

X-ray-induced photo-chemistry of metal sites within biological molecules is a concern for X-ray absorption spectroscopy, X-ray crystallography and other techniques in which samples are illuminated with X-rays. The effects of X-ray-induced photo-chemistry are reviewed and the methods used to mitigate these in X-ray absorption spectroscopy are outlined.
Keywords: X-ray absorption spectroscopy, extended X-ray absorption fine structure, X-ray absorption near edge, photo-reduction, metalloproteins, metalloenzymes
Abstract
As synchrotron light sources and optics deliver greater photon flux on samples, X-ray-induced photo-chemistry is increasingly encountered in X-ray absorption spectroscopy (XAS) experiments. The resulting problems are particularly pronounced for biological XAS experiments. This is because biological samples are very often quite dilute and therefore require signal averaging to achieve adequate signal-to-noise ratios, with correspondingly greater exposures to the X-ray beam. This paper reviews the origins of photo-reduction and photo-oxidation, the impact that they can have on active site structure, and the methods that can be used to provide relief from X-ray-induced photo-chemical artifacts.
1. Introduction
Metal ion cofactors are responsible for the most biologically significant and industrially and environmentally relevant chemistry catalyzed by living systems. Examples range from the nitrogenase enzymes, which use a Mo—Fe—S cluster to catalyze conversion of dinitrogen to ammonia (Howard & Rees, 2005 ▶); and the photosynthetic oxygen evolving complex with a CaMn4 cluster (Yano et al., 2006 ▶); to the bacterial benzoylcoenzyme A reductases which use a tungsten-containing active site to catalyze Birch-type de-aromatization reactions, in what is the most reducing biochemistry currently known (Kung et al., 2010 ▶). It has been estimated that metalloproteins constitute close to 30% of the genomic output of most organisms. Despite their importance, much remains unknown concerning the roles that metals play in biology; in support of this, analyses of archaeabacterial and eubacterial microorganisms indicate that the microbial metalloproteome is largely uncharacterized (Cvetkovic et al., 2010 ▶). An elucidation of the exact nature of the active-site structure is key to understanding catalytic mechanism, and in many cases a combination of protein structural methods and X-ray absorption spectroscopy (XAS) techniques have proved useful in determination of structure (e.g. Pushie & George, 2011 ▶; Pushie et al., 2012 ▶). Similarly, XAS has been used in combination with other spectroscopic techniques to study metal-containing drugs in cells and tissues or changes in the biochemistry of metals (Aitken et al., 2010 ▶, 2011 ▶; Carter et al., 2010 ▶). XAS also can be used directly on protein gels to determine active-site structure of metals in individual protein bands (Finney et al., 2010 ▶). The X-rays used for spectroscopy and for crystallography are ionizing radiation and thus have the ability to photo-reduce metal active sites within proteins (e.g. Penner-Hahn et al., 1989 ▶; George et al., 1999 ▶; Ascone et al., 2003 ▶; Levina et al., 2005 ▶; Yano et al., 2005 ▶; Corbett et al., 2007 ▶; Antonyuk & Hough, 2011 ▶). The purpose of this review is to discuss the phenomenon of photo-reduction, its implications for metalloprotein active-site structure determination, and the methods used to minimize the extent of photo-reduction in XAS experiments.
2. Materials and methods
XAS measurements at the Stanford Synchrotron Radiation Lightsource (SSRL) were conducted with the SPEAR3 storage ring containing 100 or 200 mA at 3.0 GeV. Hard X-ray data were collected using the structural molecular biology XAS beamlines 9-3 and 7-3. Both beamline 9-3 and 7-3 are equipped with a rhodium-coated vertical collimating mirror upstream of the Si(220) monochromator; beamline 9-3 has an additional downstream rhodium-coated bent-cylindrical focusing mirror. Harmonic rejection was accomplished by setting the cut-off angle of the mirrors to an appropriate energy. Incident and transmitted X-rays were monitored using gas ionization chambers and X-ray absorption was measured as the primary fluorescence excitation spectrum using an array of 30 germanium detectors. During data acquisition samples were maintained at a temperature of approximately 10 K using an Oxford Instruments CF1204 helium cryostat. Experiments at the sulfur K-edge were carried out on SSRL beamlines 6-2 or 4-3 as previously described (Gnida et al., 2007 ▶; Hackett et al., 2012 ▶). Data reduction and analysis employed the EXAFSPAK suite of computer programs (http://ssrl.slac.stanford.edu/exafspak.html) and was carried out as previously described (George et al., 1996 ▶).
Experiments at the Australian Synchrotron (AS) (200 mA, 3.0 GeV) were performed on the 1.9 T wiggler X-ray Absorption Spectroscopy beamline (Glover et al., 2007 ▶), using a Si(111) monochromator. Harmonic rejection was performed by setting the cut-off angle of the mirror to an appropriate energy. Incident and transmitted X-rays were monitored using gas ionization chambers and X-ray absorption was measured as the primary fluorescence excitation spectrum using a 100-pixel germanium detector (Eurisys). During data acquisition samples were maintained at room temperature for V samples and a temperature of approximately 12 K using a closed-cycle helium cryostat for protein solutions.
Experiments at the bending-magnet Australian National Beamline Facility (ANBF; beamline 20B) at the Photon Factory (300 mA, 2.5 GeV) were performed using a Si(111) monochromator. The beamline was detuned 50% for harmonic rejection. Incident and transmitted X-rays were monitored using gas ionization chambers and X-ray absorption was measured as the primary fluorescence excitation spectrum using a 36-pixel germanium detector (Eurisys). During data acquisition samples were maintained at room temperature for V samples.
For K-edge V X-ray absorption near-edge structure (XANES) the following step sizes were employed in the 5250–5700 eV range: 10 eV below 5450 eV, 0.25 eV at 5450–5525 eV and 2 eV above 5525 eV. The energy scale was calibrated using a V foil as an internal standard [calibration energy, 5465.0 eV, corresponding to the first peak of the first derivative of the V(0) edge] (Thompson et al., 2001 ▶). Calibration, averaging and splining of XANES data were performed using the XFit software package. The spectra were normalized (using the Spline program within the XFit package) according to the method of Penner-Hahn and co-workers (Weng et al., 2005 ▶), to match the tabulated X-ray cross-section data (McMaster et al., 1969 ▶; Newville, 2005 ▶) for V [in a similar manner to that described earlier for Cr(III) XANES spectra] (Nguyen et al., 2008 ▶). This normalization technique led to consistent XANES spectra for all of the samples, regardless of the beamline used and the signal-to-noise ratio.
Na[VO4] was purchased from Aldrich (purity ≥99%). Other reagents, of analytical or higher purity grade, were purchased from Merck or Sigma-Aldrich and used without further purification. Water was purified by the Milli-Q technique. Bovine serum albumin fraction V (BSA, ∼99%, Cat. No. A3059) was purchased from Sigma and heat-inactivated and membrane-filtered newborn calf serum was purchased from Invitrogen (Cat. No. 26010-066).
For reactivity studies of vanadate with blood and its components, stock solutions of Na3[VO4] (50 mM) were freshly prepared in Ar-saturated water. Aliquots of the stock solutions were then added to the biological medium [aqueous buffer solution of BSA (0.5 mM) or heat-inactivated calf serum to the final V concentration of 1.0 mM], and the reactions were performed at 310 K under ambient air conditions for 4 h or 20 h, respectively. The reaction volumes were 0.10 ml for the BSA solution and 1.0 ml for all other solutions. After the reactions the serum samples were immediately frozen at 193 K and freeze-dried (217 K, 0.5 mbar for 20 h) for XANES spectroscopy (the freeze-dried samples were kept desiccated at 277 K for 1–2 weeks prior to the analyses).
Solutions of V complexes with BSA were passed through Micro Bio-Spin P-30 gel-filtration columns (Bio-Rad Cat. No. 732-6224; filled with crosslinked polyacrylamide gel; molecular weight cut-off, 30 kDa) by gentle centrifugation (1000g for 2 min at 295 K) to remove unbound or loosely bound V(V/IV) species (De Cremer et al., 2002 ▶; Harris et al., 1984 ▶). Prior to gel-filtration, the columns were saturated with the buffer that was used for the reactions of the V complex with BSA, according to the manufacturer’s instructions. The filtrates were then freeze-dried for XANES spectroscopy, as described above. After the spectroscopic measurements the samples were re-dissolved in 0.10 ml of Milli-Q water, and separate aliquots of these solutions were taken for the determination of total V and protein content. Total V content in the filtrates was determined by graphite furnace atomic absorption spectroscopy after digestion of the samples with 69% HNO3 (Merck, trace pure), using a Varian SpectrAA20 spectrometer with GTA96 graphite tube atomiser. A standard solution of V(IV) (1000 p.p.m. V in 1.0 M HCl, Aldrich) was used for calibration. Protein content in the samples was determined with Bradford reagent (Sigma Cat. No. B6916), using BSA as the standard, according to the manufacturer’s instructions. Myoglobin samples were prepared as reported previously (Rich et al., 1998 ▶).
3. Origins of photo-reduction
Solutions of biological molecules in XAS experiments are often dilute, and to a first approximation the radiochemical properties of water will be the most important factor in solution. Water photo-chemistry therefore will be reviewed briefly before considering the effects upon biological molecules.
3.1. Water photo-chemistry
The chemical transformations of water induced by ionizing radiation have been well studied (e.g. see Gunter, 1967 ▶; Garrett, 2005 ▶; El Omar et al., 2011 ▶). Water decomposes under ionizing radiation into a handful of major species, namely hydrated electrons (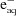 ), hydroxyl radicals (OH
), hydroxyl radicals (OH ), the water radical cation (H2O+), hydrogen radicals (H), hydroxonium ions (H3O+) and hydrogen peroxide (H2O2). In ice the free radical species such as OH have been characterized by electron paramagnetic resonance spectroscopy (e.g. Symons, 1980 ▶; Plonka et al., 2000 ▶). Ionizing radiation will cause ionization of water,
), the water radical cation (H2O+), hydrogen radicals (H), hydroxonium ions (H3O+) and hydrogen peroxide (H2O2). In ice the free radical species such as OH have been characterized by electron paramagnetic resonance spectroscopy (e.g. Symons, 1980 ▶; Plonka et al., 2000 ▶). Ionizing radiation will cause ionization of water,
The resultant radical cation can further react with water to produce hydroxonium ions and hydroxyl radicals,
These photolysis products can recombine to reconstitute water or to form new species. Thus, the hydroxyl radicals can further react to form H2O2, which in turn can engage in a series of additional reactions, e.g.
Two hydrated electrons can also combine with water to form molecular hydrogen,
The radiochemical yield of an individual chemical species is specified by its G value, which is defined as the yield per 100 eV of energy deposited in the sample. The latter is simply given by the photo-absorption cross section. G values are sensitive to a number of factors including temperature as well as the concentration and type of any radical scavengers, but typical water values for , H2, OH and H2O2 are 0.28, 0.047, 0.28 and 0.073, respectively (Choppin et al., 2002 ▶). Overall yields of reactive species are much greater at lower X-ray energies because of the much higher X-ray photo-absorption cross sections (e.g. McMaster et al., 1969 ▶; Newville, 2005 ▶), and as a consequence photo-chemical effects at low X-ray energies (e.g. below 2 keV) are particularly marked (George et al., 2008 ▶; Fong et al., 2011 ▶).
Thus, photo-ionization of water yields a number of highly reactive species that can interact with biological molecules present in solution, which may result in either photo-oxidation or photo-reduction. As far as photo-reduction is concerned, the most relevant species appear to be the hydrated electrons, mobile cavity-like structures of about 2.5 Å in radius (Kevan, 1981 ▶; Marsalek et al., 2012 ▶). Photo-oxidation can occur via reactions of OH and its degradation products such as H2O2.
Similar, but often somewhat more complex, radiochemical processes can occur with proteins, nucleic acids and other components of biological samples. Insofar as the effects of ionizing radiation on nucleic acids of living cells are concerned, it has been estimated that approximately one-third of these are due to direct photo-chemical action upon the nucleic acids, with the remainder due to mobile entities predominantly from water photo-chemistry (Hall, 1994 ▶). In many cases photo-reduction of metal sites in proteins is thought to occur well in advance of other recognized photo-damage mechanisms, such as those acting on disulfide bridges. The preference for photo-reduction probably arises, in part, from more accessible pathways along protein chains for electron transfer than oxidation processes (Levina et al., 2005 ▶).
3.2. Metal and metalloid photo-reduction and photo-oxidation
In the case of transition metal ions, by far the most commonly observed reaction is that of photo-reduction (e.g. Penner-Hahn et al., 1989 ▶; George et al., 1999 ▶; Corbett et al., 2007 ▶; Antonyuk & Hough, 2011 ▶). Photo-reduction of reducible oxy-anions such as selenate is also readily observed, even at cryogenic temperatures (Fig. 1 ▶); on modern high-brightness beamlines a pure spectrum of selenate tends to be difficult to obtain, and in many cases photo-reduction is evident and not commented upon (e.g. Misra et al., 2010 ▶). In contrast, photo-oxidation is rarely observed in XAS experiments, although it is seen in relatively low-energy measurements such as with sulfur K-edge XAS measurements of proteins (e.g. Fig. 2 ▶). Similar photo-oxidation phenomena have recently been observed in sulfur K-edge measurements of brain tissue (Hackett et al., 2012 ▶). Another example is provided by buffered aqueous solutions of arsenite [As(OH)3 at neutral pH], which will rapidly photo-oxidize in room-temperature XAS experiments to arsenate {a mixture of [As(OH)O3]2− and [As(OH)2O2]− at neutral pH; not illustrated}.
Figure 1.
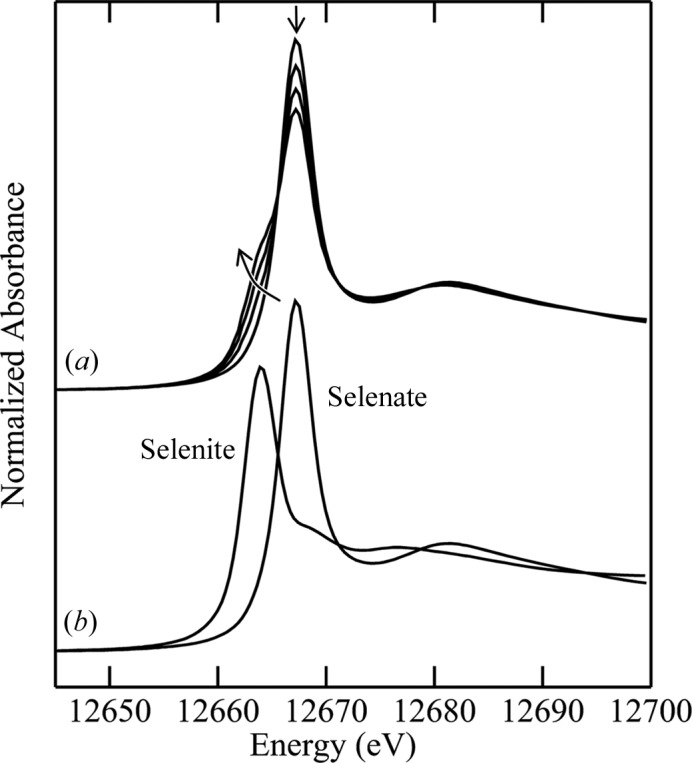
Photo-reduction of a 1 mM buffered aqueous solution of sodium selenate (50 mM HEPES/NaOH pH 7.5, 20% v/v glycerol). The sample was measured on SSRL BL9-3 at 10 K using a PIPS Si detector to monitor fluorescence. (a) Superposition of four consecutive sweeps each separated by 6 min, illustrating increasing degrees of photo-reduction on a sweep-by-sweep basis. (b) Spectra corresponding to solutions of selenate and selenite. The spectra in (a) can be fitted well using a linear combination of selenite and selenate spectra (b) (not illustrated), suggesting that the photo-chemical reaction involves only these two species.
Figure 2.
Sequential scans of the X-ray absorption near-edge region of the thiol protease papain from papaya (Carica papaya). Papain contains seven cysteine residues, six of which are involved in three cystine disulfide linkages. The spectra show the characteristic disulfide double peak close to 2470 eV, with the spectrum of oxidized forms (sulfates and sulfonates) appearing near 2480 eV with increasing exposure to the beam. Data were measured at room temperature in 1 M bis-tris buffer at pH 7.0 on SSRL beamline 6-2.
Numerous examples of metalloprotein active-site photo-reduction have been reported, both in XAS and in crystallographic experiments. Sulfite oxidase is the prototypical member of the sulfite oxidase family of molybdenum enzymes and its crystal structure was among the first of the molybdenum enzymes to be solved (Kisker et al., 1997 ▶). Comparison with previous XAS results (e.g. Cramer et al., 1981 ▶; George et al., 1996 ▶) revealed that the crystal structure was actually that of the photo-reduced enzyme (Kisker et al., 1997 ▶). Indeed, the only reported crystal structures of oxidized sulfite oxidase are of mutant enzymes possessing highly modified molybdenum redox properties (Qiu et al., 2010 ▶). In contrast, no photo-reduction has been observed in any of the reported XAS measurements of sulfite oxidase. As discussed by George et al. (1999 ▶), this is probably because the crystallographic measurements required approximately 600-fold absorbed greater radiation dose, and were carried at the much higher temperature of 95 K, compared with 10 K for the XAS. In fact, the structural changes in the active site on reduction are quite significant, changing from a cis-dioxo to a mono-oxo site with a water ligand to Mo (Harris et al., 2006 ▶), and the XAS of oxidized enzyme provided information essential to improving understanding of the catalytic mechanism of sulfite oxidase (Pushie & George, 2011 ▶).
An example of metalloprotein active-site photo-reduction in an XAS experiment has been observed for an iron-containing archaeal superoxide reductase (Clay et al., 2002 ▶) in which the kinetics were determined to a reasonable approximation as pseudo-first order (Fig. 3 ▶). As discussed by Clay et al. (2002 ▶) it seems likely that the earlier crystal structure of superoxide reductase was in fact of photo-reduced enzyme (Yeh et al., 2000 ▶). Active-site photo-reduction has also been observed in XAS of iron–sulfur proteins such as putidaredoxin (Corbett et al., 2007 ▶), in mammalian ferrochelatase (Ferreira et al., 2002 ▶) and in copper proteins (Penner-Hahn et al., 1989 ▶).
Figure 3.

Fe K-edge XAS near-edge spectra showing X-ray-induced photo-reduction of superoxide reductase from Pyrococcus furiosus (data re-plotted from Clay et al., 2002 ▶) showing (a) oxidized Fe(III) enzyme, (b) photo-reduced enzyme and (c) dithionite-reduced Fe(II) enzyme. The spectra of photo-reduced enzyme can be represented by a linear combination of Fe(III) and dithionite-reduced Fe(II) enzyme. The inset shows quantification of photo-reduction with time since start of irradiation using linear-combination fitting, indicating pseudo first-order kinetics (Clay et al., 2002 ▶). Data were measured on SSRL BL7-3.
Whether or not a particular metalloprotein will show a tendency to photo-reduction is notoriously difficult to predict. With small molecules, Mesu et al. (2006 ▶) have investigated X-ray photo-reduction of different aqueous Cu(II) species at room temperature. Although the chemistry of the solutions was not always well defined, these workers found that the photo-reduction process was influenced by a combination of factors: the counter-ions present in solution (e.g. SO4 2−, NO3 − or Cl−); the ligands bound to the copper ion; and the redox potential of the metal. Thus, no reduction was observed for Cu(II) in the presence of ammonia with SO4 2− or NO3 − as counter-ions, but reduction was observed with Cl− and Br− as counter-ions. Moreover, extensive reduction of Cu(II) in the presence of imidazole was observed irrespective of the counter-ion. In most cases, 100 mM SO4 2− provided some protection against photo-reduction, suggesting hydrated electrons are the primary reductant (Mesu et al., 2006 ▶), although a role for potential ligands to the metal also seems likely.
In a study to identify the amount of photo-reduction as a function of dose during macromolecular crystallographic measurements in order to validate the oxidation state of the Fe in the heme active site of Saccharomyces cerevisiae cytochrome c peroxidase (Bonagura et al., 2003 ▶), the single-crystal XAS facility at SSRL BL9-3 was used (Latimer et al., 2005 ▶). This facility provides the capability to measure crystallography and XAS on the same sample during the same experiment. As demonstrated in Fig. 4(a) ▶, the initial scan established the presence of Fe(IV), i.e. that the compound I state had been achieved in the crystal. However, for each ∼20 min full extended X-ray absorption fine-structure (EXAFS) scan there was a relatively rapid photo-reduction, which by ∼60 min total exposure had produced an Fe(III) oxidation state. This was corroborated by measuring the XAS of Fe(III) initial-state crystals [Fig. 4(b) ▶; note the overlap of spectra labeled Fe(IV) 40 min and Fe(III) 0 min]. This Fe(III) state was then gradually reduced to the Fe(II) state with X-ray dose/time. Indications of the change in electronic structure are demonstrated both in the change in energy for the rising edge [∼2.4 eV in Fig. 4(a) ▶] and in the change in number of transitions and their intensities and energies for the 1s → 3d pre-edge features around 7112 eV in Fig. 4 ▶ (Hedman et al., undated ▶).
Figure 4.

Fe K-edge XAS spectra of Saccharomyces cerevisiae cytochrome c peroxidase single crystals (∼300 µm × 300 µm × 200 µm) recorded at SSRL BL9-3, showing (a) gradual photo-reduction of the Fe(IV) state at t = 0 to the Fe(III) state after ∼40 min exposure, and (b) gradual photo-reduction of the Fe(III) state to Fe(II). The insets depict the Fe K-edge 1s → 3d pre-edge transitions.
4. Experimental precautions against X-ray beam-induced photo-reduction
In this section, currently used experimental precautions against photo-chemistry are discussed with an emphasis on photo-reduction as this is the major problem in collection of XAS of metalloproteins.
In most modern XAS experiments the time taken to align the sample in the X-ray beam is minimal, and the near-edge spectrum or XANES is quickly obtained in the first part of the scan, which may extend to EXAFS at very high k. Accurate energy calibration on a per-sweep basis is typically available using the simple expedient of placing a metal foil between two ion chambers following the sample (Tse et al., 2011 ▶). Examination of the sequential XANES scans often can warn that photo-reduction is taking place, and can allow determination of its extent, although absence of a change could indicate that photo-reduction is complete before the first XANES is collected (see §4.2). For new systems that are suspected to be prone to photo-damage, a reduced flux or flux density might be employed (§4.1) and then increased until the sensitivity to the beam is ascertained. In an optimal strategy, several XANES scans with careful energy calibration might be performed on any new type of biological sample at a particular beamline; the first with a minimum of exposure time and reduced flux in order to assess the shape of the unaffected XANES spectrum. Subsequently the X-ray dose could be increased gradually in order to optimize a signal-to-noise ratio compatible with the X-ray sensitivity of the sample. The comparison of the initial and final spectra after several complete sweeps will ascertain the extent of photo-damage that could otherwise affect the veracity of XANES-derived speciation and/or XANES/EXAFS structural determinations.
4.1. X-ray beam flux density
A major factor in the X-ray beam-induced chemical changes in samples is the photon flux density of the X-ray beam at the sample, here defined as photons per second per unit area at the sample. Higher flux densities will lead to more pronounced problems associated with photo-reduction. For example, ESRF ID24 produces 1012 photons s−1 in a beam spot size of 0.03 mm × 0.2 mm, whereas SSRL BL7-3 produces approximately the same flux of 1012 photons s−1 but within a much larger beam spot of approximately 2 mm × 15 mm; the flux densities of these two beamlines thus vary by about 5000 times. The available flux densities of selected beamlines world-wide are given in Table 1 ▶. From the arguments made above (§3.1) the rate of photo-reduction is expected to be approximately proportional to flux density, although this has not yet been confirmed experimentally. For measurements governed by photon statistics, signal-to-noise is expected to be proportional to the square root of the count rate, which in turn is directly proportional to both the count time and the flux density. Furthermore, if we assume that the kinetics of photo-reduction is a first-order reaction then the half-life of the photo-reduction reaction will also be inversely proportional to flux density. Thus, the strategies of simply acquiring experimental data faster and reducing the flux density are expected to be essentially equivalent in terms of signal-to-noise and extent of photo-reduction.
Table 1. Comparison of flux densities for selected XAS beamlines† at 10 keV.
| Beamline | Flux density (photons s−1 mm−2) | Aperture (V × H) (mm) | Current (mA) | GeV |
|---|---|---|---|---|
| SSRL 9-3‡ | 1.7 × 1012 | 0.4 × 3 | 100§ | 3 |
| SSRL 7-3 | 3.3 × 1010 | 2 × 15 | 100§ | 3 |
| AS XAS (focused) | 3.3 × 1014 | 0.1 × 0.3 | 200 | 3 |
| AS XAS (defocused) | 1.0 × 1013 | 0.5 × 2 | 200 | 3 |
| PF ANBF | 5.0 × 109 | 2 × 10 | 450 | 2.5 |
| APS 20BMB (focused) | 1.0 × 1012 | 0.2 × 0.5 | 100 | 7 |
| APS 20BMB (unfocused) | 2.0 × 109 | 1 × 10 | 100 | 7 |
| APS 20ID (microfocus)¶ | 4.0 × 1016 | 0.005 × 0.005 | 100 | 7 |
| ESRF ID24 | 1.7 × 1014 | 0.03 × 0.2 | 100 | 6 |
| CLS BioXAS (focused)†† | 3.4 × 1014 | 0.05 × 0.3 | 250 | 2.9 |
| CLS BioXAS (defocused)†† | 5.0 × 1012 | 0.5 × 2 | 250 | 2.9 |
| LNLS XAFS2 | 5.0 × 109 | 2 × 10 | 450 | 2.5 |
This table is not intended to provide a comprehensive review of beamlines.
Recent flux data for SSRL 9-3 are not available because this beamline is currently under repair.
SSRL now routinely operates at 450 mA and fluxes are expected to approximately scale accordingly.
We note that APS 20ID is not a standard XAS beamline. It is included here to illustrate the remarkably high flux densities that can be available on state-of-the-art third-generation infrastructure.
Calculated values presented as the CLS BioXAS beamlines are not yet operational.
At very high flux densities different chemical processes may be observed. Thus, Mesu et al. (2006 ▶) using ESRF ID24 observed quite extreme photo-chemistry such as H2 bubble formation and reduction of some Cu(II) compounds to metallic copper [Cu(0)]. The comparatively simple expedient of defocusing the X-ray beam to illuminate large samples may provide substantial relief from photo-reduction, and some modern XAS beamlines are deliberately designed to produce a large X-ray beam. For example, the only specular optic present in SSRL BL7-3 is a vertically collimating mirror prior to the X-ray monochromator. The experimental advantages of low flux densities are mitigated by the practical necessities of sample preparation and experimental design. Purified proteins at concentrations suitable for XAS usually require considerable effort to produce even small quantities, precluding large XAS sample sizes. Moreover, the use of sample cryostats (§4.2) and other end-station equipment often restricts the beam size that can be used. Thus, on SSRL BL7-3 most experiments are conducted using a beam apertured to 1 mm × 8 mm and much of the available incident X-ray beam is simply discarded. Possibly the best solution is some sort of compromise: horizontal and vertical focusing optics to produce a beam size of about 0.5 mm × 4 mm allowing for small samples.
Finally, beam attenuation may be considered. Many beamlines have built-in aluminium filters which can be switched into the flight path to attenuate the X-ray beam. Although in general discarding flux in this way is an undesirable solution, this may be of limited use for samples that are particularly prone to photo-reduction and which are available in quantity. However, metalloprotein XAS is challenging because of the dilute nature of the samples, and photon flux is important in obtaining adequate signal; hence attenuating the beam might result in inadequate signal-to-noise.
The dinuclear Cu(II) di-oxygen species reported by Hu et al. (2001 ▶) provides an example of the effects of flux density. This compound was initially investigated on SSRL BL9-3, which uses a toroidal mirror after the monochromator to provide a focused beam at the sample. On this beamline the complex of Hu et al. (2001 ▶) quantitatively reduced to the mixed-valence Cu(II)/Cu(I) complex at low temperature during preliminary alignment of the sample in the X-ray beam. Indeed, only on the lower flux density BL7-3 with attenuators in place was it possible to obtain a bona fide spectrum of the Cu(II) dinuclear species (Fig. 5 ▶).
Figure 5.

Photo-reduction of the dinuclear Cu(II) dioxygen complex of Hu et al. (2001 ▶). The spectra shown in the upper panel, a, b and c, correspond to the structures shown in the lower panel. (a) First sweep of a Cu K-edge XAS spectrum recorded on SSRL BL9-3 after about 7 min of beam exposure (the spectra did not change on subsequent sweeps). (b) Spectrum of a related Cu(II) species with similar metal coordination. (c) Spectrum of the dinuclear species recorded on the lower-flux SSRL BL7-3.
4.2. Cryogenic temperatures
The use of cryogenic temperatures is possibly the most commonly employed method to minimize the effects of photo-reduction. The use of low temperatures actually has a dual purpose, firstly to protect samples and secondly to freeze out vibrational contributions to the variance in interatomic distance (σ2), improving the EXAFS amplitudes at high k values. Modern liquid-helium flow cryostats can achieve temperatures as low as 3.5 K although at somewhat prohibitive helium flow rates. More reasonable helium flow rates will result in a sample temperature of ∼10 K. The effects of irradiation by a hard X-ray synchrotron beam for several minutes upon a frozen (glassy) solution of buffer or colorless protein [e.g. containing Zn(II) or Cu(I)] at ∼10 K are clearly seen as a dark band located at the position of the X-ray beam, ranging from brown to purple in color, depending on the sample (Fig. 6 ▶). This band is caused by color centers due to hydrated electrons trapped in the glassy ice matrix. Annealing the sample at liquid-nitrogen temperatures (77 K) for about 30 min typically causes the colored band to decrease in intensity, with most of the color having dissipated after several hours. This suggests the possibility that the hydrated electrons of the color centers might have engaged in chemistry, thereby changing the sample; these results often have been taken to suggest that liquid-nitrogen temperatures may provide less than adequate protection, although definitive studies are lacking.
Figure 6.

Color centers in a sample of Saccharomyces cerevisiae Cox11, 1 mM Cu(I) (pH 7.2 Bis-Tris-Propane, 20% v/v glycerol) following a 3 h exposure to an X-ray beam at a sample temperature of 10 K in a Cu K-edge XAS experiment (SSRL BL7-3). The profile of the beam on the sample contained in a transparent polycarbonate cuvette can be clearly seen in the lower part of the figure. Note that the apparent horizontal mis-alignment of the beam profile is due to the fact that the sample was oriented at 45° so that on the reverse side the beam is displaced towards the other end of the sample.
An example of how significant the photo-damage can be with heme proteins is illustrated by experiments performed at the focused AS XAS beamline. Early studies on the three-dimensional structure of the heme environment in deoxy- and met-myoglobin (Mb) using multiple-scattering (MS) analysis of EXAFS data were performed in the mid-1990s on BL7-3 at SSRL before the SPEAR upgrades (Rich et al., 1998 ▶). While a large number of scans were performed on the deoxy-Mb with very little apparent photo-damage at 10 K, progressive photo-reduction of Fe(III) to Fe(II) in met-Mb was apparent over many cycles. Therefore, several scans from several spots in different samples needed to be combined in order to obtain data with sufficient signal-to-noise that was also relatively free from artifacts due to photo-reduction. Using such data, three-dimensional heme structures were obtained that gave bond lengths that were quite different to those in a range of published protein crystallography studies that normally had large inherent errors in bond lengths (Rich et al., 1998 ▶). Subsequent high-resolution protein crystallography studies confirmed the accuracy of this three-dimensional structure for the deoxy-Mb and also gave similar parameters for the met-Mb as determined by MS EXAFS analysis (Levina et al., 2005 ▶). However, the bond lengths in the high-resolution met-Mb crystallography were still somewhat longer than in the MS EXAFS structure (Levina et al., 2005 ▶), which was consistent with photo-reduction in the crystallography measurements. The sensitivity of met-heme proteins to photo-reduction is apparent at the third-generation focused XAS beamlines at SSRL (9-3) and AS (XAS), where there is generally very substantial photo-reduction changes between the first and second scans at SSRL and, in the case of the AS XAS beamline, virtually complete photo-reduction of met-Mb before reaching the edge on the first scan. This is illustrated in Fig. 7 ▶ where the XANES of the first scan from met-Mb obtained at the AS at 10 K is intermediate between those reported for five-coordinate deoxy- and six-coordinate met-Mb obtained in the absence of photo-damage at SSRL (Rich et al., 1998 ▶). The met-Mb XANES obtained at the AS then remains virtually the same in subsequent scans, which shows that photo-reduction was virtually complete before reaching the edge on the first scan. The fact that the photo-reduction produced an Fe(II) species that had an XANES that was different to the deoxy-form is likely to be due to the fact that the experiments were performed at 10 K and the axial ligand was probably locked in place in the frozen solution to form a six-coordinate metastable Fe(II) heme protein, rather than the five-coordinate deoxy structure. This is also consistent with the high-resolution X-ray crystallography results where, by analogy with the XAS experiments reported here, complete photo-reduction would have occurred during the X-ray crystallography to produce a metastable Fe(II) protein that retained the six-coordinate structure of the Fe(III) form, but would have had longer bond lengths than in the met-Mb structure determined by EXAFS in the absence of photo-reduction. These results highlight the extreme caution that needs to be taken in using X-ray crystallography and third-generation focused XAS beamlines to determine accurate structures of Fe(III) proteins. Very good XAS data could be obtained from Fe(III) forms of heme proteins on the XAS beamline at the AS, but this required the beam to be defocused and the intensity attenuated by aluminium foil until photo-reduction became manageable. These sort of experimental considerations need to be taken into account if accurate structural information is required from heme proteins and other proteins containing Fe(III) or higher oxidation states.
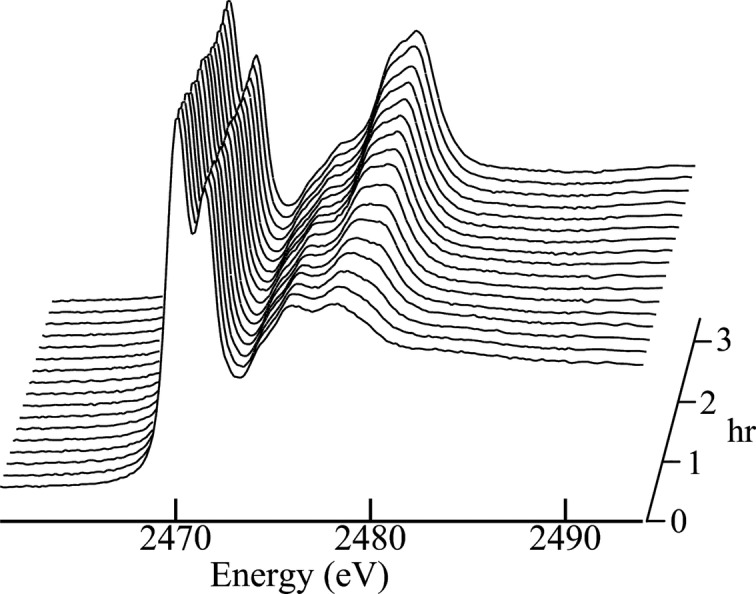
Figure 7.
X-ray absorption near-edge spectra of deoxy- and met-myoglobin compared with met-myoglobin data from the Australian Synchrotron (AS). The blue and red lines show the spectra of deoxy-myoglobin and met-myoglobin, respectively. These data were recorded in 1996 on BL7-3 at SSRL at 10 K. These are compared with the first (solid black line) and third (broken black) scans of met-myoglobin prepared in the same way but recorded on the XAS beamline at the AS, showing substantial and almost complete photo-reduction to a metastable Fe(II) complex during the first scan.
One caveat with the use of low temperatures in these experiments that always needs to be considered is when spin-equilibrium systems are present. A number of met-heme proteins, for instance, exist in a spin-equilibrium in which the high-spin Fe(III) dominates in an aqueous environment at room temperature, but at low temperature the low-spin form dominates. This causes a very substantial change in the XANES and EXAFS as a result of considerable shortening of the Fe(III)–ligand bonds and a change in axial distal coordination from an aqua ligand to a histidine (Levina et al., 2005 ▶).
4.3. Sample translation
For biological samples, averaging of multiple sweeps is needed to obtain adequate signal-to-noise ratios. One frequently employed method for minimizing the effects of photo-reduction is to translate the sample following each individual sweep so that the beam illuminates an unexposed part of the sample with each sweep. Data acquisition codes such as the SSRL program XAS-Collect (George, 2000 ▶) have the capability to move the sample stage automatically to pre-determined locations. In some cases especially large samples (∼10 mm × 20 mm) have been used to allow acquisition of adequate statistics using this method, and Fig. 8 ▶ shows an example of this for aqueous solutions of a Cu(II) peptide complex.
Figure 8.
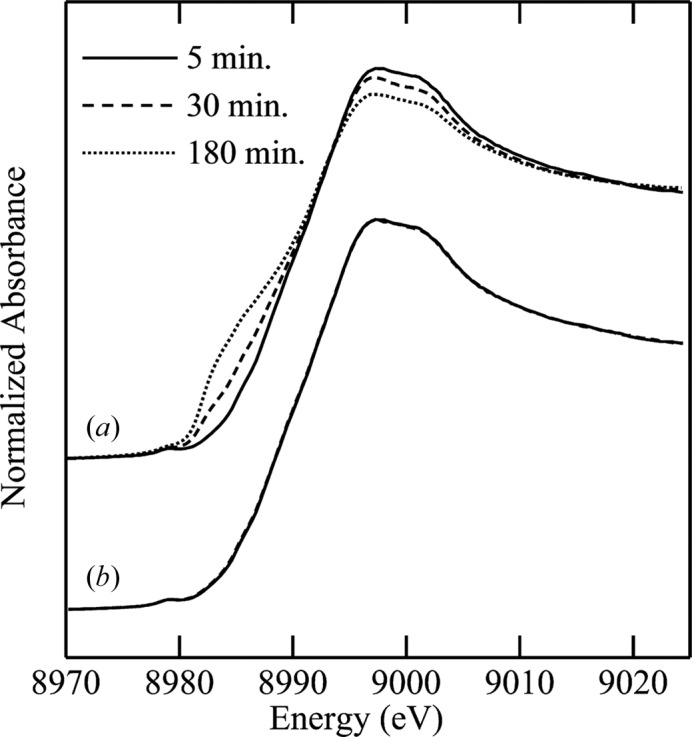
Effects of sample translation on the XAS spectrum of a Cu(II) peptide complex. (a) A sequence of spectra at the given time intervals showing clear evidence of increasing photo-reduction at the longer exposures. (b) The same time sequence for an identical sample, but with sample translation following each sweep. No significant photo-reduction is observed in (b). Data were collected on SSRL BL7-3.
4.4. X-ray exposure
A photon shutter can be positioned before the sample but after the I 0 ion chamber and used to shut off the X-ray beam when fluorescence data are not being acquired. The system in use at SSRL is a simple pneumatic shutter, which closes during monochromator rotation and at other times when fluorescence data on protein samples are not being collected. This effectively eliminates most non-productive exposure of the sample to the X-ray beam, for example during beamline alignment. Similar simple shutter systems are known to be either planned (e.g. the Canadian Light Source BioXAS beamlines) or already in place (e.g. the AS XAS beamline) at other synchrotron light facilities world-wide.
4.5. Free-radical scavengers
One possible method for mitigating the effects of X-ray exposure is the addition of high concentrations of species that are known to scavenge free radicals from the solutions being studied. As discussed in §3.2, sulfate and nitrate can scavenge hydrated electrons, and provide protection from photo-reduction, at least in liquid solution. The use of cryogenic temperatures (see §4.2) generally requires a glassing agent to be present in high concentrations to prevent artifacts arising from X-ray diffraction by ice crystals. This is usually glycerol, ethylene glycol, dimethylsulfoxide or sucrose; all of these are highly effective hydroxyl radical scavengers, and as a side-product may modify the photo-reduction process. It has been shown that γ-radiolysis of aqueous-glycerol glasses at 77 K produced mobile electrons that were capable of reducing the diferric site of Escherichia coli protein R2 of ribonucleoside diphosphate reductase and of Themiste zostericola methemerythrin, whereas this was not the case for glycols (Davydov et al., 1994 ▶). The polyethylene glycol often used in protein crystallography experiments should also serve to modify photo-reduction by X-rays (Barker et al., 2009 ▶).
A novel method of protecting sensitive proteins has been reported by Ascone et al. (2000 ▶). Proteins that were known to be sensitive to X-ray damage, namely oxy forms of human hemoglobin and hemocyanins from several species, were embedded in a glassy sucrose matrix by freezing and lyophilizing sucrose/protein solutions. This procedure eliminated most of the water with only 5–8% of the total weight of the protein remaining. The integrity of the protein and active-site structures were verified by using resonance Raman, infra-red, fluorescence and XAS spectroscopies (Ascone et al., 2000 ▶). The sucrose matrix was found to provide excellent protection against photo-damage, allowing for longer exposure to the X-ray beam. For example, XANES spectra recorded at a low-flux DCI source (LURE France), at room temperature, indicated that whelk (Rapana venosa) hemocyanin in solution was partially photo-reduced after 1 h of exposure (Ascone et al., 2000 ▶). In contrast, spectra of hemocyanins in a sucrose matrix did not show any modification after 5 h under the same conditions (Sabatucci et al., 2002 ▶). The dual effects of the loss of the water and hence of its photo-chemically active species, with free-radical scavenging by sucrose are likely responsible for this stability. Many proteins tend to lose integrity upon long-term storage in solution, and use of a glassy sucrose matrix was found to yield samples that were more stable to long-term storage than untreated protein solutions. Similar phenomena have been observed for partially dehydrated membranous protein samples (George et al., 1989 ▶, 1993 ▶) which also show decreased tendency for photo-reduction and increased stability on long-term storage (George et al., unpublished).
4.6. Freeze-drying of biological fluids, cells and tissues
Freeze-drying of protein samples may have several advantages described below but this method should be used with caution as it may induce conformational modifications or even denaturation of proteins in solution (Chang et al., 1996 ▶). Moreover, when a labile water molecule is bound to the metal, the process of freeze-drying may actually change the metal coordination, as demonstrated by EXAFS measurements of ovotransferrin (Hasnain et al., 1987 ▶). With the above caveats, if biological samples are freeze-dried then the formation of radicals from the hydrolysis of water (§3.1) can be virtually eliminated.
Although photo-chemical reactions can occur with other groups or directly with the absorbing atom within a sample, most freeze-dried samples are stable enough with respect to photo-damage to have their XAS recorded in an air atmosphere at room temperature with minimal photo-damage on the first scan. This is illustrated in Fig. 9 ▶ where successive K-edge V XANES of the products of vanadate with BSA or serum from heat-inactivated calf serum are presented, which are from a larger study of the speciation of V anti-diabetic complexes in the blood (McLeod et al., 2012 ▶). While there is little evidence for photo-damage within experimental noise for experiments performed at the bending-magnet ANBF beamline, for the third-generation insertion-device beamlines (9-3 at SSRL and XAS at AS) small but significant photo-damage had occurred by the second scan. The same is evident for a variety of freeze-dried biological samples containing Cr and V (Harris et al., 2005 ▶; Levina et al., 2007a ▶; Nguyen et al., 2008 ▶), which are examples of transition elements that are more sensitive to photo-damage than heavier transition elements, such as Mo(VI) (Levina et al., 2007b ▶), for the reasons discussed in §3.1. In a variety of such studies the first scan on the insertion-device beamlines corresponded well with those at the ANBF, which enabled the insertion-device beamlines to be used in speciation studies of the more dilute samples in both bulk cells and with microprobes in single cells (Harris et al., 2005 ▶; Levina et al., 2007a ▶; Nguyen et al., 2008 ▶; McLeod et al., 2012 ▶).
Figure 9.

Comparison of photo-reduction at various synchrotron sources in V K-edge XAS of freeze-dried biological samples: first and second X-ray absorption near-edge scans for V(V/IV) species in biological media (neat freeze-dried samples, 295 K). The reaction products of Na3[VO4] (1.0 mM) with undiluted calf serum or with BSA solution (0.50 mM in HEPES-buffered saline, pH 7.4) were prepared as described in §2. Designations of the synchrotron sources: ANBF is the Australian National Beamline at the Photon Factory, KEK, Tsukuba, Japan; AS is the XAS beamline at the Australian Synchrotron, Melbourne, Australia; and SSRL is the Stanford Synchrotron Radiation Lightsource BL7-3, Menlo Park, USA.
Apart from the above advantages, freeze-drying also has two other distinct advantages. First, the increase in the concentrations of absorbers is important for the sensitivity of the experiments on dilute biological samples. Second, the ability of the samples to be recorded at room temperature removes the need to use cryostats and, hence, the absorption of X-rays by the window materials, which both reduces the intensity of the beam reaching the samples and increases the attenuation of the fluorescence signal reaching the detector. All of these factors are very important in recording XAS spectra with sufficiently high signal-to-noise ratios to obtain speciation from dilute Cr and V concentrations in biological samples (Levina et al., 2007a ▶; Nguyen et al., 2008 ▶; McLeod et al., 2012 ▶).
One aspect that always needs to be considered with freeze-drying is the ability to change the speciation and/or structures of the metal-containing species in the samples. In the above examples this was checked by comparing the XANES from freeze-dried and snap-frozen cell pellets at low temperature; no differences were observed within the experimental noise of the XAS experiments.
4.7. Electrochemical XAS
Another way to circumvent photo-damage, even in aqueous solution at room temperature, is the use of electrochemical XAS cells, which have been used to record XAS spectra with redox proteins that were very sensitive to photo-damage. This has included XAS from the redox sensitive-non-metal, S donors in S clusters (Anxolabéhère-Mallart et al., 2001 ▶) and the metal centers redox proteins (Ascone et al., 1999 ▶, 2005 ▶). This has also been very effective, for instance, in examining various oxidation states of Cr and V catecholato complexes, which have been examined in aqueous solution at ∼293 K without any noticeable photo-reduction and/or photo-oxidation in multiple scans. By maintaining the potential of the working electrode at a value that will re-oxidize photo-reduced species and re-reduce photo-oxidized species, quite stable XANES and EXAFS of a range of oxidation states could be obtained over long periods of time (Levina et al., 2004 ▶; Milsmann et al., 2006 ▶).
4.8. Chemical redox systems
Finally, a variant to electrochemistry, i.e. using a redox couple that is chemically unreactive relative to the active site, but which acts as an oxidant for the solvated electrons, has also been proven a successful method to mitigate photo-reduction. This has been shown in particular for XAS measurements of metalloproteins at low energies (at the S K-edge around 2.5 keV), where the experiments often are performed at 277 K since the use of lower-temperature cryostats present an experimental challenge in the form of absorbing windows. For example, a ten-fold excess of K3[Fe(CN)6] in phosphate buffer was used to maintain the oxidation state of the red copper site in Nitrosomonas europaea nitrosocyanin (Basumallick et al., 2005 ▶) and the blue copper site of Anabaena variabilis plastocyanin (Hansen et al., 2006 ▶) to complete measurements of full data sets without any photo-damage.
5. Conclusion
As synchrotron light sources increase their capabilities, X-ray absorption spectroscopy beamlines are providing beams of increasing flux and increasing flux density. X-ray-induced photo-chemistry, and in particular photo-reduction of metal sites, is expected to become increasingly important in XAS studies of biological samples. A combined approach using low temperatures, sample motion, limited X-ray exposure and rapid scanning can provide some relief from photo-reduction.
Acknowledgments
Work at the University of Saskatchewan was supported by the Natural Sciences and Engineering Research Council of Canada, the Canadian Institutes for Health Research (CIHR) and the Saskatchewan Health Research Foundation (SHRF). GNG and IJP are Canada Research Chairs. MJP, KN and MJH are Fellows in the CIHR Training grant in Heath Research Using Synchrotron Techniques (CIHR-THRUST). MJP is a CIHR Postdoctoral Fellow and is also supported by SHRF. MJH is supported by a SHRF Postdoctoral Fellowship and by the CIHR/Heart and Stroke Foundation of Canada Team in Synchrotron Medical Imaging (Nichol, PI). Further support was available from PrioNet Canada. Portions of this work were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the US Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources (5P41RR001209) and the National Institute of General Medical Sciences (8 P41 GM103393). The research at the University of Sydney was supported by Australian Research Council (ARC) Discovery grants (DP0208409, DP0774173 and DP0984722), ARC Professorial Fellowships (DP0208409 and DP0984722) to PAL, and an ARC Linkage Infrastructure, Equipment and Facilities (LIEF) Program grant (LE0346515) for the 36-pixel Ge detector at ANBF. The Australian research at SSRL was also supported by the International Synchrotron Access Program (ISAP) grants of the Australian Synchrotron. As such we acknowledge travel funding provided by the ISAP managed by the Australian Synchrotron. The ISAP is an initiative of the Australian Government being conducted as part of the National Collaborative Research Infrastructure Strategy. We acknowledge the LIEF program of the ARC for financial support (proposal number LE0989759) and the High Energy Accelerator Research Organization (KEK) in Tsukuba, Japan, for operations support. IA is supported by the Centre National de la Recherche Scientifique (CNRS). We thank Professor Thomas L. Poulos and Dr Irina F. Sevrioukova for the protein crystals in the radiation damage study. We thank Michael Cheah for the assistance with XAS experiments at ANBF and Matthew Latimer for those at SSRL.
References
- Aitken, J. B., Carter, E. A., Eastgate, H., Hackett, M. J., Harris, H. H., Levina, A., Lee, Y.-C., Chen, C.-I., Lai, B., Vogt, S. & Lay, P. A. (2010). Radiat. Phys. Chem. 79, 176–184.
- Aitken, J. B., Levina, A. & Lay, P. A. (2011). Curr. Top. Med. Chem. 11, 553–571. [DOI] [PubMed]
- Antonyuk, S. V. & Hough, M. A. (2011). Biochim. Biophys. Acta, 1814, 778–784. [DOI] [PubMed]
- Anxolabéhère-Mallart, E., Glaser, T., Frank, P., Aliverti, A., Zanetti, G., Hedman, B., Hodgson, K. O. & Solomon, E. I. (2001). J. Am. Chem. Soc. 123, 5444–5452. [DOI] [PubMed]
- Ascone, I., Cognigni, A., Giorgetti, M., Berrettoni, M., Zamponi, S. & Marassi, R. (1999). J. Synchrotron Rad. 6, 384–386. [DOI] [PubMed]
- Ascone, I., Meyer-Klaucke, W. & Murphy, L. (2003). J. Synchrotron Rad. 10, 16–22. [DOI] [PubMed]
- Ascone, I., Sabatucci, A., Bubacco, L., Di Muro, P. & Salvato, B. (2000). Eur. Biophys. J. 29, 391–397. [DOI] [PubMed]
- Ascone, I., Zamponi, S., Cognigni, A., Marmocchi, F. & Marassi, R. (2005). Electrochim. Acta, 50, 2437–2443.
- Barker, A. I., Southworth-Davies, R. J., Paithankar, K. S., Carmichael, I. & Garman, E. F. (2009). J. Synchrotron Rad. 16, 205–216. [DOI] [PubMed]
- Basumallick, L., Sarangi, R., DeBeer George, S., Elmore, B., Hooper, A. B., Hedman, B., Hodgson, K. O. & Solomon, E. I. (2005). J. Am. Chem. Soc. 127, 3531–3544. [DOI] [PubMed]
- Bonagura, C. A., Bhaskar, B., Shimizu, H., Li, H., Sundaramoorthy, M., McRee, D. E., Goodin, D. B. & Poulos, T. L. (2003). Biochemistry, 42, 5600–5608. [DOI] [PubMed]
- Carter, E. A., Rayner, B. S., McLeod, A. I., Wu, L. E., Marshall, C. P., Levina, A., Aitken, J. B., Witting, P. K., Lai, B., Cai, Z., Vogt, S., Lee, Y. C., Chen, C. I., Tobin, M. J., Harris, H. H. & Lay, P. A. (2010). Mol. Biosyst. 6, 1316–1322. [DOI] [PubMed]
- Chang, B. S., Beauvais, R. M., Dong, A. & Carpenter, J. F. (1996). Arch. Biochem. Biophys. 331, 249–258. [DOI] [PubMed]
- Choppin, G., Liljenzin, J.-O. & Rydberg, J. (2002). Radiochemistry and Nuclear Chemistry, 3rd ed. Wobern: Butterwort-Heinemann.
- Clay, M. D., Jenney, F. E., Hagedoorn, P. L., George, G. N., Adams, M. W. & Johnson, M. K. (2002). J. Am. Chem. Soc. 124, 788–805. [DOI] [PubMed]
- Corbett, M. C., Latimer, M. J., Poulos, T. L., Sevrioukova, I. F., Hodgson, K. O. & Hedman, B. (2007). Acta Cryst. D63, 951–960. [DOI] [PubMed]
- Cramer, S. P., Wahl, R. & Rajagopalan, K. V. (1981). J. Am. Chem. Soc. 103, 7721–7727.
- Cvetkovic, A., Menon, A. L., Thorgersen, M. P., Scott, J. W., Poole, F. L., Jenney, F. E., Lancaster, W. A., Praissman, J. L., Shanmukh, S., Vaccaro, B. J., Trauger, S. A., Kalisiak, E., Apon, J. V., Siuzdak, G., Yannone, S. M., Tainer, J. A. & Adams, M. W. (2010). Nature (London), 466, 779–782. [DOI] [PubMed]
- Davydov, R., Kuprin, S., Gräslund, A. & Ehrenberg, A. (1994). J. Am. Chem. Soc. 116, 11120–11128.
- De Cremer, K., Van Hulle, M., Chéry, C., Cornelis, R., Strijckmans, K., Dams, R., Lameire, N. & Vanholder, R. (2002). J. Biol. Inorg. Chem. 7, 884–890. [DOI] [PubMed]
- El Omar, A. K., Schmidhammer, U., Jeunesse, P., Larbre, J. P., Lin, M., Muroya, Y., Katsumura, Y., Pernot, P. & Mostafavi, M. (2011). J. Phys. Chem. A, 115, 12212–12216. [DOI] [PubMed]
- Ferreira, G. C., Franco, R., Mangravita, A. & George, G. N. (2002). Biochemistry, 41, 4809–4818. [DOI] [PubMed]
- Finney, L., Chishti, Y., Khare, T., Giometti, C., Levina, A., Lay, P. A. & Vogt, S. (2010). ACS Chem. Biol. 5, 577–587. [DOI] [PubMed]
- Flemming Hansen, D., Gorelsky, S. I., Sarangi, R., Hodgson, K. O., Hedman, B., Christensen, H. E., Solomon, E. I. & Led, J. J. (2006). J. Biol. Inorg. Chem. 11, 277–285. [DOI] [PubMed]
- Fong, Y. Y., Visser, B. R., Gascooke, J. R., Cowie, B. C., Thomsen, L., Metha, G. F., Buntine, M. A. & Harris, H. H. (2011). Langmuir, 27, 8099–8104. [DOI] [PubMed]
- Garrett, B. C. et al. (2005). Chem. Rev. 105, 355–390. [DOI] [PubMed]
- George, G. N., Cramer, S. P., Frey, T. G. & Prince, R. C. (1993). Biochim. Biophys. Acta, 1142, 240–252. [DOI] [PubMed]
- George, G. N., Garrett, R. M., Prince, R. C. & Rajagopalan, K. V. (1996). J. Am. Chem. Soc. 118, 8588–8592.
- George, G. N., Pickering, I. J. & Kisker, C. (1999). Inorg. Chem. 38, 2539–2540.
- George, G. N., Prince, R. C. & Cramer, S. P. (1989). Science, 243, 789–791. [DOI] [PubMed]
- George, M. J. (2000). J. Synchrotron Rad. 7, 283–286. [DOI] [PubMed]
- George, S. J., Fu, J., Guo, Y., Drury, O. B., Friedrich, S., Rachfuss, T., Volkers, P. I., Peters, J. C., Scott, V., Brown, S. D., Thomas, C. M. & Cramer, S. P. (2008). Inorg. Chim. Acta, 361, 1157–1165.
- Glover, C., McKinlay, J., Clift, M., Barg, B., Boldeman, J., Ridgway, M., Foran, G., Garrett, R., Lay, P. & Broadbent, A. (2007). Am. Inst. Phys. 882, 884–886.
- Gnida, M., Sneeden, E. Y., Whitin, J. C., Prince, R. C., Pickering, I. J., Korbas, M. & George, G. N. (2007). Biochemistry, 46, 14735–14741. [DOI] [PubMed]
- Gunter, T. E. (1967). J. Chem. Phys. 46, 3818–3829.
- Hackett, M. J., Smith, S. E., Paterson, P. G., Nichol, H., Pickering, I. J. & George, G. N. (2012). ACS Chem. Neurosci. 3, 178–185. [DOI] [PMC free article] [PubMed]
- Hall, E. (1994). Radiobiology for the Radiologist, 4th ed. Philadelphia: J. B. Lippincott.
- Harris, H. H., George, G. N. & Rajagopalan, K. V. (2006). Inorg. Chem. 45, 493–495. [DOI] [PubMed]
- Harris, H. H., Levina, A., Dillon, C. T., Mulyani, I., Lai, B., Cai, Z. & Lay, P. A. (2005). J. Biol. Inorg. Chem. 10, 105–118. [DOI] [PubMed]
- Harris, W. R., Friedman, S. B. & Silberman, D. (1984). J. Inorg. Biochem. 20, 157–169. [DOI] [PubMed]
- Hasnain, S. S., Evans, R. W., Garratt, R. C. & Lindley, P. F. (1987). Biochem. J. 247, 369–375. [DOI] [PMC free article] [PubMed]
- Hedman, B., Corbett, M. A., Latimer, M. J., Poulos, T. L., Seviroukova, I. F. & Hodgson, K. O. (undated). Unpublished data.
- Howard, J. B. & Rees, D. C. (2005). Proc. Natl Acad. Sci. USA, 103, 17088–17093. [DOI] [PMC free article] [PubMed]
- Hu, Z. B., George, G. N. & Gorun, S. M. (2001). Inorg. Chem. 40, 4812–4813. [DOI] [PubMed]
- Kevan, L. (1981). Acc. Chem. Res. 14, 138–145.
- Kisker, C., Schindelin, H., Pacheco, A., Wehbi, W. A., Garrett, R. M., Rajagopalan, K. V., Enemark, J. E. & Rees, D. C. (1997). Cell, 91, 1–20. [DOI] [PubMed]
- Kung, J. W., Baumann, S., von Bergen, M., Müller, M., Hagedoorn, P. L., Hagen, W. R. & Boll, M. (2010). J. Am. Chem. Soc. 132, 9850–9856. [DOI] [PubMed]
- Latimer, M. J., Ito, K., McPhillips, S. E. & Hedman, B. (2005). J. Syncrotron. Rad. 12, 23–27. [DOI] [PubMed]
- Levina, A., Armstrong, R. S. & Lay, P. A. (2005). Coord. Chem. Rev. 249, 141–160.
- Levina, A., Foran, G. J., Pattison, D. I. & Lay, P. A. (2004). Angew. Chem. Int. Ed. Engl. 43, 462–465. [DOI] [PubMed]
- Levina, A., Harris, H. H. & Lay, P. A. (2007a). J. Am. Chem. Soc. 129, 1065–1075. [DOI] [PubMed]
- Levina, A., McLeod, A. I., Seuring, J. & Lay, P. A. (2007b). J. Inorg. Biochem. 101, 1586–1593. [DOI] [PubMed]
- McLeod, A. I., Aitken, J. B., Gasparini, S., Glover, C. J., Johannessen, B., De Silva, W. G. M., Levina, A. & Lay, P. A. (2012). In preparation.
- McMaster, W. H., Kerr Del Grande, N., Mallett, J. H. & Hubbell, J. H. (1969). Compilation of X-ray Cross Sections. National Technical Information Services L-3, US Department of Commerce, USA.
- Marsalek, O., Uhlig, F., VandeVondele, J. & Jungwirth, P. (2012). Acc. Chem. Res. 45, 23–32. [DOI] [PubMed]
- Mesu, J. G., Beale, A. M., de Groot, F. M. & Weckhuysen, B. M. (2006). J. Phys. Chem. B, 110, 17671–17677. [DOI] [PubMed]
- Milsmann, C., Levina, A., Harris, H. H., Foran, G. J., Turner, P. & Lay, P. A. (2006). Inorg. Chem. 45, 4743–4754. [DOI] [PubMed]
- Misra, S., Peak, D. & Niyogi, S. (2010). Metallomics, 2, 710–717. [DOI] [PubMed]
- Newville, M. (2005). Compilation of X-ray Cross Sections. University of Chicago, Chicago, IL, USA.
- Nguyen, A., Mulyani, I., Levina, A. & Lay, P. A. (2008). Inorg. Chem. 47, 4299–4309. [DOI] [PubMed]
- Penner-Hahn, J. E., Murata, M., Hodgson, K. O. & Freeman, H. C. (1989). Inorg. Chem. 28, 1826–1832.
- Plonka, A., Szajdzinska-Pietek, E., Bednarek, J., Hallbrucker, A. & Mayer, E. (2000). Phys. Chem. Chem. Phys. 2, 1587–1593.
- Pushie, M. J. & George, G. N. (2011). Coord. Chem. Rev. 255, 1055–1084.
- Pushie, M. J., Zhang, L., Pickering, I. J. & George, G. N. (2012). Biochim. Biophys. Acta, 1817, 938–947. [DOI] [PubMed]
- Qiu, J. A., Wilson, H. L., Pushie, M. J., Kisker, C., George, G. N. & Rajagopalan, K. V. (2010). Biochemistry, 49, 3989–4000. [DOI] [PubMed]
- Rich, A. M., Armstrong, R. S., Ellis, P. J., Freeman, H. C. & Lay, P. A. (1998). Inorg. Chem. 37, 5743–5753.
- Sabatucci, A., Ascone, I., Bubacco, L., Beltramini, M., Muro, D. & Salvato, B. (2002). J. Biol. Inorg. Chem. 7, 120–128. [DOI] [PubMed]
- Symons, M. C. R. (1980). J. Chem. Soc. Chem. Commun. pp. 675–676.
- Thompson, A. C., Attwood, D. T., Gullikson, E. M., Howells, M. R., Kortright, J. B., Robinson, A. L., Underwood, J. H., Kim, K.-J., Kirz, J., Lindau, I., Pianetta, P., Winick, H., Williams, G. P. & Scofield, J. H. (2001). X-ray Data Booklet, 2nd ed. University of California, Berkeley, CA, USA.
- Tse, J. J., George, G. N. & Pickering, I. J. (2011). J. Synchrotron Rad. 18, 527–529. [DOI] [PMC free article] [PubMed]
- Weng, T.-C., Waldo, G. S. & Penner-Hahn, J. E. (2005). J. Synchrotron Rad. 12, 506–510. [DOI] [PubMed]
- Yano, J., Kern, J., Irrgang, K. D., Latimer, M. J., Bergmann, U., Glatzel, P., Pushkar, Y., Biesiadka, J., Loll, B., Sauer, K., Messinger, J., Zouni, A. & Yachandra, V. K. (2005). Proc. Natl Acad. Sci. USA, 102, 12047–12052. [DOI] [PMC free article] [PubMed]
- Yano, J., Kern, J., Sauer, K., Latimer, M. J., Pushkar, Y., Biesiadka, J., Loll, B., Saenger, W., Messinger, J., Zouni, A. & Yachandra, V. K. (2006). Science, 314, 821–825. [DOI] [PMC free article] [PubMed]
- Yeh, A. P., Hu, Y., Jenney, F. E., Adams, M. W. & Rees, D. C. (2000). Biochemistry, 39, 2499–2508. [DOI] [PubMed]