich structures such as stress fibers, lamellipodia, filopodia and focal adhesion complexes.1
While drugs that directly modulate actin have not progressed to the stage of use as clinical cancer therapeutics, actin inhibitors have demonstrated activity in many in vitro cancer models. In addition, actin inhibitors have been extremely useful in elucidating the role of actin in cellular processes. Natural products, in particular, have been a rich source of actin inhibitors, and many have been co-crystallized with actin.2 The cytochalasins and phalloidins bind to F-actin,2 while G-actin modulators include latrunculins, mycalolide B, jasplakinolide, dolastatin 11, aplyronine A, swinholide A and bistramide A.3 Of the latter group, bistramide A is unique in its binding mode to actin. Bistramide A binds and sequesters G-actin and thus inhibits actin filament formation, thereby promoting filament disruption.4 A high-resolution X-ray crystallographic structure of the bistramide A-actin complex showed its binding site overlaps only slightly with that of other G-actin inhibitors. 5 Thus, bistramide A is a novel actin inhibitor worthy of further investigation.
Stereochemically complex natural products provide an opportunity to discover novel biologically relevant molecules. Variation of chirality at selected stereocenters presents a method for diversifying natural products as library templates or scaffolds.6 Each member of a stereoisomer library should provide a unique conformational profile that could have a substantial effect on the biological activity. It is well documented that a conformation of a molecule often modulates specific interactions with its biological target, either to enhance or attenuate biological activity. The so-called stereostructure/activity relationships (SSAR) have only recently gained attention with respect to complex natural products.6b Our group has developed and explored the utility of chiral organosilane reagents to prepare stereochemically well-defined and diversified intermediates for natural product synthesis. Having access to stereochemical iterations of these building blocks presents the opportunity to construct multiple conformations of complex natural products. In this article, we report preparation and use of these chiral reagents to synthesize, in solution phase, a focused 35-member library of the marine metabolite bistramide A 1.1, and the associated biology of these new compounds.
In 1988, Verbist and co-workers isolated the highly potent antiproliferative agent bistramide A, 1.1, from the marine ascidian Lissoclinum bistratum.7 Four additional congeners of this class of natural products were reported by the same research group in 1994.8 The structure of bistramide A was originally proposed to be a 19-membered macrolactam9 but later revised to a linear carbon framework (Scheme 1).10 The skeletal architecture of bistramide A consists of a substituted tetrahydropyran and a spiroketal subunit joined at the center by a γ-amino acid linker. Subsequent spectroscopic and computational studies allowed for accurate prediction of relative and absolute configurations of bistramides.11 Kozmin reported the first total synthesis of bistramide A in 2004, which confirmed the proposed stereostructure of the natural product.12 To date, there are several reports detailing fragment,11b,13 degradative11a,14 and total syntheses15 of bistramides A-D and K.
Scheme 1.

Structures of bistramides A–D and K.
In addition to the structural9,10,11 and synthetic challenges posed by this family of natural products, numerous reports describing their diverse bioactivity have recently emerged.16 Bistramides were shown to exhibit various biological activities, encompassing neurotoxic,7b antiproliferative16d and cytotoxic properties.7b,8 Specifically, bistramide A displayed activity against a number of tumor cell lines including KB, P388, P388/dox, B16, HT29, and NSCLC-N6 cell lines with an IC50 values in the 0.03–0.32 µg/ml range.16a The antiproliferative activity of bistramide A was initially hypothesized to be due to selective activation of protein kinase C-δ.17 In 2006, however, Kozmin and co-workers obtained a crystal structure of the natural product bound to a single monomeric unit of G-actin.4,5,18 Actin is a 43 kDa protein consisting of four subdomains. ATP and Mg2+ bind within a deep cleft between subdomains 4 and 2 and polymerization occurs asymmetrically at the pointed and barbed-ends of the monomer with 5-times greater polymerization occurring at the pointed end. For natural products known to target actin directly, the barbed-end and the ATP-binding domain are the primary sites of interaction. Bistramide A belongs to the class of natural products that induce disassembly of microfilaments (or prevent its formation) by interfering with the barbed-end of the actin monomer (inset, Figure 1). Within the bistramide A binding pocket of actin, an intimate hydrogen bonding network was observed between the protein and the central amino acid portion of the molecule. In contrast to bistramide A, however, most barbed-end targeting molecules such as reidispongiolides,19 sphinxolides,19 trisoxazoles20 and others21 have very few polar contacts within the actin pocket. The cross-sectional representation of the 1 and 3 subdomains of G-actin shows the difference in binding between bistramide A and other natural products (Figure 1). The binding of most actin targeting natural products relies mainly on hydrophobic interactions between their respective macrocycles and a shallow patch on the side of actin.3a Bistramide A, on the other hand, inserts deep into the pocket and displays multiple polar contacts; thus allowing for a unique binding mode among barbed-end targeting natural products.22
Figure 1.

A schematic drawing of the actin monomer showing the major subdomains (inset). Cross-section representation of 1 and 3 subdomains of G-actin. The red portion of the molecules insert into the “cleft” of 1 and 3 subdomains of actin. The atoms of the natural products involved in polar contacts with the protein are boxed.
With a detailed model in hand, the present study examines the biological and conformational effects of specific stereoisomers of bistramide A.23 These epimers incorporate small, subtle changes into a highly organized system, allowing us to determine the importance of each polar contact as well as how the stereochemical environment associated with these contacts defines changes in biological activity and conformation within the actin pocket. The notable differences in the way bistramide A binds to actin through a highly ordered hydrogen bonding network and few stereostructural activity relationship (SSAR) studies for this specific class of molecules supports the relevance of this study. By systematically altering the stereochemical environment of this highly-conserved portion of the molecule, a framework is provided for designing potentially more potent analogs of bistramide A.
Our initial interest was focused on reported points of hydrogen bonding (C13, C13-N, C15, C18, C18-N, C39) of the molecule and how subtle stereochemical changes would affect the overall activity of bistramide A. Next, we considered the stereochemical environment around the tetrahydropyran where hydrophobic interactions are thought to play an important role between bistramide A and the actin pocket. A recent account has explored these potential binding sites through computational measurements and highlighted the importance of tetrahydropyran fragment in regards to binding and cytotoxicity.24 A greater understanding of such molecular systems may allow for design and synthesis of more efficacious anti-cancer agents. Herein, we report the synthesis and the associated biological activity of 35 stereoisomers of the bistramide A planar structure.
Results/Discussion
Retrosynthetic Analysis
Recently, we reported a total synthesis of bistramide A that utilizes 21 steps (longest linear sequence) from readily available reagents.15b The strategy was highlighted by the use of three different chiral crotylsilane reagents to construct all principle fragments [(6R,9S,11R)-6, (15S,16R)-7 and (39S)-8] and simultaneously install 8 of 11 stereogenic centers. Following our reported synthesis, we decided to make several stereochemical iterations (35) of the natural product in order to conduct SSAR studies. While synthesis of this many stereoisomers of a complex natural product might seem daunting, the highly convergent nature of the synthesis together with ready access to a wide range of chiral organosilane reagents greatly simplified the strategic plan. Accordingly, disconnection at the amide bonds provided three fragments of varying size and complexity possessing different synthetic challenges (Scheme 2). The eight stereoisomers of the pyran 6 were envisioned to come from (E)- and (Z)-crotylsilane reagents 9 and 10. This is the first example of using one methodology to access all possible stereoisomers of a 2,5,6-substituted tetrahydropyran system. The four isomers of the central amino acid portion 7 were anticipated to arise from Lewis acid-mediated additions using chiral (E)-silane reagents 11. The spiroketal fragment, at the present time, was envisioned to remain stereostructurally intact except for the C39 alcohol. Enantioselective reduction of the precursor α,β-unsaturated ketone with CBS reagent allowed for formation of the unnatural C39-epimer.
Scheme 2.

Retrosynthetic analysis of bistramide stereoisomers 1.1–1.36.
Preparation of All Stereochemical Permutations of C1-C13 Fragment 6
Recent accounts from our group detailed formal [4+2]-annulations of enantioenriched allyl- and crotylsilanes with aldehydes to form highly functionalized dihydropyran systems.25 This methodology was applied to the synthesis of complex natural products, including (−)-apicularen A, (+)-leucascandrolide A, callipeltoside A, GEX 1A, (+)-neopeltolide and most recently kendomycin.26 To synthesize bistramide A, the then currently available silane reagents did not allow direct access to the C9-C11-cis configuration of the pyran moiety. This can be explained by inspecting the proposed mechanism of (E)-anti-crotylsilane ent-10b (Scheme 3). The absolute stereochemical outcome of the annulation reaction has been proposed to occur via an intramolecular anti-SE’mode of addition. 27
Scheme 3.

Proposed Mechanism for Formation of 2,6-trans-5,6-trans dihydropyran 12.
The crotylsilane reagent ent-10b will react with an aldehyde in the presence of a Lewis acid, forming a mixed acetal that eventually leads to generation of an oxocarbenium ion I. This reactive intermediate, if placed in a chair-like transition state with the silyl group in a pseudoaxial orientation, allows for the most effective orbital σC-Si–π overlap and stabilization of the adjacent positive charge (β-effect). Electrophilic cyclization and elimination of silicon group provides the 2,6-trans-5,6-trans-dihydropyran 12 as the major diastereomer. To obtain the 5,6-cis configuration needed for bistramide A, however, the reaction would have to proceed through a boat-like conformation II. This alternate transition state suffers from a number of destabilizing interactions that provide 2,6-cis-5,6-cis-dihydropyran 13 only as the minor product.
In order to utilize organosilane-based [4+2]-annulation methodology to construct the C1-C13 portion of bistramide A, a new crotylsilane reagent was required. We reasoned, based on the proposed transition state, that changing olefin geometry from (E)- to (Z)- could effectively allow access to the desired 5,6-cis dihydropyrans (minor isomer 13 in Scheme 3). The outlined annulation approach would create a complementary reagent to our well-established methodologies.
The (E)-syn and -anti organosilane reagents 10 were obtained through various Claisen rearrangement strategies.28 Preparation of the proposed (Z)-crotylsilane reagent 9 using these rearrangement reactions was anticipated to be problematic due to potential olefin isomerization. What was required was a route that would allow access to all structural and stereochemical variations of this new reagent. Inspired by a recent review,29 we hoped epoxysilanes 15 would provide enantioenriched crotylsilanes 9. Epoxysilanes are known for their ease of preparation and serve as versatile building blocks and synthetic intermediates.30 Methods to generate these substrates in their enantioenriched form are also available.31 The general interest in epoxysilanes stems from their predictable behavior in nucleophilic oxirane openings to provide a wide range of functionalized β-hydroxysilanes.32
Preparation of cis- and trans-epoxysilanes required for synthesis of enantioenriched (Z)-crotylsilanes is depicted in Scheme 4. As anticipated, the trans-silylglycidols 15a were obtained from a modified Sharpless asymmetric epoxidation of (E)-vinyl silane 14 using (+)- or (−)-diethyl tartrate, respectively.33 The resulting epoxysilyl alcohols 15a and ent-15a were produced in 87% yield (2 steps from propargyl alcohol) and with >98% enantiomeric excess (eq. 1, Scheme 4).34 A similar multi-gram synthesis of a derivative of enantioenriched epoxysilane 15a was previously reported.35
Scheme 4.

Preparation of enantiomers of silyl glycidols.
Generation of the cis-epoxysilanes 15b and ent-15b was initially attempted with enantioselective epoxidation of the complementary (Z)-vinylsilane 16. Subjection of this silane 16 to Sharpless asymmetric epoxidation resulted in production of the desired product in less than 5% yield.36 Accordingly, we explored the possibility of an enzymatic resolution of the racemic epoxide (eq 2, Scheme 4).37 Epoxidation of vinylsilanes 16 with m-CPBA gave a racemic mixture of epoxy alcohols rac-15b in 85% yield. Several lipase enzymes were surveyed to resolve the racemate.38 Best results were obtained using Amano PS-D lipase in the presence of vinyl acetate to provide primary alcohol 15b and acetate 17 in high yields and enantiomeric excess. Base-catalyzed methanolysis of the acetate group gave the desired cis-epoxysilane ent-15b in 97% yield.39 To the best of our knowledge this is the first example of enzymatically resolved silylglycidols.
A straightforward, 3-step sequence of oxidation, oxirane opening and Lindlar reduction was used for completion of the (Z)-crotylsilane reagent 9 (Scheme 5). Primary alcohol 15a was subjected to NaIO4 and catalytic RuCl340 to give an acid, which was immediately esterified41 with DCC to provide trans-silyl glycidate 18a (72%, 2 steps). Applying the oxidation/esterification sequence to the cis-epoxide 15b proved troublesome. Upon scale-up, a significant amount of aldehyde was observed (ca. 20%) resulting in a two-step 55% isolated yield for ester 18b. Fortunately, two sequential catalytic ruthenium oxidations42 followed by esterification with DCC/DMAP afforded the silyl glycidate 18b in 66% yield over 2 steps. Both the cis- and trans-silylglycidates 18 have been prepared in >25 g quantities.
Scheme 5.

Synthesis of (Z)-crotylsilanes 9.
Regioselective epoxide ring opening with diethylpropynylaluminum30c gave 2,3-syn- and 2,3-anti-hexyne methyl esters 19a and 19b, respectively, in moderate yields and good selectivities. Minor reaction byproducts included alkyne addition to the methyl ester and chloride opening of the silyl glycidate. The latter is a common byproduct obtained from metal additions to epoxysilanes (e.g. Grignard).29 Fortunately, addition of the alkyne to the β-carbon of the silicon functionality was not detected in the described conditions. Several explanations have been put forth on the selective opening of epoxysilanes.29,43 One interesting hypothesis includes a prior coordination of nucleophile with both the α-carbon and silicon functionality,44 though more recent studies using allyl cuprates do not support this pathway.43 Alternatively, formation of a pentacoordinated silicon followed by 1,2-migration was also postulated.45 Lastly, X-ray crystal structure46 and gas phase and theoretical studies47 showed the α C-O bond (adjacent to the Si) to be longer and weaker than the β C-O bond. Our particular system has the added advantage of a Lewis basic site on the adjacent carbonyl to aid in opening via coordination of resident metal centers. Lindlar reduction of alkynes 19 followed by protection of the secondary alcohols as their TMS ethers gave the syn- and anti-(Z)-crotylsilanes 9a and 9b and their enantiomers. Each stereoisomer was prepared in >10 g quantities.
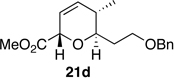
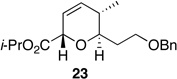
The preparation of the required 5,6-cis-dihydropyran of the C1-C13 fragment of bistramide A was explored next. Recently, we described a [4+2]-annulation of syn-(Z)-crotylsilane reagent 9a with β-benzyloxy aldehyde 20 to provide the desired 5,6-cis-pyran 21a in 66% yield (Table 1, entry 1). Extension of this methodology to the other isomer silanes would afford all possible stereochemical iterations of 2,5,6-dihydropyrans. Subjection of all syn- and anti-(Z)- and -(E)-silane reagents 9 and 10 to aldehyde 2048 under TMSOTf-catalyzed conditions afforded the eight possible stereochemical analogs of the 2,5,6-dihydropyran system (Table 1). The 2,3-syn-(Z)-crotylsilanes 9a and ent-9a provided a 2,6-trans 5,6-cis dihydropyrans 21a and ent-21a in useful yields and selectivities (entries 1 and 2). In a complementary fashion, the 2,3-anti-(Z)-crotylsilanes 9b and ent-9b gave rise to dihydropyrans 21b and ent-21b bearing all cis-2,5,6-stereochemical relationships in equally good yields and selectivities (entries 3 and 4). Similarly, the 2,3-syn-(E)-crotylsilanes 10a and ent-10a yielded 2,6-cis-5,6-trans-dihydropyrans 21c and ent-21c with excellent yields and selectivities (entries 5 and 6). The 2,3-anti-(E)-crotylsilane 10b underwent annulation to provide dihydropyran 21d in moderate yield, but with a 1:1 mixture of diastereomers (entry 7). This result, however, was not entirely surprising. A similar problem was encountered during a second generation approach to a late stage intermediate of leucascandrolide A.26c Annulations with 2,3-anti-(E)-crotylsilane 10b and protected 3-hydroxypropionaldehyde derivatives provided dihydropyran products in poor yields and selectivities. The problem was addressed through modification of the organosilane reagents to affect the resident A values of the postulated intermediate oxocarbenium ion. This analysis was loosely based on the A values of ethyl (1.1 kcal/mol) versus that of a methyl ester (1.3 kcal/mol) of a monosubstituted cyclohexane ring.49 The 0.2 kcal/mol difference between methyl and ethyl esters should translate into even higher activation energy for the isopropyl ester. Experimentally, annulation of isopropyl ester anti-(E)-crotysilanes 22 and ent-22 afforded a 2-fold increase in selectivity for the desired 2,6-trans-5,6-trans-dihydropyrans 23 and ent-23 (entries 8 and 9).
Table 1.
[4+2]-Annulation of both (Z)- and (E)-Crotylsilanes 9 and 10 with Aldehyde 20.
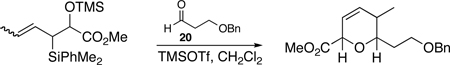 | ||||
|---|---|---|---|---|
| entry | silane | producta | yieldb | drc |
| 1 | 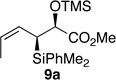 |
 |
66% | 12:1 |
| 2 | ent-9a | ent-21a | 63% | 12:1 |
| 3 |  |
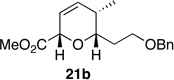 |
60% | 15:1 |
| 4 | ent-9b | ent-21b | 63% | 15:1 |
| 5 | 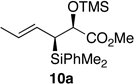 |
 |
78% | 20:1 |
| 6 | ent-10a | ent-21c | 72% | 20:1 |
| 7 |  |
 |
50% | 1:1 |
| 8 | 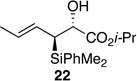 |
 |
53% | 4:1 |
| 9 | ent-22 | ent-23 | 57% | 3.5:1 |
Reactions were performed using 1.0 equiv. of silane, 1.0 equiv. of aldehyde 20 and 1.0 equiv. TMSOTf in CH2Cl2 at −50 °C.
Isolated yield after SiO2 chromatography.
Ratio determined by 1H NMR analysis of crude reaction mixtures.
The stereochemical outcome of the annulation reaction with (Z)-crotylsilanes was not entirely anticipated. In sharp contrast to the proposed mechanism of (E)-crotylsilanes (Scheme 3), the silyl group of (Z)-crotyl reagents presumably adopts a pseudo equatorial orientation. The oxocarbenium ion of ent-9b prefers the chair-like intermediate IV to give dihydropyran 13 as the major diastereomer. 50 The potential A1,3 destabilizing interactions arising from the (Z)-olefin and axial silicon in III51 prevent formation of the 2,6-trans-5,6-cis product 24.52 Though there is a turnover in selectivity about the 2,6-position between the (E)- and (Z)-crotylsilanes, the reagents remain completely complementary in their stereochemical outcome. With ample quantities of all eight stereochemical analogs of 2,5,6-dihydropyrans in hand, we were positioned to complete the synthesis of each of the desired isomers of the C1-C13 portion of bistramide A.
Each dihydropyran stereoisomer was taken through an eight-step sequence to access the diastereomeric C1-C13 fragments 6 (Scheme 7). Catalytic hydrogenation using Adam’s catalyst concomitantly reduced the double bonds and deprotected the benzyl ethers. The resulting alcohols were protected as TBDPS ethers, and the methyl esters were reduced using LiBH4. The newly generated alcohols were treated with (PhO)3P+CH3I− to afford primary iodides 25. The primary iodides were displaced with the lithium anion of 2-(1-propenyl)-1,3-dithiane to give 26 as masked α,β-unsaturated systems.53 The dithiane intermediates 26 were unveiled to give ketones 27 using a mild method developed by Panek employing Dess-Martin periodinane for the oxidative removal of thioacetals.54 Removal of the TBDPS ether was accomplished with wet HF in acetonitrile and oxidation of the resulting alcohols to carboxylic acids 6 occurred with catalytic chromium in wet acetonitrile. Completion of the eight stereoisomers of the C1-C13 tetrahydropyran fragment was accomplished in a nine-step synthetic sequence to provide the desired tetrahydropyran isomers in 20–30% overall yield.
Scheme 7.

Synthesis of Eight Stereoisomers of the C1-C13 Tetrahydropyran Fragment 6.
Synthesis of C14-C18 Subunits 7
Our initial strategy for preparation of C14-C18 fragments of bistramide analogs exploited a crotylation reaction between protected glycine aldehyde 28 and organosilane reagent (R)-11 (Scheme 8). The newly formed homoallylic alcohols 29 were thought to be attainable with syn and anti relationships about the newly formed bond. This “turnover “ in diastereoselectivity depends on the nature of Lewis acid (monodentate vs. bidentate) and the presence of a heteroatom on the aldehyde capable of providing a second contact point with a Lewis acid. The latter was showcased in the synthesis of mycalolide A where the C8-C9 anti-configuration was obtained through chelation controlled addition of silane (S)-11 with a α-nitrogen bearing aldehyde.55 Functionalization of the resulting alcohols 29 via a four-step sequence would afford the desired protected amino acids 30 in only five steps.
Scheme 8.

Initial Synthetic Approach to the C14-C18 Amino-Acid Fragment 7.
The crotylation reaction was first explored using tert-butyl carbamate (Boc) protected aldehyde 28a. Exposure of this volatile and unstable aldehyde to our standard crotylation conditions (BF3•OEt2, CH2Cl2) resulted in decomposition of both starting materials, including protodesilylation of crotylsilane reagent (R)-11. We next turned to condensation of phthalimide protected amino aldehyde 28b with the organosilane reagent (R)-11 in the presence of TiCl4. The desired homoallylic alcohol 29b was obtained in 70% yield and high diastereoselectivity (dr 20:1). This alcohol was then protected as a TBS ether and the double bond was oxidized to afford the amino acid fragment 30b in just three steps (68% yield). Unfortunately, only the syn-homoallylic alcohols were observed with aldehyde 28b when both monodentate and bidentate Lewis acids were utilized.56 Since the anti-C15-C16 stereochemistry was needed as well, an alternate protecting group was required that would be capable of providing a sufficiently active site for chelation. To that end, reaction of the Cbz-protected aldehyde 28c with silane (R)-11 afforded the desired alcohol 29c, albeit in low yield and selectivity (30% yield, dr 2:1). Modification of reaction conditions (solvents, temperatures, Lewis acids) failed to improve the selectivity and yield of this reaction. Thus, we turned our efforts toward preparation of all stereoisomers of the C14-C18 amino fragment utilizing an α-alkoxy aldehyde. In this case, a benzyl ether protecting group was employed to provide a second chelation site with a bidentate Lewis acid.57
The desired syn and anti homoallylic alcohols 33 were accessed via a two step sequence: [3+2]-annulation followed by furan opening reactions (Scheme 9).26a Use of the protected acetaldehyde 31 allowed for generation of all possible isomers of the amino acid fragment, where the primary alcohol would be later be exchanged for the desired amine functionality. Condensation of (R)-11 with α-benzyloxyacetaldehyde 31 in the presence of SnCl4 provided 2,5-anti-tetrahydrofuran 32a in 73% yield and excellent diastereoselectivity (> 20:1). Use of BF3•OEt2 as the Lewis acid promoter, on the other hand, effected formation of 2,5-cis-tetrahydrofuran ent-32b in 76% yield and 20:1 selectivity. These furans were then subjected to E2-type elimination mediated by SbCl5. The newly emerged homoallylic alcohols, 33a and ent-33b, respectively, were obtained in moderate to good yields, 62–85%, and greater than 10 g quantities. The remaining two isomers, alcohols ent-33a and 33b, were also obtained in this fashion following the same reaction pathway utilizing enantiomeric crotylsilane (S)-11.
Scheme 9.

Preparation of C15-Homoallylic Alcohols 33.
Completion of the synthesis of all isomers of the γ-amino acid fragment 7 is depicted in Scheme 10. Protection of the C15-homoallylic alcohols 33 as silyl ethers was followed by an ozonolysis/NaBH4 reduction sequence to yield primary alcohols in good yields (87–92%, 2 steps). Subsequent protection of the primary alcohols as TBS-ethers afforded the desired intermediates 34 in high yields. Cleavage of benzyl ethers (Pd/C, H2) gave primary alcohols which were subjected to Mitsunobu conditions using (PhO)2P(O)N3 to install primary azides in high yields (86–93% yields). Selective removal of the primary TBS ether was affected using CSA to provide alcohol products 35 in 87–92% yields. The required TIPS esters of the C14-C18 subunit were prepared in moderate yields via a TEMPO-mediated oxidation and protection sequence (65–89%). Reduction of the azide functionality (Pd/C, H2) gave the fully functionalized isomers of the amino-acid fragment 7 in 15–41% yield over 10 steps.
Scheme 10.

Synthesis of Four Isomers of γ-amino Acid Subunit 7.
Preparation of the left-hand side of bistramide A analogs proceeded as anticipated (Table 2). Union of eight isomers of the C1-C13 pyran fragment 6 with the four diastereomers of the γ-amino acid segments 7 was achieved in good yield using the coupling reagent PyBOP in the presence of triethylamine. The TIPS-ester fragments 36 were obtained in yields ranging between 53–81%. It is important to note that there appears to be no stereochemical dependence on the extent of conversion and the overall yield of the coupling reactions for either the pyran or the amino acid fragments. Each isomeric fragment 36 was prepared in greater than 40 mg quantities and showed little decomposition after prolonged storage at low temperature.
Table 2.
Structures of C1-C18 Isomeric Fragments 36.
|
| |||||||
|---|---|---|---|---|---|---|---|
| Coupling partners | Product | Yielda | Coupling partners | Product | Yielda | ||
| 6a | 7a | 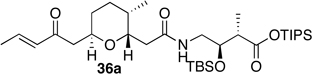 |
68 | 6c | 7a |  |
64 |
| " | ent-7a |  |
61 | " | ent-7a | 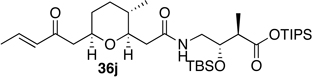 |
68 |
| " | 7b |  |
57 | " | 7b |  |
75 |
| " | ent-7b |  |
63 | " | ent-7b |  |
68 |
| ent-6a | 7a |  |
61 | ent-6c | 7a |  |
71 |
| " | ent-7a |  |
60 | " | ent-7a |  |
58 |
| " | 7b |  |
68 | " | 7b |  |
53 |
| " | ent-7b |  |
64 | " | ent-7b | 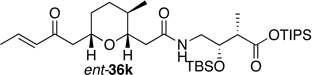 |
59 |
| 6b | 7a |  |
71 | 6d | 7a |  |
56 |
| " | ent-7a |  |
81 | " | ent-7a | 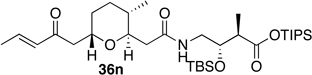 |
59 |
| " | 7b | 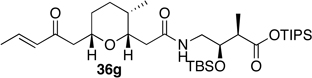 |
69 | " | 7b |  |
66 |
| " | ent-7b | 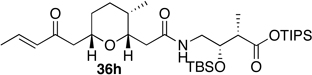 |
78 | " | ent-7b |  |
56 |
| ent-6b | 7a | 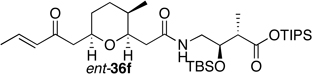 |
74 | ent-6d | 7a |  |
65 |
| " | ent-7a | 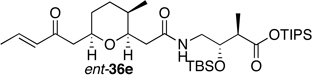 |
64 | " | ent-7a | 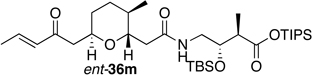 |
64 |
| " | 7b | 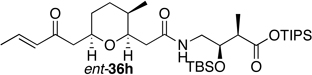 |
80 | " | 7b |  |
74 |
| " | ent-7b | 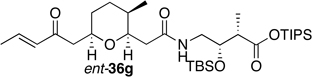 |
75 | " | ent-7b | 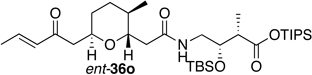 |
54 |
Isolated yields after SiO2 column chromatography.
Preparation of the Spiroketal Moiety 8
Construction of the C29-C40 segment of the spiroketal subunit utilized starting materials obtained from a chiral pool source (Scheme 11). Subjection of the commercially available (S)-1,2,4-butanetriol derivative 3758 to a Wittig olefination reaction with phosphonium salt 3859 provided olefin 39 as a mixture of geometric isomers in 70% yield (Z:E = 10:1). The exocyclic olefin was reduced and concomitant deprotection of the benzyl ether with Raney nickel yielded a primary alcohol in 78% yield. Swern oxidation of this material provided an aldehyde, which was directly converted to the α,β-unsaturated ketone 40 using the Horner-Emmons reagent, diethyl 1-methyl-2-oxopropyl phosphonate.60 The afforded ketone was enantioselectively reduced with Corey’s oxazaborolidine reagent [(R)-CBS]61 afforded the (S)-alcohol which was protected as a TBS ether 41 in 77% yield (2 steps). Reductive opening of the PMP acetal 41 with Dibal-H generated a secondary PMB ether in 98% yield. Conversion of this material to the triphenyl phosphine salt 42 was carried out through a standard two-step sequence: bromination of the primary alcohol was followed by displacement with PPh3 to afford the advanced phosphine salt partner 42.
Scheme 11.

Reaction Sequence for Synthesis of Phosphine Salt 42.a
Synthesis of the spiroketal portion of C19-C40 fragment 45 was initiated with a [4+2]-annulation employing syn-(E)-crotylsilane reagent 10a and 4-(benzyloxy)butanal (Scheme 12).62 The desired 2,6-cis-dihydropyran 43 was obtained in 97% isolated yield and high diastereoselectivity (dr 20:1). The pyran double bond was isomerized into conjugation using tetra-n-butyl ammonium hydroxide and subsequent treatment with methanolic CSA afforded the methyl glycoside (78% over 2 steps). This material was then reduced with Dibal-H to provide the intermediate aldehyde 44 which was subjected to a Wittig olefination with phosphonium salt 42. Gratifyingly, phosphorous-based olefination provided the PMB protected (Z)-alkene 45 in 86% yield and as a single geometric isomer. Incorporation of a fully functionalized C40-C32 side chain prior to spirocyclization underscores the highly convergent nature of our synthesis.
Scheme 12.

Coupling of Tetrahydropyran 44 with the Phosphine Salt 42
Our initial approach for spirocycle formation exploited a two-step sequence: deprotection of the C31-PMB group of the C28-C29 unsaturated analog 45 followed by spirocyclization (Scheme 13).63 Unmasking the alcohol, however, proved to be very difficult as deprotection under standard DDQ conditions (CH2Cl2/H2O) did not provide the free C31-secondary alcohol as hoped, but instead gave a complex mixture of compounds. Interestingly, the major product identified from this reaction was spirocycle 46, isolated in 17% yield after purification (Scheme 13).64 Attempts to optimize the spirocyclization led to 45% yield of the desired spiroketal intermediate 46.65 Selective hydrogenation of 46 with Wilkinson’s catalyst gave the required spirocycle 47 in 95% yield. However, the low yield obtained in spirocyclization prompted us to investigate alternative conditions for this transformation.
Scheme 13.

Synthesis of C19-C40 Spiroketal Fragment 49
In order to optimize the yield of the spirocyclization using DDQ, the C28-C29 olefin was reduced as it may promote premature oxonium ion formation. Selective hydrogenation of the C28-C29 olefin with Wilkinson’s catalyst provided the mono unsaturated system 48 in 75% yield. Exposure of the C28-C29 dihydro framework 48 to DDQ under aqueous or anhydrous conditions provided low yields of the desired spiroketal 47 (entries 1–3, Table 3).
Table 3.
Spiroketal Formation with the Saturated Framework 48.
 | ||
|---|---|---|
| entry | conditionsa | yieldb |
| 1 | 3 equiv DDQ, CH2Cl2/H2O (10/1) | 21 |
| 2 | 3 equiv DDQ, CH2Cl2/pH 7.0 buffer | 23 |
| 3 | 3 equiv DDQ, CH2Cl2 | <5 |
| 5 | 2 equiv DDQ, CH2Cl2, 6 equiv pyridine | 44c |
Reactions were carried out at 0 °C.
Isolated yields after SiO2 column chromatography.
Reaction was warmed to room temperature.
However, exposure of the intermediate 48 to DDQ in the presence of pyridine under anhydrous conditions provided the desired spirocycle 47 in 76% yield (entry 4).66 Warming the reaction mixture to room temperature caused significant erosion in chemical yield (entry 5). Other conditions screened for PMB deprotection/spiroketal formation include: TMSI,67 I2 in MeOH,68 BCl3•Me2S complex69 and MgBr2•Et2O.70
The role of pyridine in the reaction was not explored in detail but it may behave as a simple buffer for any adventitious acid that might cause premature formation of oxonium ion.71 Mechanistically, DDQ behaves as an electron acceptor to the local PMB group to form a charge transfer complex. Upon dissociation of this complex, the hydroquinone species 2,3-dichloro-5,6-dicyano-1,4-dihydroxybenzene (DDQH2) is generated. The DDQH2 molecule is somewhat acidic (pKa = ~5) and was considered to be the potential cause of premature oxonium ion generation under traditional conditions (water/dichloromethane).72 Additional mechanistic role of pyridine may involve production of an intermediate charge-transfer complex between pyridine and DDQ.73 Charge-transfer complexes have been documented with electron rich nitrogen heterocycles including pyrimidines and imidazoles.74
Having the desired spiroketal 47 in hand, a three step-sequence was required for completion of the C19-C40 fragment (Scheme 13). Birch reduction of the primary benzyl ether was followed by a Mitsunobu displacement of the resulting alcohol with (C6H5O)2P(O)N3. The desired primary azide was formed in 86% yield and subjected to Me3P to yield a free amine 49 via in situ hydrolysis of the phosphine imine intermediate.75
With the fully functionalized spiroketal 49 in hand, we were poised to begin construction of the complete carbon frameworks of stereochemical analogs. Accordingly, union of deprotected C1-C18 fragments 36i–l with the spirocycle 49 was accomplished using PyBOP peptide coupling reagent (Scheme 14). The resulting silyl protected analogs were obtained in 41–84% yield. Applying the previously reported TBS-deprotection conditions (PPTS, MeOH) to these stereochemical derivatives proved difficult. Significant decomposition of a subset of analogs 50.16–50.24 was observed. Closer examination of the crude reaction profile suggested that the C1-C4-α,β-unsaturated system of the isomers was affected.76 Removal of the C15- and C39-silyl ethers with HF, HF•pyridine, CSA or TBAF was unsuccessful. Interestingly, use of TBAF in THF selectively removed the C15-TBS ether without decomposition of the product. With this key observation, slight modification of the spirocycle 49 was performed prior to coupling allowing access to stereochemical analogs without incident. With that accomplished, azide 51 was treated with CSA in methanol to expose alcohol (S)-8 in 85% yield (Scheme 15). Reduction of azide (S)-8 to the primary amine with trimethylphosphine afforded the natural spiroketal fragment (S)-52. Enantioselective inversion of the allylic alcohol intermediate (S)-8 provided an additional point of stereochemical induction. The C39-hydroxyl group has been shown to have a bridged contact point with a water molecule of the Tyr143 residue of actin; this residue is also hydrogen bonded to the C18 amide of the natural product. Inversion of the C39-stereocenter was achieved in a two-step process. Oxidation of the alcohol (S)-8 with Dess-Martin periodinane afforded an α,β-unsaturated ketone (92% yield), which was reduced with (S)-CBS reagent to give the C39-epimeric alcohol (R)-8 in good yield and diastereoselectivity (81%, dr 20:1). Staudinger reaction and hydrolysis of the resulting iminophosphorane provided the unnatural spiroketal fragment (R)-52, epimeric at C39.
Scheme 14.

Coupling of acids 36 and spiroketal 49 and silyl deprotection
Scheme 15.

Preparation of C39-epimer of the Spiroketal Subunit 52
Completion of the skeletal backbones of stereochemical analogs was achieved in two steps (Scheme 16). Coupling of C1-C18 acids 36a–p and ent-36a–p with the natural spiroketal (S)-52 was mediated with PyBOP after initial treatment of TIPS-acids with TBAF. The resulting C15-protected alcohol products 53.1–53.32 were obtained in 60–80% yield. Subjection of these advanced intermediates to TBAF afforded the bistramide A as well as the desired 31 unnatural products 1.2–1.32 in good yields and greater than 3 mg quantities. On the other hand, union of (R)-spiroketal 52 with the isomers of the natural pyran 36a–d provided four additional epimers 1.33–1.36 of the natural product.
Scheme 16.

Completion of the Synthesis of Stereoisomers 1.1–1.36
Biological Evaluation of Bistramide A Stereochemical Analogs
The 35-membered stereochemical library of bistramide A was screened for cytotoxicity and disruption of cellular actin. The stereostructure activity relationships (SSAR) of bistramide A 1.1 and its derivatives (1.2–1.36) were explored using cell growth assays: renal carcinoma line UO-31 and the CNS tumor cell line SF-295 (Table 4). Using a two-day assay for cell growth inhibition of UO-31 cells, analog 1.21 was shown to be approximately twice as potent (44 nM) as the natural product 1.1 (77 nM). Interestingly, the compound 1.21 differs stereochemically from bistramide A 1.1 at the C6 and C9 positions of the pyran moiety (Figure 2). The C1-C13 portion of the bistramide A is not involved in polar contacts with the protein but was suggested to only attenuate lipophilicity of the natural product in the barbed-end binding pocket. A recent publication describes computational docking studies using the X-ray structure concerning the structural determinants of macrolide-actin binding.24 In this study, flexible docking of the bistramide A pyran fragment in the hydrophobic cleft of actin revealed at least three hydrophobic interactions between the protein and tetrahydropyran/enone portion of the molecule. The highest scoring flexibly docked structure was thus shown to have rotation around the C12-C13 bond of the natural product. Thus, the results of the computational docking experiments may help to explain the enhanced activity of analog 1.21 in the UO-31 cell line.
Table 4.
Cell-Growth Inhibition of Bistramide Stereochemical Analogs 1.1–1.36 Against UO-31 and SF-295 Cancer Cell Lines.
| compound | UO-31 (IC50 ± SEM µM) | SF-295 (IC50 ± SEM µM) |
|---|---|---|
| 1.1 | 0.071 ± 0.01 | 0.47 ± 0.2 |
| 1.2 | 5.9± 1.1 | _a |
| 1.3 | 45 ± 8 | 78.8 ± 4 |
| 1.4 | 4.6 ± 0.7 | 4.7 ± 2 |
| 1.5 | 0.62± 0.03 | 2 ± 0 |
| 1.6 | 14.8 ± 4.5 | _a |
| 1.7 | 45.3 ± 0.5 | 76.5 |
| 1.8 | 15.9 ± 7.3 | _a |
| 1.9 | 0.32 ± 0.02 | 0.2 ± 0.0 |
| 1.10 | 15.6 ± 4.5 | _a |
| 1.11 | 17.1 ± 2.2 | _a |
| 1.12 | 2.8 ± 0.4 | _a |
| 1.13 | 1.9 ± 0.2 | ~2.4b |
| 1.14 | 7.8 ± 3.5 | 77.5 ± 4 |
| 1.15 | 18.2 ± 5.8 | _a |
| 1.16 | 11 ± 1.4 | ~96b |
| 1.17 | 0.33 ± 0.02 | 3.5 |
| 1.18 | 13.9 ± 5.3 | _a |
| 1.19 | 44.7 ± 9.7 | _a |
| 1.20 | 14.5 ± 5 | ~14b |
| 1.21 | 0.044 ± 0.017 | ~3.8b |
| 1.22 | 44.7 ± 20.7 | _a |
| 1.23 | 6.2 ± 0.5 | 17.1 ± 6 |
| 1.24 | 45.1 ± 30.8 | 75.2 |
| 1.25 | 5.7 ± 0.7 | ~34b |
| 1.26 | 0.82 ± 0.07 | ~30b |
| 1.27 | 17.4 ± 6.4 | _a |
| 1.28 | 15 ± 9.9 | _a |
| 1.29 | 0.81 ± 0.08 | ~20b |
| 1.30 | 14.9 ± 0.4 | _a |
| 1.31 | 14.6 ± 10.2 | _a |
| 1.32 | 5.3 ± 1.4 | _a |
| 1.33 | 0.33 ± 0.04 | 0.34 ± 0.1 |
| 1.34 | 12 ± 1.7 | _a |
| 1.35 | 15.4 ± 9.3 | _a |
| 1.36 | 2.2 ± 0.3 | ~16b |
Compounds were not tested.
Estimated IC50 value.
Figure 2.

Representative Structures of Potent Stereochemical Analogs of Bistramide A.
Six other epimers demonstrated sub-micromolar potency against the UO-31 cell line (Table 4). Similar to the 1.21 variant, the derivatives 1.5, 1.9, 1.17, 1.25, 1.29, differ only in stereochemistry at the C6, C9 and C11 positions of the pyran moiety (Figure 2). Interestingly, compound 1.33, the C39-epimer of the natural product also showed good micromolar potency. The remainder of the stereoisomers exhibited lower activity for the UO-31 cell line, with the least potent analog 1.7 having an IC50 value of 45.3 µM (Table 4).
A subset of the compounds was also tested against the CNS tumor cell line SF-295 under the same protocol (Table 4). In general, the compounds inhibited cell growth to a lesser degree, but the relative rank potency was similar to the UO-31 cell line. Stereoisomer 1.33 showed slight better inhibition (0.34 µM) in comparison to bistramide A (0.47 µM) for the SF-295 cell line.
The effects on cell cycle were assessed for two of the most potent cell growth inhibiting compounds (1.1 and 1.33), as well as for the least active one (1.7). A dose responsive reduction of the proportion of cells in S phase and increase in G1 phase by analogs 1.1 and 1.33 was observed. These effects were demonstrated in both the UO-31 and SF-295 cell lines indicating blockade of the cell cycle by the two isomers. Stereoisomer 1.7, which was essentially inactive as an inhibitor of cell growth, also had no effect on the cell cycle. As such, cell cycle data is presented for a relatively potent library member, the natural product (bistramide A) and, as well as a very weak binding member of the collection. The essential point is that potent members of the library appear to block the cell cycle in a manner consistent with actin inhibition, while weak members of the collection do not.
The inhibition of cell cycle by bistramide A 1.1 and analog 1.33 was confirmed with confocal microscopy examination of FITC-phalloidin stained cells. At concentrations that inhibited cell growth and cell cycle progression, the actin cytoskeleton was disrupted as indicated by the loss of fluorescence and alteration of the cytoskeletal structure. These effects were consistent with both cell lines.
Finally, the natural product 1.1 and C6, C9-stereochemical analog 1.21 were subjected to the NCI 60-cell assay. The obtained results provide further insight into the action of bistramide A and its epimers. Stereoisomer 1.21 was approximately twice as potent as the natural product 1.1, with mean GI50 values of 9 nM and 18 nM, respectively. The patterns of cell growth inhibition were highly correlated (Pearson coefficient of 0.81 at the GI50 level and 0.81 at the TGI level of response). The most sensitive panels were the renal cancer cell lines and the CNS cancer cell lines. The difference between the least and most sensitive cell lines was between 2 and 2.5 log units at the GI50 level of response, and approximately 3.5 log units at the TGI level. This data was compared to other compounds known to bind to and interfere with actin (Table 5). With the notable exception of aplyronine A, the data for all other natural products showed significant, albeit modest, correlations in 60-cell patterns to the data for compounds 1.1 and 1.21.
Table 5.
Pearson Correlation Coefficients Between Compounds 1.1 and 1.21 and other known Actin-Binding Natural Products. Correlations were obtained for the GI50 level of response.
| Name | NSC | GI50 to 1.1 | GI50 to 1.21 |
|---|---|---|---|
| aplyronine A | 687160 | −0.11 | −0.03 |
| cytochalasin A | 174119 | 0.55 | 0.42 |
| cytochalasin B | 107658 | 0.55 | 0.49 |
| cytochalasin D | 209835 | 0.71 | 0.63 |
| cytochalasin E | 175151 | 0.71 | 0.66 |
| cytochalasin H | 305222 | 0.69 | 0.56 |
| cytochalasin Q | 675858 | 0.69 | 0.49 |
| dolastatin 11 | 606195 | 0.37 | 0.45 |
| jasplakinolide | 613009 | 0.34 | 0.41 |
| latrunculin A | 613011 | 0.58 | 0.44 |
| mycalolide C,D mixture | V4827 | 0.28 | 0.50 |
| mycalolide E | V4828 | 0.30 | 0.50 |
| swinholide H | 685570 | 0.49 | 0.61 |
Biological Activity: Experimental
Cytotoxicity assays
A tetrazolium dye [2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide; XTT]-based colorimetric assay was used in 96-well plates to measure inhibition of the proliferation/survival of tumor cell lines in vitro.77 All test compounds were formulated in DMSO and applied to cells such that the final DMSO concentration was ≤ 0.1%.
Cell cycle analysis
SF-295 and UO-31 cells were harvested from flasks at 75–85% confluency, counted and plated into 35-mm dishes at a density of approximately 1 × 106 cells/dish, and allowed to re-attach overnight. Compounds were prepared as a 5 mM stock solution in DMSO and applied to cells at final concentrations of 1, 0.1 and 0.001 µM. A control dish was treated with vehicle (DMSO) alone. Cells were incubated for 24 h in the continuous presence of the various treatments, at which times they were rinsed with PBS, harvested by trypsinization, resuspended in complete medium, pelleted by centrifugation, rinsed once with PBS, and resuspended in 1 mL of Krishan’s buffer (0.1% sodium citrate, 0.02 mg/ml RNAse, 0.3% NP-40 and 50 mg/ml of propidium iodide (PI) at pH 7.4). Cells were analyzed for relative DNA content by flow cytometry using a Becton Dickinson FASCan (Becton Dickinson Immunocytometry Systems, San Jose, CA) equipped with a DDM using CELLQuest software. The analysis was done using MODFIT LT 2.0 (Verity Software House, Inc., Topsham, ME).
Confocal images
UO-31 and SF-295 cell monolayers were established on glass coverslips and incubated overnight with the compounds. After treatment, the cells were fixed in 3.7% formaldehyde, permeabalized with 0.1% Triton X-100 and stained with fluorescein phalloidin (Molecular Probes, Invitrogen Corporation, Carlsbad, CA) at a final concentration of 165 nM. After staining, slides were rinsed with PBS, and mounted in a DAPI containing mounting medium (Molecular Probes). Confocal images were acquired using a 63× oil objective (Zeiss UV510, Oberkochen, Germany).
NCI 60-Cell Assay
The NCI 60-cell assay was conducted as previously described.78 Compounds were tested first at a single 10−5 M dose, then at two separate times in full dose response format in five ten-fold dilutions. Compare analyses were performed as previously described.79
Conclusions
We have shown that employing a unified strategy, the creation of a library of selected stereoisomers of the natural product bistramide A, is viable providing the reaction methodology is sufficiently robust. We have learned that the generation of unique biological information on stereostructure-activity relationships (SSAR) is a consequence of such an approach. In the present case, we have sampled a small number of stereoisomers (35 of the 2048 possible compounds), selected where crystallographic studies have indicated that the chirality might be critical for binding to actin. Our synthetic strategy was highlighted by preparation of the stereochemical variants of the principle fragments with three structurally and stereochemically different enantioenriched organosilane reagents. The compounds were screened for growth inhibition against UO-31 and SF-295 cell lines as well as for binding to actin. Six steroisomers (1.5, 1.9, 1.17, 1.29 and 1.33) inhibited growth in the micromolar range, while compound 1.21 was shown to be the most potent in the UO-31 cell line assay. Stereochemical analog 1.21 was shown to be approximately twice as potent than the natural bistramide A 1.1 with mean GI50 values of 9 nM and 18 nM, respectively in the NCI 60 cell assay. Given that stereoisomer libraries are generally difficult to achieve by solution phase synthesis, is a testament to the versatility of chrial silane-based bond construction methodology. Enabling, enhanced facility in asymmetric synthesis and more efficient modular tactics in convergent assembly make it possible to elucidate the role of each chiral center in bioactivity. With bistramide A, the correlation to crystallographic data validated the approach, but it should be possible to empirically probe stereochemical factors in the absence of a target as well. For compounds with large numbers of chiral centers, it may not be desirable to synthesize all possible stereoisomers, but an iterative approach which samples several centers in each stage to guide successive syntheses should facilitate a complete analysis of SSAR, when coupled with high throughput biological testing methods.
Experimental Section
The following experimental section is representative and describes the complete details of the convergent synthesis of bistramide A stereoisomer 1.21.
(2S,5R,6S)-methyl 6-(2-(benzyloxy)ethyl)-5-methyl-5,6-dihydro-2H-pyran-2-carboxylate (ent-21c)
To a solution of 3-(benzyloxy)propanal 20 (1.87 g, 11.4 mmol) and (E)-syn crotylsilane ent-10a (2.00 g, 5.70 mmol) in methylene chloride (120 mL) at −50 °C was added trimethylsilyl trifluoromethanesulfonate (2.08 mL, 11.4 mmol) dropwise. Reaction was stirred for 24 hours at −50 °C before it was quenched with saturated NaHCO3 and warmed to room temperature. The layers were separated and the aqueous layer was extracted with methylene chloride (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by column chromatography (silica, 10% EtOAc/hexanes) afforded dihydropyran ent-21c in 72% yield. −116.7° (c 2.0, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.33-7.25 (m, 5H), 5.73 (tq, J = 2.0, 10.2, 22.6 Hz, 2H), 4.67 (m, 1H), 4.50 (q, J = 12.0, 24.0 Hz, 1H), 3.75 (s, 3H), 3.72- 3.61 (m, 2H), 3.32 (dt, J = 2.4, 9.6 Hz, 1H), 2.21 (m, 1H), 2.08-2.00 (m, 1H), 1.84-1.75 (m, 1H), 0.93 (d, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 170.6, 138.6, 134.0, 128.3, 127.6, 127.4, 133.3, 77.3, 74.7, 72.9, 66.7, 52.3, 34.0, 33.2, 17.1; IR (neat) νmax: 2958, 2870, 1736, 1263, 1097, 700 cm−1; HRMS (CI, NH3) m/z calc’d for C17H22O4 [M+23]+ 313.1416, found 313.1354.
(2S,5R,6S)-methyl 6-(2-hydroxyethyl)-5-methyltetrahydro-2H-pyran-2-carboxylate
To a solution of pyran ent-21c (0.880 g, 3.01 mmol) in methanol (19 mL) was added PtO2 (0.171 g, 25 mol%). The reaction flask was then placed under an atmosphere of hydrogen and stirred for 12 hours. The heterogeneous mixture was filtered over Celite®, washed with methanol and concentrated in vacuo. Purification by flash chromatography (silica, 50% EtOAc/hexanes) provided 95% of desired product as a clear oil. −60.1° (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 3.94 (dd, J = 2.6, 11.8 Hz, 1H), 3.76 (m, 2H), 3.69 (s, 3H), 3.21 (dt, J = 2.8, 9.8 Hz, 1H), 1.97- 1.80 (m, 3H), 1.73 (m, 1H), 1.55 (dq, J = 3.8, 13.0, 25.0 Hz, 1H), 1.43 (m, 1H), 1.22 (dt, J = 3.8, 13.0, 25.2 Hz, 1H), 0.79 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 171.5, 84.7, 76.1, 61.3, 52.0, 34.7, 34.6, 32.0, 28.8, 17.3; IR (neat) νmax: 3460, 2954, 2929, 2876, 1753, 1439, 1212, 1088 cm−1; HRMS (CI, NH3) m/z calc’d for C10H19O4 [M+1]+ 203.1283, found 203.1286.
(2S,5R,6S)-methyl-6-(2-(tert-butyldiphenylsilyloxy)ethyl)-5-methyltetrahydro-2H-pyran-2-carboxylate
To a solution of alcohol (2.90 g, 14.3 mmol) in N,N-dimethylformamide (22.0 mL) at 0 °C was added 1H-imidazole (1.20 g, 17.9 mmol) followed by tert-butylchlorodiphenylsilane (4.10 mL, 15.8 mmol) dropwise. Reaction was allowed to warm up to room temperature on its own and was stirred for 16 hours at room temperature. Reaction mixture was then diluted with water and extracted with Et2O (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 3% EtOAc/hexanes) provided 99% of desired product as a clear oil. −34.1° (c 2.0, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.64 (m, 4H), 7.36 (m, 5H), 3.91 (dt, J = 4.8, 9.6 Hz, 1H), 3.84 (dd, J = 2.2, 11.8, 1H), 3.76 (m, 1H), 3.72 (s, 3H), 3.17 (dt, J = 2.2, 9.4 Hz, 1H), 2.00- 1.81 (m, 3H), 1.67- 1.53 (m, 2H), 1.40 (m, 1H), 1.23 (m, 1H), 1.02 (s, 9H), 0.81 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 172.1, 135.56, 135.55, 134.1, 134.0, 129.47, 129.44, 127.55, 127.53, 80.2, 60.0, 51.9, 35.7, 34.6, 32.7, 29.4, 26.9, 19.2, 17.7; IR (neat) νmax: 2955, 2930, 2856, 1428, 1112, 702 cm−1; HRMS (CI, NH3) m/z calc’d for C26H36O4Si [M]+ 440.2383, found 440.2334.
(2S,5R,6S)-6-(2-(tert-butyldiphenylsilyloxy)ethyl)-5-methyltetrahydro-2H-pyran-2-yl)methanol
To a solution of ester (1.24 g, 28.1 mmol) in diethyl ether (25.0 mL) at 0 °C was added lithium tetrahydroborate (1.23 g, 5.60 mmol) in one portion. Reaction mixture was allowed to warm to room temperature and stirred for additional 2 hours. Reaction was quenched by addition of water and stirred until bubbling ceased. Layers were separated and the aqueous layer was extracted with Et2O (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 10% EtOAc/hexanes) provided 88% of desired product as a clear oil. −26.0° (c 1.1, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.66 (m, 4H), 7.38 (m, 6H), 3.78 (m, 2H), 3.50 (dt, J = 3.0, 8.8 Hz, 1H), 3.43-3.30 (m, 2H), 3.12 (dt, J = 2.0, 9.6 Hz, 1H), 1.96 (m, 1H), 1.78 (m, 2H), 1.54 (m, 1H), 1.45 (m, 1H), 1.34-1.11 (m, 2H), 1.03 (s, 9H), 0.80 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 135.5, 134.0, 129.5, 127.6, 79.8, 77.5, 66.3, 60.3, 36.1, 35.1, 32.4, 27.6, 26.9, 19.2, 17.7; IR (neat) νmax: 3448, 3069, 2931, 2858, 1428, 1102, 703 cm−1; HRMS (CI, NH3) m/z calc’d for C25H38O3Si [M+23]+ 414.2593, found 414.2560.
tert-butyl(2-((2S,3R,6S)-6-(iodomethyl)-3-methyltetrahydro-2H-pyran-2-yl)ethoxy) diphenylsilane (ent-25c)
To a solution of alcohol (3.60 g, 8.72 mmol) and 2,6-lutidine (2.38 mL, 20.6 mmol) in N,N-dimethylformamide (70.0 mL) at 0 °C was added methyltriphenoxyphosphonium iodide (8.48 g, 18.8 mmol) in one portion. Reaction stirred for 1 hour at 0 °C before diethyl ether and water were added. Layers were separated and aqueous layer was extracted with diethyl ether (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 3% EtOAc/hexanes) provided ent-25c in 79% as a clear oil. −3.9° (c 1.5, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.66 (m, 4H), 7.35 (m, 5H), 3.85 (m, 2H), 3.23 (m, 1H), 3.12 (dt, J = 2.0, 9.6 Hz, 1H), 3.05 (d, J = 5.6 Hz, 2H), 1.95 (m, 1H), 1.83-1.74 (m, 2H), 1.53 (m, 2H), 1.30-1.16 (m, 2H), 1.04 (s, 9H), 0.80 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 135.8, 134.4, 129.7, 127.8, 80.6, 77.7, 60.6, 36.3, 35.0, 32.9, 31.9, 27.2, 19.5, 17.7, 10.0; IR (neat) νmax: 3068, 2931, 2857, 1465, 1097, 702 cm−1; HRMS (CI, NH3) m/z calc’d for C25H35IO2Si [M+23]+ 545.1349, found 545.1363.
tert-butyl(2-((2S,3R,6S)-3-methyl-6-((2-((E)-prop-1-enyl)-1,3-dithian-2-yl)methyl)tetrahydro-2H-pyran-2-yl)ethoxy)diphenylsilane (ent-26c)
To a solution of 2-((E)-1-propenyl)-[1,3]-dithiane (2.02 g, 12.6 mmol) and hexamethylphosphoramide (3.30 mL, 18.9 mmol) in tetrahydrofuran (50.0 mL) at −78 °C was added tert-butyllithium in pentane (1.70 M, 7.43 mL) dropwise. Reaction mixture was stirred for 1 hour at −78 °C before a solution of iodide ent-25c (3.30 g, 6.32 mmol) in tetrahydrofuran (50.0 mL) was added dropwise. Reaction mixture was allowed to stir for 1 hour at −78 °C and then was warmed to 0 °C and stirred for an additional one hour. Reaction was quenched by addition of water and extracted with diethyl ether (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 1% EtOAc/hexanes) provided ent-26c in 90% as a clear oil. −7.9° (c 2.5, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.65 (m, 4H), 7.37 (m, 6H), 5.80 (m, 1H), 5.43 (d, J = 15.2 Hz, 1H), 3.83 (m, 2H), 3.38 (m, 1H), 2.99 (dt, J = 2.0, 9.6 Hz, 1H), 2.76 (m, 1H), 2.67-2.56 (m, 3H), 2.09 (dd, J = 6.8, 14.8 Hz, 1H), 1.91-1.75 (m, 4H), 1.70 (m, 1H), 1.64 (d, J = 6.4 Hz, 3H), 1.55 (m, 2H), 1.35-1.13 (m, 3H), 1.02 (s, 9H), 0.76 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 135.6, 134.3, 133.7, 129.4, 128.4, 127.5, 80.4, 74.3, 61.4, 53.1, 48.1, 36.3, 34.8, 33.2, 33.1, 27.1, 27.0, 26.9, 25.2, 19.2, 17.8, 17.5; IR (neat) νmax: 3068, 2928, 2856, 1428, 1087, 703 cm−1; HRMS (CI, NH3) m/z calc’d for C32H46O2S2Si [M]+ 554.2708, found 554.2720.
(E)-1-((2S,5R,6S)-6-(2-(tert-butyldiphenylsilyloxy)ethyl)-5-methyltetrahydro-2H-pyran-2-yl)pent-3-en-2-one (ent-27c)
To a solution of dithiane ent-26c (100 mg, 0.180 mmol) in a mixture of acetonitrile (4.00 mL), methylene chloride (0.500 mL), and water (0.500 mL) was added Dess-Martin periodinane in methylene chloride (0.30 M, 1.20 mL) dropwise. Reaction was allowed to stir at room temperature for 5 hours before ethyl acetate and water were added. Layers were separated and water was extracted with ethyl acetate (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 2–5% EtOAc/hexanes) provided ent-27c in 60% as a clear oil. −22.7° (c 1.1, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 7.64 (m, 4H), 7.34 (m, 6H), 6.76 (m, 1H), 6.07 (d, J = 15.6 Hz, 1H), 3.79-3.63 (m, 3H), 3.09 (dt, J = 1.6, 9.2 Hz, 1H), 2.71 (A of ABX, J = 6.0, 14.8 Hz, 1H), 2.46 (B of ABX, J = 6.0, 15.2 Hz, 1H), 1.91 (m, 1H), 1.79 (d, J = 6.8 Hz, 3H), 1.74-1.63 (m, 2H), 1.50 (m, 1H), 1.22 (m, 3H), 1.02 (s, 9H), 0.78 (d, J = 6.0 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 198.7, 143.0, 135.5, 134.1, 132.5, 129.4, 127.5, 80.2, 74.3, 60.4, 46.6, 36.2, 35.0, 32.8, 32.2, 29.7, 26.9, 19.2, 18.2; IR (neat) νmax: 3068, 2954, 2857, 1672, 1632, 1431, 1108, 703 cm−1; HRMS (CI, NH3) m/z calc’d for C29H40O3Si [M+23]+ 487.2644, found 487.2681.
(E)-1-((2S,5R,6S)-6-(2-hydroxyethyl)-5-methyltetrahydro-2H-pyran-2-yl)pent-3-en-2-one
To a solution of ent-27c (100 mg, 0.215 mmol) in a nalgene vial in acetonitrile (10.0 mL) at room temperature was added hydrofluoric acid (1.00 mL) dropwise. Reaction was allowed to stir for 3 hours at room temperature before it was quenched by slow addition of saturated NaHCO3. The aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 50% EtOAc/hexanes) provided product in 95% as a clear oil. −25.3° (c 2.9, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 6.85 (dq, J = 6.8, 13.6 Hz, 1H), 6.11 (ddd, J = 1.6, 3.2, 15.8 Hz, 2H), 3.81 (m, 1H), 3.71 (dt, J = 1.6, 4.4 Hz, 2H), 3.20 (dt, J = 2.8, 9.6 Hz, 1H), 2.79 (A of ABX, J = 8.0, 15.6 Hz, 1H), 2.51 (B of ABX, J = 4.8, 15.6 Hz, 1H), 1.88 (dd, J = 1.8, 7.0 Hz, 3H), 1.84 (m, 1H), 1.76 (dq, J = 2.8, 6.8 Hz, 1H), 1.66-1.52 (m, 2H), 1.40-1.17 (m, 3H), 0.79 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 198.3, 143.6, 132.5, 84.6, 74.3, 61.5, 46.0, 35.0, 34.7, 32.5, 31.8, 18.3, 17.6; IR (neat) νmax: 3447, 2924, 2873, 1670, 1631, 1441, 1085, 973 cm−1; HRMS (CI, NH3) m/z calc’d for C13H23O3 [M+1]+ 227.1647, found 227.1670.
2-((2S,3R,6S)-3-methyl-6-((E)-2-oxopent-3-enyl)tetrahydro-2H-pyran-2-yl)ethanoic acid (ent-6c)
To a solution of alcohol (45.0 mg, 0.199 mmol) in acetonitrile (1 ml) and water (15 µL) mixture at 0 °C was added CrO3/H5IO6 stock solution (500 µL) dropwise. Reaction stirred at 0 °C for 1/2 hour before another equivalent of CrO3/H5IO6 stock solution (500 µL) was added dropwise and reaction stirred for 1/2 hour at 0°C. Reaction was quenched by addition of Na2HPO4 (60mg/1ml H2O), diluted with ethyl ether and stirred for 30 minutes at room temperature. Layers were separated and the aqueous layer was extracted with ethyl ether (3×). Combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 50% EtOAc/hexanes) provided ent-6c in 80% as a clear oil. −35.1° (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 6.84 (dq, J = 7.0, 13.8, 15.6 Hz, 1H), 6.10 (dq, J = 1.6, 3.2, 15.6 Hz, 1H), 3.86 (m, 1H), 3.42 (dt, J = 3.0, 9.4 Hz, 1H), 2.80 (A of ABX, J = 7.6, 15.6 Hz, 1H), 2.67 (A of ABX, J = 2.8, 15.6 Hz, 1H), 2.57 (B of ABX, J = 5.0, 15.6 Hz, 1H), 2.38 (B of ABX, J = 9.6, 15.6 Hz, 1H), 1.87 (dd, J = 1.6, 6.8 Hz, 3H), 1.79 (m, 1H), 1.68 (m, 1H) 1.41-1.20 (m, 3H), 0.83 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 75 MHz): δ 198.2, 174.5, 143.8, 132.2, 79.9, 74.4, 45.9, 38.7, 34.8, 32.1, 31.6, 18.3, 17.4; IR (neat) νmax: 2929, 1713, 1631, 1438, 1194, 1082, 971 cm−1; HRMS (CI, NH3) m/z calc’d for C13H20O4 [M+23]+ 263.1259, found 263.1249.
(2S,3R)-triisopropylsilyl 3-(tert-butyldimethylsilyloxy)-2-methyl-4-(2-((2S,3R,6S)-3-methyl-6-((E)-2-oxopent-3-enyl)tetrahydro-2H-pyran-2-yl)ethanamido)butanoate (ent-36j)
A solution of azide (1.00 mg, 0.233 mmol) in tetrahydrofuran (10.0 mL) was treated with palladium 10 wt.% on activated carbon (5.0 mg). Reaction flask was flushed with argon and hydrogen atmospheres sequentially. Reaction was then placed under an atmosphere of hydrogen and stirred for 12 hours. The heterogeneous mixture was filtered over Celite®, rinsed with tetrahydrofuran, and concentrated in vacuo. The resulting thick oil of 7a was used in the next step without further purification.
To a solution of the crude amine 7a in methylene chloride (10.0 mL) was added crude acid ent-6c (50.0 mg, 0.208 mmol), PyBOP (119 mg, 0.229 mmol), and triethylamine (34.8 µL, 0.250 mmol). Reaction mixture was allowed to stir for 24 hours at room temperature. Upon completion of the reaction, diethyl ether was added. The organic layer was washed sequentially with water, saturated sodium bicarbonate, and brine solutions. The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 20% EtOAc/hexanes) provided product ent-36j in 71% as a clear oil. −9.5° (c 1.0, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 6.81 (m, 2H), 6.06 (dd, J = 1.6, 16.0 Hz, 1H), 4.07 (q, J = 6.4, 10.4 Hz, 1H), 3.79 (m, 1H), 3.48 (quint, J = 6.8, 13.2 Hz, 1H), 3.29 (dt, J = 1.8, 9.2 Hz, 1H), 3.17 (dt, J = 5.4, 13.6 Hz, 1H), 2.77 (A of ABX, J = 7.0, 16.8 Hz, 1H), 2.63 (m, 1H), 2.56 (B of ABX, J = 5.8, 16.8 Hz, 1H), 2.49 (A of ABX, J = 2.0, 15.6 Hz, 1H), 2.20 (B of ABX, J = 9.2, 15.6 Hz, 1H), 1.84 (dd, J = 1.6, 6.8 Hz, 3H), 1.73 (m, 1H), 1.62 (m, 1H), 1.24 (m, 6H), 1.13 (d, J = 7.6 Hz, 3H), 1.03 (d, J = 7.6 Hz, 18 H), 0.82 (s, 9H), 0.79 (d, J = 6.4 Hz, 3H), 0.04 (s, 3H), 0.03 (s, 3H); 13C NMR (CDCl3, 75 MHz): δ 197.6, 173.7, 171.3, 142.9, 132.1, 80.6, 73.7, 71.9, 46.1, 45.1, 42.6, 40.5, 34.6, 32.3, 31.5, 25.8, 18.2, 18.0, 17.8, 17.7, 12.8, 11.8, −4.5, −4.9; IR (neat) νmax: 3361, 2932, 2867, 1674, 1464, 1194, 1085, 837 cm−1; HRMS (CI, NH3) m/z calculated for C33H64O6NSi2 [M+1]+ 626.4272, found 626.4299.
(2S,5S,E)-7-((2S,6S,8R,9S)-8-(3-azidopropyl)-9-methyl-1,7-dioxaspiro[5.5]undecan-2-yl)-3,5-dimethylhept-3-en-2-ol ((S)-8)
To a solution of 51 (50.0 mg, 0.0986 mmol) in a mixture of methylene chloride (1.0 mL) and methanol (1.0 mL) at 0 °C was added 10-camphorsulfonic acid (7.0 mg, 0.03 mmol). Reaction was allowed to stir at 0 °C for 2 hours before it was quenched with saturated solution of NaHCO3. The aqueous layer was extracted with dichloromethane (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by column chromatography (silica, 15% EtOAc/hexanes) affords alcohol (S)-8 in 85% as a clear oil. −36.9° (c 0.3, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 5.16 (d, J = 9.2 Hz, 1H), 4.17 (q, J = 6.0, 12.4 Hz, 1H), 3.41 (br. t, 1H), 3.30 (m, 2H), 3.14 (t, J = 9.6 Hz, 1H), 2.33 (m, 1H), 1.91 (m, 1H), 1.75 (m, 2H), 1.60 (s, 3H), 1.58-1.28 (m, 15H), 1.23 (d, J = 6.4 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 137.2, 131.4, 95.5, 74.1, 73.4, 69.2, 51.9, 36.0, 35.4, 35.0, 34.1, 33.6, 31.9, 31.3, 30.3, 27.9, 25.5, 21.6, 21.0, 19.2, 18.0, 11.8; IR (neat) νmax: 3407, 2932, 2868, 2095, 1457, 1225, 1096, 985 cm−1; HRMS (CI, NH3) m/z calc’d for C22H39O3N3 [M+23]+ 416.2889, found 416.2913.
(S,E)-7-((2S,6S,8R,9S)-8-(3-azidopropyl)-9-methyl-1,7-dioxaspiro[5.5]undecan-2-yl)-3,5-dimethylhept-3-en-2-one
To a solution of (S)-8 (38.0 mg, 0.0966 mmol) in methylene chloride (9.70 mL) at 0 °C was added Dess-Martin periodinane (0.552 mL, 0.35 M in methylene chloride) dropwise. Reaction was allowed to warm up to room temperature over 1 hour and was diluted with ethyl acetate. The organic layer was washed with saturated solution of sodium bicarbonate followed by sodium chloride solution. The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by column chromatography (silica, 10% EtOAc/hexanes) gives product as a colorless glass (35.0 mg, 0.0894 mmol, 92.5%). +37.8° (c 0.8, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 6.37 (d, J = 9.6 Hz, 1H), 3.43 (m, 1H), 3.31 (d quint, 2H), 3.13 (dt, J = 2.4, 9.6 Hz, 1H), 2.55 (m, 1H), 2.30 (s, 3H), 1.92 (m, 1H), 1.80 (m, 1H), 1.76 (s, 3H), 1.71 (m, 1H), 1.62-1.44 (m, 8H), 1.36 (m, 7H), 1.13 (dq, J = 3.6, 12.8, 25.2 Hz, 1H), 1.03 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 200.2, 149.2, 136.3, 95.5, 74.2, 69.0, 51.9, 36.0, 35.4, 34.9, 34.2, 33.7, 32.9, 31.3, 30.3, 27.8, 25.5, 25.4, 20.0, 19.1, 18.0, 11.4; IR (neat) νmax: 2932, 2869, 2095, 1670, 1457, 1096, 986 cm−1; HRMS (CI, NH3) m/z calc’d for C22H37O3N3 [M+23]+ 414.2733, found 414.2733.
(2R,5S,E)-7-((2S,6S,8R,9S)-8-(3-azidopropyl)-9-methyl-1,7-dioxaspiro[5.5]undecan-2-yl)-3,5-dimethylhept-3-en-2-ol ((R)-8)
To a solution of ketone (13.0 mg, 0.0332 mmol) in toluene (2.00 mL) at −78 °C was added (S)-CBS (39.8 µL, 0.0398 mmol, 1.0 M in toluene) dropwise. After 5 minutes, catecholborane (66.4 uL, 0.0664 mmol, 1.0 M in toluene) was added dropwise and the reaction mixture was allowed to stir at −78 °C for 24 hours. Reaction was quenched by addition of methanol (0.070 mL) and was allowed to warm up to room temperature and was stirred for one hour. The mixture was diluted with ethyl acetate and saturated solution of sodium bicarbonate. The aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by column chromatography (silica, 15% EtOAc/hexanes) affords alcohol (R)-8 as a colorless glass (13.0 mg, 0.0330 mmol, 99%). +39.6° (c 0.3, CHCl3); 1H NMR (CDCl3, 400 MHz): δ 5.16 (d, J = 9.2 Hz, 1H), 4.17 (q, J = 6.0, 12.4 Hz, 1H), 3.41 (br. t, 1H), 3.30 (m, 2H), 3.14 (t, J = 9.6 Hz, 1H), 2.33 (m, 1H), 1.91 (m, 1H), 1.75 (m, 2H), 1.60 (s, 3H), 1.58-1.28 (m, 15H), 1.23 (d, J = 6.4 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz): δ 137.2, 131.4, 95.5, 74.1, 73.4, 69.2, 51.9, 36.0, 35.4, 35.0, 34.1, 33.6, 31.9, 31.3, 30.3, 27.9, 25.5, 21.6, 21.0, 19.2, 18.0, 11.8; IR (neat) νmax: 3407, 2932, 2868, 2095, 1457, 1225, 1096, 985 cm−1; HRMS (CI, NH3) m/z calc’d for C22H39O3N3 [M+23]+ 416.2889, found 416.2913.
(2S,3R)-3-hydroxy-N-(3-((2R,3S,6S,8S)-8-((3S,6S,E)-6-hydroxy-3,5-dimethylhept-4-enyl)-3-methyl-1,7-dioxaspiro[5.5]undecan-2-yl)propyl)-2-methyl-4-(2-((2S,3R,6S)-3-methyl-6-((E)-2-oxopent-3-enyl)tetrahydro-2H-pyran-2-yl)ethanamido)butanamide (1.21)
To a solution of azide (S)-8 (5.00 mg, 0.0127 mmol) in a mixture of tetrahydrofuran (0.900 mL) and water (0.300 mL) at room temperature was added trimethyl phosphine (63.5 µL, 1.0 M in tetrahydrofuran) dropwise. Reaction was allowed to stir for 1 hour and was diluted with saturated solution of sodium chloride. The aqueous layer was extracted with ethyl ether (3×). The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The crude, clear oil was used directly in the coupling step without further purification.
To a solution of TIPS acid ent-36j (8.00 mg, 0.0128 mmol) in THF (1.30 mL) at 0 °C was added TBAF (14.7 µL, 1.00 M in THF) dropwise. Reaction was stirred for 1/2 h before it was diluted with ethyl acetate. The organic layer was washed with 0.01 M HCl and brine solutions. The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. The crude acid was used in the coupling step without further purification.
To a solution of C1-C18 acid and C19-C40 amine (S)-52 (6.00 mg, 0.0163 mmol) in methylene chloride (1.60 mL) at room temperature was added PyBOP (9.34 mg, 0.0180 mmol) and triethylamine (3.18 µL, 0.0228 mmol). Reaction mixture was allowed to stir for 2 hours at room temperature before it was concentrated in vacuo. Purification by column chromatography (silica, 80% EtOAc/hexanes) affords coupled product 53.1 as a colorless oil (10.0 mg, 0.0122 mmol, 74.8%, 2 steps).
To a solution of 53.1 (10.0 mg, 0.0122 mmol) in tetrahydrofuran (1.20 mL) at 0 °C was added TBAF (14.6 µL, 1.00 M in THF) dropwise. Reaction was stirred for 2 hours at 0 °C before it was diluted with ethyl ether. The organic layer was washed with 0.1 M HCl and brine solutions sequentially. The combined organic layers were dried with magnesium sulfate, filtered, and concentrated under reduced pressure. Purification by flash chromatography (silica, 100% EtOAc) provided product 1.21 in 63% (3 steps) as a clear oil. +18.5° (c 0.6, CHCl3); 1H NMR (C6D6, 400 MHz): δ 7.66 (t, J = 5.6 Hz, 1H), 7.24 (obs. t, 1H), 6.59 (dq, J = 6.8, 13.6, 16.0 Hz, 1H), 5.88 (dd, J = 1.6, 16.0 Hz, 1H), 5.37 (dd, J = 0.8, 8.0 Hz, 1H), 5.27 (d, J = 6.0 Hz, 1H), 4.11 (q, J = 6.4, 12.8 Hz, 1H), 3.89 (m, 1H), 3.81 (t, J = 4.8 Hz, 1H), 3.72 (m, 1H), 3.58 (m, 2H), 3.42 (t, J = 9.4 Hz, 1H), 3.27 (m, 1H), 3.03 (t, J = 9.8 Hz, 1H), 2.47 (m, 3H), 2.23 (B of ABX, J = 10.0, 16.4 Hz, 1H), 2.00 (m, 3H), 1.85-0.85 (m, 25H), 1.65 (s, 3H), 1.43 (dd, J = 1.6, 6.8 Hz, 3H), 1.33 (d, J = 6.8 Hz, 3H), 1.26 (d, J = 6.4 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.4 Hz, 3H), 0.53 (d, J = 6.4 Hz, 3H); 13C NMR (C6D6, 75.0 MHz): δ 197.5, 175.4, 172.7, 143.4, 138.4, 132.0, 130.5, 95.5, 80.4, 74.9, 74.2, 73.3, 72.8, 69.4, 45.9, 45.3, 43.2, 40.2, 39.5, 36.6, 36.0, 35.4, 34.7, 34.6, 34.1, 32.5, 32.3, 31.7, 31.5, 30.6, 28.5, 26.3, 22.3, 21.4, 19.8, 18.2, 17.9, 17.5, 16.0, 12.6; IR (neat) νmax: 3328, 2928, 2868, 1645, 1549 cm−1; HRMS (CI, NH3) m/z calculated for C40H68N2NaO8 [M+Na]+ m/z calculated for 727.4873, found: 727.4865.
Supplementary Material
Figure 3.

Cell-Growth Inhibition of Stereochemical Analogs 1.1–1.10 Against Renal Carcinoma Cell Line UO-31.
Figure 4.

Cell Cycle Analysis of the UO-31 Cell Line for the Most Potent Cell Growth Inhibiting Isomers 1.1 and 1.33 and Least Active Isomer 1.7.
Figure 5.

Cell Cycle Analysis of the SF-295 Cell Line for the Most Potent Cell Growth Inhibiting Analogs 1.1 and 1.33 and Least Active Stereochemical Compound 1.7.
Figure 6.

Confocal Microscopy Using Phalloidin Staining Images of UO-31 Cells. Bistramide 1.1 causes disruption of F-actin in dose dependent manner, while least active epimer 1.7 does not.
Figure 7.

Confocal Microscopy Using Phalloidin Staining Images of SF-295 Cells. Bistramide 1.1 and stereochemical analog 1.33 causes disruption of F-actin in dose dependent manner, while least active epimer 1.7 does not.
Scheme 6.

Possible mechanism for (Z)-crotylsilane ent-9b.
Acknowledgments
Financial support for this research is obtained from NIH CA56304. JSP is grateful to Amgen, Johnson & Johnson, Merck Co., Novartis, Pfizer and GSK for financial support. IEW acknowledges a Novartis Graduate Fellowship. JTL acknowledges an ACS Graduate Fellowship (sponsored by Bristol-Myers Squibb) and Merck Co. Graduate Fellowship. Supported by the Intramural Research Program of the National Cancer Institute, NIH, Center for Cancer Research. Flow cytometry and cell cycle analysis was done by Kathleen Noer, Roberta Matthai and Samantha Bauchiero of the CCR-Frederick Flow Cytometry Core, SAIC-Frederick, Inc. We thank the Developmental Therapeutics Program, NCI, for 60-cell testing. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400.
Footnotes
Supporting Information Available: Complete spectroscopic and analytical data including copies 1H- and 13C-NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nobes CD, Hall A. Cell. 1995;81:53. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 2.Cooper JA. J. Cell Biol. 1987;105:1473. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Allingham JS, Klenchin VA, Rayment I. Cell Mol. Life Sci. 2006;63:2119. doi: 10.1007/s00018-006-6157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Saito S, Watabe S, Ozaki H, Kigoshi H, Yamada K, Fusetani N, Karaki H. J. Biochem. 1996;120:552. doi: 10.1093/oxfordjournals.jbchem.a021449. [DOI] [PubMed] [Google Scholar]; (c) Bubb MR, Spector I, Bershadsky AD, Korn ED. J. Biol. Chem. 1995;270:3463. doi: 10.1074/jbc.270.8.3463. [DOI] [PubMed] [Google Scholar]
- 4.Statsuk AV, Bai R, Baryza JL, Verma VA, Hamel E, Wender PA, Kozmin SA. Nat. Chem. Biol. 2005;1:383. doi: 10.1038/nchembio748. [DOI] [PubMed] [Google Scholar]
- 5.Rizvi SA, Tereshko V, Kossiakoff AA, Kozmin SA. J. Am. Chem. Soc. 2006;128:3882. doi: 10.1021/ja058319c. [DOI] [PubMed] [Google Scholar]
- 6.(a) Zhang Q, Lu H, Richard C, Curran DP. J. Am. Chem. Soc. 2004;126:36. doi: 10.1021/ja038542e. [DOI] [PubMed] [Google Scholar]; (b) Curran DP, Zhang Q, Richard C, Lu H, Gudipati V, Wilcox CS. J. Am. Chem. Soc. 2006;128:9561. doi: 10.1021/ja061801q. [DOI] [PubMed] [Google Scholar]; (c) Dandapani S, Jeske M, Curran DP. J. Org. Chem. 2005;70:9447. doi: 10.1021/jo051526a. [DOI] [PubMed] [Google Scholar]
- 7.(a) Gouiffès D, Moreau S, Helbecque N, Bernier JL, Hénichart JP, Barbin Y, Laurent D, Verbist JF. Tetrahedron. 1988;44:451. [Google Scholar]; (b) Gouiffès D, Juge M, Grimaud N, Welin L, Sauviat MP, Barbin Y, Laurent D, Roussakis C, Henichart JP, Verbist JF. Toxicon. 1988;26:1129. doi: 10.1016/0041-0101(88)90297-8. [DOI] [PubMed] [Google Scholar]
- 8.Biard J-F, Roussakis C, Kornprobst J-M, Gouiffès-Barbin D, Verbist J-F, Cotelle P, Foster MP, Ireland CM, Debitus C. J. Nat. Prod. 1994;57:1336. doi: 10.1021/np50112a002. [DOI] [PubMed] [Google Scholar]
- 9.Degnan BM, Hawkins CJ, Lavin MF, McCaffrey EJ, Parry DL, Watters DJ. J. Med. Chem. 1989;32:1354. doi: 10.1021/jm00126a035. [DOI] [PubMed] [Google Scholar]
- 10.Foster MP, Mayne CL, Dunkel R, Pugmire RJ, Grant DM, Kornprobst JM, Verbist JF, Biard JF, Ireland CM. J. Am. Chem. Soc. 1992;114:1110. [Google Scholar]
- 11.(a) Solladié G, Baudet C, Biard JF. Tetrahedron Lett. 2000;41:7747. [Google Scholar]; (b) Gallagher PO, McErlean CSP, Jacobs MF, Watters DJ, Kitching W. Tetrahedron Lett. 2002;43:531. [Google Scholar]; (c) Wipf P, Uto Y, Yoshimura S. Chem. Eur. J. 2002;8:1670. doi: 10.1002/1521-3765(20020402)8:7<1670::aid-chem1670>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (d) Zuber G, Goldsmith M-R, Hopkins TD, Beratan DN, Wipf P. Org. Lett. 2005;7:5269. doi: 10.1021/ol052154v. [DOI] [PubMed] [Google Scholar]
- 12.Statsuk AV, Liu D, Kozmin SA. J. Am. Chem. Soc. 2004;126:9546. doi: 10.1021/ja046588h. [DOI] [PubMed] [Google Scholar]
- 13.Lowe JT, Panek JS. Org. Lett. 2005;7:3231. doi: 10.1021/ol050982i. [DOI] [PubMed] [Google Scholar]
- 14.Bauder C, Biard J-F, Solladié G. Org. Biomol. Chem. 2006;4:1860. doi: 10.1039/b603767d. [DOI] [PubMed] [Google Scholar]
- 15.(a) Yadav JS, Chetia L. Org. Lett. 2007;9:4587. doi: 10.1021/ol702095n. [DOI] [PubMed] [Google Scholar]; (b) Lowe JT, Wrona IE, Panek JS. Org. Lett. 2007;9:327. doi: 10.1021/ol062957y. [DOI] [PubMed] [Google Scholar]; (c) Crimmins MT, DeBaillie AC. J. Am. Chem. Soc. 2006;128:4936. doi: 10.1021/ja057686l. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wipf P, Hopkins TD. Chem. Commun. 2005:3421. doi: 10.1039/b505100b. [DOI] [PubMed] [Google Scholar]
- 16.(a) Johnson WEB, Watters DJ, Suniara RK, Brown G, Bunce CM. Biochem. Biophys. Res. Commun. 1999;260:80. doi: 10.1006/bbrc.1999.0854. [DOI] [PubMed] [Google Scholar]; (b) Gautret P, Le Pape P, Biard JF, Menard D, Verbist JF, Marjolet M. Acta Parasitol. 1998;43:50. [Google Scholar]; (c) Pusset J, Maillere B, Debitus C. J. Nat. Toxins. 1996;5:1. [Google Scholar]; (d) Riou D, Roussakis C, Robillard N, Biard JF, Verbist JF. Biol. Cell. 1993;77:261. doi: 10.1016/s0248-4900(05)80196-0. [DOI] [PubMed] [Google Scholar]; (e) Sauviat MP, Verbist JF. Gen. Physiol. Biophys. 1993;12:465. [PubMed] [Google Scholar]; (f) Sauviat MP, Chesnais JM, Choukri N, Diacono J, Biard JF, Verbist JF. Cell Calcium. 1993;14:301. doi: 10.1016/0143-4160(93)90051-7. [DOI] [PubMed] [Google Scholar]; (g) Sauviat MP, Gouiffès-Barbin D, Ecault E, Verbist JF. Biochim. Biophys. Acta, Biomembranes. 1992;1103:109. doi: 10.1016/0005-2736(92)90063-r. [DOI] [PubMed] [Google Scholar]
- 17.Griffiths G, Garrone B, Deacon E, Owen P, Pongracz J, Mead G, Bradwell A, Watters D, Lord J. Biochem. Biophys. Res. Commun. 1996;222:802. doi: 10.1006/bbrc.1996.0830. [DOI] [PubMed] [Google Scholar]
- 18.Rizvi SA, Courson DS, Keller VA, Rock RS, Kozmin SA. Proc. Natl. Acad. Sci. U.S.A. 2008;105:4088. doi: 10.1073/pnas.0710727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allingham JS, Zampella A, D’Auria MV, Rayment I. Proc. Natl. Acad. Sci. USA. 2005;102:14527. doi: 10.1073/pnas.0502089102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klenchin VA, Allingham JS, King R, Tanaka J, Marriott G, Rayment I. Nat. Struct. Biol. 2003;10:1058. doi: 10.1038/nsb1006. [DOI] [PubMed] [Google Scholar]
- 21.(a) Hirata K, Muraoka S, Suenaga K, Kuroda T, Kato K, Tanaka H, Yamamoto M, Takata M, Yamada K, Kigoshi H. J. Mol. Biol. 2006;356:945. doi: 10.1016/j.jmb.2005.12.031. [DOI] [PubMed] [Google Scholar]; (b) Klenchin VA, King R, Tanaka J, Marriott G, Rayment I. Chem. Biol. 2005;12:287. doi: 10.1016/j.chembiol.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 22.For a review see: Yeung KS, Paterson I. Angew. Chem. Int. Ed. 2002;41:4632. doi: 10.1002/anie.200290057. Spector I, Braet F, Shochet NR, Bubb MR. Microsc. Res. Tech. 1999;47:18. doi: 10.1002/(SICI)1097-0029(19991001)47:1<18::AID-JEMT3>3.0.CO;2-E. Fenteany G, Zhu S. Curr. Top. Med. Chem. 2003;3:593. doi: 10.2174/1568026033452348.
- 23.For structural activity relationship studies on bistramide A, see reference 18.
- 24.Melville JL, Moal IH, Baker-Glenn C, Shaw PE, Pattenden G, Hirst JD. Biophys. J. 2007;92:3862. doi: 10.1529/biophysj.106.103580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H, Panek JS. J. Am. Chem. Soc. 2000;122:9836. [Google Scholar]
- 26.(a) Huang H, Panek JS. Org. Lett. 2004;6:4383. doi: 10.1021/ol0480325. [DOI] [PubMed] [Google Scholar]; (b) Su Q, Panek JS. J. Am. Chem. Soc. 2004;126:2425. doi: 10.1021/ja037957x. [DOI] [PubMed] [Google Scholar]; (c) Su Q, Dakin L, Panek JS. J. Org. Chem. 2007;72:2. doi: 10.1021/jo0610412. [DOI] [PubMed] [Google Scholar]; (d) Zhang Y, Panek JS. Org. Lett. 2007;9:3141. doi: 10.1021/ol701427k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Youngsaye W, Lowe JT, Pohlki F, Ralifo P, Panek JS. Angew. Chem. Int. Ed. 2007;46:9211. doi: 10.1002/anie.200704122. [DOI] [PubMed] [Google Scholar]; (f) Lowe JT, Panek JS. Org. Lett. 2008;10:3813. doi: 10.1021/ol801499s. [DOI] [PubMed] [Google Scholar]
- 27.Masse CE, Panek JS. Chem. Rev. 1995;95:1293. [Google Scholar]
- 28.(a) Sparks MA, Panek JS. J. Org. Chem. 1991;56:3431. [Google Scholar]; (b) Panek JS, Yang M. J. Org. Chem. 1991;56:5755. [Google Scholar]; (c) Panek JS, Clark TD. J. Org. Chem. 1992;57:4323. [Google Scholar]
- 29.Hudrlik PF, Hudrlik AM. Advances in Silicon Chemistry. Vol. 2. Greenwich, CT: JAI Press; 1993. p. 1. [Google Scholar]
- 30.(a) Chakraborty TK, Laxman P. Tetrahedron Lett. 2003;44:4989. [Google Scholar]; (b) Hudrlik PF, Gebreselassie P, Tafesse L, Hudrlik AM. Tetrahedron Lett. 2003;44:3409. [Google Scholar]; (c) Kim K, Okamoto S, Takayama Y, Sato F. Tetrahedron Lett. 2002;43:4237. [Google Scholar]; (d) Whitham GH. Sci. Synth. 2002;4:633. [Google Scholar]; (e) Babudri F, Fiandanese V, Marchese G, Punzi A. Tetrahedron. 2001;57:549. [Google Scholar]; (f) Chakraborty TK, Reddy GV. Tetrahedron Lett. 1990;31:1335. [Google Scholar]
- 31.For examples of Sharpless asymmetric epoxidation to generate epoxysilanes see: Chauret DC, Chong JM, Ye Q. Tetrahedron: Asymmetry. 1999;10:3601. Kobayashi Y, Ito T, Yamakawa I, Urabe H, Sato F. Synlett. 1991;811 Takeda Y, Matsumoto T, Sato F. J. Org. Chem. 1986;51:4728. For an example of Shi epoxidation see: Heffron TP, Jamison TF. Org. Lett. 2003;5:2339. doi: 10.1021/ol0347040. For examples of kinetic resolutions see: Carlier PR, Mungall WS, Schroder G, Sharpless KB. J. Am. Chem. Soc. 1988;110:2978. Kitano Y, Matsumoto T, Sato F. J. Chem. Soc., Chem. Comm. 1986;17:1323. For substrate directed epoxidations using VO(acac)2 see: Kobayashi Y, Uchiyama H, Kanbara H, Sato F. J. Am. Chem. Soc. 1985;107:5541.
- 32.(a) Adam J-M, de Fays L, Laguerre M, Ghosez L. Tetrahedron. 2004;60:7325. [Google Scholar]; (b) Landais Y, Mahieux C, Schenk K, Surange SS. J. Org. Chem. 2003;68:2779. doi: 10.1021/jo026774a. [DOI] [PubMed] [Google Scholar]; (c) Ohi H, Inoue S, Iwabuchi Y, Irie H, Hatakeyama S. Synlett. 1999;11:1757. [Google Scholar]; (d) Fleming I, Barbero A, Walter D. Chem. Rev. 1997;97:2063. doi: 10.1021/cr941074u. [DOI] [PubMed] [Google Scholar]; (e) Chauret DC, Chong JM. Tetrahedron Lett. 1993;34:3695. [Google Scholar]; (f) Russell AT, Procter G. Tetrahedron Lett. 1987;28:2041. [Google Scholar]; (g) Tamao K, Nakajo E, Ito Y. J. Org. Chem. 1987;52:4412. [Google Scholar]; (h) Hudrlik PF, Peterson D, Rona RJ. J. Org. Chem. 1975;40:2263. [Google Scholar]
- 33.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamune H, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765. [Google Scholar]
- 34. Lowe JT, Youngsaye W, Panek JS. J. Org. Chem. 2006;71:3639. doi: 10.1021/jo060134g. The enantiomeric excess was determined to be 98% for both 15a and ent-15a as observed using Chiralcel OD 99/1 (hexanes/IPA) at 1 ml/min. (tR 35:92 for 15a and 45.17 for ent-15a).
- 35.For preparation of (2R,3R)-3-(triphenylsilyl)-2,3-epoxypropane-l-ol see: Raubo P, Wicha J. Tetrahedron: Asymmetry. 1995;6:577.
- 36.Elevated temperatures (−20, 0 and 25 °C) under the described conditions for the (E)-vinylsilane did not provide the desired epoxide.
- 37.A Shi epoxidation of 16 was also considered, however literature precedent showed attenuated ee’s for similar systems, see: reference 32d for more information.
- 38.Lipase AK “Amano” 20, Lipase PS “Amano” and Lipase PS-D “Amano” were initially screened. Lipase PS-D was found to be superior.
- 39.The enantiomeric excess was determined to vary between 94–96% for both 15b and ent-15b as observed using Chiralcel OD 85/15 (hexanes/IPA) at 1 ml/min (tR 5:03 for 15b and 7.08 for ent-15b).
- 40.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J. Org. Chem. 1981;46:3936. [Google Scholar]
- 41.Decomposition of cis and trans epoxide acids was noticed upon prolonged storage or column chromatography.
- 42.During the course of oxidation, the reaction solution became a dark green color with a black suspension, suggesting an inactive catalytic system. In order to drive the reaction to completion, catalyst loading was increased and extended reaction times were explored with little success. Only a reset of the catalytic system provided full conversion.
- 43.Hudrlik PF, Ma D, Bhamidipati RS, Hurdlik AM. J. Org. Chem. 1996;61:8655. [Google Scholar]
- 44.(a) Eisch JJ, Galle JE. J. Org. Chem. 1976;41:2615. [Google Scholar]; (b) Eisch JJ, Trainor JT. J. Org. Chem. 1963;28:2870. [Google Scholar]
- 45.(a) Davis AP, Hughes GJ, Lowndes PR, Robbins CM, Thomas EJ, Whitham GH. J. Chem. Soc. Perkin Trans. 1. 1981:1934. [Google Scholar]; (b) Eisch JJ, Chiu CS. J. Organomet. Chem. 1988;358:C1. [Google Scholar]
- 46.Siriwardane U, Chu SSC, Buynak JD. Acta Crystallogr., Sect. C : Cryst. Struct. Commun. 1989;C45:531. [Google Scholar]
- 47.Fristad WE, Bailey TR, Paquette LA, Gleiter R, Boehm MC. J. Am. Chem. Soc. 1979;101:4420. [Google Scholar]
- 48.Cloarec J-M, Charette AB. Org. Lett. 2004;6:4731. doi: 10.1021/ol047997l. [DOI] [PubMed] [Google Scholar]
- 49.Smith MB, March J. Advan. Org. Chem. 5th Ed. p. 174. [Google Scholar]
- 50.It has been suggested that silicon prefers an axial orientation for both electronic and steric reasons but may still eliminate even without optimal orbital overlap. For a detailed discussion and relevant examples see: Lambert JB. Tetrahedron. 1990;46:2677.
- 51.Intramolecular silyl-modified Sakurai condensations of vinylsilanes suggest allylic strain plays a role in stereochemical outcome: Markó IE, Dobbs AP, Scheirmann V, Chellé F, Bayston DJ. Tetrahedron Lett. 1997;38:2899.
- 52.It is conceivable that an oxonia-Cope rearrangement could play a role in the stereochemical outcome of these reactions, however no byproducts from this pathway were observed. For an example of oxonia-Cope rearrangements in allyl systems, see: Roush WR, Dilley GJ. Synlett. 2001:955.
- 53.(a) Corey EJ, Seebach D. Angew. Chem., Int. Ed. 1965;4:1075. [Google Scholar]; (b) Gröbel BT, Seebach D. Synthesis. 1977:357. [Google Scholar]
- 54.Langille NF, Dakin LA, Panek JS. Org. Lett. 2003;5:575. doi: 10.1021/ol027518n. [DOI] [PubMed] [Google Scholar]
- 55.Liu P, Panek JS. J. Am. Chem. Soc. 2000;122:1235. [Google Scholar]
- 56.The homoallylic substrates were converted to 1,1-dimethyl acetonides in a three step sequence.
- 57.(a) Panek JS, Yang M. J. Am. Chem. Soc. 1991;113:9868. [Google Scholar]; (b) Panek JS, Beresis R. J. Org. Chem. 1993;58:809. [Google Scholar]
- 58.Blakemore PR, Kim SK, Schulze VK, White JD, Yokochi AFT. J. Chem. Soc., Perkin Trans. 2001;1:1831. [Google Scholar]
- 59.Gaeta FCA, Lehman de Gaeta LS, Kogan TP, Or YS, Foster C, Czarniecki M. J. Med. Chem. 1990;33:964. doi: 10.1021/jm00165a013. [DOI] [PubMed] [Google Scholar]
- 60.Altenbach HJ, Korff R. Tetrahedron Lett. 1981;22:5175. [Google Scholar]
- 61.Corey EJ, Helal CJ. Angew. Chem. Int. Ed. 1998;37 doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z. 1986 and references found therein. [DOI] [PubMed] [Google Scholar]
- 62.Cloarec J-M, Charette AB. Org. Lett. 2004;6:4731. doi: 10.1021/ol047997l. [DOI] [PubMed] [Google Scholar]
- 63.Fénézou JP, Gauchet-Prunet J, Julia M, Pancrazi A. Tetrahedron Lett. 1998;29:3667. [Google Scholar]
- 64.For a previous example of spirocyclization in the presence of DDQ see: Paterson I, Chen DY-K, Franklin AS. Org. Lett. 2002;4:391. doi: 10.1021/ol017082w.
- 65.Anhydrous conditions resulted in no reactions while using phosphate buffer (pH 7) provided 19% yield of the desired spirocycle.
- 66.Conditions found in Table 3 were also applied to 44 with little success.
- 67.Gordon DM, Danishefsky SJ. J. Am. Chem. Soc. 1992;114:659. [Google Scholar]
- 68.Vaino AR, Szarek WA. Synlett. 1995:1157. [Google Scholar]
- 69.Congreve MS, Davison EC, Fuhry MAM, Holmes AB, Payne AN, Robinson RA, Ward SE. Synlett. 1993:663. [Google Scholar]
- 70.Onoda T, Shirai R, Iwasaki S. Tetrahedron Lett. 1997;38:1443. [Google Scholar]
- 71.The use of collidine as a buffer (against DDQH2) in the DDQ oxidation of silyl enol ethers has also been noted, see: Fleming I, Paterson I. Synthesis. 1979:736. Siewert J, Textor A, Grond S, von Zezschwitz P. Chem. Eur. J. 2007;13:7424. doi: 10.1002/chem.200601688. Banwell MG, Hockless DCR, McLeod MD. New J. Chem. 2003;27:50. Fevig TL, Elliott RL, Curran DP. J. Am. Chem. Soc. 1988;110:5064.
- 72.Akutagawa T, Saito G. Bull. Chem. Soc. Jpn. 1995;68:1753. [Google Scholar]
- 73.(a) Bhattacharya A, DiMichele LM, Dolling UH, Grabowski EJJ, Grenda VJ. J. Org. Chem. 1989;54:6118. [Google Scholar]; (b) Bhattacharya A, DiMichele LM, Dolling UH, Douglas AW, Grabowski EJJ. J. Am. Chem. Soc. 1988;110:3318. [Google Scholar]; (c) Wallace DJ, Gibb AD, Cottrell IF, Kennedy DJ, Brands KMJ, Dolling UH. Synthesis. 2001;12:1784. [Google Scholar]
- 74.Charge-transfer complexes with other nitrogen heterocycles including imidazoles and pyrimidines have been reported, see: Salman HMA, Rabie UM, Abd-Alla EM. Can. J. Anal. Sci. Spectrosc. 2004;49:1. Boraei AAA. Spectrochim. Acta, Part A. 2002;58:1895. doi: 10.1016/s1386-1425(01)00655-2. Khashaba PY, El-Shabouri SR, Emara KM, Mohamed AM. J. Pharm. Biomed. Anal. 2000;22:363. doi: 10.1016/s0731-7085(99)00280-0.
- 75.For a review of the Staudinger reaction see: Gololobov YG, Kasukhin LF. Tetrahedron. 1992;48:1353.
- 76.The unknown product was obtained as a mixture of isomers with a loss of C2-C3 olefin. We were unable to separate the mixture for characterization.
- 77.Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Cancer Res. 1988;48:4827. [PubMed] [Google Scholar]
- 78.Monks A, Scudiero DA, Johnson GS, Paull KD, Sausville EA. Anticancer Drug Des. 1997;12:533. [PubMed] [Google Scholar]
- 79.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. J. Natl. Cancer Inst. 1989;81:1088. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.