

Abstract
Large-scale cancer genomics efforts are identifying hundreds of somatic genomic alterations in glioblastoma (GBM). Distinguishing between active driver and neutral passenger alterations requires functional assessment of each gene; therefore, integrating biological weight of evidence with statistical significance for each genomic alteration will enable better prioritization for downstream studies. Here, we demonstrate the feasibility and potential of in vitro functional genomic screens to rapidly and systematically prioritize high-probability candidate genes for in vivo validation. Integration of low-complexity gain- and loss-of-function screens designed on the basis of genomic data identified 6 candidate GBM oncogenes, and RINT1 was validated as a novel GBM oncogene based on its ability to confer tumorigenicity to primary nontransformed murine astrocytes in vivo. Cancer genomics-guided low-complexity genomic screens can quickly provide a functional filter to prioritize high-value targets for further downstream mechanistic and translational studies.
Keywords: functional genomics, glioblastoma, oncogene, oncogenomics
Glioblastoma (GBM) is the most common and aggressive primary brain tumor, and median survival is a mere 14 months from diagnosis.1,2 GBMs are highly resistant to chemotherapeutics, and surgical resection provides only temporary relief prior to inevitable recurrence. Beyond well-known signature genetic alterations in the phosphatidylinositol 3-kinase (PI3K) and epidermal growth factor receptor (EGFR) signaling pathways,3,4 genomic characterization of the GBM oncogenome has revealed significant heterogeneity in the profiles of genetic alterations harbored by any single tumor.4 Although few novel mutations are highly recurrent in GBM, hundreds of low-frequency events have been identified,4 some of which likely represent true cancer drivers.5,6 Therefore, in addition to statistical significance, experimental demonstration of cancer-relevant functions is required to prove the biological importance of these genomic candidates.
The challenge of functionally validating the large number of candidate oncogenes identified via cancer genomics efforts necessitates the use of genetic screening platforms that offer a high-throughput means of systematically assaying the functions of hundreds, even thousands, of genes simultaneously. Historically, genetic screens using cDNA or shRNA libraries have been effectively leveraged in the discovery of many known cancer genes. Here, we used cancer genomic data from primary GBM specimens to design focused cDNA and shRNA libraries for use in gain- and loss-of-function screens. The integration of cancer genomics data with these functional genomics approaches led to the identification of RINT1 as a novel amplified and transforming GBM oncogene.
Materials and Methods
Anchorage-Independent Growth
All cell lines were maintained in DMEM or RPMI (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Cell Generation) and 1% penicillin/streptomycin unless otherwise noted. For 96-well anchorage-independent growth screens, expression constructs were arrayed and transfected into 293T cells in 96-well plates. Viral supernatants were collected and used to infect LN215 and LNZ308 cells in 96-well plates in the presence of 8 µg/mL polybrene (Millipore). Mean cell number per well was determined, and the infected LN215 and LNZ308 cells were then seeded in 96-well soft agar assays. Soft agar plates were monitored for 4 weeks for the appearance of colonies. Because of the low background of these cell lines in this assay, all wells containing colonies >10 cells were considered to be positive. All other soft agar assays were performed in triplicate in 6-well plates with 10 000 cells seeded per well in regular medium containing 0.4% low-melting agarose on bottom agar containing 0.7% low-melting agarose in regular medium. After 2 weeks, these plates were stained with Iodonitrotetrazolium Chloride (Sigma), and the colonies were manually counted.
Arrayed shRNA Screens
The RNAi Consortium (TRC) at the Broad Institute generated a library of 473 pLKO.1 lentivirally delivered shRNAs targeting 85 genes recurrently amplified in GBM (Supplementary material, Table S4).7 Viral supernatants for individual shRNA constructs were then produced in 293T cells in 96-well format and used to infect LN215, LN229, LNZ308, and LN382 human glioma cells in quadruplicate 384-well plates. The day after infection, 2 replicate plates were selected with puromycin to measure infection efficiency, and 5 days after selection, all plates were assayed for relative cell number using a luminometric assay in which luminescence is directly proportional to cell number (CellTiterGlo; Promega). To rank gene hits, the screen results were analyzed on a per-gene basis, and the effects of individual genes were compared between cell lines with and without the given amplification, using methodology previously used to identify TBK1 as synthetically lethal with mutant KRAS.8 In brief, the relative cell number of each shRNA construct in each cell line was first normalized using the median and maximum absolute deviation of a collection of 89 control shRNA constructs in the same cell line. The shRNAs were next mapped across cell lines and ranked according to the magnitude of their differential cell proliferation score (i.e., the difference of their means in each phenotype: with the gene amplified vs. control). After the shRNAs were ranked using this approach, an enrichment score was computed for each gene based on the distribution of its shRNAs in the list. This enrichment score was computed using a 2-sample statistic based on the likelihood ratio9 and is representative of both the extremeness of the differential cell proliferation scores of shRNAs targeting a given gene and of their consistency. The lower the shRNAs of a given gene appeared in the list, the higher their (negative) enrichment score and the greater the decrease in relative cell number that knockdown of the gene produced between cell lines with and without the amplification. To account for the fact that different genes are targeted by different numbers of shRNAs in the library, we normalized each enrichment score with the use of random permutations of an shRNA set of the same size. This permutation test also provided nominal P values for reach gene enrichment score. The end result is a list of genes sorted by their normalized enrichment scores and false discovery rate.
Proliferation Assay
To assay growth after RINT1 knockdown, 1000 cells per well were seeded into 96-well plates. The next day, cell seeding was assessed using CellTiterGlo, and experimental cells were infected with lentiviral shRNA constructs. Infected cells received fresh medium 24 h after infection and, 24 h later, were switched to media with or without puromycin; viability was scored 6 days later.
Tumorigenicity Studies
For in vivo tumorigenicity assays, 106 cells were mixed with 50% matrigel (Fisher), transplanted subcutaneously into flanks of Ncr nude mice (Taconic), and observed for tumor development. Each subline was injected bilaterally in 5 mice, and time to tumor development was monitored for each injection site. Tumor development was defined as the point at which tumor volume exceeded a minimum of 20 mm3, as determined by the equation l · w · h · π/6 (l = length, w = width, h = height). In cases in which an animal was euthanized prior to tumor development, any tumor-free injection sites were censored from the analysis. All animal experiments were approved by Harvard's Institutional Animal Care and Use Committee under Protocol No. 04-136.
Plasmids
Open reading frames of 78 recurrently amplified genes were obtained from the Human ORFeome Collection and Open Biosystems. The Gateway recombination system was used to transfer these cDNAs into the pLenti6/V5-DEST (Invitrogen), pLenti6.3/V5-DEST, and pBabe-puro-Gateway-HA expression vectors. For the loss of function screens, we used constructs targeting amplified genes from the TRC shRNA library.7 Lentiviruses and retroviruses were produced in 293T cells, and target cells were infected at 48 h and 72 h after transfection in the presence of 8 µg/mL polybrene. Infected cells were selected with 2 µg/mL puromycin for 3–5 days or 2.5–5 µg/mL blasticidin (Invitrogen) for 6 days before assaying expression. Lentivirally delivered shRNA constructs targeting GFP and RINT1 were obtained from TRC. Sequences are available from their Web site (www.broadinstitute.org/trc).
Genomic Analyses
The sample set used for aCGH was previously described3 but included specimens from 28 pathologically verified stage IV gliomas and 18 established human GBM cell lines. The criteria for inclusion in the minimal common region analysis required an amplicon to be present in at least 2 samples (at least 1 of which was a tumor), be less than 2 Mb in size, and to have a mean log2 amplitude >1. A total of 20 recurrent amplicons containing 112 known and predicted genes met these criteria. Validation of copy number amplifications was performed using data generated by TCGA from primary GBM specimens (July 28, 2011 Firehose run of the Broad Genome Data Analysis Center [http://gdac.broadinstitute.org]) and from primary low-grade glioma specimens (February 17, 2012 Firehose run of the Broad Genome Data Analysis Center). Genes with copy number log2 ratio values >0.3 were designated to be in regions of copy number gain. The Pearson correlation coefficient was used to assess the association of copy number gain and the corresponding mRNA expression, and the statistical significance was evaluated using the t statistic: 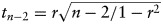 (r = Pearson correlation, n = number of samples).
(r = Pearson correlation, n = number of samples).
Immunohistochemistry and Immunoblot Analyses
Normal adult brain and primary GBM specimens were preserved in 4% paraformaldehyde and embedded in Optimum Cutting Temperature medium prior to sectioning. Antigen retrieval was performed in Citrate with steaming, and the specimens were probed with anti-RINT1 antibody (Sigma). Visualization was performed using the EnVision+ (DAB) detection system (Dako, Carpenteria), and the specimens were counterstained with hematoxylin. For immunoblot analyses, whole cell extracts were resolved using 4%–12% Bis-Tris gradient gels (Invitrogen) and transferred to PVDF membrane (Millipore) before being probed with antibodies targeting RINT1 or Vinculin (H-10, Santa Cruz Biotechnologies).
Statistical Analyses
Differences in tumor-free survival were assessed using the log rank test in GraphPad Prism software. Frequency of co-amplification of genomic loci was assessed using Fisher's exact test with a maximum false discovery rate of 5%. All other statistical differences were assessed using 2-tailed, unpaired t tests.
Results and Discussion
Whole genome copy number analysis by array-based CGH (aCGH) previously identified recurrent alterations in the tumor genomes of patients with GBM.3 Although many known GBM-relevant oncogenes and tumor suppressors were identified using this approach, a number of these statistically significant recurrently altered regions did not contain known cancer-relevant genes. On the basis of our dataset of 28 primary GBM specimens and 18 human GBM cell lines,3 we performed a minimal common region (MCR) analysis to define 20 regions of recurrent amplification containing a total of 112 known and predicted genes (Supplementary material, Table S1).
To determine the functional relevance of the candidate oncogenes resident in these regions of amplification, we constructed cDNA and shRNA libraries to interrogate the biological activities of all genes for which reagents were available. To identify candidate oncogenes capable of driving transformation, open reading frames (ORFs) representing 78 of the 112 amplified genes were cloned into a lentiviral expression plasmid (Supplementary material, Table S2). We then performed an arrayed gain-of-function screen in 96-well format to identify ORFs capable of promoting anchorage-independent growth in soft agar in 2 human GBM cell lines (LN215 and LNZ308) chosen for their low background activity in this assay. Both LN215 and LNZ308 are wild type for CDKN2A and mutant for TP53; LNZ308 cells are also mutant for PTEN, and LN215 cells are wild type at this locus.10 Visual scoring of colonies with >10 cells in this primary screen identified 17 ORFs capable of conferring anchorage-independent growth in at least one of these cell lines (Supplementary material, Table S3).
In parallel, hypothesizing that driver alterations lead to oncogene addiction in established cancer cells, we performed an arrayed RNAi loss-of-function screen in 4 human GBM cell lines chosen for their representative genetic backgrounds: LN215, LN229, LN382, and LNZ308. All 4 lines carry mutations in TP53, but in contrast to LN215 and LNZ308, LN229 and LN382 cells are both null for CDKN2A; LN229 cells are wild type for PTEN, and LN382 cells carry a truncating point mutation in PTEN.10 A library of 473 lentivirally delivered shRNA vectors targeting 85 of the amplified genes (median of 5 shRNAs per gene; Supplementary material, Table S4) was used to transduce individual shRNAs in an arrayed fashion, and any shRNA reducing relative proliferation by >1.5 standard deviations below the mean of the control shRNAs was considered to be a hit. Using this approach, 25 genes were identified as either the top scorers in the cell lines that harbored amplifications of the candidate gene or were the strongest scoring genes of their amplicon across all cell lines screened (Supplementary material, Table S5).
By combining the results of these gain-of-function and loss-of-function screens, we identified 6 candidate oncogenes (DYNC1I1, GLYCTK, NUP155, PLAGL2, RINT1, and TWF2) that were both capable of promoting anchorage-independent growth and required for the viability and/or proliferation of GBM cancer cell lines (Fig. 1A). The veracity of this prioritized list was supported by our recent work demonstrating that PLAGL2 is indeed a GBM oncogene that promotes self-renewal and suppresses differentiation of precursor cells, in part, through activation of Wnt/β-catenin signaling.11
Fig. 1.

Integration of low-complexity genetic screens identified candidate GBM oncogenes. (A) Venn diagram summarizing the results of independent primary genetic screens. Six genes were found to promote anchorage-independent growth and to be required for the proliferation of human GBM cell lines. (B) Summary of copy number changes and correlation to mRNA expression data for the 6 candidates in 484 primary GBM specimens. RINT1 was amongst the most frequently targeted for copy number gain (log2 > 0.3) and exhibited the greatest degree of copy number-correlated overexpression. (C) RINT1 is expressed in astrocytes and glioblastoma. (a) Immunohistochemical staining for RINT1 in normal adult human brain shows strong expression in reactive astrocytes in the cortex and subcortical white matter (brown stain), while staining in human GBM tumors shows a range of expression from strong diffuse cytoplasmic (b) to low cytoplasmic levels in a smaller percentage of cells (c).
As a first step in validation, we sought to confirm the human relevance of these 6 prioritized candidates in a larger cohort of human GBM samples. Specifically, we leveraged the dataset generated by The Cancer Genome Atlas on >480 primary GBM tumors for which there were both copy number and mRNA expression data (July 28, 2011 Firehose run of the Broad GDAC [http://gdac.broadinstitute.org]). On the basis of the rationale that amplification of a true oncogenic driver would be expected to result in a concomitant increase in mRNA expression level, we calculated the correlation of copy number to mRNA expression level across 484 primary GBM specimens characterized by the TCGA (Fig. 1B). RINT1 demonstrated the highest degree of copy number–correlated overexpression in this dataset. Furthermore, the RINT1 locus (located on 7q22) exhibited copy number gains in 68% of patients (log2 > 0.3; 343 of 501 samples) in the TCGA GBM dataset, and 15.4% of patients (77 of 501 samples) exhibited copy number gains greater than would be expected from trisomy in a homogeneous tumor cell population (log2 > 0.6). Furthermore, in a recent cross-tumor analysis of copy number alterations in >3100 tumor specimens,12 RINT1 was found to be significantly amplified (q = 0.13), strongly suggesting that RINT1 is recurrently targeted for amplification in human tumors. Furthermore, as a complementary test of GBM tumor relevance, we performed immunohistochemical staining of RINT1 in specimens of normal adult brain and primary GBM tumors (Fig. 1C). Although RINT1 expression was only observed in reactive astrocytes of the cortex and subcortical white matter of normal brain, primary GBM specimens exhibited a range of RINT1 expression levels in the majority of tumor cells, thus confirming that RINT1 is expressed in primary GBM tumors. Taken together, these data supported the prioritization of RINT1 for functional validation in vitro and in vivo.
To verify and extend the primary screen results, we next confirmed that RINT1 overexpression promoted anchorage-independent growth in 3 independent GBM cell lines, LNZ308, Hs683, and U343 cells (Fig. 2A). Complementing these gain-of-function in vitro studies, we also validated the requirement of RINT1 in maintaining viability of established human GBM cells. Here, 3 independent shRNAs targeting RINT1 used in the primary screen were obtained, and relative suppression by each shRNA was assessed in LN340 cells by immunoblot (Fig. 2B left panel). The 2 shRNAs with the strongest phenotype, sh234 and sh1052, each of which significantly decreased RINT1 protein levels, were then assessed for their impact on proliferation and viability in a panel of 5 GBM cell lines, including the 4 cell lines tested in the original loss-of-function screen. These cell lines were selected for their range of RINT1 copy number and expression status: LN340, LN382, and LNZ308 exhibit copy number gains of RINT1 (log2 > 0.3), whereas LN229 and GB1 do not have amplification of this locus but maintain expression of RINT1 protein (Supplementary material, Fig. S1). As shown in Fig. 2B, the viability of all 5 tested GBM cell lines, independent of RINT1 copy number, was significantly reduced by at least 1 of the shRNAs targeting RINT1. Examination of these cells confirmed that introduction of shRNAs that decrease RINT1 protein levels significantly decreased the growth of these cells, whereas shRNAs that do not affect RINT1 protein levels have no effect on growth.
Fig. 2.

Expression of RINT1 promotes anchorage-independent growth and is required for the viability of human GBM cell lines. (A) Overexpression of RINT1 promoted soft agar colony formation in three human GBM cell lines. Each experiment was performed in triplicate and repeated at least twice, and a representative experiment is shown. (B) Left panel: Three unique shRNAs targeting RINT1 were assessed for their ability to down-regulate RINT1 protein expression in LN340 cells relative to uninfected cells (Parental) or cells infected with a nontargeting shRNA (shNTC) or an shRNA targeting GFP (shGFP). Right panel: The two shRNAs exhibiting the greatest effect on RINT1 protein expression (sh234 and sh1052) were infected in a panel of human GBM cell lines and the relative viability of each line was determined 7 days later using an ATP-based luminescence assay. The mean relative viability (±SD) of triplicate wells of each cell line is plotted. The experiment shown is representative of triplicate experiments. *P < .05, **P < .005.
Mindful of the limitations of in vitro assays in established human GBM cells, we next tested the sufficiency of RINT1 to promote in vivo tumorigenesis in primary nontransformed astrocytes isolated from genetically engineered mouse models. Specifically, we overexpressed RINT1 in primary immortalized murine p16Ink4a/Arf−/−; Pten−/− (Fig. 3A) and p16Ink4a/Arf−/− (Fig. 3B) astrocytes prior to subcutaneous transplantation in immunodeficient mice. Overexpression of GFP did not significantly alter tumor latency or penetrance relative to parental astrocytes, and PLAGL2 served as a positive control.11 As shown in Fig. 3A, both PLAGL2- and RINT1-overexpressing p16Ink4a/Arf−/−; Pten−/− astrocytes formed subcutaneous tumors with significantly shorter latency and higher penetrance than was observed for either the parental astrocytes alone or astrocytes overexpressing GFP (P = .002 for RINT1 vs GFP; P < .0001 for PLAGL2 vs GFP). Overexpression of RINT1 also significantly promoted tumor formation (P = .002) in p16Ink4a/Arf−/− astrocytes (Fig. 3B), indicating that loss of Pten was not required for RINT1 to promote tumorigenesis. Taken together, these gain-of-function and loss-of-function assays in both mouse and human cell systems in vitro and in vivo validated RINT1 as a novel GBM oncogene that is capable of transforming primary murine astrocytes and is required for viability of established human GBM cells.
Fig. 3.

RINT1 promoted in vivo tumor formation in primary murine astrocytes. Kaplan–Meier plots of tumor-free injection sites. (A) p16Ink4A/Arf−/−;Pten−/− primary nontransformed murine astrocytes were transduced with GFP, RINT1, or PLAGL2 and injected subcutaneously in nude mice. Overexpression of either RINT1 or PLAGL2 significantly promoted tumor formation (P = .002 and P < .0001, respectively). (B) p16Ink4A/Arf−/− primary murine astrocytes were transduced with GFP or RINT1 and injected subcutaneously in nude mice. RINT1 significantly promoted tumor formation in these cells (P = .002). Each experiment was performed at least twice, and a representative experiment is shown. Representative photomicrographs (10× magnification) of hematoxylin and eosin–stained sections of GFP- and RINT1-overexpressing tumors are included in the lower panels.
To better understand the genetic context in which RINT1 gain occurs, we assessed the frequency of RINT1 alterations among tumors of the 4 transcriptomal subtypes previously defined by TCGA.13 Overexpression and copy number gain of RINT1 is observed significantly more frequently in classical-type tumors from the TCGA sample set, although the degree of overrepresentation of RINT1 alterations was less than that observed for EGFR (Supplementary Material, Fig. S2). Although statistically significant, it is unlikely that this degree of enrichment of RINT1 alterations is sufficient to drive the mRNA expression profile that defines classical-type tumors. Furthermore, among the signature GBM genetic alterations, amplification of EGFR (P = 1.28 × 10−16) or MET (P = 1.06 × 10−65) significantly correlated with increased RINT1 copy number. Although this observation does not prove that these oncogenic receptor tyrosine kinases functionally cooperate with RINT1 in primary tumors, the possibility of functional interaction should be explored in future studies.
Advances in high-resolution microarrays and massively parallel sequencing have made it possible to characterize cancer genomes and their evolution at an unprecedented level.4,14,15 To translate such genomic discoveries into tangible therapeutic and diagnostic end points in the clinic, extensive and time-consuming functional studies are required to differentiate driver from passenger mutations. Here, we have demonstrated the feasibility of using low-complexity screens to rapidly apply a biological filter to a set of candidate oncogenes defined by evidence of genomic copy number gains and, through this approach, identified RINT1 as a novel GBM oncogene.
RINT1 (or RAD50 interactor 1), as its name suggests, was identified from a yeast-2-hybrid experiment to find proteins interacting with RAD50.16 It has since been shown to play complex roles in the G2/M checkpoint, telomere elongation, maintenance of centrosome integrity, and vesicle trafficking between the endoplasmic reticulum and Golgi apparatus.16–19 Although some of these studies have suggested a role as a tumor suppressor, our data demonstrate that RINT1 functions as an oncogene in GBM. Furthermore, the absence of somatic mutations or gene fusions involving RINT1 validates our approach of identifying candidate oncogenes based just on copy number alterations and consequent mRNA expression changes. Delineating the mechanisms and pathways through which RINT1 promotes GBM tumorigenesis will be necessary as a next step to identify new points of therapeutic intervention for this devastating disease.
Supplementary Material
Funding
S. N. Q. was supported by a fellowship from the Canadian Institutes of Health Research. M. G. C. was supported by National Institutes of Health grants K08NS062907 and K12CA090354, an AACR-NBTF Fellowship in memory of Bonnie Brooks, and a Trudy Bettiker/American Brain Tumor Foundation Fellowship. L. C., W. C. H., and K. L. L. are supported by grants from the Ben and Catherine Ivy Foundation, the Snyder Medical Foundation, the Sontag Foundation, the Goldhirsh Foundation, and National Institutes of Health grants PO1CA095616, U01CA141508, U24CA143845, and RC2CA148268.
Supplementary Material
Acknowledgments
We thank members of the Chin, Hahn, and DePinho laboratories for helpful discussions. Authors Steven N. Quayle and Milan G. Chheda contributed equally to this work.
Conflict of interest statement. None declared.
References
- 1.Van Meir EG, Hadjipanayis CG, Norden AD, et al. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60(3):166–193. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Wiedemeyer R, Brennan C, Heffernan TP, et al. Feedback circuit among INK4 tumor suppressors constrains human glioblastoma development. Cancer Cell. 2008;13(4):355–364. doi: 10.1016/j.ccr.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318(5853):1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 6.Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moffat J, Grueneberg DA, Yang X, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124(6):1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 8.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J. Powerful goodness-of-fit tests based on the likelihood ratio. J R Stat Soc Series B Stat Methodol. 2002;64(2):281–294. [Google Scholar]
- 10.Ishii N, Maier D, Merlo A, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9(3):469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng H, Ying H, Wiedemeyer R, et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell. 2010;17(5):497–509. doi: 10.1016/j.ccr.2010.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463(7283):899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding L, Ellis MJ, Li S, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464(7291):999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao J, Liu CC, Chen PL, Lee WH. RINT-1, a novel Rad50-interacting protein, participates in radiation-induced G(2)/M checkpoint control. J Biol Chem. 2001;276(9):6105–6111. doi: 10.1074/jbc.M008893200. [DOI] [PubMed] [Google Scholar]
- 17.Kong LJ, Meloni AR, Nevins JR. The Rb-related p130 protein controls telomere lengthening through an interaction with a Rad50-interacting protein, RINT-1. Mol Cell. 2006;22(1):63–71. doi: 10.1016/j.molcel.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 18.Arasaki K, Taniguchi M, Tani K, Tagaya M. RINT-1 regulates the localization and entry of ZW10 to the syntaxin 18 complex. Mol Biol Cell. 2006;17(6):2780–2788. doi: 10.1091/mbc.E05-10-0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin X, Liu CC, Gao Q, et al. RINT-1 serves as a tumor suppressor and maintains Golgi dynamics and centrosome integrity for cell survival. Mol Cell Biol. 2007;27(13):4905–4916. doi: 10.1128/MCB.02396-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.