

Abstract
Diynes and cyanamides undergo an iron-catalyzed [2+2+2] cycloaddition to form highly substituted 2-aminopyridines in an atom-efficient manner that is both high yielding and regioselective. This system was also used to cyclize two terminal alkynes and a cyanamide to afford a 2,4,6-trisubstituted pyridine product regioselectively.
Introduction
The introduction of complex substitution patterns onto pyridine rings can require multiple synthetic steps. Alternatively, metal catalyzed [2+2+2] cycloadditions can create highly substituted pyridines in a single step from simple starting materials.1 This remarkably powerful method allows for the efficient and regioselective construction of pyridines in addition to bi- and tricyclic-fused pyridine ring systems. Despite the abundance and low cost of iron salts, only a few examples of iron-catalyzed pyridine formation exist.2
We recently developed the first general iron-catalyzed method for pyridine synthesis.3 In our initial report, the strong tendency for iron to cyclotrimerize alkynes was overcome using alkynenitrile substrates along with a unique bis(aldimino)pyridine ligand (L1) (eq 1).4 Shortly after our initial report, Wan and co-workers published an iron-catalyzed synthesis of pyridines from diynes and nitriles (eq 2).5 Unfortunately, relatively high catalyst loadings were required in both systems. In addition, these systems either gave pyridines in moderate yields (former) or required ten to twenty molar equivalents of nitrile (latter).6 Although these reports are an important step forward, an efficacious iron-catalyzed route to pyridines that utilizes low catalyst loading yet affords high yield of pyridine remains scarce.
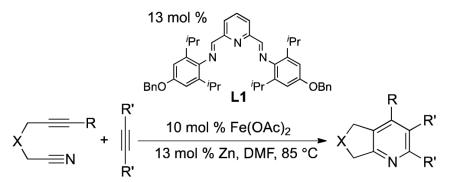 |
(1) |
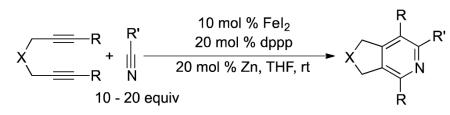 |
(2) |
Attempts to react unactivated nitriles with diynes using our system afforded only traces of pyridine products. Reactions with electron deficient nitriles were also unsuccessful. Our recent work with cyanamides suggested that they could be suitable partners in Fe-catalyzed cycloaddition chemistry as they appeared to be more reactive in Ni-catalyzed cycloadditions.7 Thus, 2-aminopyridines could potentially be prepared by our iron bisiminopyridine catalyst without requiring a large excess of cyanamide or having to tether the cyanamide to an alkyne.
Variations on the 2-aminopyridine core have been studied extensively due to their potential as medicinally useful compounds.8 2-Aminopyridines are also a central structural motif in α-carboline natural products9 as well as in a variety chromophores,10 pharmacophores,11 OLED’s,12 and inorganic ligands.13 These broadly applicable compounds are commonly synthesized by Buchwald-Hartwig type aminations,14 nucleophilic aromatic substitutions,15 and multi-component condensations.16 Such methods are useful for the creation of simple 2-aminopyridines, but more complex substitution patterns require additional synthetic manipulations. For more complex aminopyridines, Co, Ni, Rh, and Ti catalysts and a photocatalytical system have all been used to mediate [2+2+2] cycloadditions.17 However, in most of these studies (i.e., Ni, Rh, and Ti), only a single example of a [2+2+2] cycloaddition with a cyanamide was demonstrated. In an effort to improve the conditions and expand the scope of these reactions, we recently developed a Ni/NHC-catalyzed system for the [2+2+2] formation of 2-aminopyridines from diynes and cyanamides.7 Although this system was high yielding and regioselective, the low cost and environmentally-benign nature of iron compares favorably to nickel. Additionally we felt that highly reactive cyanamides presented an opportunity to improve on iron-catalyzed pyridine formation. Herein, we report the successful development of an iron catalyst for the cycloaddition of diynes and cyanamides to afford 2-aminopyridines.
Results and Discussion
We were delighted when our first attempt to make 2-aminopyridine 3a from diyne 1a and 2 equivalents of cyanamide 2a using our original catalyst system (10 mol % Fe(OAc)2, 13 mol % Zn and 13 mol % L1 in DMF) afforded 2-aminopyridine 3a in 69% isolated yield at 85 °C (eq 3). Furthermore, the use of a simpler ligand (L2) led to an increase in isolated yield (85%).18 Further optimization led to conditions employing 5 mol % catalyst loading of inexpensive FeCl2, 10 mol % Zn and 10 mol % L2 in benzene over 4 hours at room temperature. Evaluation of other ligands (N-heterocyclic carbenes, phosphines, and pyridyl diimines provided no product.19
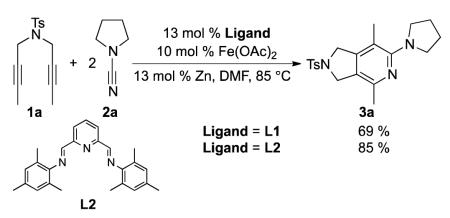 |
(3) |
Unfortunately, the mild conditions that worked for the model substrates (i.e., 1a and 2a) were not amenable to other diynes. Although increasing the reaction temperature to 70 °C led to good reactivity of the substrates, diyne dimerization accounted for as much as half of substrate conversion. An excess of cyanamide (10 equiv) led to no reactivity suggesting that cyanamide binding out-competes alkyne binding at high concentrations. Gratifyingly, slow addition of diyne over the course of 3 hours solved this problem and provided a general method for coupling a variety of diynes. Slow addition of the diyne also allowed us to decrease the cyanamide:diyne ratio to 1.2:1 with no deleterious effects on the yield.
Control studies to establish the necessity of iron for the reaction were carried out. Importantly, removing any part of the catalytic system renders the reaction ineffective. Additionally, (L2)FeBr2 was independently synthesized and used directly as an all-in-one catalyst precursor to provide a 97 % NMR yield (eq 4). That is, the combination of (L2)FeBr2 and Zn dust provided a catalytically competent system. No reaction occurred in the absence of zinc dust.20
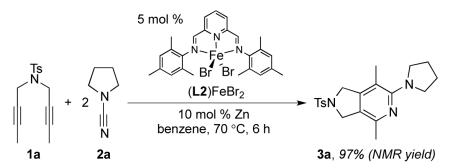 |
(4) |
A broad range of diynes and cyanamides were successfully converted to their respective 2-aminopyridines (Table 1). The protected nitrogen backbone diyne 1a and malonate backbone diyne 1b both afforded products (3a and 3b) in excellent yield (entries 1-2). The challenging terminal diyne 1c provided 3c in good yield (entry 3). However, other difficult substrates such as phenyl and TMS substituted diynes (1d and 1e respectively, entries 4-5) were unreactive. The lack of Thorpe-Ingold effect in substrates 1f and 1g did not effect cycloaddition as products 3f and 3g were both obtained in 84% yield (entries 6-7). Notably, entries 1 and 6 demonstrate that heteroatom tethers, which were problematic in the previous iron systems,3,5 are well tolerated by this system. Diyne 1h successfully provided the 6,6-bicyclic pyridine (3h) in good yield (entry 8). High yields of 2-aminopyridine were obtained with methyl-phenyl (2d) and dimethyl (2b) substituted cyanamides substrates (entries 9 and 11). Surprisingly, cycloaddition of diethyl cyanamide 2c afforded a lower yield of product (entry 10). Cyclic cyanamides N-cyanopiperidine (2e) and N-cyanomorpholine (2f) reacted readily (entries 12-13). Despite the apparent negative effect of cyanamide sterics in entry 10, the large dibenzazepinyl cyanamide provided 3n in good yield (entry 14). This result compares well with the 19% yield afforded by cobalt.17c Attempts to react (dimethylamino)acetonitrile with diyne 1b were unsuccessful, which may indicate that the inherent electronic structure of cyanamides, not a chelation effect, is the source of their high reactivity in this system.21 To demonstrate the synthetic utility of this methodology, entry 1 was repeated on a 1 mmol scale leading to a comparable yield of 94 % (eq 5).
Table 1.
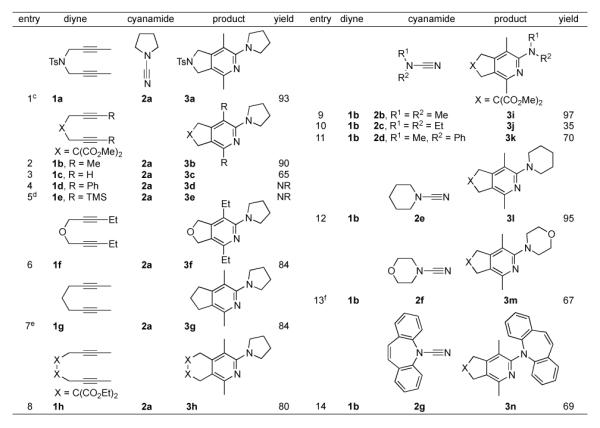
|
5 mol % FeCl2, 10 mol % L2, 10 mol % Zn dust, diyne (0.4 M), cyanamide (0.48 M), benzene, 70 °C.
3 h slow addition of diyne unless otherwise noted.
TS = p-toluenesulfonyl.
TMS = trimethylsilyl.
5 h slow addition of substrate.
10 mol % FeCl2, 20 mol % L2, 20 mol % Zn dust.
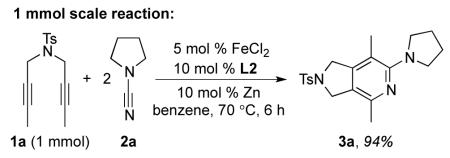 |
(5) |
For future synthetic application, an understanding of regioselectivity of unsymmetrical coupling partners is paramount. Diynes 1i-k were evaluated under our optimized conditions and the results are summarized in Table 2. In each of these cases, good yields of 2-aminopyridine products were obtained. What is surprising, however, is that each of the reactions shows a strong preference to place the larger substituent proximal to the pyridine nitrogen. For example, the cycloaddition of H, Me substituted diyne 1i and cyanamide 2b provided an 85:15 ratio of 3o and 3o’ in 72% combined yield (entry 1). A similar product ratio was obtained in the cycloaddition of a bulkier diyne (1j, entry 2) as well as aryl/alkyl diyne (1k, entry 3). These regioselectivity trends provide an alternative to nickel-catalyzed systems where the larger substituent is placed at the 3-position of the 2-aminopyridine ring.7 Additionally, this Fe-catalyzed method is milder than the cobalt-catalyzed system which is primarily limited to terminal alkynes.17c Notably, the regioselectivity is the reverse of that observed in the previous Fe-catalyzed system involving diynes and nitriles wherein the larger alkyne substituent was placed ortho to the nitrile substituent.5 To verify that this trend is a result of the catalyst and not the type of nitrile, we reacted diyne 3q with dimethyl cyanamide 2b under the catalyst developed by Wan and co-workers (eq 6). The FeI2/dppp system provided the product in a 63% combined yield but with 3q’ as the major regioisomer in a 10:90 (3q, 3q’) ratio, the opposite selectivity of our system.
Table 2.
Unsymmetrical Diynes
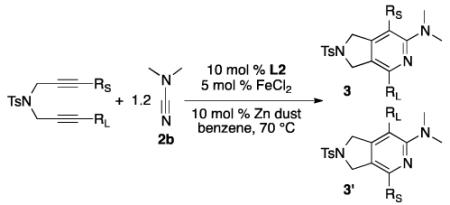
| entry | substrate | product | yielda(3:3′)b |
|---|---|---|---|
| 1 | 1i, RS = H, RL = Me | 3o | 72(85:15) |
| 2 | 1j, RS = Me, RL = tBu | 3p | 80(86:14) |
| 3 | 1k, RS = Me, RL = Ph | 3q | 67(88:12) |
Yields reported as a combination of both regiosiomers.
Product ratios determinted by 1H NAR
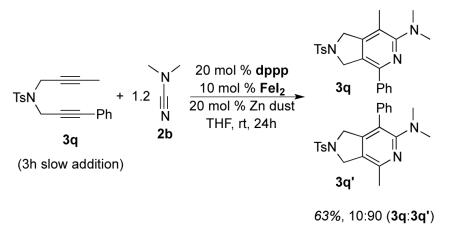 |
(6) |
The fully intermolecular [2+2+2] cycloaddition of alkynes and nitriles to form pyridines is a challenging reaction. To date, intermolecular reactions with cyanamides are limited to cobalt catalysts, and these systems provide a mixture of products when using unsymmetrical internal alkynes.22 Furthermore, no reports of [2+2+2] cycloadditions involving aryl acetylenes and cyanamides exist. Terminal aryl acetylenes are particularly challenging substrates since these tend to undergo rapid oligomerization to provide unwanted side products.1,23 Despite this potential pitfall, as well as the possibility of forming multiple regioisomers, we found the reaction of two equivalents of alkyne 4a and one equivalent of cyanamide 2b afforded 5a as a single regioisomer in 90 % yield (eq 4). This mild and efficient route to 2-amino-4,6-aryl pyridines may provide an alternative to multi-step syntheses of biologically active compounds24 and is the topic of further investigation in our lab.
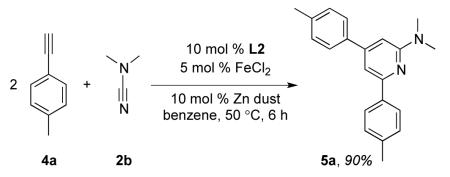 |
(7) |
The contrast in regioselectivity between our FeCl2/L2 system and the FeI2/dppp system is the first example of ligand-dependent regioselectivity in metal-catalyzed [2+2+2] pyridine formation. The regioselectivity observed in our Fe system mirrors that observed in Co-catalyzed protocols. As such, this suggests that the L2/Fe-catalyzed cycloaddition of cyanamides follows a similar mechanism to that of the well-established mechanism for Co-catalyzed pyridine formation.25 Following in-situ ligand coordination and reduction by zinc, a reduced Fe catalyst binds the diyne and facilitates oxidative coupling of the two alkyne units to form a ferracyclopentadiene (Scheme 2). Insertion of cyanamide and reductive elimination subsequently afford the pyridine product. Interestingly, the FeI2/dppp catalyst system, which follows the regioselectivity patterns of nickel, has been proposed to undergo a mechanism involving initial oxidative coupling between an alkyne and a nitrile instead.5 This is in agreement with the proposed mechanism for nickel-catalyzed pyridine formation.26 The regioselectivity results of this study, combined with what is known about cobalt and nickel systems, may indicate that iron is capable of following either mechanistic pathway and is dependent on ligand choice. Studies to gain further insight into the mechanistic difference between these two Fe based systems are currently underway.
Conclusions
The iron catalyzed [2+2+2] cycloaddition of alkynes and cyanamides is a high yielding, atom efficient and regioselective method to obtain 2-aminopyridines. Iron catalyzed pyridine synthesis is no longer limited by alkynenitrile substrates or large excesses of nitrile. We are continuing our efforts to expand this chemistry to other nitrile substrates.
Experimental
General Experimental
All reactions were conducted under an atmosphere of N2 using standard Schlenk techniques or in a N2 filled glove-box unless otherwise noted. Benzene was dried over neutral alumina under N2 using a Grubbs type solvent purification system. Iron Chloride (99.95% purity) was purchased from Alfa Aesar. Diynes 1a,27 1b-c,28 1d,29 1e,30 1f,31 1g,32 1h,33 1i,34 and 1j-k35 were prepared from known literature procedures. Liquid cyanamides were degassed using three sequential freeze-pump-thaw cycles. Slow addition of diyne was performed using a syringe pump 22 from a disposable 1 mL syringe with a 6 inch stainless steel 24 gauge needle. The needle was dried overnight at 160 °C and was fitted to the syringe while hot. The syringe/needle joint was then wrapped tightly with Teflon tape.
1H and 13C Nuclear Magnetic Resonance spectra of pure compounds were acquired at 400 and 100 MHz, unless otherwise noted. All spectra are referenced to a singlet at 7.27 ppm for 1H and to the center line of a triplet at 77.23 ppm for 13C. The abbreviations s, d, dd, dt, dq, td, t, q, and quint stand for singlet, doublet, doublet of doublets, doublet of triplets, doublet of quartets, triplet of doublets, triplet, quartet, and quintet, respectively. All 13C NMR spectra were proton decoupled. Gas Chromatography was performed using the following conditions: initial oven temperature: 100 °C; temperature ramp rate 10 °C/min.; final temperature: 300 °C held for 12 minutes; detector temperature: 250 °C.
General Procedure for the Cycloaddition
In a nitrogen filled glove box, 5 mol % FeCl2, 10 mol % L2 and benzene was added to a vial. The mixture was stirred for 10 to 15 minutes at which time 1.2 equivalents of cyanamide 2 (2.7 M in benzene) and 10 mol % Zn dust was added. The vial was then capped with a Teflon lined septum screw cap and removed from the glove box. The vial was stirred in a 70 °C oil bath and a solution of diyne 1 (0.49 M in benzene) was slowly added to the vial over 3 hours (unless otherwise noted) via syringe pump. (The final concentration of the diyne after addition was 0.4 M.)
Purification Procedure A
For 2-aminopyridines with an Rf > 0.3 (20 % ethyl acetate in hexanes) L2 (Rf = 0.51, 20% ethyl acetate in hexanes) may co-elute with and contaminate the final product. This method was devised to avoid this issue. Once the reaction was complete, as determined by GC, the crude mixture was stirred in aqueous HCl for 10 minutes to protonate the 2-aminopyridine product. The aqueous layer was collected and the organic layer was further extracted with 4 × 15 mL portions of aqueous HCl. The aqueous extracts were collected then a saturated aqueous NaHCO3 solution was carefully added until the pH >7 causing the product to precipitate. The aqueous layer was then extracted with 3 × 25 mL portions of diethyl ether. The organic extracts were collected, dried over Na2SO4, filtered and the solvent was removed in vacuo. The crude product was then purified via silica gel flash chromatography.
Purification Procedure B
For 2-aminopyridines with Rf < 0.3 (20 % ethyl acetate in hexanes). Once the reaction was complete, as determined by GC, the crude mixture was purified via flash silica gel chromatography. The product often contained unreacted cyanamide at this point so the product was stirred in aqueous HCl for 10 minutes. The aqueous layer was collected and the organic layer was further extracted with 4 × 15 mL portions of aqueous HCl. The aqueous extracts were collected then a saturated aqueous NaHCO3 solution was carefully added until the pH >7 causing the product to precipitate. The aqueous layer was then extracted with 3 × 25 mL portions of diethyl ether. The organic extracts were collected, dried over Na2SO4, filtered and the solvent was removed in vacuo.
Synthesis of 4,7-dimethyl-6-(pyrrolidin-1-yl)-2-tosyl-2,3-dihydro-1H-pyrrolo[3,4-c]pyridine (3a)
Compound 3a was prepared using the general procedure with 2a (42 mg, 4.4 × 10−1 mmol), FeCl2 (2.3 mg, 1.8 × 10−2 mmol), L2 (13.4 mg, 3.6 × 10−2 mmol), and zinc (2.4 mg, 3.6 × 10−2 mmol) in 418 μL of benzene. Diyne 1a (100 mg, 3.6 × 10−1 mmol), dissolved in 490 μL benzene was added over 3 h at 70 °C. The reaction was stirred an additional 1 h for a total reaction time of 4 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 10 % ethyl acetate in hexanes followed by an acid/base extraction with 3M HCl to yield 3a (126 mg, 93 %) as a white solid. Rf = 0.19 (20 % ethyl acetate in hexanes). 1H and 13C NMR spectral data was compared with known literature values.7a
Synthesis of dimethyl 1,4-dimethyl-3-(pyrrolidin-1-yl)-5H-cyclopenta[c]pyridine-6,6-(7H)-dicarboxylate (3b)
Compound 3b was prepared using the general procedure with 2a (49 mg, 5.1 × 10−1 mmol), FeCl2 (2.7 mg, 2.1 × 10−2 mmol), L2 (16 mg, 4.2 × 10−2 mmol), and zinc (2.8 mg, 4.2 × 10−2 mmol) in 560 μL of benzene. Diyne 3b (100 mg, 4.2 × 10−1 mmol), dissolved in 498 μL benzene, was added over 3 h at 70 °C. The reaction mixture was stirred for an additional 3 h for a total reaction time of 6 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 20 % ethyl acetate in hexanes followed by an acid/base extraction with 1M HCl to yield 3b (127 mg, 90 %) as a yellowish solid. Rf = 0.27 (20 % ethyl acetate in hexanes). 1H and 13C NMR spectral data was compared with known literature values.7a
Synthesis of dimethyl 3-(pyrrolidin-1-yl)-5H-cyclopenta[c]pyridine-6,6-(7H)-dicarboxylate (3c)
Compound 3c was prepared using the general procedure with 2a (55 mg, 5.7 × 10−1 mmol), FeCl2 (3.0 mg, 2.4 × 10−2 mmol), L2 (18 mg, 4.8 × 10−2 mmol), and zinc (3.1 mg, 4.8 × 10−2 mmol) in 194 μL of benzene. Diyne 1c (99 mg, 4.8 × 10−1 mmol), dissolved in 1 mL benzene, was added over 3 h at 70 °C. The reaction was stirred at 70 °C for an additional 15h for a total reaction time of 18 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 2 % methanol in dichloromethane followed by an acid/base extraction with 1M HCl to yield 3c (94 mg, 65 %) as a white solid. Rf = 0.01 (20 % ethyl acetate in hexanes). 1H and 13C NMR spectral data was compared with known literature values.7a
Synthesis of 4,7-diethyl-6-(pyrrolidin-1-yl)-1,3-dihydrofuro[3,4-c]pyridine (3f)
Compound 3f was prepared using the general procedure with 2a (36 mg, 3.8 × 10−1 mmol), FeCl2 (2.0 mg, 1.6 × 10−2 mmol), L2 (12 mg, 3.2 × 10−2 mmol), and zinc (2.1 mg, 3.2 × 10−2 mmol) in 292 μL of benzene. Diyne 1f (47 mg, 3.2 × 10−1 mmol), dissolved in 497 μL benzene, was added over 5 h at 70 °C. The reaction was stirred an additional 1 h for a total reaction time of 6 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure A with an acid/base extraction using 1M HCl then silica gel flash chromatography using 5 % ethyl acetate in hexanes (500 mL), then 10 % ethyl acetate in hexanes (500 mL) to yield 3f (65 mg, 84 %) as a yellow oil. Rf = 0.52 (20 % ethyl acetate in hexanes). 1H (300 MHz, CDCl3): δ (ppm) 1.14 (t, J = 7.7 Hz, 3H), 1.23 (t, J = 7.5 Hz, 3H), 1.93 (quint, J = 3.3 Hz, 4H), 2.51-2.60 (m, 4H), 3.52 (quint, J = 3.2 Hz, 4H), 5.05 (s, 4H). 13C (75 MHz, CDCl3) δ (ppm) 158.0, 150.3, 149.8, 122.9, 116.2, 72.8, 72.4, 50.3, 29.2, 25.8, 23.0, 13.8, 12.5. IR (cm−1): 2966, 2932, 2869, 1761, 1600, 1428, 1381, 1343, 1311, 1228, 1143, 1053, 906, 783. HRMS (ESI) m/z calcd. for C15H23N2O [M+H]+ 247.1810, found 247.1814.
Synthesis of 1,4-dimethyl-3-(pyrrolidin-1-yl)-6,7-dihydro-5H-cyclopenta[c]pyridine (3g)
Compound 3g was prepared using the general procedure with 2a (55 mg, 5.7 × 10−1 mmol), FeCl2 (3.0 mg, 2.4 × 10−2 mmol), L2 (18 mg, 4.8 × 10−2 mmol), and zinc (3.1 mg, 4.8 × 10−2 mmol) in 429 μL of benzene. Diyne 1g (57 mg, 4.8 × 10−1 mmol), dissolved in 765 μL benzene, was added over 5 h at 70 °C. The reaction was stirred an additional 2 h for a total reaction time of 7 hours. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure A with an acid/base extraction using 1M HCl then silica gel flash chromatography using 5 % ethyl acetate in hexanes (500 mL), then 10 % ethyl acetate in hexanes (500 ml) to yield 3g (86 mg, 84 %) as a yellow oil. Rf = 0.49 (20 % ethyl acetate in hexanes). 1H (300 MHz, CDCl3): δ (ppm) 1.89-1.92 (m, 4H), 2.06, (quint, J = 7.4 Hz, 2H), 2.17 (s, 4H), 2.33 (s, 3H), 2.80 (t, J = 7.5 Hz, 4H), 3.44 (t, J = 6.5 Hz, 4H). 13C (75 MHz, CDCl3) δ (ppm) 154.5, 147.4, 129.1, 129.0, 114.3, 50.4, 32.3, 30.6, 25.6, 24.8, 22.0, 16.0. IR (cm−1): 2953, 2868, 1595, 1425, 1347, 1204, 1137, 1063, 938, 858, 756. HRMS (ESI) m/z calcd. for C14H21N2 [M+H]+ 217.1705, found 217.1709.
Synthesis of tetraethyl-1,4-dimethyl-3-(pyrrolidin-1-yl)isoquinoline-6,6,7,7(5H,8H)-tetracarboxylate (3h)
Compound 3h was prepared using the general procedure with 2a (18 mg, 1.9 × 10−1 mmol), FeCl2 (1.0 mg, 0.8 × 10−2 mmol), L2 (5.8 mg, 1.5 × 10−2 mmol), and zinc (1.0 mg, 1.5 × 10−2 mmol) in 100 μL of benzene. Diyne 1h (67 mg, 1.6 × 10−1 mmol) dissolved in 300 μL benzene was added over 3 h at 70 °C. The reaction mixture was stirred an additional 3 h for a total reaction time of 6 h. After the reaction was complete (reaction monitored by GC), the crude product was isolated with only silica gel flash chromatography (no acid/base extraction) using 10 % ethyl acetate in hexanes (200 mL), then 15 % ethyl acetate in hexanes (400 mL) to yield 3h (65 mg, 80 %) as a yellow oil. Rf = 0.19 (20 % ethyl acetate in hexanes). 1H (400 MHz, CDCl3): δ (ppm) 1.23 (td, J1 = 3.2, J2 = 3.2, J3 = 3.2 Hz, 12H), 1.86-1.92 (m, 4H), 2.12 (s, 3H), 2.34 (s, 3H), 3.30 (s, 2H), 3.36 (m, 6H), 4.13-4.25 (m, 8H). 13C (100 MHz, CDCl3) δ (ppm) 170.2, 170.1, 157.9, 150.2, 141.7, 118.2, 115.8, 62.2, 62.0, 61.9, 57.4, 57.1, 50.4, 33.1, 31.8, 25.5, 22.3, 14.8, 13.9. IR (cm−1): 2981, 2937, 2871, 2361, 1735, 1571, 1429, 1367, 1326, 1270, 1241, 1202, 1096, 1052, 942, 864, 784, 703, 650, 614, 580. HRMS (ESI) m/z calcd. for C28H27N2O4 455.1971, found 455.1969.
Synthesis of dimethyl 3-(dimethylamino)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (3i)
Compound 3i was prepared using the general procedure B with 2b (20 mg, 2.8 × 10−1 mmol), FeCl2 (1.5 mg, 1.2 × 10−2 mmol), L2 (8.8 mg, 2.4 × 10−2 mmol), and zinc (1.6 mg, 2.4 × 10−2 mmol) in 103 μL of benzene. Diyne 1b (56 mg, 2.4 × 10−1 mmol) dissolved in 489 μL benzene was added over 3 h at 70 °C. The reaction mixture was stirred an additional 2 h for a total reaction time of 5 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 5 % ethyl acetate in hexanes (500 mL), then 10 % ethyl acetate in hexanes (500 mL), then 20 % ethyl acetate in hexanes (500 mL) followed by an acid/base extraction with 1M HCl to yield 3i (71 mg, 97 %) as a yellow oil. Rf = 0.27. 1H and 13C NMR spectral data was compared with known literature values.7a
Synthesis of dimethyl-3-(diethylamino)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (3j)
Compound 3j was prepared using the general procedure with 2c (22 mg, 2.8 × 10−1 mmol), FeCl2 (1.5 mg, 1.2 × 10−2 mmol), L2 (8.8 mg, 2.4 × 10−2 mmol), and zinc (1.6 mg, 2.4 × 10−2 mmol) in 103 μL of benzene. Diyne 1b (56 mg, 2.4 × 10−1 mmol) dissolved in 489 μL benzene was added over 3 h at 70 °C. The reaction mixture was stirred an additional 2 h for a total reaction time of 5 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 10 % ethyl acetate in hexanes followed by an acid/base extraction with 1M HCl to yield 3j (28 mg, 35 %) as a yellow oil. Rf = 0.27 (20 % ethyl acetate in hexanes). 1H and 13C NMR spectral data was compared with known literature values.7a
Synthesis of dimethyl 1,4-dimethyl-3-(methyl(phenyl)amino)-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (3k)
Compound 3k was prepared using the general procedure with 2d (22 mg, 1.8 × 10−1 mmol), FeCl2 (1.0 mg, 7.5 × 10−2 mmol), L2 (5.5 mg, 1.5 × 10−2 mmol), and zinc (1.0 mg, 1.5 × 10−2 mmol) in 65 μL of benzene. Diyne 1b (35 mg, 1.5 × 10−1 mmol) dissolved in 310 μL benzene was added over 3 h at 70 °C. The reaction mixture was stirred an additional 2 hours for a total reaction time of 5 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 5 % ethyl acetate in hexanes (250 mL), then 10 % ethyl acetate in hexanes (250 mL), 20 % ethyl acetate in hexanes (500 mL) followed by an acid/base extraction with 1M HCl to yield 3k (39 mg, 70 %) as a yellowish oil. Rf = 0.16 (20 % ethyl acetate in hexanes). 1H and 13C NMR spectral data was compared with known literature values.7a
Synthesis of dimethyl-1,4-dimethyl-3-(piperidin-1-yl)-5H-cyclopenta[c]-pyridine-6,6-(7H)-dicarboxylate (3l)
Compound 3l was prepared using the general procedure with 2e (31 mg, 2.8 × 10−1 mmol), FeCl2 (1.5 mg, 1.2 × 10−2 mmol), L2 (8.8 mg, 2.4 × 10−2 mmol), and zinc (1.6 mg, 2.4 × 10−2 mmol) in 121 μL of benzene. Diyne 1b (56 mg, 2.4 × 10−1 mmol) dissolved in 471 μL benzene was added over 3 h at 70 °C. The reaction mixture was stirred an additional 2 hours for a total reaction time of 5 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 5 % ethyl acetate in hexanes (250 mL), then 10 % ethyl acetate in hexanes (250 mL), and 20 % ethyl acetate and hexanes (250 mL) followed by an acid/base extraction with 1M HCl to yield 3l (78 mg, 97 %) as a yellowish oil. Rf = 0.27 (20 % ethyl acetate in hexanes). 1H (400 MHz, CDCl3): δ (ppm) 1.58 (q, J = 7.2 Hz, 2H) 1.68 (quint, J = 6 Hz, 4H), 2.14 (s, 3H), 2.33 (s, 3H), 3.0 (t, J = 4.8 Hz, 4H), 3.48 (s, 2H), 3.50 (s, 2H), 3.77 (s, 6H). 13C (100 MHz, CDCl3) δ (ppm) 172.3, 161.8, 150.2, 148.3, 127.6, 118.3, 59.7, 53.3, 51.7, 40.1, 38.9, 26.6, 24.9, 21.9, 14.5. IR (cm−1): 2930, 2851, 1738, 1586, 1432, 1371, 1266, 1199, 1163, 1114, 1062, 1028, 963, 858, 610. HRMS (ESI) m/z calcd. for C19H27N2O4 [M+H]+ 347.1971, found 347.1980.
Synthesis of dimethyl-1,4-dimethyl-3-morpholino-5H-cyclopenta[c]pyridine-6,6-(7H)-dicarboxylate (3m)
Compound 3m was prepared using the general procedure with 2f (48 mg, 4.7 × 10−1 mmol), FeCl2 (2.7 mg, 2.1 × 10−2 mmol), L2 (16 mg, 4.2 × 10−2 mmol), and zinc (2.8 mg, 4.2 × 10−2 mmol) in 200 μL of benzene. Diyne 2b (50 mg, 2.1 × 10−1 mmol) dissolved in 400 μL benzene was added over 3 h at 70 °C. The reaction mixture was stirred an additional 2 h for a total reaction time of 5 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure A with an acid/base extraction using 1M HCl then silica gel flash chromatography using 20 % ethyl acetate in hexanes (200 mL), then 30 % ethyl acetate in hexanes (400 mL) to yield 3m (49 mg, 67 %) as a yellowish oil. Rf = 0.38 (20 % ethyl acetate in hexanes). 1H and 13C NMR spectral data was compared with known literature values.7a
Synthesis of dimethyl-3-(5H-dibenzo[b,f]azepin-5-yl)-1,4-dimethyl-5H-cyclopenta[c]pyridine-6,6(7H)-dicarboxylate (3n)
Compound 3n was prepared using the general procedure with 2g (83 mg, 4.7 × 10−1 mmol), FeCl2 (2.68 mg, 2.1 × 10−2 mmol), L2 (15.6 mg, 4.2 × 10−2 mmol), and zinc (2.8 mg, 4.2 × 10−2 mmol) in 121 μL of benzene. Diyne 1b (50 mg, 2.1 × 10−1 mmol) dissolved in 471 μL benzene was added over 3 h at 70 °C. The reaction mixture was stirred an additional 19 h for a total reaction time of 22 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 10 % ethyl acetate in hexanes followed by an acid/base extraction with 1M HCl to yield 3n (133 mg, 69 %) as a red oil. Rf = 0.29 (20 % ethyl acetate in hexanes). 1H (400 MHz, CDCl3): δ (ppm) 1.78 (s, 3H), 2.44 (s, 3H), 3.44 (s, 2H), 3.54 (s, 2H), 3.75 (s, 6H), 6.85 (s, 2H), 7.07 (dt, J = 7.2 Hz, 0.8 Hz, 2H), 7.16 (dd, J = 8 Hz, 1.6 Hz, 2H), 7.22 (dd, J = 8 Hz, 1.6 Hz, 2H), 7.71 (dd, J = 8 Hz, 0.8 Hz, 2H). 13C (100 MHz, CDCl3) δ (ppm) 172.2, 156.1, 151.7, 148.4, 148.1, 135.1, 132.4, 129.6, 128.8, 127.3, 124.7, 120.4, 59.8, 53.3, 40.3, 39.0, 22.1, 14.9. IR (cm−1): 3020, 2953, 2923, 2854, 2361, 1737, 1589, 1482, 1432, 1340, 1266, 1200, 1163, 1113, 1060, 950, 922, 865, 794, 767, 735, 662, 559. HRMS (ESI) m/z calcd. for C28H27N2O4 [M+H]+ 455.1971, found 455.1982.
Synthesis of N,N,4-trimethyl-2-tosyl-2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6-amine (3o)
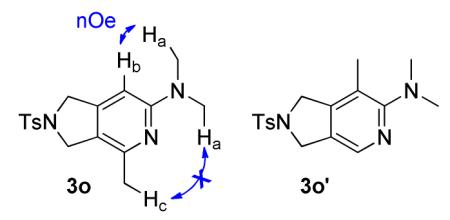 Compounds 3o and 3o’ were prepared using the general procedure with 2b (13 mg, 1.9 × 10−1 mmol), FeCl2 (1.0 mg, 7.9 × 10−
2 mmol), L2 (5.8 mg, 1.6 × 10−2 mmol), and zinc (1.0 mg, 1.6 × 10−2 mmol) in 93 μL of benzene. Diyne 1i (41 mg, 1.6 × 10−1 mmol), dissolved in 303 μL benzene was added over 3 h at 70 °C. The reaction was stirred an additional 5 h for a total reaction time of 8 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 20 % ethyl acetate in hexanes followed by an acid/base extraction with 3M HCl to yield 3o and 3o’ (38 mg, 72 %, 85:15) as a white solid. Rf = 0.11 (20 % ethyl acetate in hexanes). 3o: 1H (400 MHz, CDCl3): δ (ppm) 2.26 (s, 3H), 2.42 (s, 3H), 3.02 (s, 6H), 4.45 (s, 2H), 4.51 (s, 2H), 6.12 (s, 1H), 7.32 (d, J = 8, 2H), 7.77 (d, J = 8, 2H). 13C (100 MHz, CDCl3) δ (ppm) 159.4, 150.6, 147.2, 143.9, 134.0, 130.0, 118.2, 96.4, 53.9, 51.9, 38.4, 22.3, 21.7. MP 176-178 °C. 2-D NOESY (500 MHz, CDCl3): Hb (6.12 ppm) correlates to Ha (4.51 ppm). Hc (2.56 ppm) shows no correlation to Ha. IR (cm−1): 2918, 2849, 1614, 1579, 1503, 1408, 1342, 1309, 1159, 1099, 815, 669, 579, 543. HRMS (ESI) m/z calcd. for C17H22N3O2S [M+H]+ 332.1433, found 332.1433.
Compounds 3o and 3o’ were prepared using the general procedure with 2b (13 mg, 1.9 × 10−1 mmol), FeCl2 (1.0 mg, 7.9 × 10−
2 mmol), L2 (5.8 mg, 1.6 × 10−2 mmol), and zinc (1.0 mg, 1.6 × 10−2 mmol) in 93 μL of benzene. Diyne 1i (41 mg, 1.6 × 10−1 mmol), dissolved in 303 μL benzene was added over 3 h at 70 °C. The reaction was stirred an additional 5 h for a total reaction time of 8 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 20 % ethyl acetate in hexanes followed by an acid/base extraction with 3M HCl to yield 3o and 3o’ (38 mg, 72 %, 85:15) as a white solid. Rf = 0.11 (20 % ethyl acetate in hexanes). 3o: 1H (400 MHz, CDCl3): δ (ppm) 2.26 (s, 3H), 2.42 (s, 3H), 3.02 (s, 6H), 4.45 (s, 2H), 4.51 (s, 2H), 6.12 (s, 1H), 7.32 (d, J = 8, 2H), 7.77 (d, J = 8, 2H). 13C (100 MHz, CDCl3) δ (ppm) 159.4, 150.6, 147.2, 143.9, 134.0, 130.0, 118.2, 96.4, 53.9, 51.9, 38.4, 22.3, 21.7. MP 176-178 °C. 2-D NOESY (500 MHz, CDCl3): Hb (6.12 ppm) correlates to Ha (4.51 ppm). Hc (2.56 ppm) shows no correlation to Ha. IR (cm−1): 2918, 2849, 1614, 1579, 1503, 1408, 1342, 1309, 1159, 1099, 815, 669, 579, 543. HRMS (ESI) m/z calcd. for C17H22N3O2S [M+H]+ 332.1433, found 332.1433.
Synthesis of 4-(tert-butyl)-N,N,7-trimethyl-2-tosyl-2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6-amine (3p)
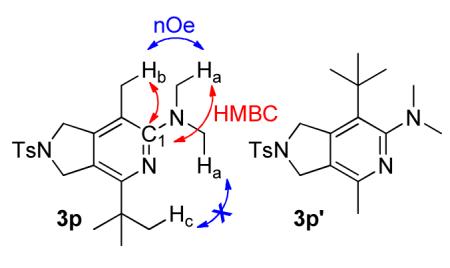 Compounds 3p and 3p’ were prepared using the general procedure with 2b (13 mg, 1.9 × 10−1 mmol), FeCl2 (1.0 mg, 7.9 × 10−2 mmol), L2 (5.8 mg, 1.6 × 10−2 mmol), and zinc (1.0 mg, 1.6 × 10−2 mmol) in 59 μL of benzene. Diyne 1j (50 mg, 1.6 × 10−1 mmol), dissolved in 336 μL benzene was added over 3 h at 70 °C. The reaction was stirred an additional 3 h for a total reaction time of 6 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 10 % ethyl acetate in hexanes followed by an acid/base extraction with 3M HCl to yield 3p and 3p’ (49 mg, 80 %, 88:12) as a white solid. Rf = 0.27 (20 % ethyl acetate in hexanes). 3p: 1H (500 MHz, CDCl3): δ (ppm) 1.29 (s, 9H), 2.10 (s, 3H), 2.42 (s, 3H), 2.79 (s, 6H), 4.24 (s, 2H), 2.74 (s, 2H), 7.34 (d, J = 8 Hz, 2H), 7.80 (d, J = 8 Hz, 2H). 13C (125 MHz, CDCl3) δ (ppm) 160.2, 157.6, 148.0, 143.9, 134.0, 123.1, 127.8, 121.0, 114.6, 53.6, 52.3, 42.3, 38.6, 29.5, 21.7, 14.9. MP 101-102 °C. 2-D NOESY (800 MHz, CDCl3): Ha (2.84 ppm) correlates to Hb (2.09 ppm). Hc (1.28 ppm) does not correlate to Ha. HMBC (800 MHz, CDCl3): C (161.1 ppm) couples with Ha (2.84 ppm) and Hb (2.09 ppm). IR (cm−1): 3633, 2954, 2866, 2792, 2361, 1726, 1599, 1566, 1478, 1453, 1416, 1392, 1351, 1315, 1251, 1204, 1165, 1099, 1066, 955, 930, 816, 772, 737, 711, 665, 607, 570, 548, 513. HRMS (ESI) m/z calcd. for C21H30N3O2S [M+H]+ 388.2059, found 388.2069.
Compounds 3p and 3p’ were prepared using the general procedure with 2b (13 mg, 1.9 × 10−1 mmol), FeCl2 (1.0 mg, 7.9 × 10−2 mmol), L2 (5.8 mg, 1.6 × 10−2 mmol), and zinc (1.0 mg, 1.6 × 10−2 mmol) in 59 μL of benzene. Diyne 1j (50 mg, 1.6 × 10−1 mmol), dissolved in 336 μL benzene was added over 3 h at 70 °C. The reaction was stirred an additional 3 h for a total reaction time of 6 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 10 % ethyl acetate in hexanes followed by an acid/base extraction with 3M HCl to yield 3p and 3p’ (49 mg, 80 %, 88:12) as a white solid. Rf = 0.27 (20 % ethyl acetate in hexanes). 3p: 1H (500 MHz, CDCl3): δ (ppm) 1.29 (s, 9H), 2.10 (s, 3H), 2.42 (s, 3H), 2.79 (s, 6H), 4.24 (s, 2H), 2.74 (s, 2H), 7.34 (d, J = 8 Hz, 2H), 7.80 (d, J = 8 Hz, 2H). 13C (125 MHz, CDCl3) δ (ppm) 160.2, 157.6, 148.0, 143.9, 134.0, 123.1, 127.8, 121.0, 114.6, 53.6, 52.3, 42.3, 38.6, 29.5, 21.7, 14.9. MP 101-102 °C. 2-D NOESY (800 MHz, CDCl3): Ha (2.84 ppm) correlates to Hb (2.09 ppm). Hc (1.28 ppm) does not correlate to Ha. HMBC (800 MHz, CDCl3): C (161.1 ppm) couples with Ha (2.84 ppm) and Hb (2.09 ppm). IR (cm−1): 3633, 2954, 2866, 2792, 2361, 1726, 1599, 1566, 1478, 1453, 1416, 1392, 1351, 1315, 1251, 1204, 1165, 1099, 1066, 955, 930, 816, 772, 737, 711, 665, 607, 570, 548, 513. HRMS (ESI) m/z calcd. for C21H30N3O2S [M+H]+ 388.2059, found 388.2069.
Synthesis of N,N,7-trimethyl-4-phenyl-2-tosyl-2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6-amine (3q)
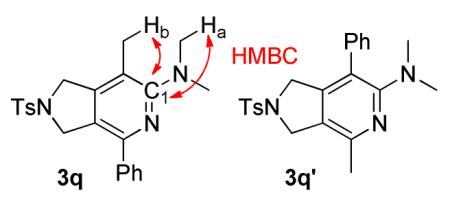 Compounds 3q and 3q’ were prepared using the general procedure with 2b (13 mg, 1.9 × 10−1 mmol), FeCl2 (1.0 mg, 7.9 × 10−2 mmol), L2 (5.8 mg, 1.6 × 10−2 mmol), and zinc (1.0 mg, 1.6 × 10−2 mmol) in 93 μL of benzene. Diyne 1k (53 mg, 1.6 × 10−1 mmol), dissolved in 302 μL benzene was added over 3 h at 70 °C. The reaction was stirred an additional 3 h for a total reaction time of 6 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 10 % ethyl acetate in hexanes followed by an acid/base extraction with 3M HCl to yield 3q and 3q’ (43 mg, 67 %, 86:14) as a white solid. Rf = 0.14 (20 % ethyl acetate in hexanes). 3q: 1H (400 MHz, CDCl3): δ (ppm) 2.18 (s, 3H), 2.42 (s, 3H), 2.87 (s, 6H), 4.53 (s, 2H), 4.82 (s, 2H), 7.28-7.47 (m, 5H), 7.76 (dd, J = 6.5, 7.5 Hz, 4H). 13C (125 MHz, CDCl3) δ (ppm) 161.9, 148.2, 147.1, 144.0, 139.4, 134.0, 130.1, 128.8, 128.7, 127.9, 127.8, 122.5, 116.9, 53.7, 52.9, 42.2, 21.7, 15.3. HMBC (800 MHz, CDCl3): C1 (165.2 ppm) couples with Ha(2.86 ppm) and Hb (2.17 ppm). MP 176-177 °C. IR (cm−1): 2924, 2854, 2361, 1733, 1595, 1491, 1458, 1400, 1349, 1161, 1098, 1065, 913, 816, 753, 703, 673, 614, 581, 548. HRMS (ESI) m/z calcd. for C23H26N3O2S [M+H]+ 408.1746, found 408.1752. A crystal of 3q’ suitable for x-ray analysis was grown by slow evaporation from an ether solution (see supporting information).
Compounds 3q and 3q’ were prepared using the general procedure with 2b (13 mg, 1.9 × 10−1 mmol), FeCl2 (1.0 mg, 7.9 × 10−2 mmol), L2 (5.8 mg, 1.6 × 10−2 mmol), and zinc (1.0 mg, 1.6 × 10−2 mmol) in 93 μL of benzene. Diyne 1k (53 mg, 1.6 × 10−1 mmol), dissolved in 302 μL benzene was added over 3 h at 70 °C. The reaction was stirred an additional 3 h for a total reaction time of 6 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure B with silica gel flash chromatography using 10 % ethyl acetate in hexanes followed by an acid/base extraction with 3M HCl to yield 3q and 3q’ (43 mg, 67 %, 86:14) as a white solid. Rf = 0.14 (20 % ethyl acetate in hexanes). 3q: 1H (400 MHz, CDCl3): δ (ppm) 2.18 (s, 3H), 2.42 (s, 3H), 2.87 (s, 6H), 4.53 (s, 2H), 4.82 (s, 2H), 7.28-7.47 (m, 5H), 7.76 (dd, J = 6.5, 7.5 Hz, 4H). 13C (125 MHz, CDCl3) δ (ppm) 161.9, 148.2, 147.1, 144.0, 139.4, 134.0, 130.1, 128.8, 128.7, 127.9, 127.8, 122.5, 116.9, 53.7, 52.9, 42.2, 21.7, 15.3. HMBC (800 MHz, CDCl3): C1 (165.2 ppm) couples with Ha(2.86 ppm) and Hb (2.17 ppm). MP 176-177 °C. IR (cm−1): 2924, 2854, 2361, 1733, 1595, 1491, 1458, 1400, 1349, 1161, 1098, 1065, 913, 816, 753, 703, 673, 614, 581, 548. HRMS (ESI) m/z calcd. for C23H26N3O2S [M+H]+ 408.1746, found 408.1752. A crystal of 3q’ suitable for x-ray analysis was grown by slow evaporation from an ether solution (see supporting information).
Synthesis of N,N-dimethyl-4,6-di-p-tolylpyridin-2-amine (5a)
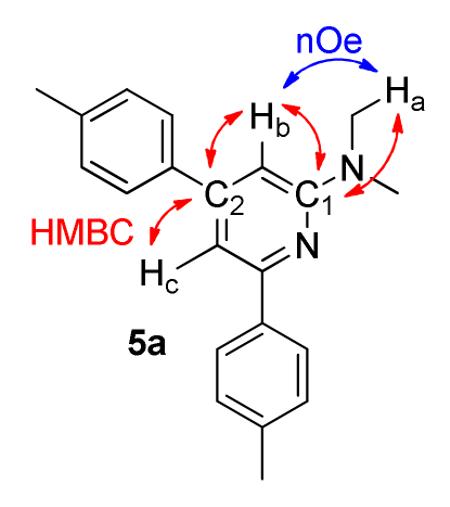 Compound 5a was prepared using the general procedure with 2b (22 mg, 3.2 × 10−1 mmol), FeCl2 (2.0 mg, 1.6 × 10−2 mmol), L2 (11.7 mg, 3.2 × 10−2 mmol), and zinc (2.1 mg, 3.2 × 10−2 mmol) in 108 μL of benzene. 4-ethynyltoluene (73 mg, 6.3 × 10−
1mmol), dissolved in 287 μL benzene was added over 3 h at 50 °C. The reaction was stirred an additional 2 h for a total reaction time of 5 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure A with an acid/base extraction using 1M HCl then silica gel flash chromatography using 5 % ethyl acetate in hexanes (500 mL to yield 5a (86 mg, 90 %) as a white solid. Rf = 0.57 (20 % ethyl acetate in hexanes). 1H-NMR (500 MHz, CD2Cl2) δ (ppm) 2.41 (d, J = 4.0 Hz, 6H), 3.20 (s, 6H), 6.67 (s, 1H), 7.24 (s, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 7.5 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H), 8.0 (d, J = 7.0 Hz, 2H). 13C (125 MHz, CDCl3) δ (ppm) 159.9, 155.8, 150.7, 138.51, 138.48, 137.8, 137.7, 129.7, 129.3, 127.2, 127.0, 107.1, 102.2, 38.3, 21.5, 21.4. 2-D NOESY (800 MHz, CDCl3): Ha (3.24 ppm) correlates with Hb (6.66 ppm). HMBC (800 MHz, CDCl3): C1 (159.6 ppm) couples with Ha (3.24 ppm) and Hb (6.66 ppm). C2 (137.6 ppm) couples with Hb (6.66 ppm) and Hc (7.30 ppm). MP 99 °C. IR (cm−1): 3026, 2920, 1599, 1543, 1511, 1416, 1401, 1250, 1181, 1114, 986, 836, 807, 602, 559. HRMS (ESI) m/z calcd. for C21H23N2 [M+H]+ 303.1861, found 303.1864.
Compound 5a was prepared using the general procedure with 2b (22 mg, 3.2 × 10−1 mmol), FeCl2 (2.0 mg, 1.6 × 10−2 mmol), L2 (11.7 mg, 3.2 × 10−2 mmol), and zinc (2.1 mg, 3.2 × 10−2 mmol) in 108 μL of benzene. 4-ethynyltoluene (73 mg, 6.3 × 10−
1mmol), dissolved in 287 μL benzene was added over 3 h at 50 °C. The reaction was stirred an additional 2 h for a total reaction time of 5 h. After the reaction was complete (reaction monitored by GC), the product was isolated as described in Purification Procedure A with an acid/base extraction using 1M HCl then silica gel flash chromatography using 5 % ethyl acetate in hexanes (500 mL to yield 5a (86 mg, 90 %) as a white solid. Rf = 0.57 (20 % ethyl acetate in hexanes). 1H-NMR (500 MHz, CD2Cl2) δ (ppm) 2.41 (d, J = 4.0 Hz, 6H), 3.20 (s, 6H), 6.67 (s, 1H), 7.24 (s, 1H), 7.26 (d, J = 8.0 Hz, 2H), 7.30 (d, J = 7.5 Hz, 2H), 7.56 (d, J = 8.5 Hz, 2H), 8.0 (d, J = 7.0 Hz, 2H). 13C (125 MHz, CDCl3) δ (ppm) 159.9, 155.8, 150.7, 138.51, 138.48, 137.8, 137.7, 129.7, 129.3, 127.2, 127.0, 107.1, 102.2, 38.3, 21.5, 21.4. 2-D NOESY (800 MHz, CDCl3): Ha (3.24 ppm) correlates with Hb (6.66 ppm). HMBC (800 MHz, CDCl3): C1 (159.6 ppm) couples with Ha (3.24 ppm) and Hb (6.66 ppm). C2 (137.6 ppm) couples with Hb (6.66 ppm) and Hc (7.30 ppm). MP 99 °C. IR (cm−1): 3026, 2920, 1599, 1543, 1511, 1416, 1401, 1250, 1181, 1114, 986, 836, 807, 602, 559. HRMS (ESI) m/z calcd. for C21H23N2 [M+H]+ 303.1861, found 303.1864.
Ligand Synthesis
 L1,3
L2,36 and L429 were synthesized according to the literature methods.
L1,3
L2,36 and L429 were synthesized according to the literature methods.
Synthesis of 4-(methoxy)-2,6-dimethylaniline
 4-(methoxy)-2,6-dimethylaniline was prepared using a similar literature procedure.30 In a nitrogen glove box, a 20 mL scintillation vial was filled with CuI (353.8 mg, 1.86 mmol) 3,4,7,8-tetramethyl-1,10-phenanthroline (877.4 mg, 3.71 mmol), Cs2CO3 (21.5g, 111.4 mmol), and 4-iodo-2,6-dimethyl aniline4a (9.17 g, 37.1 mmol). The vial was sealed with a rubber septum, removed from the glove box then evacuated and backfilled with Argon three times. Toluene (14 mL) was added and the mixture was stirred at 80 °C for 20 minutes. Methanol (3.6 mg, 111.4 mmol) was added and the rubber septum was quickly replaced with a vial cap. The reaction was stirred for 24 hours at 80 °C then cooled to room temperature, filtered through a silica gel plug and flushed with 150 mL of ethyl acetate. The resulting solution was reduced in vacuo and purified via silica gel flash chromatography with 10% ethyl acetate in hexanes to yield 4-(methoxy)-2,6-dimethylaniline (3.90 g, 51%) as a blue solid. Rf = 0.12 (20 % ethyl acetate in hexanes). 1H NMR (300 MHz, CDCl3): δ (ppm) 2.20 (s, 6H), 3.23 (s, 2H), 3.75 (s, 3H), 6.58 (s, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 152.23, 134.61, 123.4, 114.1, 55.9, 18.2. MP 38 °C. IR (cm−1): 3449, 3371, 1923, 2835, 1604, 1490, 1378, 1328, 1300, 1243, 1192, 1150, 1064, 949, 854, 728, 599. HRMS (ESI) m/z calcd. for C9H14NO [M+H]+ 152.1075, found 152.1083.
4-(methoxy)-2,6-dimethylaniline was prepared using a similar literature procedure.30 In a nitrogen glove box, a 20 mL scintillation vial was filled with CuI (353.8 mg, 1.86 mmol) 3,4,7,8-tetramethyl-1,10-phenanthroline (877.4 mg, 3.71 mmol), Cs2CO3 (21.5g, 111.4 mmol), and 4-iodo-2,6-dimethyl aniline4a (9.17 g, 37.1 mmol). The vial was sealed with a rubber septum, removed from the glove box then evacuated and backfilled with Argon three times. Toluene (14 mL) was added and the mixture was stirred at 80 °C for 20 minutes. Methanol (3.6 mg, 111.4 mmol) was added and the rubber septum was quickly replaced with a vial cap. The reaction was stirred for 24 hours at 80 °C then cooled to room temperature, filtered through a silica gel plug and flushed with 150 mL of ethyl acetate. The resulting solution was reduced in vacuo and purified via silica gel flash chromatography with 10% ethyl acetate in hexanes to yield 4-(methoxy)-2,6-dimethylaniline (3.90 g, 51%) as a blue solid. Rf = 0.12 (20 % ethyl acetate in hexanes). 1H NMR (300 MHz, CDCl3): δ (ppm) 2.20 (s, 6H), 3.23 (s, 2H), 3.75 (s, 3H), 6.58 (s, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 152.23, 134.61, 123.4, 114.1, 55.9, 18.2. MP 38 °C. IR (cm−1): 3449, 3371, 1923, 2835, 1604, 1490, 1378, 1328, 1300, 1243, 1192, 1150, 1064, 949, 854, 728, 599. HRMS (ESI) m/z calcd. for C9H14NO [M+H]+ 152.1075, found 152.1083.
Synthesis of 4-(methoxy)-2,6-diisopropylaniline
 4-(methoxy)-2,6-diisopropylaniline was prepared using a similar literature procedure.37 In a nitrogen glove box, a 20 mL scintillation vial was filled with CuI (91.9 mg, 0.48 mmol) 3,4,7,8-tetramethyl-1,10-phenanthroline (228 mg, 0.95 mmol), Cs2CO3 (5.58 g, 28.9 mmol), and 4-iodo-2,6-diisopropyl aniline4a (2.93 g, 9.65 mmol). The vial was sealed with a rubber septum, removed from the glove box then evacuated and backfilled with Argon three times. Toluene (5 mL) was added and the mixture was stirred at 80 °C for 20 minutes. Methanol (869 mg, 28.9 mmol) was added and the rubber septum was quickly replaced with a vial cap. The reaction was stirred for 24 hours at 80 °C then cooled to room temperature, filtered through a silica gel plug and flushed with 150 mL of ethyl acetate. The resulting solution was reduced in vacuo and purified via silica gel flash chromatography with 10% ethyl acetate in hexanes to yield 4-(methoxy)-2,6-diisopropylaniline (1.83 g, 91%) as a blue oil. Rf = 0.28 (20 % ethyl acetate in hexanes). 1H NMR (300 MHz, CDCl3): δ (ppm) 1.32 (d, J = 6.5 Hz, 12H), 3.01 (quint J = 7.5 Hz, 2H), 3.50 (s, 2H), 3.82 (s, 3H), 6.70 (s, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 152.9, 134.4, 134.0, 108.8, 55.7, 28.3, 22.6. IR (cm−1): 3459, 3382, 2961, 2871, 2832, 2361, 1600, 1466, 1435, 1383, 1348, 1311, 1242, 1219, 1172, 1123, 1103, 1042, 939, 865, 760, 669. HRMS (ESI) m/z calcd. for C13H22NO [M+H]+ 208.1701, found 208.1700.
4-(methoxy)-2,6-diisopropylaniline was prepared using a similar literature procedure.37 In a nitrogen glove box, a 20 mL scintillation vial was filled with CuI (91.9 mg, 0.48 mmol) 3,4,7,8-tetramethyl-1,10-phenanthroline (228 mg, 0.95 mmol), Cs2CO3 (5.58 g, 28.9 mmol), and 4-iodo-2,6-diisopropyl aniline4a (2.93 g, 9.65 mmol). The vial was sealed with a rubber septum, removed from the glove box then evacuated and backfilled with Argon three times. Toluene (5 mL) was added and the mixture was stirred at 80 °C for 20 minutes. Methanol (869 mg, 28.9 mmol) was added and the rubber septum was quickly replaced with a vial cap. The reaction was stirred for 24 hours at 80 °C then cooled to room temperature, filtered through a silica gel plug and flushed with 150 mL of ethyl acetate. The resulting solution was reduced in vacuo and purified via silica gel flash chromatography with 10% ethyl acetate in hexanes to yield 4-(methoxy)-2,6-diisopropylaniline (1.83 g, 91%) as a blue oil. Rf = 0.28 (20 % ethyl acetate in hexanes). 1H NMR (300 MHz, CDCl3): δ (ppm) 1.32 (d, J = 6.5 Hz, 12H), 3.01 (quint J = 7.5 Hz, 2H), 3.50 (s, 2H), 3.82 (s, 3H), 6.70 (s, 2H). 13C NMR (125 MHz, CDCl3): δ (ppm) 152.9, 134.4, 134.0, 108.8, 55.7, 28.3, 22.6. IR (cm−1): 3459, 3382, 2961, 2871, 2832, 2361, 1600, 1466, 1435, 1383, 1348, 1311, 1242, 1219, 1172, 1123, 1103, 1042, 939, 865, 760, 669. HRMS (ESI) m/z calcd. for C13H22NO [M+H]+ 208.1701, found 208.1700.
Synthesis of (N,N’E,N,N’E)-N,N’-(pyridine-2,6-diylbis(methanylylidene))bis(4-methoxy-2,6-dimethylaniline) (L3)
 L3 was prepared from the similar literature procedure36 with 4-methoxy-2,6-methyl aniline (1.54 g, 10.2 mmol) and 2,6-pyridinedicarboxaldehyde (673 mg, 4.98 mmol) and 10 drops of glacial acetic acid in 15 mL 100% ethanol. The reaction was stirred overnight at room temperature. The mixture was then cooled to 0 °C, filtered and rinsed with cold 100% ethanol to yield L3 (1.9 g, 95 %)as a yellow solid. Rf = 0.37 (20 % ethyl acetate in hexanes). 1H NMR (300 MHz, CDCl3): δ (ppm) 2.21, (s, 12H), 3.81 (s, 6H), 6.67 (s, 4H), 7.97 (t, J = 7.8 Hz, 1H), 8.37-8.40 (m, 4H). 13C NMR (75 MHz, CDCl3): δ (ppm) 163.4, 156.4, 154.8, 143.9, 137.4, 128.8, 122.8, 113.7, 55.53, 19.0. MP 190 °C. IR (cm−1): 3044, 2924, 2863, 2300, 1608, 1545, 1518, 1491, 1459, 1421, 1388, 1263, 1226, 1157, 1034, 996, 948, 879, 818, 736, 638, 573, 516. HRMS (ESI) m/z calcd. for C25H28N3O2 [M+H]+ 402.2182, found 402.2189.
L3 was prepared from the similar literature procedure36 with 4-methoxy-2,6-methyl aniline (1.54 g, 10.2 mmol) and 2,6-pyridinedicarboxaldehyde (673 mg, 4.98 mmol) and 10 drops of glacial acetic acid in 15 mL 100% ethanol. The reaction was stirred overnight at room temperature. The mixture was then cooled to 0 °C, filtered and rinsed with cold 100% ethanol to yield L3 (1.9 g, 95 %)as a yellow solid. Rf = 0.37 (20 % ethyl acetate in hexanes). 1H NMR (300 MHz, CDCl3): δ (ppm) 2.21, (s, 12H), 3.81 (s, 6H), 6.67 (s, 4H), 7.97 (t, J = 7.8 Hz, 1H), 8.37-8.40 (m, 4H). 13C NMR (75 MHz, CDCl3): δ (ppm) 163.4, 156.4, 154.8, 143.9, 137.4, 128.8, 122.8, 113.7, 55.53, 19.0. MP 190 °C. IR (cm−1): 3044, 2924, 2863, 2300, 1608, 1545, 1518, 1491, 1459, 1421, 1388, 1263, 1226, 1157, 1034, 996, 948, 879, 818, 736, 638, 573, 516. HRMS (ESI) m/z calcd. for C25H28N3O2 [M+H]+ 402.2182, found 402.2189.
Synthesis of (N,N’E,N,N’E)-N,N’-(pyridine-2,6-diylbis(methanylylidene))bis(4-(benzyloxy)-2,6-diisopropylaniline) (L5)
L5 was prepared from the similar literature procedure36 with 4-N methoxy-2,6-diisoproply aniline (807 mg, 3.89 mmol) and 2,6-N N pyridinedicarboxaldehyde (263 mg, 1.95 mmol) and 5 drops of MeO OMe glacial acetic acid in 5 mL 100% ethanol. The reaction was stirred L5 overnight at room temperature. The mixture was then cooled to 0 °C, filtered and rinsed with cold 100% ethanol to yield L3 (891 mg, 89 %) as a yellow solid. Rf = 0.61 (20 % ethyl acetate in hexanes). 1H NMR (500 MHz, CDCl3): δ (ppm) 1.20 (d, J = 6.7 Hz, 24H), 3.03 (quint, J = 6.9 Hz, 4H), 3.85 (s, 6H), 6.74 (s, 4H), 7.99 (t, J = 7.8 Hz, 1H), 8.36-8.40 (m, 4H). 13C NMR (125 MHz, CDCl3): δ (ppm) 163.4, 157.0, 154.7, 142.1, 139.1, 137.5, 122.8, 108.8, 55.5, 28.4, 23.7. MP 181 °C. IR (cm−1): 3046, 2961, 2871, 2836, 1638, 1601, 1582, 1462, 1383, 1327, 1289, 1238, 1198, 1169, 1125, 1107, 1076, 1039, 990, 942, 866, 763, 739, 625, 526. HRMS (ESI) m/z calcd. for C33H44N3O2 [M+H]+ 514.3434, found 5143.3441.
Synthesis of (L2)FeBr2
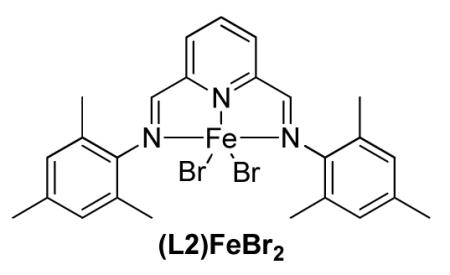 Adapted from the literature procedure.19b In a nitrogen filled glove box, L2 N (109 mg, 5.1 × 10−1 mmol), FeBr2 (111 mg, 5.1 × 10−1 mmol), and 3.0 mL of THF were combined in a vial and stirred at room temperature overnight. Pentane was added to the mixture creating a green precipitate. This green solid was filtered and rinsed with pentane then dried in vacuo yiedling a dark green solid (299.3 mg, >99 %). A crystal suitable for x-ray analysis was grown from slow diffusion of pentane into a solution of (L2)FeBr2 in THF. Elemental Analysis calculated: C, 51.31 H, 4.65 N, 7.18 found: C, 51.59 H, 4.88 N, 7.19.
Adapted from the literature procedure.19b In a nitrogen filled glove box, L2 N (109 mg, 5.1 × 10−1 mmol), FeBr2 (111 mg, 5.1 × 10−1 mmol), and 3.0 mL of THF were combined in a vial and stirred at room temperature overnight. Pentane was added to the mixture creating a green precipitate. This green solid was filtered and rinsed with pentane then dried in vacuo yiedling a dark green solid (299.3 mg, >99 %). A crystal suitable for x-ray analysis was grown from slow diffusion of pentane into a solution of (L2)FeBr2 in THF. Elemental Analysis calculated: C, 51.31 H, 4.65 N, 7.18 found: C, 51.59 H, 4.88 N, 7.19.
Supplementary Material
Scheme 1.

Proposed mechanism.
Acknowledgment
The NIH (GM076125) and NSF (0911017) are acknowledged for financial support. We thank Dr. J. Muller and Dr. Atta Arif for providing HRMS and X-ray crystallographic data, respectively.
Footnotes
Supporting Information Available Experimental details, 1H and 13C NMR spectra, and crystal structure data for (L2)FeBr2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1(a).For selected reviews on [2+2+2] cycloadditions, see: Varella JA, Saá C. Chem. Rev. 2003;103:3787–3801. doi: 10.1021/cr030677f. Kotha S, Brahmachary E, Lahiri K. Eur. J. Org. Chem. 2005:4741–4767. Chopade PR, Louie J. Adv. Synth. Catal. 2006;348:2307–2327. Heller B, Hapke M. Chem. Soc. Rev. 2007;36:1085–1094. doi: 10.1039/b607877j. Shibata T, Tsuchikama K. Org. Biomol. Chem. 2008;6:1317–1323. doi: 10.1039/b720031e. Varela JA, Saá C. Synlett. 2008;17:2571–2578. Galan BR, Rovis T. Angew. Chem. Int. Ed. 2009;48:2830–2834. doi: 10.1002/anie.200804651. Leboeuf D, Gandon V, Malacria M. Handbook of Cyclization Reactions. 2010;1:367–405. Wang C, Wan B. Chin. Sci. Bull. 2012;57:2338–2351. Agenet N, Buisine O, Slowinski F, Gandon V, Aubert C, Malacria M. Org. React. 2007;68:1–302.
- 2(a).Schmidt U, Zenneck U. J. Organomet. Chem. 1992;440:187–190. [Google Scholar]; (b) Knoch F, Kremer F, Schmidt U, Zenneck U. Organometallics. 1996;15:2713–2719. [Google Scholar]; (c) Ferré K, Toupet L, Guerchais V. Organometallics. 2002;21:2578–2580. [Google Scholar]
- 3.D’Souza, B. R, Lane TK, Louie J. Org. Lett. 2011;13:2936–2939. doi: 10.1021/ol2009939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4(a).Selected examples of iron catalyzed alkyne cyclotrimerization: tom Dieck H, Diercks R. Angew. Chem. Int. Ed. 1983;22:1138–1146. Breschi C, Piparo L, Pertici P, Caporusso AM, Vitulli G. J. Organomet. Chem. 2000;607:57–63. Saino N, Kogure D, Okamoto S. Org. Lett. 2005;7:3065–3067. doi: 10.1021/ol051048q. Saino N, Kogure D, Kase K, Okamoto S. J. Organomet. Chem. 2006;691:3129–3136. Liu Y, Yan X, Yang N, Xi C. Catal. Commun. 2011;12:489–492..
- 5.Wang C, Li X, Wu F, Wan B. Angew. Chem. Int. Ed. 2011;50:7162–7166. doi: 10.1002/anie.201102001. [DOI] [PubMed] [Google Scholar]
- 6.The large excess of nitrile presumably favors pyridine formation over diyne dimerization.
- 7(a).Stolley RM, Maczka MT, Louie J. Eur. J. Org. Chem. 2011:3815–3824. doi: 10.1002/ejoc.201100428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kumar P, Prescher S, Louie J. Angew. Chem. Int. Ed. 2011;50:10694–10698. doi: 10.1002/anie.201104475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8(a).Kamal A, Reddy JS, Ramaiah MJ, Dastagiri D, Bharathi EV, Sagar MVP, Pushpavalli SNCVL, Ray P, Pal-Bhadra M. Med. Chem. Commun. 2010;8:355–360. [Google Scholar]; (b) Hilton S, Naud S, Caldwell JJ, Boxall K, Burns S, Anderson VE, Antoni L, Allen CE, Pearl LH, Oliver AW, Aherne GW, Garrett MD, Collins I. Bioorg. Med. Chem. 2010;18:707–718. doi: 10.1016/j.bmc.2009.11.058. [DOI] [PubMed] [Google Scholar]; (c) Garcin ED, Arvai AS, Rosenfeld RJ, Kroeger MD, Crane BR, Andersson G, Andrews G, Hamley PJ, Mallinder PR, Nicholls DJ, St-Gallay SA, Tinker AC, Gensmantel NP, Mete A, Cheshire DR, Connolly S, Stuehr DJ, Aberg A, Wallace AV, Tainer JA, Getzoff ED. Nature Chem. Biol. 2008;4:700–707. doi: 10.1038/nchembio.115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Takahashi Y, Furukawa K, Ishibashi M, Kozutsumi D, Ishiyama H, Kobayashi J, Ohizumi Y. Eur. J. Pharmacol., Mol. Pharmacol. Sect. 1995;288:285–293. doi: 10.1016/0922-4106(95)90040-3. [DOI] [PubMed] [Google Scholar]; (e) Peczyska-Czoch W, Pognan F, Kaczmarek L, Boratynski J. J. Med. Chem. 1994;37:3503–3510. doi: 10.1021/jm00047a008. [DOI] [PubMed] [Google Scholar]
- 9(a).Kim J, Shin-ya S, Furihata K, Hayakawa Y, Seto H. Tetrahedron Lett. 1997;38:3431–3434. [Google Scholar]; (b) Alajarin M, Molina P, Vidal A. J. Nat. Prod. 1997;60:747–748. [Google Scholar]
- 10.Toshiki M, Cheon J, Tsuchiya G, Araki K. J. Chem. Soc. Perkin Trans. 2. 2002:862–865. [Google Scholar]
- 11.Burchak ON, Mugherli L, Ostuni M, Lacapere JJ, Balakirev MY. J. Am. Chem. Soc. 2011;133:10058–10061. doi: 10.1021/ja204016e. [DOI] [PubMed] [Google Scholar]
- 12.Motoyama T, Sasabe H, Seino Y, Takamatsu J, Kido J. Chem. Lett. 2011;40:306–308. [Google Scholar]
- 13.Horie H, Koyama I, Kurahashi T, Matsubara S. Chem. Commun. 2011;47:2658–2660. doi: 10.1039/c0cc04061d. [DOI] [PubMed] [Google Scholar]
- 14(a).Shen Q, Hartwig JF. Org. Lett. 2008;10:4109–4112. doi: 10.1021/ol801615u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maiti D, Buchwald SL. J. Am. Chem. Soc. 2009;131:17423–17429. doi: 10.1021/ja9081815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15(a).Thomas S, Roberts S, Pasumansky L, Gamsey S, Singaram B. Org. Lett. 2003;5:3867–3870. doi: 10.1021/ol035430j. [DOI] [PubMed] [Google Scholar]; (b) Londregan AT, Jennings S, Wei L. Org. Lett. 2010;12:5254–5257. doi: 10.1021/ol102301u. [DOI] [PubMed] [Google Scholar]
- 16(a).Salem MAI, Madkour HMF, Soliman ESA, Mahmoud NFH. Heterocycles. 2000;53:1129. [Google Scholar]; (b) Teague SJ. J. Org. Chem. 2008;73:9765–9766. doi: 10.1021/jo801303v. [DOI] [PubMed] [Google Scholar]
- 17(a).Cobalt: Heller B, Reihsig J, Schulz W, Oehme G. Appl. Organomet. Chem. 1993;7:641–646. Fatland AW, Eaton BE. Org. Lett. 2000;2:3131–3133. doi: 10.1021/ol006327m. Boñaga LVR, Zhang H-C, Maryanoff BE. Chem. Commun. 2004:2394–2395. doi: 10.1039/b410012c. Hapke M, Kral K, Fischer C, Spannenberg A, Gutnov A, Redkin D, Heller B. J. Org. Chem. 2010;75:3993–4003. doi: 10.1021/jo100122d. Garcia P, Evanno Y, Gorge P, Sevrin M, Ricci G, Malacria M, Aubert C, Gandon V. Org. Lett. 2011;13:2030–2033. doi: 10.1021/ol200417p. Garcia P, Evanno Y, Gorge P, Sevrin M, Ricci G, Malacria M, Aubert C, Gandon V. Chem. Eur. J. 2012;18:4337–4344. doi: 10.1002/chem.201103906.Nickel: Bönnemann H. Angew. Chem. Int. Ed. Engl. 1985;24:248–262. Rhodium: Tanaka K, Suzuki N, Nishida G. Eur. J. Org. Chem. 2006:3917–3922. Titanium: Tanaka R, Yuza A, Watai Y, Suzuki D, Takayama Y, Sato F, Urabe H. J. Am. Chem. Soc. 2005;127:7774–7780. doi: 10.1021/ja050261e. Photocatalytically: Heller B, Sundermann B, Buschmann H, Drexler H, You J, Holzgrabe U, Heller E, Oehme G. J. Org. Chem. 2002;67:4414–4412. doi: 10.1021/jo011032n.
- 18.Additional PDAI class ligands were screened and are included in the Supporting Information.
- 19(a).Studies involving iron bis(imino)pyridine and bis(aldimino)pyridine complexes: Bart SC, Chłopek K, Bill E, Bouwkamp MW, Lobkovsky E, Neese F, Weighardt K, Chirik PJ. J. Am. Chem. Soc. 2006;128:13901–13912. doi: 10.1021/ja064557b. Russell SK, Milsmann C, Lobkovsky E, Weyhermüller T, Chirik P. J. Inorg. Chem. 2011;50:3159–3169. doi: 10.1021/ic102186q.
- 20.See Supplementary Information for details of control experiments.
- 21(a).Yamamoto Y, Kinpara K, Nishiyama H, Itoh K. Adv. Synth. Catal. 2005;347:1913–1916. [Google Scholar]; (b) Yamamoto Y, Kinpara K, Ogawa R, Nishiyama H, Itoh K. Chem. Eur. J. 2006;12:5618–5631. doi: 10.1002/chem.200600176. [DOI] [PubMed] [Google Scholar]
- 22(a).Cobalt: Heller B, Sundermann B, Buschmann H, Drexler H-H, You J, Holzgrabe U, Heller E, Oehme G. J. Org. Chem. 2002;67:4414–4422. doi: 10.1021/jo011032n. Heller B, Reihsig J, Schulz W, Oehme G. Appl. Organomet. Chem. 1993;7:641–646. Pietro D, Ingrosso G, Lucherini A, Vanacore D. J. Mol. Catal. 1987;41:261–270. Rhodium: Cioni P, Civersi P, Ingrosso G, Lucherini A, Ronca P. J. Mol. Catal. 1987;40:337–357.
- 23.Liu Y, Yan X, Yang N, Xi C. Cat. Commun. 2011;12:489–492. [Google Scholar]
- 24.Henke BR, Drewry DH, Jones SA, Stewart EL, Weaver SL, Wiethe RW. Bioorg. Med. Chem. Lett. 2001;11:1939–1942. doi: 10.1016/s0960-894x(01)00321-3. [DOI] [PubMed] [Google Scholar]
- 25(a).Yamazaki H, Wakatsuki Y. Tetrahedron Lett. 1973;14:3383–3384. [Google Scholar]; (b) Bönnemann H. Angew. Chem., Int. Ed. Engl. 1985;24:248–262. [Google Scholar]; (c) Diercks R, Eaton BE, Gürtzgen S, Jalisatgi S, Matsger AJ, Radde RH, Vollhardt KPC. J. Am Chem. Soc. 1998;120:8247–8248. [Google Scholar]; (d) Dazinger G, Torres-Rodrigues M, Kirchner K, Calhorda MJ, Costa PJ. J. Organomet. Chem. 2006;691:4434–4445. [Google Scholar]; (e) Dahy AA, Yamada K, Koga N. Organometallics. 2009;28:3636–3649. [Google Scholar]; (f) Dahy AA, Koga N. J. Organomet. Chem. 2010;695:2240–2250. [Google Scholar]
- 26(a).Takahashi T, Tsai FY, Kotora M. J. Am. Chem. Soc. 2000;122:4994–4995. [Google Scholar]; (b) Takahashi T, Tsai FY, Li Y, Wang H, Kondo Y, Yamanaka M, Nakajima K, Kotora M. J. Am. Chem. Soc. 2002;124:5059–5067. doi: 10.1021/ja017507+. [DOI] [PubMed] [Google Scholar]; (c) Eisch JJ, Ma X, Han KI, Gitua JN, Kruger C. Eur. J. Inorg. Chem. 2001:77–88. [Google Scholar]
- 27.Nishida M, Shiga H, Mori M. J. Org. Chem. 1998;63:8606–8608. [Google Scholar]
- 28.Atkinson RS, Grimshire MJ. J. Chem. Soc. Perkin Trans. 1. 1986:1215–1217. [Google Scholar]
- 29.Hu Y, Sun Y, Hu J, Zhu T, Yu T, Zhao Q. Chem. Asian J. 2011;6:797–800. doi: 10.1002/asia.201000692. [DOI] [PubMed] [Google Scholar]
- 30.Ishizaki M, Hoshino O. Tetrahedron. 2000;56:8813–8819. [Google Scholar]
- 31.Tanaka K, Takeishi K, Noguchi K. J. Am. Chem. Soc. 2006;128:4586–4587. doi: 10.1021/ja060348f. [DOI] [PubMed] [Google Scholar]
- 32.Han Y, Harlan J, Stoessel P, Frost BJ, Norton JR, Miller S, Bridgewater B, Xu Q. Inorg. Chem. 2001;40:2942–2952. doi: 10.1021/ic0100931. [DOI] [PubMed] [Google Scholar]
- 33.Tekavec TN, Louie J. J. Org. Chem. 2008;73:2641–2648. doi: 10.1021/jo702508w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Apte S, Radetich B, Shin S, RajanBabu TV. Org. Lett. 2004;6:4053–4056. doi: 10.1021/ol048265w. [DOI] [PubMed] [Google Scholar]
- 35.Trost BM, Rudd MT. J. Am. Chem. Soc. 2003;125:11516–11517. doi: 10.1021/ja036410f. [DOI] [PubMed] [Google Scholar]
- 36.Britovsek GJP, Bruce M, Gibson VC, Kimberley BS, Maddox PJ, Mastroianni S, McTavish SJ, Redshaw C, Solan GA, Stroemberg S, White AJP, Williams DJ. J. Am. Chem. Soc. 1999;121:8728–8740. [Google Scholar]
- 37.Altman RA, Shafir A, Choi A, Lichtor PA, Buchwald SL. J. Org. Chem. 2008;73:284. doi: 10.1021/jo702024p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.