

Abstract
Vascular endothelial growth factor (VEGF) is a critical regulator of angiogenesis. Inhibiting the VEGF–VEGF receptor (R) signal transduction pathway in glioblastoma has recently been shown to delay progression, but the relative benefit and mechanisms of response and failure of anti-VEGF therapy and VEGFR inhibitors are not well understood. The purpose of our study was to evaluate the relative effectiveness of VEGF sequestration and/or VEGFR inhibition on orthotopic tumor growth and the mechanism(s) of treatment resistance. We evaluated, not only, the effects of anti-VEGF therapy (bevacizumab), anti-VEGFR therapy (sunitinib), and the combination on the survival of mice bearing orthotopic gliomas, but also the differential effects of the treatments on tumor vascularity, cellular proliferation, mesenchymal and stem cell markers, and myeloid cell infiltration using flow cytometry and immunohistochemistry. Bevacizumab significantly prolonged survival compared with the control or sunitinib alone. Both antiangiogenic agents initially reduced infiltration of macrophages and tumor vascularity. However, multitargeted VEGFR inhibition, but not VEGF sequestration, rapidly created a vascular gradient and more rapidly induced tumor hypoxia. Re-infiltration of macrophages was associated with the induction of hypoxia. Combination treatment with bevacizumab and sunitinib improved animal survival compared with bevacizumab therapy alone. However, at the time of tumor progression, a significant increase in CD11b+/Gr1+ granulocyte infiltration was observed, and tumors developed aggressive mesenchymal features and increased stem cell marker expression. Collectively, our results demonstrate a more prolonged decrease in tumor vascularity with bevacizumab than with sunitinib, associated with a delay in the development of hypoxia and sustained reduction of infiltrated myeloid cells.
Keywords: angiogenesis, bevacizumab, glioblastoma, myeloid cell, VEGF
The growth of highly aggressive glioblastomas depends on the formation of new blood vessels. Vascular endothelial growth factor (VEGF), a potent angiogenic, plays a central role in mediating the phenotype of glioblastoma. VEGF mediates endothelial cell migration, proliferation, and survival1 and is highly expressed in glioblastoma.2 In addition to directly promoting angiogenesis via its effects on endothelial cells, VEGF and the closely related placental growth factor (PlGF) are myeloid cell chemokines known to be important for attracting VEGF receptor (R)1 monocytes to tumors.3 Recent experimental evidence suggests that elimination of VEGFR1 signaling in bone marrow–derived cells significantly decreases glioma growth and vascularization4 and that VEGFR1-expressing myeloid cells play an important role in sustaining glioma angiogenesis.
Although “tumor escape” from anti-VEGF therapy involves activation of other growth factors important for angiogenesis, laboratory data suggest that bone marrow–derived cells may play a critical role in tumor escape from antiangiogenic therapy.5,6 In the early phases of antiangiogenic therapy, tumor oxygenation improves through the process of vascular normalization.7 However, with prolonged antiangiogenic treatment, tumors develop progressive hypoxia, which may be central to promoting tumor resistance to therapy and ultimately tumor progression.5,8 Du et al.5 recently showed that tumor hypoxia is a driving force behind the release of stromal cell–derived factor 1α, which attracts myeloid cells from the bone marrow to gliomas. Other circulating chemokines, such as granulocyte colony-stimulating factor, attract CD11b+/Gr1+ cells, which may also mediate tumor refractoriness to anti-VEGF therapy.9,10 It is not known whether the development of tumor resistance is delayed by therapeutic modulation of angiogenesis to minimize the development of hypoxia and decrease the infiltration of myeloid cells.
Therapeutic strategies to sequester VEGF or block endothelial-associated VEGFRs (mainly VEGFR2) are undergoing intensive investigation as treatments for glioblastoma. Clinical reports with bevacizumab (a monoclonal antibody to human VEGF-A) in patients with recurrent glioblastoma have confirmed the potential efficacy of blocking VEGF by demonstrating an impressive radiographic response rate and an improvement in progression-free survival.11,12 Although there are many potential benefits of antiangiogenic therapy in patients with glioblastoma (including reducing cerebral edema via removal of VEGF and thus allowing a reduction in corticosteroid use), controversy still exists regarding the impact of anti-VEGF therapy on tumor growth and patient survival. Of note, VEGFR-targeted therapy using agents such as sunitinib13 and cediranib14 has not shown an equal ability to improve progression-free survival or duration of response compared with anti-VEGF therapy.
We sought to characterize the changes in tumor vascularity, cell proliferation, and the microenvironment, including the formation of hypoxia and infiltration of different myeloid cell populations, to determine whether these factors could account for the differential outcomes observed with anti-VEGF and VEGFR inhibitor therapies. The purpose of this study was to determine potential mechanisms for the difference in benefit of anti-VEGF therapy and VEGFR inhibitors in gliomas and to explore potential mechanisms of treatment failure. In the present study, we compared the effect of the monoclonal antibody bevacizumab (anti-VEGF) and sunitinib (a VEGFR inhibitor and colony stimulating factor–1 receptor [CSF-1R] inhibitor)15 on animal survival in an orthotopic glioma model. We also evaluated the combination of bevacizumab and sunitinib to modulate the recruitment of myeloid cells of the monocyte/macrophage lineage.
Materials and Methods
Cell Lines, Reagents, and Treatment
Human glioblastoma cell line U87MG was obtained from the American Type Culture Collection and was tested and authenticated via short tandem repeats fingerprinting by the Brain Tumor Center of The University of Texas MD Anderson Cancer Center. U87 cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. Sunitinib (Pfizer) was suspended in carboxymethyl cellulose buffered to pH 4.2. Bevacizumab (Genentech) was dissolved in phosphate buffered saline (PBS) immediately prior to i.p. injection.
Animal Xenografts
For in vivo experiments, U87 glioblastoma cells (5 × 05 cells) were implanted intracranially into nude mice.16 Beginning 5 days after implantation, bevacizumab (10 mg/kg) was administered to the animals by i.p. injection twice a week; sunitinib (50 mg/kg) was administered by oral gavage daily Monday through Friday; and the combination was administered as described. One cohort of 10 animals was treated continuously and followed up for survival. A separate cohort of 6 animals per group was treated continuously until the designated time point, and the tumors from these mice were extracted at 2, 4, and 6 weeks for pharmacodynamic analyses. Control animals for these cohorts were treated with PBS by i.p. injection or carboxymethyl cellulose vehicle by oral gavage. When the mice developed signs and symptoms of advanced tumors, they were euthanized, and their brains were removed and processed for analysis. The institutional animal care and use committee of MD Anderson Cancer Center approved all of the experiments in our study.
Immunohistochemistry
Tissues were fixed in 4% paraformaldehyde for 24 hours, embedded in paraffin, sectioned serially (4 μm), and stained with hematoxylin and eosin (Sigma-Aldrich). For immunohistochemical stains, slides were deparaffinized and subjected to graded rehydration. After blocking in 5% serum and antigen retrieval (citrate buffer, pH 6.0), we incubated the slides with the primary antibodies overnight at 4°C. After the slides were washed in PBS with Tween 20, the primary antibody reactions were detected using the Vectastain ABC Kit (Vector Laboratories) with the respective secondary antibody. Immunohistochemical analysis of the slides was performed for microvascular density (von Willebrand factor/factor VIII, A0082, diluted 1:500; DAKO), cell proliferation (Ki-67, code M 7240, diluted 1:80; DAKO), hypoxia (carbonic anhydrase [CA]9, NB100-417, diluted 1:1000; Novus Biological), the macrophage marker CD68 (M0814, diluted 1:2000; DAKO), vimentin (V9.1, diluted 1:900; DAKO), smooth muscle actin (1A4, diluted 1:80 000; Sigma-Aldrich), and e-cadherin (HECD-1, diluted 1:100; Invitrogen).
For double immunofluorescence studies, tissue sections were blocked with 10% fetal bovine serum following heat-induced antigen retrieval and then incubated with anti–tumor growth factor (TGF)–β (code ab66043, diluted 1:50; Abcam) or zinc finger E-box-binding homeobox 2 (ZEB2; nbp1-82991, diluted 1:50; Novus), F4/80 (RB6-8C5, 108401, diluted 1:50; Biolegend), and anti-nestin (AB5922,diluted 1:1000; Millipore). Texas Red fluorescein isothiocyanate (FITC)–conjugated secondary antibodies and green FITC–conjugated antibodies (Invitrogen) were used for 1hour at room temperature.
Flow Cytometry
Whole blood obtained by means of retro-orbital bleed and terminal cardiac puncture was collected in vials containing ethylenediaminetetraacetic acid. Infiltrated myeloid cells were isolated from tumors using a Miltenyi Biotech Neural-Cell Dissociation Kit. Cells were washed in PBS and then incubated with primary antibody for 1 hour at room temperature. The following conjugated antibodies were used to detect myeloid cell lineages in each tumor (n ≥ 6 per group): VEGFR1-allophycocyanin (APC) and CD45-PerCP-Cy5 (BD Biosciences); Syto 16 (Invitrogen); CD11b-APC-Cy7, F4/80-FITC, CSF-1R-PE, and Gr1-PE-Cy7 (Ebiosciences); and matched isotype control antibodies. After washing cells twice with PBS, we performed flow cytometry using a FACSCanto Flow Cytometer (BD Biosciences) and analyzed acquired data with FlowJo software (Tree Star) with gate analysis designed to remove cellular debris. Isotype antibodies for each marker were used as the markers' controls. Myeloid cell populations in the tumors were normalized to total numbers of viable nucleated CD45+ cells.
Quantitative Image Analysis by Computer-Assisted Microscopy
The evaluation of stains for vascularity and hypoxia was performed by a pathologist without knowledge of the treatment group. Computer-assisted microscopy was used to capture the immunostaining images. Images were taken with an Axioskop 40 microscope (Zeiss) equipped with Zeiss AxioVision Release 4.2 software. The intensity and area of sinusoidal endothelial staining were quantitatively measured using the Image-Pro Plus system version 7.0 (Media Cybernetics). The tissue sample from each animal was measured at a magnification of 100×. The images were then imported into the Image-Pro Plus software, where they were calibrated to a known area of measurement. Positively stained endothelial cells or endothelial cell clusters (clearly separate from tumor cells or other connective tissue elements) were counted, regardless of size or shape. Immunohistochemical and immunofluorescent staining was selected using the color selection function, and the area/density (intensity) measurement functions were used as described previously17 to calculate the respective blood vessel (factor VIII) and hypoxia (CA9) density values. Five high-power fields were selected randomly in 3 regions of the tumor: the peripheral/invading edge and the middle center regions. Area density measurements were not influenced by tumor size.
Cell proliferation was determined using expression of Ki-67 antigen at 400× magnification. The labeled cell count was determined in 5 high-power fields for each tumor area. Ki-67 proliferation was recorded as the number of proliferating cells in each high-power field.
Real-Time PCR
Total RNA was extracted from tumor-bearing mouse brain tissue using the RNeasy Mini Kit coupled with DNase treatment (Qiagen) and reverse transcribed with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Each cDNA was analyzed in triplicate using a real-time TaqMan probe (Applied Biosystems). Quantitative PCR analysis was performed on a chromo 4 sequence-detection system (Bio-Rad). Relative quantification of mRNA levels was performed using the comparative cycle threshold (Ct) method, with glyceraldehyde phosphate dehydrogenase as the reference gene and the formula 2−ΔΔCt.
Statistical Analysis
All statistical analyses were performed with GraphPad (InStat) software for Windows. Survival analysis was performed using the Kaplan–Meier method, and cohorts were compared using the log-rank test. All other data were compared using an unpaired 2-tailed Student's t test. Summary statistics for continuous data are expressed as mean ± SEM. The relationship between intensity of immunohistochemical marker and number of myeloid cells was determined using a nonparametric Spearman rank correlation. P < .05 was considered statistically significant.
Results
Anti-VEGF Antibody but not VEGFR Inhibitor Therapy Prolongs Animal Survival
The clinical utility of antiangiogenic therapy for the treatment of glioblastoma is unquestioned, but the relative effectiveness of sequestering the ligand (VEGF) vs inhibiting VEGFR is not well understood. To examine this question, we first performed a survival study in our U87 orthotopic glioma model. As reported previously,8 we observed a significantly longer survival in animals treated with bevacizumab twice a week than in controls (median survival of 46.5 days for bevacizumab-treated mice vs 25.5 days for controls, P < 0.001). However, there was no statistically significant difference in survival in sunitinib-treated mice compared with controls. Median survival for sunitinib-treated mice was 28 days compared with 25.5 days for controls (Fig. 1A). In our orthotopic mouse model, targeting the VEGF ligand prolonged animal survival longer than blocking VEGFR.
Fig. 1.

Anti-VEGF but not VEGF receptor inhibitor therapy prolongs survival in an orthotopic glioma xenograft model. (A) Kaplan–Meier graph showing improved survival in nude mice with U87 tumors treated with bevacizumab (Bev) or sunitinib compared with untreated controls. (B) Kaplan–Meier graph showing improved survival in nude mice with U87 tumors treated with bevacizumab (Bev) + sunititnib compared with bevacizumab alone.
Reports have suggested that antiangiogenic agents used in combination may improve outcome by targeting multiple angiogenesis pathways.18,19 In addition to inhibiting VEGFR2, sunitinib inhibits the myeloid cell–associated receptor CSF-1R.15 Because bone marrow cells are potential mediators of escape from antiangiogenic therapy,6 we hypothesized that adding sunitinib to bevacizumab would prolong the duration of animal survival. In separate experiments, we evaluated how bevacizumab + sunitinib affected the duration of animal survival. Bevacizumab + sunitinib showed significantly prolonged survival compared with bevacizumab alone (Fig. 1B; 43.5 days vs 63.5 days for combination therapy, P = .046).
VEGFR Inhibition Produces Greater Reduction in Central Tumor Vascularity than Does Anti-VEGF Therapy
To determine whether the difference in treatment efficacy between VEGF ligand sequestration and VEGFR-targeted antiangiogenic therapy was related to relative effects on tumor vasculature, we measured changes in tumor microvascular density over time. Figure 2A shows representative photomicrographs of each tumor area at 2, 4, and 6 weeks for mice in all treatment groups.
Fig. 2.

VEGFR inhibition produces greater reduction in central tumor vascularity than does anti-VEGF therapy. (A) Representative light microscopy images showing immunohistochemical detection of factor VIII (brown staining) in U87 tumors from control, bevacizumab (bev), sunitinib, and bev + sunitinib treated animals (200×) at 2, 4, and 6 weeks. (B) Bar graph demonstrating the average percentage of change in fractional area of factor VIII staining in central (c), middle (m), and peripheral (p) tumor locations in all treatment groups. *P < .05 compared with untreated controls; #P < .05 compared with sunitinib; ♦ P < .05 compared with the same treatment group at either 2 or 4 weeks. (C) Percent change in vascular density comparing the central vs peripheral tumor regions at each time point for each treatment group. *P < .05 compared with untreated controls; #P < .05 compared with sunitinib; +P < .05 compared with bev treatment. (D) Glioma cell proliferation using Ki-67 analysis at each time point. *P < .05 compared with untreated controls; #P < .05 compared with sunitinib; + P < .05 compared with the same treatment group at any of the 4.
At 2 weeks, quantitative analysis demonstrated a significant reduction in tumor vascularity in the central, middle, and peripheral tumor regions in all treatment groups compared with controls (P < .05). The decrease in vascularity ranged from approximately20% to 51%.
At 4 weeks, the sunitinib-treated tumors had escaped from therapy, ultimately leading to the animals' death. In these tumors, there was a significantly greater reduction in vascularity in the central region than in the peripheral region. This vascular gradient was absent in the bevacizumab and combination therapy groups. Alternatively, bevacizumab was better able to inhibit vascularity in the periphery of the tumor. Furthermore, the bevacizumab and combination groups had a sustained and uniform reduction in vascularity across all 3 tumor areas at 4 weeks, in contrast to the control and sunitinib groups. In the former 2 groups, the 50%–73% decrease in vascularity was significant compared with that seen in the control and sunitinib groups (P < .05).
At 6 weeks, as the bevacizumab and combination groups developed resistance and the tumors began to progress, there was a 63%–74% greater increase in vascularity in the middle and peripheral regions than had been observed in their respective treatment groups at 4 weeks (P < .05). Treatment failure in these tumors was associated with the establishment of a vascular gradient, as was observed in the control and sunitinib treatment groups at 4 weeks. This relative change in vessel density in the central region compared with the peripheral region is summarized graphically in Figure 2C. These data suggest that sunitinib therapy does not sustain control of vascular proliferation at the periphery but does create a larger gradient between the central and peripheral tumor regions.
Finally, changes in glioma cell proliferation were explored in the 3 tumor regions (Fig. 2D). In general, changes in cell proliferation mirrored the changes in tumor vascularity. Tumor areas with lower vessel density had less tumor cell proliferation. At 2 weeks, the bevacizumab and combination cohorts had statistically lower levels of proliferation (P < .05) than did controls. Only the peripheral region of the sunitinib-treated tumors had a significant reduction compared with controls. As the controls and sunitinib-treated tumors began to escape at 4 weeks, there was a concomitant increase in cell proliferation, whereas the bevacizumab and combination groups sustained a decrease in cell proliferation.
Of note, the bevacizumab and combination groups demonstrated a significant increase in cell proliferation in the peripheral region only at 6 weeks compared with the results at 4 weeks. This delayed increase in cell proliferation could have been due to the fact that median animal survival was longer in these groups (beyond the 6-week time point at which these measurements were made) or potentially because of the pattern of tumor escape. The longer-surviving animals in both the bevacizumab and combination groups had evidence of tumor invasion (Fig. 6), a pattern not observed in the control animals. These results indicate that tumor invasion could have contributed to animal morbidity in the absence of a rapidly proliferating tumor.
Fig. 6.


Antiangiogenic therapy induces mesenchymal changes to glioma tumor tissue in vivo. (A) Tumors treated with antiangiogenic therapy induced aggressive (invasive) behavior and histologic characteristics characteristic of mesenchymal tumors (see white arrows). Expression of mesenchymal markers vimentin and smooth muscle actin (SMA) are increased following antiangiogenic therapy, whereas e-cadherin expression decreases. (B) Tumors treated with bevacizumab and sunitinib show a significant increase in the expression of the glioma stem cell markers nestin and SOX2. (C) Expression levels of TGF beta and F4/80 in U87 glioblastoma xenograft tumors in response to antiangiogenic therapy. Tissue slides from xenografts were stained with anti-TGF beta (green) and anti-F4/80 (red) antibodies as described in “Materials and Methods.” The bar graph represents the percentage of TGF beta-positive cells in each condition under ×200. (D) The expression of ZEB2 and nestin in U87 glioblastoma xenograft tumors in response to antiangiogenic therapy. Xenografts tissue was stained with anti-ZEB2 (green) and anti-nestin (red) antibodies as described in “Materials and Methods.” The bar graph represents the percentage of ZEB2-positive cells for each treatment condition under ×200.
VEGFR Inhibition Induces Greater Tumor Hypoxia than Does Anti-VEGF Therapy
The relative impacts of VEGF sequestration and VEGFR inhibition on the induction of glioma hypoxia are not known. Therefore, the development of a vascular gradient at earlier time points in the sunitinib-treated groups owing to a greater reduction in central vascularity at the time of treatment failure led us to examine the impact of this gradient on the induction of tumor hypoxia, a known stimulant of resistance. Figure 3A shows representative photomicrographs of the hypoxia marker CA9. At the earliest time point (2 weeks), there was little evidence of tumor hypoxia in all groups (Fig. 3B). However, at 4 weeks, hypoxia had increased by 24% and 43% in the control and sunitinib groups, respectively, compared with the results at 2 weeks. Although higher than the 2-week levels, the CA9 staining levels in the bevacizumab and combination treatment groups were significantly lower than in either the control group or the sunitinib group. CA9 staining had increased significantly at 6 weeks in the bevacizumab and combination treatment groups compared with the 4-week levels from the same groups. Figure 3B summarizes the fractional area of CA9 staining at each time point for all treatment groups. The degree of hypoxia in each of the different tumor areas correlated with a decrease in vessel density in that region (data not shown).
Fig. 3.
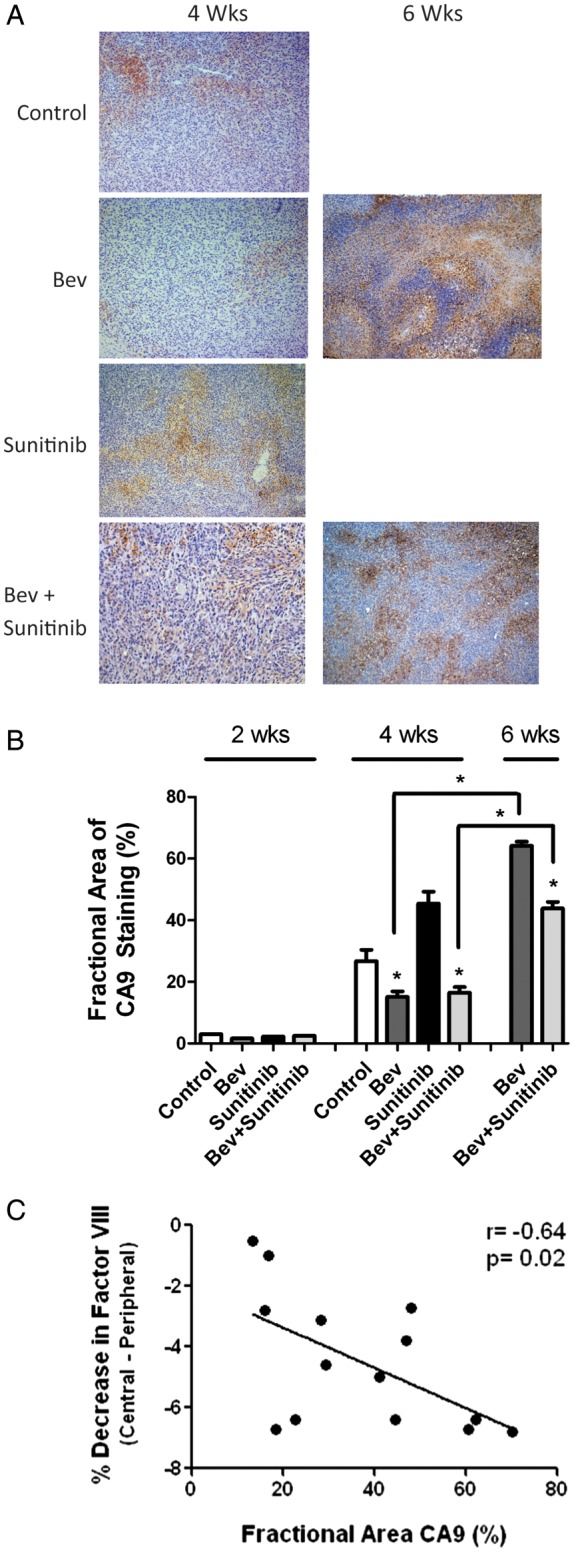
VEGFR inhibition induces greater tumor hypoxia than anti-VEGF therapy (A) Representative light microscopy image at 200× magnification showing immunohistochemical detection of carbonic anhydrase 9 (CA9; brown nuclear staining) demonstrating regions of tumor hypoxia in the 4 treatment groups at 4 and 6 weeks. (B) Bar graph depicting the quantification of fractional staining of CA9. *P < .05 compared with controls. (C) An inverse correlation between factor VIII staining and fractional area of CA9 staining was seen for all tumor treatment groups.
An inverse correlation existed between the magnitude of the difference in central and peripheral vessel density and the level of CA9 staining. That is, tumors with a larger vascular gradient between central and peripheral vessel densities had greater levels of hypoxia (Fig. 3C). The degree of hypoxia in the bevacizumab and combination treatment groups at 6 weeks was similar to the amount of hypoxia observed in the sunitinib-treated tumors at 4 weeks, suggesting that hypoxia-mediated escape mechanisms might occur in these groups at different times, depending on the acuity or degree of change in vessel density. These results indicate that one difference between the relative efficacies seen in VEGFR inhibition compared with VEGF ligand sequestration may be related to the relative degree of vascular inhibition.
Antiangiogenic Agents Decrease CD11b+/F4/80+/Gr1− Myeloid Cell Recruitment to Tumors
VEGF, PlGF, and other cytokines are known to attract bone marrow–derived cells to tumors, in part via VEGF-mediated signaling through VEGFR1 located on some myeloid cells.20 We used flow cytometry and immunohistologic staining to analyze the time course of changes in CD11b+/F4/80+/Gr1– myeloid cell recruitment in our orthotopic glioma model in controls and in mice treated with antiangiogenic agents at 2, 4, and 6 weeks. Figure 4A presents representative flow cytometry data showing the time course of changes in tumor-infiltrated CD11b+/F4/80+ cells with the percentage of Gr1+ cells pseudocolored in green. Quantitative analysis (Fig. 4B, upper panel) showed that at the early (2-week) time point, sunitinib, bevacizumab, and combination therapy resulted in 52%, 70%, and 47% reductions, respectively, in CD11b+/F4/80+/Gr1– cells compared with controls (all statistically significant, P < .05). No statistical differences were noted between the anti-VEGF treatment groups. In the control and sunitinib groups, which had tumor progression by 4 weeks, there was a significant increase in myeloid cell recruitment of 43% and 80%, respectively, compared with recruitment at 2 weeks (P < .05 for both groups).
Fig. 4.

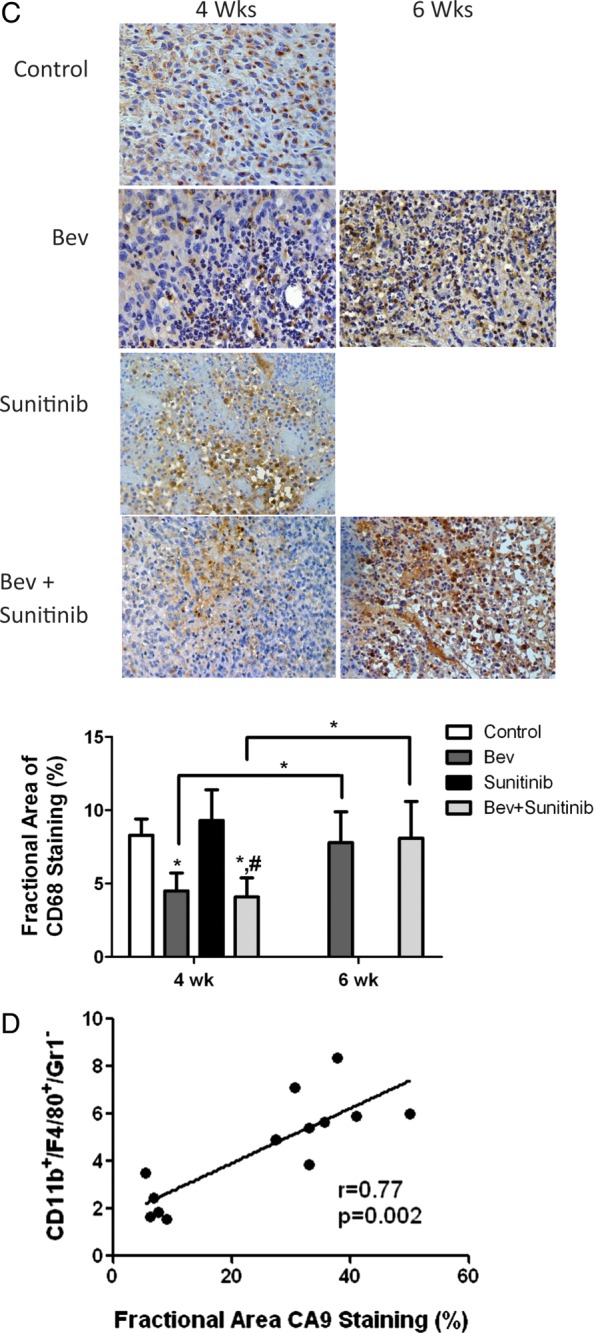
Antiangiogenic agents modulate CD11b+/F4/80+/Gr1− myeloid cell recruitment to tumors. (A) Representative flow cytometry analyses from tumor at 4 and 6 weeks in each group. (B) Upper panel, bar graph demonstrating the average percentage of CD11b+/F4/80+/Gr1- myeloid cells at each time point for the 4 treatment groups. Lower panel, bar graph showing average percentage of CD11b+/Gr1- cells expressing VEGFR1+. (C) Top, representative photomicrographs of CD68 macrophage staining at the corresponding time points. Bottom, bar graph of fractional area of CD68 staining. *P < .05 compared with controls; #P < .05 compared with sunitinib treated animals. (D) Positive correlation between number of CD11b+/F4/80+/Gr1- macrophages using flow cytometry and fractional area of CA9 staining.
Of note, at 4 weeks there was a significantly greater decrease in CD11b+/F4/80+/Gr1– cells in the combination treatment group than in the bevacizumab group (2.2% vs 1.1%, P < .05), potentially related to greater inhibition of myeloid cell infiltration owing to the CSF-1R inhibition by sunitinib. As with the control and sunitinib groups at 4 weeks, the bevacizumab and combination therapy groups had significant increases in myeloid cell infiltration of 52% and 64%, respectively, at 6 weeks compared with infiltration at 4 weeks (P < .05 for both groups). There was a trend toward a greater decrease in myeloid cell infiltration in the combination therapy group than in the bevacizumab group, although this difference was not statistically significant.
Although multiple chemokines may attract bone marrow–derived cells to tumors,21 it is possible that VEGF and PlGF are prominent mediators of myeloid cell recruitment to gliomas via activation of VEGFR1 on myeloid cells. We performed a detailed analysis of the percentage of CD11b+/F4/80+/Gr1– cells expressing VEGFR1, as well as the monocyte/macrophage receptor CSF-1R, in each treatment group. Figure 4B (lower panel) shows that approximately60%–80% of infiltrated myeloid cells expressed both VEGFR1 and CSF-1R. There was no statistical difference between the number of cells with VEGFR1/CSF-1R expression in any of the treatment groups at different time points.
The expression of macrophages within tumors at 4 and 6 weeks was confirmed with immunohistochemical staining of tumor tissue using CD68, a pan-macrophage marker (see Fig. 4C). Changes in macrophage expression by immunohistochemistry were similar to those in the quantitative flow analyses. The fractional area of CD68 staining was significantly smaller at 4 weeks in the bevacizumab and combination therapy groups than it was in the control and sunitinib groups. As described previously, CD68 staining was higher in the bevacizumab and combination therapy groups at 6 weeks than it was at 4 weeks.
Myeloid cells are known to be attracted to regions of tumor hypoxia, where they are thought to be important in mediating resistance to antiangiogenic therapy.22,23 Our results showed a strong association between the infiltration of myeloid cells and the degree of tumor hypoxia. Figure 4D shows that combined analysis of all treatment groups at 4 and 6 weeks demonstrated a strong correlation between the fractional area of CA9 staining and the numbers of infiltrated CD11b+/F4/80+/Gr1– cells (correlation r = 0.77, P = .002). Figure 4C shows what appears to be a large number of CD68+ cells around the areas of necrosis, although their localization was not restricted to these areas.
Tumor Resistance to Anti-VEGF Therapy Is Associated with Infiltration of CD11b+/Gr1+ Cells
Although tumor resistance to antiangiogenic therapy is associated with the recruitment of CD11b+/F4/80+/Gr1– cells, we also evaluated the infiltration of other bone marrow–derived cells. In particular, we were interested in exploring the recruitment of CD11b+/Gr1+ cells, given their role in mediating refractoriness to anti-VEGF therapy in other tumor models.9 Figure 5A shows the flow cytometry plots for CD11b+/F4/80– cells with pseudocoloring of Gr1+ cells from representative samples in the different treatment groups. Quantitative analysis of this cell population (Fig. 5B) demonstrated that there was little change in Gr1+ cells at 2 or 4 weeks in the different treatment groups. However, there was a significant increase in CD11b+/Gr1+ cells in the bevacizumab and combination therapy groups at 6 weeks compared with the same groups at 4 weeks.
Fig. 5.

Antiangiogenic agents modulate CD11b+/F4/80-/Gr1+ myeloid cell recruitment to tumors. (A) Representative flow cytometry analyses from tumor at 4 and 6 weeks in each group. (B) Bar graph demonstrating the average percentage of CD11b+/F4/80-/Gr1+ myeloid cells at each time point for the 4 treatment groups; *, P < 0.05.
Antiangiogenic Therapy Induces Mesenchymal Changes to Glioma Tumor Tissue In Vivo
Tumors extracted from animals after the experiments were completed had striking histologic changes compared with the controls. Hematoxylin and eosin staining revealed that tumors in the sunitinib, bevacizumab, and combination therapy groups had multiple areas containing spindle-shaped cells and whorls of tumor cells characteristic of a sarcoma or gliosarcoma histology (Fig. 6A, left column). These mesenchymal features increased with survival time. Consistent with a more mesenchymal phenotype, these tumors also had sheets of invading tumor cells (Fig. 6A, white arrows). Given the association between hypoxia and mesenchymal transformation,24,25 we performed immunohistochemical analysis of mesenchymal markers to further characterize changes within tumors following prolonged antiangiogenic treatment. Figure 6A shows that the antiangiogenic therapy groups had increased vimentin and smooth muscle actin staining and decreased e-cadherin staining compared with control tumors, all consistent with a mesenchymal transformation of these treated tumors.
Furthermore, there was an increase in stem cell marker expression in tumors treated with anti-VEGF therapy. RNA isolated from tumors extracted from bevacizumab- and sunitinib-treated animals was evaluated for expression of stem cell markers nestin and Sox2 using real-time PCR. Tumors had 2.5- and 88.0-fold increases in nestin and Sox2 expression, respectively, following prolonged (6-week) treatment with bevacizumab compared with untreated control tumors and 4.2- and 8.5-fold increases in these markers for sunitinib-treated tumors (94 weeks). Taken together, these results indicate that anti-VEGF therapy induces a phenotypic shift toward a more aggressive mesenchymal phenotype, consistent with the invasive and treatment-resistant behavior of these tumors. An increase in or selection for stem cells may contribute to this aggressive phenotype.
Both TGF-β and ZEB2 contribute to epithelial mesenchymal transition (EMT) in other solid tumor types. To investigate whether these mediators of EMT in other tumors might be responsible for the mesenchymal transition induced by antiangiogenic therapy in glioma, we co-cultured U87 cells with macrophage and performed human EMT gene array analysis (SABiosciences). We found that the expression levels of TGF-β and ZEB2 were significantly higher in U87 cells co-cultured with macrophages compared with U87 cells alone (data not shown). To determine whether these changes were relevant in our in vivo model, we evaluated TGF-β and ZEB2 expression levels in vivo by immunofluorescent staining and found that the expressions of TGF-β (Fig. 6C) and ZEB2 (Fig. 6D) were markedly increased in xenograft U87 glioma when mice were treated with bevacizumab or sunitinib or bevacizumab + sunitinib compared with tumors in the control group.
Discussion
This study sought to determine the relative effect of sequestering VEGF vs inhibiting VEGFR in an orthotopic glioma model and to determine the potential mechanisms for differential sensitivity to these 2 agents. VEGF sequestration with bevacizumab or bevacizumab + sunitinib significantly prolonged animal survival, whereas sunitinib monotherapy did not. Although all treatment groups showed early decreases in tumor vascularity, these decreases were not sustained in the sunitinib-treated group. Our findings were that sunitinib-treated animals developed a vascular gradient that may have been related to a greater effect of bevacizumab in reducing tumor vascularity at the tumor periphery or a greater reduction in central vascularity in VEGF tyrosine kinase–treated animals. The resultant central tumor hypoxia in turn potentially accelerated resistance mechanisms such as infiltration of bone marrow–derived cells. A strong correlation was observed between the decrease in tumor vascularity and a reduction in CD11b+/F4/80+/Gr1– myeloid cell infiltration in all treatment groups. However, VEGF sequestration imparted a more uniform decrease in tumor vascularity, less hypoxia, and a more sustained decrease in myeloid cell infiltration. At the time of progression, tumors were infiltrated by CD11b+/Gr1+ myeloid cells and became more invasive with features of mesenchymal differentiation and increased expression of stem cell markers.
Both anti-VEGF antibodies and VEGFR inhibitors are currently being used to treat glioblastoma. Anti-VEGF therapy only sequesters VEGF, leaving multiple alternate pro-angiogenic pathways to continue vascular proliferation unchecked. Multitargeted tyrosine kinase inhibitors have been developed that can block multiple pathways and have been shown to decrease tumor vascularity more than anti-VEGF therapy alone.19,26,27 Paradoxically, anti-VEGF therapy has demonstrated a more durable benefit in clinical applications than multitargeted VEGFR tyrosine kinase inhibitors either alone or in combination with chemotherapy. The reasons for this observation are unknown, but it has been shown in preclinical models that high-dose sunitinib (at the same doses used in this study; 40 mg/kg/d) prevents drug delivery of temozolomide to orthotopic brain tumors, suggesting that the most efficacious dose of these agents has not been completely worked out.28
By measuring tumor vascularity in 3 distinct regions of the tumor, we were able to show that the multitargeted agent sunitinib more rapidly promotes the development of a vascular gradient with a greater loss of tumor vessels in the central portion of the tumor. Our results show a significant association between a decrease in central tumor vascularity and the development of hypoxia in the different anti-VEGF groups. This association was observed at earlier time points in the sunitinib-treated animals than in the bevacizumab-treated animals. The greater decrease in central tumor vascularity in the VEGFR-inhibitor group may have been related to greater vessel pruning in the central tumor regions. Although the reasons that bevacizumab created a more uniform decrease in vascularity are not known, it could be due to less potent inhibition of signaling downstream of VEGFR2, less penetration of the drug into the tumor to exert its effects, longer half-life, or the drug's effects on infiltrating bone marrow cells. Bevacizumab sequesters only human (tumor-derived) VEGF, whereas a VEGFR inhibitor will block both human and mouse VEGFRs. In our bevacizumab-treated animals, the mouse microenvironment could have secreted mouse VEGF and thus prevented excessive hypoxia. Some evidence exists that elimination of all VEGF from tumors may paradoxically promote tumor growth.29 Additional study is needed to determine the level of VEGF required for preventing or delaying this unwanted effect, but higher levels of inhibition may be detrimental by promoting tumor resistance.
Recent data suggest that blockade of VEGFR1 signaling by either ligand sequestration or receptor inhibition can decrease tumor infiltration by bone marrow–derived cells. In a preclinical model of metastatic glioblastoma, blockade of VEGFR1 led to a partial decrease in bone marrow–derived cell infiltration inside and around the growing metastatic nodule.30,31 Kerber et al.4 demonstrated that elimination of VEGFR1 signaling in bone marrow cells significantly reduced glioma vascularity and tumor growth. Our current study shows that antiangiogenic therapy reduces infiltration of CD11b+/F4/80+/Gr1– myeloid cells into orthotopic gliomas. Our results indicate that these bone marrow–derived cells express mostly CSF-1R and VEGFR1 and are likely to be derived from macrophages. Anti-VEGF therapy appeared to be more effective in sustaining a decrease in myeloid cell infiltration and in controlling tumor vascularity, possibly by not inducing hypoxia. These factors may have contributed to the slower tumor growth and the survival advantage seen in the bevacizumab-only and combination therapy groups.
The impact of antiangiogenic agents on tumor growth is controversial and has not yet been clearly defined. Antiangiogenic agents may slow tumor growth by eliminating tumor blood vessels and thus reducing blood flow to the tumor.32 Antiangiogenic agents may also enhance the effectiveness of radiation and chemotherapy via vessel normalization.7 We propose that one of the mechanisms by which antiangiogenic agents decrease tumor vascularity and delay tumor growth in gliomas is by decreasing myeloid cell recruitment and infiltration into the tumor. Myeloid cells, such as macrophages and neutrophils, are known to promote tumor proliferation, angiogenesis, and invasion via secretion of multiple growth factors, such as matrix metalloproteinases, interleukin-6, and interleukin-8.21 A decrease in infiltrated myeloid cells may indirectly decrease glioma proliferation, vascularity, and invasion by removing these myeloid cell–secreted factors. Furthermore, in a recent clinical trial, we showed that early decreases in circulating CD14+ myeloid cells correlated with response to antiangiogenic therapy in patients with recurrent glioblastoma.33 Thus, pro-angiogenic and pro-tumorigenic myeloid cells appear to play a central role in glioma angiogenesis, and changes in the recruitment of these cells may serve as a biomarker of response to antiangiogenic therapy.
Resistance to antiangiogenic therapy is thought to be mediated, in part, by bone marrow–derived cells attracted to hypoxic tumors.5,6,8 In our model, the mice treated with sunitinib showed earlier development of hypoxia. In our experience, this effect was not limited to sunitinib. We have observed this phenomenon with other VEGFR inhibitors in this model (data not shown). Here, we observed an increase in CD11b+/F4/80+/Gr1– cells at the time of tumor progression in the control and sunitinib-treated animals at 4 weeks, as well as an increase in CD11b+/Gr1+ cells at 6 weeks in the groups treated with bevacizumab only. The strong correlation between the number of infiltrated myeloid cells and the level of hypoxia further supports the idea that hypoxia is an important initiator of myeloid cell infiltration. These data suggest that multiple bone marrow–derived cell types may be involved in resistance to therapy and that targeting only one bone marrow–derived cell type may not prevent resistance.
We used sunitinib in combination with bevacizumab to block the infiltration of CSF-1R+ cells.15 In the combination therapy group, there was a significant improvement in animal survival and a significant decrease in CD11b+/F4/80+/Gr1– myeloid cells at 4 weeks compared with the group treated with bevacizumab alone. At the later time points, this effect was persistent, although it did not reach statistical significance. Thus, multiple antiangiogenic agents or agents used to block CSF-1R+ cells may provide better suppression of tumor growth through inhibition of bone marrow–derived cell infiltration. Of note, the combination of these 2 antiangiogenic agents did not further promote tumor hypoxia at 4 weeks. Although the reason for this is unknown, we observed that at each time point evaluated, the tumors in the combination group were smaller in diameter than those in the other treatment groups (data not shown). This finding is in agreement with the lower proliferation rate measured in the combination therapy group. Thus, the reduction in tumor vasculature and concomitant reduction in cell proliferation may have limited the ability of the tumor to outgrow its vascular supply and thus reduced the induction of hypoxia.
Some antiangiogenic agents have been shown to promote tumor growth and metastasis.34,35 Although multiple mechanisms may be at work, it is possible that induction of hypoxia promotes a mesenchymal phenotype. Recently, sunitinib was shown to promote mesenchymal transformation in renal cell carcinoma, and this effect appeared to be reversible.36 For the first time, we demonstrate that chronic antiangiogenic therapy can promote expression of mesenchymal markers in glioma tumors in vivo, as demonstrated by a loss of e-cadherin and an increase in vimentin, smooth muscle actin, and ZEB2 expression. Loss of e-cadherin decreases cell adhesion and can promote cell invasion,37,38 as seen in these tumors. This phenomenon, potentially related to the induction of hypoxia,39,40 could underlie the enhanced invasiveness and significant resistance of tumors in patients receiving chronic antiangiogenic therapy. Furthermore, our results suggest that TGF-β is highly expressed in glioma following treatment with antiangiogenic therapy, and this mesenchymal shift could be inhibited by TGF-β inhibitors such as LY2109761.
In summary, our results have many implications for clinical practice. Anti-VEGF therapy appears to be more effective than targeting VEGFR. We suggest that one mechanism for the beneficial effect of antiangiogenic therapy is its ability to disrupt recruitment of bone marrow–derived cells to gliomas. This effect can be transient if excessive pruning promotes tumor hypoxia. Antiangiogenic therapy may promote tumor invasion by switching gliomas to develop a mesenchymal phenotype and by selecting for glioma cells expressing stem cell markers. Our findings emphasize the need to better understand the mechanisms by which antiangiogenic agents influence tumor progression and the obvious need to target pathways involved in the development of resistance, such as myeloid cells.
Funding
This work was supported in part by an American Society of Clinical Oncology (ASCO) Career Development Award and a grant from the Martha G. Williams Brain Tumor Research Fund (both to J. F. G.). The MD Anderson Cancer Center is supported in part by a Core grant (CA16672).
Conflict of interest statement. Genentech consultant/advisory board, J. F. G. and J. V. H.
References
- 1.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 2.Takano S, Yoshii Y, Kondo S, et al. Concentration of vascular endothelial growth factor in the serum and tumor tissue of brain tumor patients. Cancer Res. 1996;56:2185–2190. [PubMed] [Google Scholar]
- 3.Loges S, Schmidt T, Carmeliet P. “Antimyeloangiogenic” therapy for cancer by inhibiting PlGF. Clin Cancer Res. 2009;15:3648–3653. doi: 10.1158/1078-0432.CCR-08-2276. [DOI] [PubMed] [Google Scholar]
- 4.Kerber M, Reiss Y, Wickersheim A, et al. Flt-1 signaling in macrophages promotes glioma growth in vivo. Cancer Res. 2008;68:7342–7351. doi: 10.1158/0008-5472.CAN-07-6241. [DOI] [PubMed] [Google Scholar]
- 5.Du R, Lu KV, Petritsch C, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13:206–220. doi: 10.1016/j.ccr.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 8.Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15:4589–4599. doi: 10.1158/1078-0432.CCR-09-0575. [DOI] [PubMed] [Google Scholar]
- 9.Shojaei F, Wu X, Malik AK, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nature Biotech. 2007;25:911–920. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 10.Shojaei F, Wu X, Qu X, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106:6742–6747. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–4740. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 12.de Groot JF, Yung WK. Bevacizumab and irinotecan in the treatment of recurrent malignant gliomas. Cancer J. 2008;14:279–285. doi: 10.1097/PPO.0b013e3181867bd6. [DOI] [PubMed] [Google Scholar]
- 13.Neyns B, Sadones J, Chaskis C, et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol. 2011;103:491–501. doi: 10.1007/s11060-010-0402-7. [DOI] [PubMed] [Google Scholar]
- 14.Batchelor TT, Mulholland P, Neyns B, et al. The efficacy of cediranib as monotherapy and in combination with lomustine compared with lomustine alone in patients with recurrent glioblastoma: a phase III randomized study. Neuro-oncol. 2010;12:iv69–iv78. [Google Scholar]
- 15.Murray LJ, Abrams TJ, Long KR, et al. SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metas. 2003;20:757–766. doi: 10.1023/b:clin.0000006873.65590.68. [DOI] [PubMed] [Google Scholar]
- 16.Lal S, Lacroix M, Tofilon P, et al. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg. 2000;92:326–333. doi: 10.3171/jns.2000.92.2.0326. [DOI] [PubMed] [Google Scholar]
- 17.Chebib I, Shabani-Rad MT, Chow MS, et al. Microvessel density and clinicopathologic characteristics in hepatocellular carcinoma with and without cirrhosis. Biomark Insights. 2007;2:59–68. [PMC free article] [PubMed] [Google Scholar]
- 18.Bergers G, Song S, Meyer-Morse N, et al. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–1295. doi: 10.1172/JCI17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hashizume H, Falcon BL, Kuroda T, et al. Complementary actions of inhibitors of angiopoietin-2 and VEGF on tumor angiogenesis and growth. Cancer Res. 2010;70:2213–2223. doi: 10.1158/0008-5472.CAN-09-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer C, Mazzone M, Jonckx B, et al. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 21.Murdoch C, Muthana M, Coffelt SB, et al. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8:618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 22.Dirkx AE, Oude Egbrink MG, Wagstaff J, et al. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol. 2006;80:1183–1196. doi: 10.1189/jlb.0905495. [DOI] [PubMed] [Google Scholar]
- 23.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104:2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 24.Imai T, Horiuchi A, Wang C, et al. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol. 2003;163:1437–1447. doi: 10.1016/S0002-9440(10)63501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang MH, Wu KJ. TWIST activation by hypoxia inducible factor-1 (HIF-1): implications in metastasis and development. Cell Cycle. 2008;7:2090–2096. doi: 10.4161/cc.7.14.6324. [DOI] [PubMed] [Google Scholar]
- 26.Erber R, Thurnher A, Katsen AD, et al. Combined inhibition of VEGF and PDGF signaling enforces tumor vessel regression by interfering with pericyte-mediated endothelial cell survival mechanisms. Faseb J. 2004;18:338–340. doi: 10.1096/fj.03-0271fje. [DOI] [PubMed] [Google Scholar]
- 27.Polverino A, Coxon A, Starnes C, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res. 2006;66:8715–8721. doi: 10.1158/0008-5472.CAN-05-4665. [DOI] [PubMed] [Google Scholar]
- 28.Zhou Q, Gallo JM. Differential effect of sunitinib on the distribution of temozolomide in an orthotopic glioma model. Neuro-oncology. 2009;11:301–310. doi: 10.1215/15228517-2008-088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vecchiarelli-Federico LM, Cervi D, Haeri M, et al. Vascular endothelial growth factor–a positive and negative regulator of tumor growth. Cancer Res. 2010;70:863–867. doi: 10.1158/0008-5472.CAN-09-3592. [DOI] [PubMed] [Google Scholar]
- 30.Dawson MR, Duda DG, Fukumura D, et al. VEGFR1-activity-independent metastasis formation. Nature. 2009;461:E4. doi: 10.1038/nature08254. discussion E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dawson MR, Duda DG, Chae SS, et al. VEGFR1 activity modulates myeloid cell infiltration in growing lung metastases but is not required for spontaneous metastasis formation. PLoS One. 2009;4:e6525. doi: 10.1371/journal.pone.0006525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Groot JF, Piao Y, Tran H, et al. Myeloid biomarkers associated with glioblastoma response to anti-VEGF therapy with aflibercept. Clin Cancer Res. 2011;17:4872–4881. doi: 10.1158/1078-0432.CCR-11-0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ebos JM, Lee CR, Cruz-Munoz W, et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paez-Ribes M, Allen E, Hudock J, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–231. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hammers HJ, Verheul HM, Salumbides B, et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: evidence from a xenograft study. Molec Cancer Ther. 2010;9:1525–1535. doi: 10.1158/1535-7163.MCT-09-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson EW, Torri J, Sabol M, et al. Oncogene-induced basement membrane invasiveness in human mammary epithelial cells. Clin Exp Metastasis. 1994;12:181–194. doi: 10.1007/BF01753886. [DOI] [PubMed] [Google Scholar]
- 38.Behrens J, Mareel MM, Van Roy FM, et al. Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J Cell Biol. 1989;108:2435–2447. doi: 10.1083/jcb.108.6.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim WY, Perera S, Zhou B, et al. HIF2alpha cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest. 2009;119:2160–2170. doi: 10.1172/JCI38443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu X, Kang Y. Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res. 2010;16:5928–5935. doi: 10.1158/1078-0432.CCR-10-1360. [DOI] [PMC free article] [PubMed] [Google Scholar]