

Abstract
The repertoire of gastrointestinal (GI) symptoms is finite; however, the etiologies and mechanisms underlying symptom generation and perception are diverse and, in many cases, unknown. This review examines the clinical and experimental evidence exploring the putative relationship between gluten sensitivity (GS) and the generation of GI symptoms. It explores the hypothesis that, in a proportion of patients, GS causes functional bowel disorder (FBD)-like symptoms. We propose a model for investigating and understanding the induction of GI symptoms and dysfunction by gluten in FBD and organic disease. We hypothesize that, even in the absence of fully developed celiac disease, gluten can induce symptoms similar to FBD. We discuss the hypothesis that GS and post-infectious irritable bowel syndrome (IBS) provide two triggers that can explain at least part of the spectrum that constitutes IBS, further advancing an understanding of the role of mucosal responses to luminal factors in FBDs. We propose that the animal model of GS in human leukocyte antigen (HLA)-DQ8 mice allows investigation of mucosal pathophysiological changes that occur before the onset of full-blown inflammation in a GS host. A better understanding of how gluten can cause symptoms in sensitive individuals will illuminate the interaction between host genotype, diet, and intestinal microbiota in generating one of the most common GI conditions.
INTRODUCTION
The repertoire of gastrointestinal (GI) symptoms is finite; however, the etiologies and mechanisms underlying symptom generation and perception are diverse and, in many cases, unknown. Symptoms, symptom complexes, and symptom characteristics are rarely, if ever, diagnostic. Functional bowel disorders (FBD), such as irritable bowel syndrome (IBS), and functional dyspepsia (FD) are, by definition, conditions that are accompanied by GI symptoms for which no mechanistic cause has been identified. This review examines the clinical and experimental evidence exploring the putative relationship between gluten sensitivity (GS) and the generation of GI symptoms. It explores the hypothesis that, in a proportion of patients, GS causes FBD-like symptoms. We propose a model for investigating and understanding the induction of GI symptoms and dysfunction by gluten in FBD and organic disease. We hypothesize that, even in the absence of fully developed celiac disease, gluten can induce symptoms similar to FBD. A better understanding of how gluten can cause symptoms in sensitive individuals will illuminate the interaction between host genotype, diet, and intestinal microbiota in generating one of the most common GI conditions.
DEFINITIONS OF CELIAC DISEASE AND IRRITABLE BOWEL SYNDROME
Celiac disease is a condition traditionally characterized by chronic inflammation of the proximal small intestine resulting in villous atrophy and malabsorption that can develop in genetically susceptible individuals ingesting gluten, the storage proteins of wheat, barley, and rye. e prevalence of celiac disease in the United States (1) and Canada (2) is as high as 0.5–1% , similar to earlier estimates outside North America (3-6).
In contrast to celiac disease, IBS is a clinical syndrome defined, in the most recent Rome III consensus (7), by the presence of abdominal pain or discomfort, at least 3 days per month in the last 3 months, and two or more other symptom features: improvement with defecation, association with a change in stool frequency, and association with a change in stool form or appearance. Other GI symptoms, such as bloating and distension, are also considered to be consistent with a diagnosis of FBDs such as IBS. With a prevalence of about 10% , IBS is one of the more common GI disorders in our society, imposing a very high economic burden in North America (8).
OVERLAP BETWEEN IBS AND CELIAC DISEASE
Although IBS is a collection of a diverse grouping of symptom-defined syndromes, celiac disease may also present with a wide spectrum of both GI and non-GI symptoms, many of which bear a large degree of overlap with IBS (9). Other studies show a combined 4.5% prevalence of celiac disease in patients clinically diagnosed with IBS when Rome I and II criteria are used (10-12). A systematic review and meta-analysis has concluded that celiac disease, as diagnosed by positive serology and positive biopsy, was four-fold more prevalent among patients with a clinical presentation of IBS than in non-IBS populations (13). Finally, it has been reported that testing for celiac disease in patients with diarrhea-predominant IBS is cost-effective if the prevalence of celiac disease is above 1% (14). Not only do the symptoms of IBS and celiac disease overlap, but epidemiological studies also suggest a greater than by chance association (4–5-fold increased risk), especially in patients seeking health care (15-17).
By convention, a patient with confirmed celiac disease is no longer considered to have IBS. Despite this, it has never been determined whether celiac disease and IBS cannot coexist, and there is no reason to think that a diagnosis of celiac disease necessarily precludes a diagnosis of IBS. As a matter of fact, Fasano et al. (1) have concluded that about 3% of patients with a “clinical” presentation of IBS were subsequently diagnosed with celiac disease.
In this review, we would like to focus on the emerging concept that gluten-induced pathophysiology may constitute an underlying factor in symptom generation in a proportion of patients with IBS-like symptoms.
WHAT IS “GLUTEN SENSITIVITY”?
Both the pathological and clinical spectra of celiac disease can vary considerably from severe to subtle, and clinically significant symptoms are not necessarily restricted to the individuals who manifest classic mucosal injury (18,19). Throughout this review, we define “gluten-sensitivity” as a condition of some morphological, immunological, or functional disorder that responds to gluten exclusion. The concept of GS incorporates a variety of pathological, immunological, and clinical scenarios that have been described recently . These include gluten-sensitive diarrhea, immunological mucosal response to gluten in family members of celiac disease, persistently positive specific serology for celiac disease in the absence of defined enteropathy, and subtle immunopathological changes in the intestine exposed to gluten. These minimal immunopathological changes can include increased intraepithelial lymphocytosis, increased IgA deposition in the intestinal villi, changes in the microvillus border, and perhaps an increase in secreted antibodies directed against gliadin. Typically, these disorders occur in individuals who carry the same human leukocyte antigen (HLA) genotypes associated with celiac disease, DQ2 and DQ8. One of the earliest appreciations of this part of the spectrum of GS was in family members of celiac disease who, despite not having villous atrophy, often had evidence of immune responsiveness to gluten, such as increased rectal response to gluten in child siblings of patients with celiac disease (20). is supports the concept that GS, an abnormal immune response to gluten, can occur in the absence of the enteropathy that characterizes celiac disease (19-21). On the other hand, a study has determined that eight out of ten patients with symptoms suggestive of celiac disease showed minimal mucosal lesions (Marsh I or II); both symptoms and incipient mucosal lesions improved after a gluten-free diet. All of these patients carried the genetic markers HLA-DQ2 or DQ8 associated with celiac disease (22). Another study investigating the role of histology in the diagnosis of GS has determined that 12 out of 28 patients with intraepithelial lymphocytosis had GS on the basis of a symptomatic response to a gluten-free diet (23). At its earliest stage, GS may manifest with anti-gliadin antibodies in jejunal aspirate in the absence of any histological changes (24) or IgA deposits in duodenal biopsies (25). GS also includes subjects who, despite the absence of circulating antibodies to tissue transglutaminase (tTG), show a mucosal lymphocytic infiltrate after gluten challenge (20,26). Without villous atrophy, patients with a symptomatic response to a gluten-free diet (GFD) that does not show tTG serological responses characterizing celiac disease are diagnosed as “IBS gluten sensitive,” particularly in the presence of genetic markers for celiac disease (Figure 1). A study investigated 102 patients with diarrhea-predominant IBS who had normal biopsy or increased intraepithelial cell counts and negative serology for celiac disease. In all, 58% of IBS patients carrying the HLA-DQ2 alleles also had positive IgA anti-gliadin or anti-TTG antibodies in the duodenal aspirate and higher IELs, compared with just 15% of IBS patients with negative HLA-DQ2 status (27). A subsequent study identified “IBS gluten-sensitive” patients based on symptomatic responses to gluten challenge or withdrawal and concluded that responders to a GFD are more likely to carry HLA-DQ2 (28). There are individuals who have infiltration of the surface epithelial layer of the duodenum with lymphocytes (intraepithelial lymphocytes), also called lymphocytic duodenosis (LD). A majority of subjects (50–80%) who have LD do not have celiac disease or even belong to the spectrum of GS, and yet the role of LD in these symptomatic patients is uncertain (29,30). Why LD occurs in the non-gluten-sensitive individuals is unknown, but it may be a reaction to some other luminal antigen such as Helicobacter pylori (22). Clinical management of GS patients is uncertain, as there are to date no clear therapeutic guidelines. Gluten-sensitive individuals, who have IBS symptoms and IgG antibodies to gluten, are orphans, living in no man’s land, acknowledged neither by functional disease specialists nor by celiac disease specialists.
Figure 1.

The spectrum of celiac disease includes subjects with genetic predisposition and sensitivity to gluten, celiac disease, and its complications. It is unknown whether a portion of gluten-sensitive subjects without celiac disease will ever progress in the severity of the spectrum or constitute a separate entity. Latent celiac disease is part of the spectrum, but differs from potential in that, with exposure to gluten, it will progress to celiac disease. GFD, gluten-free diet; Ttg Ab, tissue transglutaminase antibody.
MECHANISMS UNDERLYING SYMPTOM GENERATION IN IBS
Psychological factors are considered to be important to the pathogenesis of IBS (31) , although the occurrence of IBS under some circumstances, such as after acute enteric infections, suggests that peripheral mechanisms are important, in the absence of psychological triggers (32-37). Neuromotor dysfunction has been proposed as a mechanism for symptom generation in a proportion of patients with IBS (for a review, see Spiller (38) ). Abnormal small intestinal motor patterns seem to be slightly increased from about 30% in healthy controls to about 40% in patients with IBS (39). Colonic high-amplitude propagated pressure waves have been reported to be twice as frequent in patients with IBS compared with controls (40). There is now considerable evidence that persistent low-grade inflammation plays an important role in the pathogenesis of IBS (33-35). The question arises as to what drives low-grade intestinal inflammation in patients with functional GI disorders. Up to 30% of patients develop persistent low-grade inflammation and IBS symptoms after a GI infection (32-36) and the role of small intestinal bacterial overgrowth has been raised (37). The role of other factors in the genesis of low-grade inflammation and IBS or IBS-like symptoms remains a matter of conjecture. However, there are numerous reports of food-induced aggravation of IBS and improvement after dietary exclusion (41-44). Zar et al. (45) reported the presence of IgG4 antibodies to wheat in 60% of patients with IBS compared with 27% of healthy controls. Another study explored the effect of dietary restriction based on the presence of circulating IgG antibodies against nutrients. The treatment diet excluded foods to which the patient had IgG antibodies. The sham diet eliminated the same number of foods, but not those to which patients had IgG antibodies. Patients adhering to the true diet experienced 10% greater reduction in symptom severity than did those in the sham diet. Moreover, the reintroduction of the food they had been asked to eliminate worsened symptoms in 83% of patients in the true diet compared with 31% of patients in the sham diet. These results suggest that an immune reaction to specific foods may occur in some IBS patients. The mechanisms are unknown, but they differ from classical (Type I-mediated) food allergy (46).
Thus, there are reasonable grounds for thinking that an immune-mediated response to specific dietary constituents, such as gluten, may be responsible for the generation of IBS-like symptoms in susceptible individuals. Patients with IBS frequently report exacerbation or triggering of symptoms by the ingestion of specific foods that are often implicated. However, controlled trials of elimination or rechallenge have often not objectively confirmed the patients’ suspicion (47). The relevance or importance of this mechanism in the overall population of individuals with IBS-like symptoms is unknown. On the one hand, it is not known what proportion of the normal, symptom-free population has elevated food-related IgG antibodies. It may be that food-related antibodies mediate symptoms only in a proportion of individuals, either because of the type of immune response or because of other factors, such as increased visceral sensitivity in susceptible individuals. On the other hand, it is not known why individuals with IBS symptoms and elevated gluten-related IgG antibodies do not report, more frequently, that wheat or gluten contributes to their symptoms. In the latter case, it is possible that the ubiquity and diversity of gluten-containing foods permeating the western diet make it very difficult for such individuals to identify gluten specically as a cause of their symptoms. In addition, there may well be genetic, physiological, and adaptive differences between individuals with respect to their responses to different nutrients (48), and given the heterogeneity of IBS-like symptoms, subsets of patients may be identified in whom the symptoms are aggravated by a particular and immunologically distinct food component, such as gluten (Figure 2).
Figure 2.

Putative factors underlying the pathophysiology of gastrointestinal (GI) symptoms.
PUTATIVE MECHANISMS TO EXPLAIN IBS-LIKE SYMPTOMS IN GS INDIVIDUALS
As with IBS, the pathogenetic mechanisms underlying symptom generation in GS patients (without malabsorption and classic celiac disease) are poorly understood. e linkage of GS and IBS is highly controversial as the assumption is being made that IBS and immune-mediated food intolerance are two distinct biological entities. However, IBS as a clinical descriptor can reflect a number of biological entities. It is possible that, in a proportion of patients presenting with IBS symptoms, GS and indeed other food intolerances provide a mechanism for symptom generation (Figure 3). Can GS contribute to the generation of gut neuromotor abnormalities, a proposed mechanism of symptom generation in functional GI disorders? Both submucosal and myenteric intrinsic afferent neurons are cholinergic in rodents and humans (49,50). Abnormal systemic parasympathetic and sympathetic functions as well as intestinal dysmotility have been reported in fully evolved celiac disease (51,52). Sensory transducer cells such as enterochromaffin cells in the gut epithelium release 5-HT that activate intrinsic sub-mucosal and myenteric afferent neurons (53,54). There is strong functional and electrophysiological evidence that myenteric intrinsic primary afferent neurons are involved in mechano- and chemosensitive responses in the GI tract (55-57). Reduced expression of serotonin reuptake transporter (SERT), required to terminate the action of 5-HT in the gut, can be induced during inflammatory conditions such as ulcerative colitis (58). Several studies in patients with IBS have detected low-grade inflammatory changes involving CD3+ cells, mast cells (59-61) , and reduced mucosal expression of SERT (58). Excess serotonin production in response to a carbohydrate meal has also been described and may be a mechanism for dyspeptic symptoms in celiac disease (62). Prolonged 5-HT activity, owing to reduced SERT activity, would be expected to lead to activation of afferent pathways, increased acetylcholine (Ach) release, and activation of peristaltic and secretory reflexes, which may affect gut function and symptom generation. Induction of increased ACh release by gluten provides a putative mechanism to explain the presence of symptoms in a proportion of patients with gluten-sensitive-IBS. As yet, these studies have not been carried out in GS patients without celiac disease.
Figure 3.
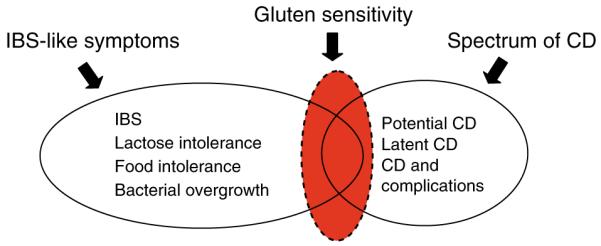
A clinical presentation of irritable bowel syndrome (IBS) does not constitute a specific diagnosis. Gluten sensitivity (GS) may be one of the underlying mechanisms for symptom generation and may not necessarily belong to the spectrum of celiac disease (CD).
Can GS drive low-grade inflammation in the absence of overt celiac disease and intestinal atrophy? The intestinal mucosa is a portal for dietary and microbial antigens, and exposure to triggering environmental factors in a genetically predisposed host may contribute to abnormal immune responses (63,64). Thus, altered barrier function could be a critical step in facilitating the host responses that underlie the clinical manifestations of the GS spectrum. In active celiac disease, intestinal permeability is elevated and correlates with the degree of mucosal atrophy (65). Gliadin induces intercellular tight junction (TJ) dysfunction through the release of zonulin, the mammalian counterpart of microbial zonula occludens toxin (zot), (66) leading to a sustained opening of TJ in celiac disease but not in healthy epithelium. Others have suggested an increased transcellular uptake of gliadin, pointing at an abnormal intestinal transport of IgA-gliadin complexes through overexpression of the transferrin receptor CD71 in celiac disease (67). Despite studies suggesting that variants in TJ genes may contribute to the pathogenesis of celiac disease, the phenotypic changes described above were only detected in active celiac disease, arguing against a constitutive epithelial defect. It has been suggested that in celiac patients, once immunotoxic gliadin peptides have penetrated the mucosa through open TJ or by a transcellular route, it stimulates lamina propria macrophages. Interaction of gliadin with macrophages elicits a MyD88-dependent but TLR2- and TLR4-independent release of zonulin through a proinflammatory cytokine milieu (68). Peritoneal macrophages have been shown to release chemokines such as RANTES and TNF-α when stimulated by gliadin peptide fragments but not when stimulated by other food proteins (69). Human monocyte-derived dendritic cells (DCs) stimulated by gliadin have also been shown to release chemokines and cytokines, mainly IL-6, IL-8, MCP-1, MCP-2, macrophage-derived chemokine, and RANTES (70). It is still unclear whether intestinal barrier dysfunction in GS hosts is controlled by a direct effect of gliadin on TJs or by the subsequent activation of innate immune cells. Interestingly, increased intestinal permeability has also been described in healthy rst-degree relatives of patients with celiac disease (71) and in patients without celiac disease but with biliary cirrhosis (72) , an autoimmune disorder that associates with celiac disease on the basis of HLA haplotypes (73). In non-celiac disease subjects, monocytes from HLA-DQ2+, but not HLA-DQ2-individuals, spontaneously release IL-8. Monocytes from HLA-DQ2+ persons release two- to threefold more IL-8 than do monocytes from HLA-DQ2 persons (74). Moreover, co-incubation of gliadin with IFN-γ further enhances the upregulation in the surface density and percentage of cells expressing T cell co-stimulatory molecules CD 80, CD 86, CD 83, and CD 40, suggesting a synergistic effect of gliadin and IFN-γ on innate immune cell activation (74). These findings suggest that in non-celiac disease subjects who are HLA-DQ2+, innate cells present a state of heightened activation compared with subjects who are HLA-DQ2−. The immunomodulatory effects of gluten may, in part, extend to other cereals that induce IL-10 from the peripheral blood monocytes through a TLR4-dependent mechanism that likely mimics LPS effects (75). Taken together, the results are consistent with the hypothesis that gliadin can induce minimal mucosal changes manifesting as increased intestinal permeability and innate immune cell activation.
LESSONS LEARNED USING A TRANSGENIC MOUSE MODEL OF GS
In non-transgenic mice, sensitization with gluten fails to trigger a T-cell-mediated atrophic lesion (76-79). As HLA DQ2 and DQ8 molecules play a pivotal role in celiac disease, models for GS using transgenic mice that express only human class II (DQ8) and CD4 molecules (DQ8/HCD4 4) have been developed (80). HLA transgenic mice represent a step closer to the human system than conventional mice, because the MHC molecules in these transgenic mice are the same ones involved in human disease. T cells from sensitized HLA-DQ8 and HLA-DQ8/HCD4 mice show increased proliferation when incubated with gliadin. We have shown that both arms of the immune system, innate and adaptive, are activated in DQ8 and DQ8/HCD4 when sensitized to gliadin and that these responses are not observed in transgenic negative controls (C57BL6), haplotype controls (HLA-DQ6), or DQ8 mice sensitized with bovine serum albumin (81). However, even in the presence of DQ8 genes, immunization with gliadin does not lead to villous atrophy (81,82). The lack of significant atrophic enteropathy may indicate that factors in addition to genetic susceptibility and gliadin exposure are required for full expression of disease (73).
This model provides insight into the mechanisms underlying the relationship between GS and functional changes that could explain the development of GI symptoms. Gluten sensitization of DQ8 mice increased ACh release by the myenteric plexus after in vitro nerve stimulation. ACh is a major neurotransmitter involved in the control of gut motility, secretomotor function, and intestinal permeability (83,84). Release of ACh by the myenteric plexus is similar in GS and control DQ8 mice after sodium chloride (KCl) stimulation, suggesting a functional alteration in the release of the neurotransmitter rather than a change in production or storage (81). The increased ACh release is accompanied by hypercontractility in vitro (81) and dysmotility in vivo , characterized by the increased incidence of retroperistalsis and gastric emptying. The precise mechanisms underlying the induction of increased ACh release in gluten-sensitive mice remain to be elucidated, but it could be because of the altered sensing pathways of nutrients in sensitized individuals.
Gliadin-sensitization and oral challenge causes impaired barrier function in DQ8 mice (81,85,86). It is likely that the integrity of the intestinal barrier is a critical determinant of the exposure of the mucosal immune system to dietary and microbial antigens, and of the subsequent triggering of effector responses, including those that occur in the absence of villous atrophy. Experiments on GS DQ8/HCD4 mice suggest that alteration of the intestinal barrier by chronic low-dose indomethacin administration leads to more severe epithelial cell injury, characterized by altered mitochondrial and TJ structure. The results also show the induction of intestinal dysbacteriosis and loss of systemic ignorance to commensal bacteria in GS mice treated with indomethacin (85). Clinical studies have reported the presence of serological responses to bacterial and microbial antigens in patients with celiac disease (87-89). A recent epidemiological study has determined that the consumption of non-steroidal anti-inflammatory drugs (NSAIDs) is a risk factor for the development of IBS (90). The results raise the hypothesis that the effect of GS on symptom generation in a genetically predisposed host may be potentiated by environmental factors that affect the intestinal barrier and promote low-grade inflammation and intestinal cell injury.
Thus, by combining the clinical observations that support the gluten-sensitive induction of GI symptoms and syndromes in humans along with our mechanistic studies in gluten-sensitized humanized mice, we propose a model tested in HLA-DQ8 mice that reproduces the mucosal alterations reported in individuals with mild GS rather than classic celiac disease. The model includes genetic susceptibility, low-grade mucosal inflammatory changes, and allows for investigation of putative triggers, in addition to gluten, such as NSAIDs or infection, barrier dysfunction, and microbial antigens.
IMPLICATIONS FOR THE MANAGEMENT OF IBS-LIKE SYMPTOMS IN GS PATIENTS
Avoidance diets are difficult and can pose significant socioeconomic challenges for patients. In addition, some countries that reimburse patients for gluten-free food require biopsy confirmation of celiac disease to justify government subsidy for gluten-free food (16,17). Hence, it is crucial that a rational approach to the prescription of a GFD be undertaken (Table 1). In symptomatic patients with minimal enteropathy and specific serology for celiac disease, a GFD trial period can be considered. In those patients with minimal enteropathy but negative serology, a number of approaches are reasonable. HLA genotyping could be performed, and if they carry the at-risk haplotypes DQ2 or DQ8 and do not have another readily treatable cause, a trial of a GFD with careful follow-up and possibly rebiopsy after a 3–6 month period to document recovery could be useful. Ultimate proof of GS may require a re-challenge and biopsy, though many patients will decline the challenge. Alternatively, patients may elect to be followed up to see whether progression will occur as may be common in those tTG individuals (Figure 4). Those patients with increased IELs but negative serology and lacking the celiac disease-associated HLA types are less likely to derive benefit but, anecdotally, may realize some subjective benefit (30).
Table 1.
Proposed management of patients with IBS-like symptoms and minimal histology
| Symptom | LD | HLA type | Serology | Treatment |
|---|---|---|---|---|
| IBS | + | + | + | + |
| IBS | + | + | + | Trial |
| IBS | + | − | − | Seek other cause |
| IBS | − | + | + | Treat or follow |
| IBS | − | + | − | Follow or trial of GFD |
| IBS | − | − | − | No GFD |
| None | + | + | + | Treat or follow |
| None | + | − | − | Other cause |
GFD, gluten-free diet; HLA, human leukocyte antigen; IBS, irritable bowel syndrome; LD, lymphocytic duodenosis.
Figure 4.
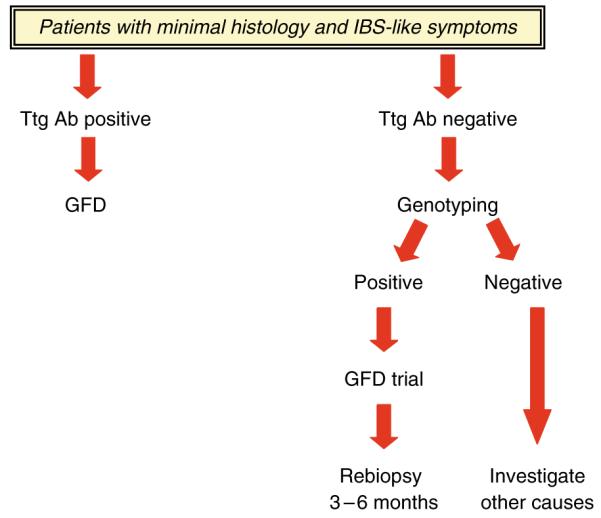
Algorithm for the management of patients with IBS-like symptoms and minimal histological change. GFD, gluten-free diet; IBS, irritable bowel syndrome; Ttg Ab, tissue transglutaminase antibody.
SUMMARY AND CONCLUSIONS
This review is intended to highlight the relationship between celiac disease, IBS, and GS, as well as to emphasize a number of specific issues:
“GS” is defined by one or more of a variety of immunological, morphological, or symptomatic manifestations that may also be shared by celiac disease and IBS.
Although GS and classic celiac disease may share a common etiology, gluten sensitivity does not fulfill diagnostic criteria for celiac disease.
Although GS and IBS may share common symptomatology, GS by definition is not IBS.
Successful therapeutic options have been difficult to develop in IBS because of the lack of pharmacological targets and even a conceptualized framework for the underlying pathogenetic mechanisms.
The role of subtle persistent inflammation in causing the symptoms of post-infectious IBS has served a prototypical model for linking gut immune responses to symptoms in IBS, but likely only represents a subgroup of IBS patients.
Recent clinical observations support GS as a mechanism underlying the generation of GI symptoms that would otherwise be considered diagnostic of IBS.
The animal model of GS in HLA-DQ8 mice reproduces the effects reported in individuals with mild GS rather than classic celiac disease, as the neuromotor and barrier abnormalities occur in the absence of mucosal atrophy.
The animal model of GS in HLA-DQ8 mice allows investigation of mucosal pathophysiological changes that occur before the onset of full-blown inflammation in a GS host.
Gluten sensitivity and post-infectious IBS provide two triggers that can explain at least part of the spectrum that constitutes IBS, further advancing an understanding of the role of mucosal responses to luminal factors in FBDs.
Footnotes
CONFLICT OF INTEREST
Guarantor of the article: Elena F. Verdu, MD, PhD.
Specific author contributions: Elena F. Verdu, main writer; D. Armstrong and J. Murray, scientific discussions and major editing.
Financial support: None.
Potential competing interests: None.
REFERENCES
- 1.Fasano A, Berti I, Gerarduzzi T, et al. Prevalence of celiac disease in at-risk and not-at-risk groups in the United States: a large multicenter study. Arch Intern Med. 2003;163:286–92. doi: 10.1001/archinte.163.3.286. [DOI] [PubMed] [Google Scholar]
- 2.Cranney A, Zarkadas M, Graham ID, et al. The Canadian Celiac Health Survey. Dig Dis Sci. 2007;52:1087–95. doi: 10.1007/s10620-006-9258-2. [DOI] [PubMed] [Google Scholar]
- 3.Catassi C, Ratsch JM, Fabiani E, et al. Coeliac disease in the year: exploring the iceberg. Lancet. 2000;343:200–3. doi: 10.1016/s0140-6736(94)90989-x. C.G.D.S. [DOI] [PubMed] [Google Scholar]
- 4.Csizmadia CG, Mearin ML, von Blomberg BME, et al. An iceberg of child-hood coeliac disease in the Netherlands. Lancet. 1999;353:813–4. doi: 10.1016/S0140-6736(99)00243-3. [DOI] [PubMed] [Google Scholar]
- 5.Weile I, Grodzinsky E, Skogh T, et al. High prevalence rates of adult silent celiac disease, as seen in Sweden, must be expected in Denmark. APMIS. 2001;109:745–50. doi: 10.1034/j.1600-0463.2001.d01-141.x. [DOI] [PubMed] [Google Scholar]
- 6.Gomez JC, Selvaggio GS, Viola M, et al. Prevalence of celiac disease in Argentina: screening of an adult population in the La Plata area. Am J Gastroenterol. 2001;96:2700–4. doi: 10.1111/j.1572-0241.2001.04124.x. [DOI] [PubMed] [Google Scholar]
- 7.Longstreth GF, Thompson WG, Chey WD, et al. Functional bowel disorders. Gastroenterology. 2006;130:1480–91. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- 8.Boivin M. Socioeconomic impact of irritable bowel syndrome in Canada. Can J Gastroenterol. 2001;15(Suppl B):8B–11B. doi: 10.1155/2001/401309. [DOI] [PubMed] [Google Scholar]
- 9.Holt R, Darnley SE, Kennedy T, et al. Screening for celiac disease in patients with clinical diagnosis of irritable bowel syndrome. Gastroenterology. 2001;120(Suppl 1) AB4064. [Google Scholar]
- 10.Sanders DS, Patel D, Stephenson TJ, et al. A primary care cross-sectional study of undiagnosed adult coeliac disease. Eur J Gastroenterol Hepatol. 2003;15:407–13. doi: 10.1097/00042737-200304000-00012. [DOI] [PubMed] [Google Scholar]
- 11.Sanders DS, Carter MJ, Hurlstone DP, et al. Association of adult coeliac disease with irritable bowel syndrome: a case-control study in patients fulfilling ROME II criteria referred to secondary care. Lancet. 2001;358:1504–8. doi: 10.1016/S0140-6736(01)06581-3. [DOI] [PubMed] [Google Scholar]
- 12.Shahbazkhani B, Forootan M, Merat S. Coeliac disease presenting with symptoms of irritable bowel syndrome. Aliment Pharmacol Ther. 2003;18:231–5. doi: 10.1046/j.1365-2036.2003.01666.x. [DOI] [PubMed] [Google Scholar]
- 13.Ford AC, Chey WD, Talley NJ, et al. Utility of diagnostic tests for celiac disease in irritable bowel syndrome: systematic review and meta-analysis. Gut. 2008;57(suppl II):A75. doi: 10.1001/archinternmed.2009.22. [DOI] [PubMed] [Google Scholar]
- 14.Spiegel BM, DeRosa VP, Gralnek IM, et al. Testing for celiac sprue in irritable bowel syndrome with predominant diarrhea: a cost-effectiveness analysis. Gastroenterology. 2004;126:1721–32. doi: 10.1053/j.gastro.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 15.Locke GR, III, Murray JA, Zinsmeister AR, et al. Celiac disease serology in irritable bowel syndrome and dyspepsia: a population-based case-control study. Mayo Clin Proc. 2004;79:476–82. doi: 10.4065/79.4.476. [DOI] [PubMed] [Google Scholar]
- 16.Hallert C, Grännö C, Hulten S, et al. Living with coeliac disease: controlled study of the burden of illness. Scand J Gastroenterol. 2002;37:39–42. doi: 10.1080/003655202753387338. [DOI] [PubMed] [Google Scholar]
- 17.Hallert C, Grännö C, Grant C, et al. Quality of life of adult coeliac patients treated for 10 years. Scand J Gastroenterol. 1998;33:933–8. doi: 10.1080/003655298750026949. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson A, Arranz E, OMahony S. Clinical and pathological spectrum of coeliac disease: active, silent, latent, potential. Gut. 1993;34:150–1. doi: 10.1136/gut.34.2.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Troncone R, Paparo F, Mazzarella G, et al. e spectrum of gluten sensitivity; Proc 8th Int. Symp. on Coeliac disease; Naples, Italy. April 1999. [Google Scholar]
- 20.Troncone R, Franzese A, Mazzarella G, et al. Gluten sensitivity in a subset of children with insulin dependent diabetes mellitus. Am J Gastroenterol. 2003;98:590–5. doi: 10.1111/j.1572-0241.2003.07301.x. [DOI] [PubMed] [Google Scholar]
- 21.Marsh MN. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’) Gastroenterology. 1992;102:330–54. Review. [PubMed] [Google Scholar]
- 22.Kaukinen K, Maki M, Partanen J, et al. Celiac disease without villous atrophy: revision of criteria called for. Dig Dis Sci. 2001;46:879–87. doi: 10.1023/a:1010729207320. [DOI] [PubMed] [Google Scholar]
- 23.Mino M, Lauwers GY. Role of lymphocytic immunophenotyping in the diagnosis of gluten-sensitive enteropathy with preserved villous architecture. Am J Surg Pathol. 2003;27:1237–42. doi: 10.1097/00000478-200309000-00007. [DOI] [PubMed] [Google Scholar]
- 24.O’Mahony S, Vestey JP, Ferguson A. Similarities in intestinal humoral immunity in dermatitis herpetiformis without enteropathy and in coeliac disease. Lancet. 1990;335:1487–90. doi: 10.1016/0140-6736(90)93029-o. [DOI] [PubMed] [Google Scholar]
- 25.Salmi TT, Collin P, Jarvinen O, et al. Immunoglobulin A autoantibodies against transglutaminase 2 in the small intestinal mucosa predict forthcoming coeliac disease. Aliment Pharmacol Ther. 2006;24:541–52. doi: 10.1111/j.1365-2036.2006.02997.x. [DOI] [PubMed] [Google Scholar]
- 26.Niveloni S, Dezi R, Pedreira S, et al. Gluten sensitivity in patients with primary biliary cirrhosis. Am J Gastroenterol. 1998;93:404–8. doi: 10.1111/j.1572-0241.1998.00404.x. [DOI] [PubMed] [Google Scholar]
- 27.Wahnschaffe U, Ullrich R, Riecken EO, et al. Celiac disease-like abnormalities in a subgroup of patients with irritable bowel syndrome. Gastroenterology. 2001;121:1329–38. doi: 10.1053/gast.2001.29572. [DOI] [PubMed] [Google Scholar]
- 28.Wahnschaffe U, Schulzke JD, Zeitz M, et al. Predictors of clinical response to gluten-free diet in patients diagnosed with diarrhea-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol. 2007;5:844–50. doi: 10.1016/j.cgh.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 29.Kakar S, Nehra V, Murray JA, et al. Significance of intraepithelial lymphocytosis in small bowel biopsy samples with normal mucosal architecture. AJG. 2003;98:2027–33. doi: 10.1111/j.1572-0241.2003.07631.x. [DOI] [PubMed] [Google Scholar]
- 30.Vande Voort JL, Murray JA, Lahr BD, et al. Lymphocytic duodenosis and the spectrum of celiac disease. Am J Gastroenterol. 2009;104:142–8. doi: 10.1038/ajg.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drossman DA. Review article: an integrated approach to the irritable bowel syndrome. Alim Pharmacol Ther. 1999;13:3–14. doi: 10.1046/j.1365-2036.1999.0130s2003.x. [DOI] [PubMed] [Google Scholar]
- 32.Rodrigues LA, Ruigomez A. Increased risk of irritable bowel syndrome after bacterial gastroenteritis: cohort study. BMJ. 1999;318:565–6. doi: 10.1136/bmj.318.7183.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spiller RC, Jenkins D, Thornley JP, et al. Increased rectal mucosal enteroendocrine cells, T lymphocytes and increased gut permeability following acute Campylobacter enteritis and in postdysenteric irritable bowel syndrome. Gut. 2000;47:804–11. doi: 10.1136/gut.47.6.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neal KR, Hebden J, Spiller R. Prevalence of gastrointestinal symptoms 6 months after bacterial gastroenteritis and risk factors for development of the irritable bowel syndrome: postal survey of patients. BMJ. 1997;314:779–82. doi: 10.1136/bmj.314.7083.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gwee KA, Collins SM, Read NW, et al. Increased rectal mucosal expression of interleukin 1β in recently acquired postinfectious irritable bowel syndrome. Gut. 2003;52:523–6. doi: 10.1136/gut.52.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marshall JK, Thabane M, Garg AX, et al. Walkerton Health Study Investigators Incidence and epidemiology of irritable bowel syndrome after a large waterborne outbreak of bacterial dysentery. Gastroenterology. 2006;131(2):445–50. doi: 10.1053/j.gastro.2006.05.053. [DOI] [PubMed] [Google Scholar]
- 37.Pimentel M, Chow EJ, Lin HC. Normalization of lactulose breath testing correlates with symptom improvement in irritable bowel syndrome. a double-blind, randomized, placebo-controlled study. Am J Gastroenterol. 2003;98:412–9. doi: 10.1111/j.1572-0241.2003.07234.x. [DOI] [PubMed] [Google Scholar]
- 38.Spiller R. Role of motility in chronic diarrhea. Neurogastroenterol Motil. 2006;18:1045–55. doi: 10.1111/j.1365-2982.2006.00836.x. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt T, Hackelsberger N, Widmer R, et al. Ambulatory 24-hour jejunal motility in diarrhea-predominant irritable bowel syndrome. Scand J Gastroenterol. 1996;31:581–9. doi: 10.3109/00365529609009131. [DOI] [PubMed] [Google Scholar]
- 40.Clemens CH, Samsom M, Roelofs JM, et al. Association between pain episodes and high amplitude propagated pressure waves in patients with irritable bowel syndrome. Am J Gastroenterol. 2003;98:1838–43. doi: 10.1111/j.1572-0241.2003.07541.x. [DOI] [PubMed] [Google Scholar]
- 41.Ragnarsson G, Bodemar G. Pain is temporally related to eating but not defecation in the irritable bowel syndrome (IBS) patient’s description of diarrhea, constipation and symptom variation during 6-week study. Eur J Gastroenterol Hepatol. 1998;10:415–21. doi: 10.1097/00042737-199805000-00011. [DOI] [PubMed] [Google Scholar]
- 42.Fernandez-Banares F, Esteve-pardo M, de Leon R, et al. Sugar malabsorption in functional bowel disease: clinical implications. Am J Gastroenterol. 1993;88:2044–50. [PubMed] [Google Scholar]
- 43.Atkinson W, Sheldon TA, Shaath N, et al. Food elimination based on IgG antibodies in irritable bowel syndrome: a randomized controlled trial. Gut. 2004;53:1459–64. doi: 10.1136/gut.2003.037697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zar S, Kumar D, Benson MJ. Food hypersensitivity and irritable bowel syndrome. Aliment Pharmacol Ther. 2001;15:439–49. doi: 10.1046/j.1365-2036.2001.00951.x. [DOI] [PubMed] [Google Scholar]
- 45.Zar S, Benson MJ, Kumar D. Food specific serum IgG4 and IgE titres to common food antigens in irritable bowel syndrome. Am J Gastroenterol. 2005;100:1550–7. doi: 10.1111/j.1572-0241.2005.41348.x. [DOI] [PubMed] [Google Scholar]
- 46.Shanahan F, Whorwell PJ. IgG-mediated food intolerance in irritable bowel syndrome: a real phenomenon or an epiphenomenom? Am J Gastroenterol. 2005;100:1558–9. doi: 10.1111/j.1572-0241.2005.50009.x. [DOI] [PubMed] [Google Scholar]
- 47.Burden S. Dietary treatment of irritable bowel syndrome: current evidence and guidelines for future practice. Br Dietetic Assoc. 2001;14:231–41. doi: 10.1046/j.1365-277x.2001.00284.x. [DOI] [PubMed] [Google Scholar]
- 48.Murray JA, Watson T, Clearman B, et al. Effect of a gluten-free diet on gastrointestinal symptoms in celiac disease. Am J Clin Nutr. 2004;79:669–73. doi: 10.1093/ajcn/79.4.669. [DOI] [PubMed] [Google Scholar]
- 49.Pan H, Gershon MD. Activation of intrinsic afferent pathways in submucosal ganglia of the guinea pig small intestine. J Neurosci. 2000;20:3295–309. doi: 10.1523/JNEUROSCI.20-09-03295.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brehmer A, Croner R, Dimmler A, et al. Immunohistochemical characterization of putative primary afferent sensory neurons in human small intestine. Autonom Neurosci. 2004;112:49–59. doi: 10.1016/j.autneu.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 51.Giorgetti GM, Tursi A, Iani C, et al. Assessment of autonomic function in untreated adult celiac disease. World J Gastroenterol. 2004;10:2715–8. doi: 10.3748/wjg.v10.i18.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gibbons CH, Freeman R. Autonomic neuropathy and celiac disease. J Neurol Neurosurg Psychiatry. 2005;76:579–81. doi: 10.1136/jnnp.2004.047480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galligan JJ, Pan H, Mesori E. Signalling mechanisms coupled to 5-hydroxytriptamine4 receptor-mediated facilitation of fast synaptic transmission in the guinea pig ileum myenteric plexus. Neurogastroenterol Motil. 2003;15:523–9. doi: 10.1046/j.1365-2982.2003.00428.x. [DOI] [PubMed] [Google Scholar]
- 54.Pan H, Galligan JJ. 5-HT1A and 5-HT-4 receptors mediate inhibitiion and facilitation of fast synaptic transmission in enteric neurons. Am J Physiol. 1994;266:G230–8. doi: 10.1152/ajpgi.1994.266.2.G230. [DOI] [PubMed] [Google Scholar]
- 55.Raybould HE. Visceral perception: sensory transduction in visceral afferents and nutrients. Gut. 2002;51:11–4. doi: 10.1136/gut.51.suppl_1.i11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bertrand PP, Kunze WAA, Bornstein JC, et al. Analysis of the responses of myenteric neurons in the small intestine to chemical stimulation of the mucosa. Am J Physiol. 1997;273:G422–35. doi: 10.1152/ajpgi.1997.273.2.G422. [DOI] [PubMed] [Google Scholar]
- 57.Kunze WAA. Influence of the mucosa on the excitability of myenteric neurons. Neuroscience. 1997;76:619–34. doi: 10.1016/s0306-4522(96)00408-3. [DOI] [PubMed] [Google Scholar]
- 58.Coates MD, Mahoney CR, Linden DR, et al. Molecular defects in mucosal serotonin content and decreased serotonin reuptake transporter in ulcerative colitis and IBS. Gastroenterology. 2004;126:1657–64. doi: 10.1053/j.gastro.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 59.Törnblom H, Lindberg G, Nyberg B, et al. Full-thickness biopsy of the jejunum reveals inflammation and enteric neuropathy in irritable bowel syndrome. Gastroenterology. 2002;123:1972–9. doi: 10.1053/gast.2002.37059. [DOI] [PubMed] [Google Scholar]
- 60.Bercik P, Verdu EF, Collins SM. Is irritable bowel syndrome a low-grade inflammatory bowel disease? Gastroenterol Clin North Am. 2005;34:235–45. doi: 10.1016/j.gtc.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 61.Weston AP, Biddle WL, Bhatia PS, et al. Terminal ileal mucosal mast cells in irritable bowel syndrome. Dig Dis Sci. 1993;38:1590–5. doi: 10.1007/BF01303164. [DOI] [PubMed] [Google Scholar]
- 62.Coleman NS, Foley S, Dunlop SP, et al. Abnormalities of serotonin metabolism and their relation to symptoms in untreated celiac disease. Clin Gastroenterol Hepatol. 2006;4:874–81. doi: 10.1016/j.cgh.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 63.McCole DF, Barrett KE. Varied role of the gut epithelium in mucosal homeostasis. Curr Opin Gastroenterol. 2007;23:647–54. doi: 10.1097/MOG.0b013e3282f0153b. [DOI] [PubMed] [Google Scholar]
- 64.Resta-Lenert S, Smitham J, Barrett KE. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/−mice. Am J Physiol Gastrointest Liver Physiol. 2005;289:G153–62. doi: 10.1152/ajpgi.00395.2004. [DOI] [PubMed] [Google Scholar]
- 65.Cummins AG, Thompson FM, Butler RN, et al. Improvement in intestinal permeability precedes morphometric recovery of the small intestine in coeliac disease. Clin Sci (Lond) 2001;100:379–86. [PubMed] [Google Scholar]
- 66.Fasano A, Not T, Wang W, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518–9. doi: 10.1016/S0140-6736(00)02169-3. [DOI] [PubMed] [Google Scholar]
- 67.Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med. 2008;205:143–54. doi: 10.1084/jem.20071204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas KE, Sapone A, Fasano A, et al. Gliadin stimulation of murine macrophage inflammatory gene expression and intestinal permeability are MyD88-dependent: role of the innate immune response in Celiac disease. J Immunol. 2006;176:2512–21. doi: 10.4049/jimmunol.176.4.2512. [DOI] [PubMed] [Google Scholar]
- 69.Jelínková L, Tucková L, Cinová J, et al. Gliadin stimulates human monocytes to production of IL-8 and TNF-alpha through a mechanism involving NF-kappaB. FEBS Lett. 2004;571:81–5. doi: 10.1016/j.febslet.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 70.Palová-Jelínková L, Rozková D, Pecharová B, et al. Gliadin fragments induce phenotypic and functional maturation of human dendritic cells. J Immunol. 2005;175:7038–45. doi: 10.4049/jimmunol.175.10.7038. [DOI] [PubMed] [Google Scholar]
- 71.van Elburg RM, Uil JJ, Mulder CJ, et al. Intestinal permeability in patients with coeliac disease and relatives of patients with coeliac disease. Gut. 1993;34:354–7. doi: 10.1136/gut.34.3.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feld JJ, Meddings J, Heathcote EJ. Abnormal intestinal permeability in primary biliary cirrhosis. Dig Dis Sci. 2006;51:1607–13. doi: 10.1007/s10620-006-9544-z. [DOI] [PubMed] [Google Scholar]
- 73.Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology. 1993;105:910–22. doi: 10.1016/0016-5085(93)90912-v. [DOI] [PubMed] [Google Scholar]
- 74.Cinova J, Palová-Jelínková L, Smythies LE, et al. Gliadin peptides activate blood monocytes from patients with celiac disease. J Clin Immunol. 2007;27:201–9. doi: 10.1007/s10875-006-9061-z. [DOI] [PubMed] [Google Scholar]
- 75.Yamazaki K, Murray JA, Kita H. Innate immunomodulatory effects of cereal grains through induction of IL-10. J Allergy Clin Immunol. 2008;121:172–8. doi: 10.1016/j.jaci.2007.08.031. [DOI] [PubMed] [Google Scholar]
- 76.Kozakova H, Stepankova R, Tuckova L, et al. Humoral and cellular immune responses in gluten-treated suckling or hand-fed rats. Physiol Res. 2000;49:665–72. [PubMed] [Google Scholar]
- 77.Stepankova R, Kofronova O, Tuckova L, et al. Experimentally induced gluten enteropathy and protective effect of epidermal growth factor in artificially fed neonatal rats. J Pediatr Gastroenterol Nutr. 2003;36:96–104. doi: 10.1097/00005176-200301000-00018. [DOI] [PubMed] [Google Scholar]
- 78.Troncone R, Ferguson A. Animal model of gluten induced enteropathy in mice. Gut. 1991;32:871–5. doi: 10.1136/gut.32.8.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maurano F, Siciliano RA, De Giulio B, et al. Intranasal administration of one alpha gliadin can downregulate the immune response to whole gliadin in mice. Scand J Immunol. 2001;53:290–5. doi: 10.1046/j.1365-3083.2001.00869.x. [DOI] [PubMed] [Google Scholar]
- 80.Black KE, Murray JA, David CS. HLA-DQ determines the response to exogenous wheat proteins: a model of gluten sensitivity in transgenic knockout mice. J Immunol. 2002;169:5595–600. doi: 10.4049/jimmunol.169.10.5595. [DOI] [PubMed] [Google Scholar]
- 81.Verdu EF, Huang XX, Natividad J, et al. Gliadin-dependent neuromuscular and epithelial secretory responses in the gut. Am J Physiol Gastrointest Liver Physiol. 2008;294:G217–25. doi: 10.1152/ajpgi.00225.2007. [DOI] [PubMed] [Google Scholar]
- 82.Senger S, Luongo D, Maurano F, et al. Intranasal administration of a recombinant alpha-gliadin down-regulates the immune response to wheat gliadin in DQ8 transgenic mice. Immunol Lett. 2003;88:127–34. doi: 10.1016/s0165-2478(03)00069-5. [DOI] [PubMed] [Google Scholar]
- 83.Galligan JJ. Ligand-gated ion channels in the enteric nervous system. Neurogastroenterol Motil. 2002;14:611–23. doi: 10.1046/j.1365-2982.2002.00363.x. [DOI] [PubMed] [Google Scholar]
- 84.Cameron HL, Perdue MH. Muscarinic acetylcholine receptor activation increases transcellular transport of macromolecules across mouse and human intestinal epithelium in vitro. Neurogastroenterol Motil. 2007;19:47–56. doi: 10.1111/j.1365-2982.2006.00845.x. [DOI] [PubMed] [Google Scholar]
- 85.Natividad J, Jury J, Sanz Y, et al. Role of intestinal barrier modulation by nonsteroidal anti-inflammatory drugs in gluten-induced epithelial damage in HLA-DQ8/HCD4 transgenic mice. Gastroenterology. 2008;134:A-151. abstract. [Google Scholar]
- 86.Pinier M, Verdú EF, Nasser-Eddine M, et al. Polymeric binders suppress gliadin-induced toxicity in the intestinal epithelium. Gastroenterology. 2009;136:288–98. doi: 10.1053/j.gastro.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 87.Ashorn S, Raukola H, Valineva T, et al. Serological responses to bacterial antigens in celiac disease patients. Gastroenterology. 2007;132:A1132. doi: 10.1007/s10875-008-9255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mankai A, Belhadj R, Ghedira-Besbes L, et al. Anti-Saccharomyces cerevisiae antibodies in coeliac disease. Scand J Gastroenterol. 2007;42:821–6. doi: 10.1080/00365520601154996. [DOI] [PubMed] [Google Scholar]
- 89.Granito A, Zauli D, Muratori P, et al. Anti-Saccharomyces cerevisiae and perinuclear anti-neutrophil cytoplasmic antibodies in coeliac disease before and after gluten-free diet. Aliment Pharmacol Ther. 2005;21:881–7. doi: 10.1111/j.1365-2036.2005.02417.x. [DOI] [PubMed] [Google Scholar]
- 90.Ford AC, Forman D, Bailey AG, et al. Irritable bowel syndrome: a 10-year natural history of symptoms, and factors that influence consultation behavior. AJG. 2008;103:1229–39. doi: 10.1111/j.1572-0241.2007.01740.x. [DOI] [PubMed] [Google Scholar]