

Abstract
Histone deacetylases (HDACs), a family of enzymes involved in epigenetic regulation, have been implicated in the control of synaptic plasticity, as well as learning and memory. Previous work has demonstrated administration of pharmacological HDAC inhibitors, primarily those targeted to class I HDACs, enhance learning and memory as well as long-term potentiation. However, a detailed understanding of the role of class II HDACs in these processes remains elusive. Here, we show that selective loss of Hdac4 in brain results in impairments in hippocampal-dependent learning and memory and long-term synaptic plasticity. In contrast, loss of Hdac5 does not impact learning and memory demonstrating unique roles in brain for individual class II HDACs. These findings suggest that HDAC4 is a crucial positive regulator of learning and memory, both behaviorally and at the cellular level, and that inhibition of Hdac4 activity may have unexpected detrimental effects to these processes.
Introduction
Epigenetic mechanisms control accessibility of chromatin to transcriptional factors in neurons, which in turn impact gene expression and ultimately regulate neurotransmission, synaptic plasticity, as well as disease states (Abel and Zukin, 2008; Borrelli et al., 2008; Jiang et al., 2008). A key epigenetic modification involves the posttranslational removal or addition of an acetyl group to specific lysine residues in the N terminus of histone residues, which is accomplished by histone deacetylases (HDACs) and histone acetytransferases (HATs), respectively. Alterations in histone acetylation in animal models has been implicated in learning and memory as well as synaptic plasticity (Alarcón et al., 2004; Korzus et al., 2004; Levenson et al., 2004; Vecsey et al., 2007), with HDAC inhibitors facilitating these processes in wild-type mice and mouse models of neurodegenerative diseases (Fischer et al., 2007). Studies based on pharmacological HDAC inhibitors have primarily focused on class I HDAC family members (HDAC1, HDAC2, HDAC3, and HDAC8), with recent data suggesting that brain deletion of Hdac2 facilitates memory formation and synaptic plasticity (Guan et al., 2009). However, the role of class IIa HDACs (HDAC4, HDAC5, HDAC7, and HDAC9), which act as transcriptional repressors and shuttle between the nucleus and cytoplasm in response to intracellular signaling (Haberland et al., 2009), has not been addressed in these processes.
HDAC4 and HDAC5 are highly enriched in the brain with strong expression in broad forebrain regions such as hippocampus, cortex, and amygdala (Broide et al., 2007), suggesting they may be important in underlying enhanced learning and memory as well as synaptic plasticity observed with broad HDAC inhibitors. Constitutive Hdac5−/− knock-out (KO) mice show enhanced reward to cocaine in conditioned place preference (Renthal et al., 2007). Conversely, overexpression of Hdac4 or Hdac5 attenuates the rewarding effects of cocaine in conditioned place preference (Kumar et al., 2005; Renthal et al., 2007). Collectively, these data have implicated HDAC4 and HDAC5 as negative regulators of rewarding related behavior. The maintenance of reward-related behavior likely involves alterations in synaptic plasticity, although specific roles for HDAC4 and HDAC5 in these specific processes, as well as in learning and memory related tasks remain unexplored.
The goal of this study was to investigate the potential involvement of HDAC4 and HDAC5 in learning, memory, and synaptic plasticity. Constitutive Hdac5−/− null mice are viable, while in contrast constitutive Hdac4 mice die perinatally (Vega et al., 2004). We therefore generated mice with conditional brain-specific Hdac4 deletion and show that these animals have alterations in motor coordination and anxiety, as well as significant deficits in learning tasks. The learning and memory deficits in the brain-specific Hdac4 knock-out mice were associated with significant impairments in long-term potentiation (LTP) induction in the hippocampus without alterations in basal synaptic transmission. In contrast, Hdac5−/− mice display no abnormalities in these behaviors, including learning and memory. These findings establish a surprising and key role of HDAC4, but not HDAC5, as a positive regulator of memory formation and long-term synaptic plasticity.
Materials and Methods
Generation of knock-out mice.
Constitutive Hdac4 mice die perinatally (Vega et al., 2004), precluding the ability to examine them for behavioral deficits. We therefore generated conditional brain-specific Hdac4 knock-out mice. Floxed Hdac4 mice (Hdac4loxP/loxP) were generated by flanking exon 5 with loxP sites, which resulted in an out-of-frame mutation in the Hdac4 allele (Potthoff et al., 2007). To generate mice lacking Hdac4 in the brain, mice heterozygous for the Hdac4loxP allele were mated to CaMKII-Cre transgenic mice that express Cre recombinase specifically in the forebrain beginning at ∼10 d of age (Luikart et al., 2005). The resulting mice were crossed to homozygous Hdac4 floxed mice (Hdac4loxP/loxP) to generate forebrain-specific conditional null mice (Hdac4loxP/loxP;CaMKII-Cre). The conditional Hdac4 brain-specific knock-out (Hdac4bko) mice were genotyped by PCR. The primers for detection of Hdac4 are as follows: HDAC4, forward, 5′-ATC TGC CCA CCA GAG TAT GTG-3′; HDAC4, reverse, 5′-CTT GTT GAG AAC AAA CTC CTG CAG CT-3′; and nLacZ, reverse, 5′-GAT TGA CCG TAA TGG GAT AGG TTA CG-3′. The primers for Cre recombinase are as follows: 5′-AGG TTC GTT CAC TCA TGG A-3′ and 5′-TCG ACC AGT TTA GTT ACC C-3′. The constitutive Hdac5−/− mice had been previously generated and the deletion confirmed (Chang et al., 2004). The Hdac5−/− and wt littermate controls were generated by heterozygous × heterozygous crosses.
Histology.
Mice were transcardially perfused, and the brains were removed and fixed overnight in DEPC-treated 4% paraformaldehyde. The brains were paraffin-embedded, sectioned at 5 μm, and mounted on gelatin-coated slides. For in situ hybridization, riboprobes were labeled with [α-35S]UTP using the MAXIscript in vitro transcription kit (Ambion) following the manufacturer's instructions. In situ hybridization of sectioned tissues was performed as previously described (Shelton et al., 2000). The Hdac4 riboprobe was originally generated by PCR using the following primers: forward, 5′-CAG AAG CTG CAG CAG CTC AAG-3′, and reverse, 5′-GGT GGA GAG CTC TGG TCA AGG-3′.
RNA isolation and quantitative RT-PCR.
RNA was isolated from cortex, hippocampus, and cerebellum using the RNeasy kit (QIAGEN) according to the manufacturer's instructions. Two micrograms of RNA were converted to cDNA using random primers and SuperScript III Reverse Transcriptase (Invitrogen). Quantitative analysis was performed by real-time PCR using the ABI PRISM 7000 sequence detection system with TaqMan primers (Applied Biosystems) or with SYBR Green Master Mix reagent (Applied Biosystems). To quantify the relative changes in gene expression, the comparative Ct method was used (Livak and Schmittgen, 2001). A fold change was calculated by the following equation: 2−ΔΔCt = [(ΔΔCt Hdac − Ct Gapdh)knock-out − (Ct Hdac − Ct Gapdh)wild type].
Behavioral overview.
All behavior testing was done on male mice at least 2 months of age that were littermates. The mice were on a 12 h light/dark cycle with ad libitum access to food and water. Mice were allowed to habituate in the behavior room for 1 h before all behavioral testing. All data were analyzed and scored by an observer blind to the genotype. For all experiments, data were presented as mean ± SEM, and Student's t test was used to analyze data, unless otherwise noted, with significance set as p < 0.05. All animal experimental procedures were reviewed and approved by the Institutional Animal Care and Research Advisory Committee at University of Texas Southwestern Medical Center.
Locomotor activity.
Mice were placed in a fresh home cage and locomotor activity was measured for 2 h using four photocell beams linked to computer data acquisition software (San Diego Instruments). Ambulation was scored as horizontal movement by the animal within the 2 h of testing. Data were analyzed with repeated ANOVA.
Rotarod.
Each mouse was placed on the activated rotarod (IITC Life Science), and its speed ramped up from 0 to 45 rpm in 60 s. The time for the mouse to fall off the rotarod or turn one full revolution was measured for four consecutive trials. After the test, the mouse was returned to its original cage for ∼24 h. The test was repeated for a total of four runs the next day. Data were analyzed with repeated ANOVA.
Elevated plus maze.
Mice were placed in the center of an elevated plus maze under dim lighting, and their behavior was monitored for 5 min using a video-tracking system (Ethovision 3.0). The time spent in the center, closed and open arms was determined.
Open field.
Mice were videotaped under dim lighting for their activity during 5 min in a 42 cm square open field. The time spent in the center, the broader area of the center, and borders of the open field (periphery) was determined.
Fear conditioning.
The fear conditioning paradigm was performed as described previously (Monteggia et al., 2004; Barbosa et al., 2008). Briefly, mice were placed in individual chambers (MED Associates) for 12 min during which time they received four scrambled footshocks at 0.5 mA for 2 s. The footshocks were administered at 2.5, 5, 9, and 11.5 min after placement of the animals into the chamber. After the 12 min training session, the mice were immediately removed and placed back into their home cages. Ninety minutes later, short-term memory (STM) formation was tested. The mice were placed back in the same chamber for 3 min (no shock), and their freezing behavior was assessed by an observer blind to the genotypes. To examine long-term memory (LTM) formation, the mice were placed back in the same chamber 24 h after the training, and their freezing behavior was assessed for 3 min. The freezing behavior was defined as no movement except for respiration. To assess pain sensitivity to the footshock, mice were placed back in the individual chambers for 2 min to habituate. The animals were then shocked (0.05 mA for 1 s), and their behavior was scored as no movement, flinching, or jumping. Every 30 s, the shock was increased by 0.05 mA, with a maximum shock of 0.6 mA, until the animal flinched and jumped in response to the shock.
Morris water maze.
A circular pool was filled with room temperature water to a depth of ∼12 inches. A platform (10 cm diameter) was placed in one quadrant of the pool with the top of the platform ∼2 cm below the water level. White nontoxic paint was added to enhance the contrast with the animal and to hide the location of the platform. Each day, the mice were placed in the pool and allowed to swim for 1 min to find the platform. The swim path, swim velocity, and time spent in each quadrant were obtained using automated video-tracking software from Noldus (Ethovision 2.3.19). If the mouse did not find the platform within a minute, they were gently guided or placed on the platform for 10 s, and then removed from the pool and returned to their home cage. Each animal was placed in the pool for a total of four times, starting from a different quadrant for each trial, each day for 10 d. Immediately following the training days, a probe test was conducted in which the platform is removed from the pool and each mouse was allowed to swim for 1 min to determine whether the animal had learned the location of the platform.
Slice electrophysiology.
For extracellular field recordings, 2- to 5-month-old mice were used. Mice were anesthetized by intraperitoneal Nembutal injection and decapitated. The brain was rapidly dissected, and hippocampal slices (400 μm) were collected in ice-cold dissection buffer containing the following (in mm): 212 sucrose, 3 KCl, 5 MgCl2, 0.5 CaCl2, 1 NaH2PO4, 26 NaHCO3, and 10 glucose. CA3 region was cut to avoid epileptiform activity. Slices were placed at 30°C for 2 h in artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 10 glucose. ACSF and dissection buffer were bubbled with 95% O2/5% CO2. Before recordings, slices were placed in a submersion-recording chamber, maintained at 30°C, and perfused with ACSF. Concentric, bipolar tungsten electrodes were used to activate Schaffer collateral/commissural (SC) fibers in the hippocampal CA1 region. Extracellular glass microelectrodes filled with ACSF (resistance, ∼1 MΩ) were placed in the stratum radiatum to measure field fEPSPs. For baseline recordings, slices were stimulated at 0.033 Hz for 20 min at stimulation intensities of 40–50% of the highest measured fEPSP size. LTP was induced by applying a theta burst stimulus consisting of three bursts (10 s interval), each composed of 15 trains (5 Hz) with four pulses (100 Hz). Paired-pulse facilitation (PPF) was tested by applying two pulses with interstimulus intervals ranging from 20 to 200 ms. An Axoclamp 2B amplifier (Molecular Devices) was used for experiments. Data were sampled at 5 kHz and analyzed using a program written in LabView (National Instruments). For experiments, data were presented as mean ± SEM.
Cell culture.
Primary hippocampal neurons were cultured as previously described (Kavalali et al., 1999). Briefly, whole hippocampi were isolated from homozygous Hdac4loxP/loxP, or constitutive Hdac5−/− KO mice on postnatal days 0–1. After dissection, tissues were trypsinized for 10 min at 37°C, mechanically dissociated using siliconized glass pipettes, and plated onto Matrigel-coated coverslips. A concentration of 4 μm cytosine arabinoside (Sigma-Aldrich) was added at 1 d in vitro (DIV) and reduced to a concentration of 2 μm at 4 DIV.
Lentivirus production.
Lentivirus was generated as previously described (Dittgen et al., 2004; Nelson et al., 2006; Akhtar et al., 2009). Briefly, HEK 293 cells were transfected with the expression plasmid, pFUGW or pFUGW-Cre, and two helper plasmids, Δ8.9 and vesicular stomatitis virus G-protein, at 3 μg of each DNA per 75 cm2 flask using the Fugene 6 transfection system (Roche Molecular Biochemicals). Lentivirus-containing culture medium was harvested 48 h after transfection, filtered at a 0.45 μm pore size, and immediately used for infection. Hippocampal cultures were infected at 4 DIV by adding 300 μl of viral suspension to each well containing 700 μl of media, and recordings were done at 14–17 DIV.
In vitro electrophysiology recordings.
Synaptic activity was recorded from hippocampal pyramidal neurons (in at least three independent cultures) using a whole-cell voltage-clamp technique. Data were acquired using an Axopatch 200B amplifier and Clampex 9.2 software (Molecular Devices). Recordings were filtered at 2 kHz and sampled at 200 μs. A modified Tyrode's solution containing the following (in mm): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH 7.4, was used as external bath solution, with 50 μm picrotoxin and 1 μm TTX to isolate miniature EPSCs (mEPSCs). The pipette internal solution for the voltage-clamp experiments contained the following (in mm): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 TEA-Cl, 4 Mg-ATP, 0.3 Na3GTP, pH 7.35, and 10 QX-314 (300 mOsm). Statistical analysis was performed using Student's t test. Statistical significance was defined as p < 0.05. For experiments, data were presented as mean ± SEM.
Results
Generation of brain-specific Hdac4 knock-out mice
To generate mice lacking Hdac4 specifically in forebrain neurons, we mated mice carrying a homozygous floxed Hdac4loxP/loxP allele (Potthoff et al., 2007) with mice harboring a transgene that expresses Cre recombinase under the control of the calcium/calmodulin-dependent protein kinase II promoter (CaMKII-Cre). The CaMKII-Cre transgenic mouse expresses Cre recombinase from postnatal day 10 in forebrain regions, including the neocortex and the limbic system, but not in the cerebellum (Luikart et al., 2005). Mice with homozygous deletion of Hdac4 in the forebrain (hereafter referred to as Hdac4bko mice for Hdac4 brain KO; wild-type littermate controls are represented as Hdac4wt mice) were born at expected Mendelian ratios and showed normal body weight, gross brain morphology, and life span (data not shown). Successful deletion of Hdac4 in the brain of mutant mice was validated by in situ hybridization, Western blot analysis, and quantitative RT-PCR. In Hdac4bko mice, Hdac4 mRNA expression (Fig. 1A) and protein levels (Fig. 1B) were dramatically reduced in the forebrain, including the cortex, amygdala, and hippocampus, but not in the cerebellum. To identify whether the loss of Hdac4 impacted other class II HDACs, we examined the expression of Hdac5, Hdac7, and Hdac9. We found that Hdac5, Hdac7, and Hdac9 mRNAs were not significantly altered in the hippocampus of Hdac4bko compared with littermate Hdac4wt mice, suggesting that there were no significant compensatory changes in expression of these functionally related genes (Fig. 1C). We also examined the expression of class I HDACs in the hippocampus of Hdac4bko compared with littermate Hdac4wt mice, and no significant change in Hdac1 (110 ± 15.2%), Hdac2 (124 ± 16.8%), or Hdac3 (118 ± 15.1%) mRNA was observed. Hematoxylin and eosin staining of brain sections did not reveal any detectable aberrations in brain morphology of the Hdac4bko compared with Hdac4wt mice (Fig. 1D).
Figure 1.

Generation of forebrain-specific Hdac4bko mice. A, Detection of Hdac4 transcripts by in situ hybridization in coronal sections from Hdac4wt and Hdac4bko mice at 4 months of age. B, Western blots of protein from brain lysates showed deletion of HDAC4 in the cortex and hippocampus, but not the cerebellum of Hdac4bko mice. GAPDH protein was used as a loading control. C, Expression levels of class II HDAC mRNAs (Hdac4, Hdac5, Hdac7, and Hdac9) in the hippocampus of Hdac4bko mice compared with Hdac4wt, as determined by quantitative RT-PCR. Error bars indicate ± SEM. *p < 0.05. D, Hematoxylin and eosin staining showed no obvious changes in brain morphology between 4-month-old Hdac4bko and Hdac4wt mice. CTX, Cortex; DG, dentate gyrus; Hp, hippocampus.
Hdac4bko mice display behavioral learning and memory impairments
To determine whether the loss of Hdac4 in brain impacts behavior, the Hdac4bko mice were tested in several paradigms. We examined motor coordination using the rotarod test and found the Hdac4bko mice exhibited a significant impairment relative to Hdac4wt mice (Fig. 2A). The deficits in motor coordination, with normal Hdac4 expression in the cerebellum (Fig. 1B), suggest that it is not loss of Hdac4 expression in the cerebellum that is contributing to the phenotype. In previous work, we demonstrated that this Cre driver line results in motor coordination deficits in the Rett syndrome Mecp2 mouse, without alterations in Mecp2 expression in the cerebellum, suggesting that altered gene loss independent of the cerebellum can impact this behavioral task (Gemelli et al., 2006).
Figure 2.
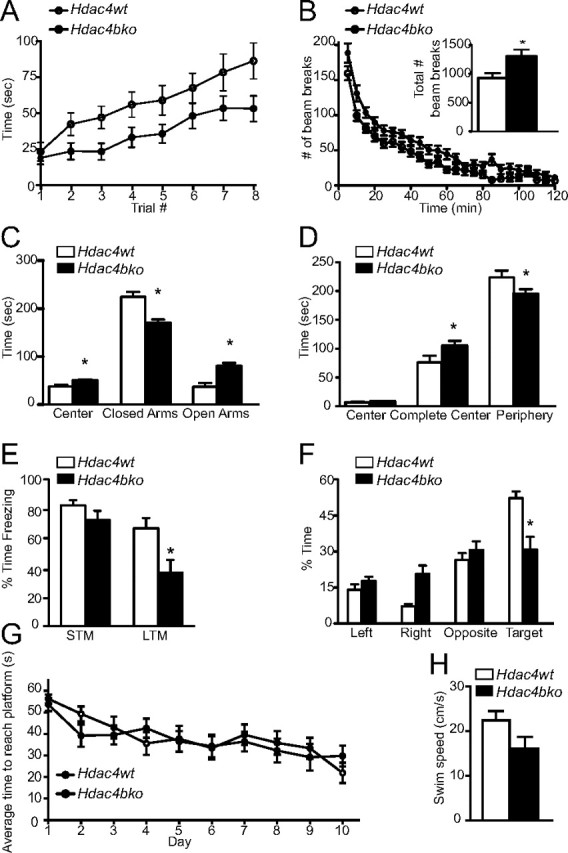
Hdac4bko mice show behavioral abnormalities including impaired memory formation. A, The Hdac4bko mice were tested on the rotarod for motor coordination. Based on repeated measures with mixed-model analysis, there were significant group effects between Hdac4bko and Hdac4wt mice (F(1,39) = 4.69; value of p = 0.0366) and trial effect (F(7,272) = 23.59; value of p < 0.0001). B, Hdac4bko mice display an increase in locomotor activity, as assessed by an increase in the number of beam breaks over 120 min. The Hdac4bko had a significant increase in the total number of beam breaks over 120 min compared with Hdac4wt mice (inset). Total locomotor activity was assessed for 120 min, and a significant total ambulation difference between the Hdac4bko and Hdac4wt mice was detected, demonstrating that the Hdac4bko mice were significantly hyperactive compared with littermate controls (*p < 0.05; inset). To better delineate the alteration in locomotor activity, the data were analyzed in 5 min increments. Based on repeated measures with mixed-model analysis, there were significant group effects between Hdac4bko and Hdac4wt mice (F(1,39) = 6.18; value of p = 0.0173) and time effect (F(23,897) = 109.44; value of p < 0.0001) further demonstrating the hyperactivity of the Hdac4bko mice. C, Hdac4bko show a significant increase in time spent in the center of the maze as well as the open arms, and a significant decrease in time spent in the closed arms (*p < 0.05; Hdac4wt, n = 18; Hdac4bko, n = 23). D, Hdac4bko were less anxious than littermate Hdac4wt mice in the open-field test. The Hdac4bko mice spent significantly more time in the complete center and less time in the periphery compared with Hdac4wt mice (*p < 0.05; Hdac4wt, n = 18; Hdac4bko, n = 23). E, The time spent freezing 90 min and 24 h after training was presented to examine STM and LTM, respectively, in the fear-conditioning paradigm. Results indicate a statistically significant decrease in LTM formation but not STM in the Hdac4bko compared with Hdac4wt mice (*p < 0.05; Hdac4wt, n = 8; Hdac4bko, n = 13). F, Hdac4bko mice are impaired in spatial learning in the Morris water maze. A probe trial was performed on day 11. Hdac4bko mice spent a similar amount of time in the four quadrants, while the Hdac4wt mice showed a preference for the target quadrant and spent significantly more time in the target quadrant (*p < 0.05; Hdac4wt, n = 11; Hdac4bko, n = 11). G, Hdac4bko mice show no differences from Hdac4wt mice in the average time to reach the platform during the 10 d training (*p < 0.05; Hdac4wt, n = 11; Hdac4bko, n = 11). H, Hdac4wt mice and Hdac4bko mice have similar swim speeds, and there were no significant differences in average swim velocity between the two groups (*p < 0.05; Hdac4wt, n = 11; Hdac4bko, n = 11). For all experiments, error bars show SEM.
Total locomotor activity was assessed for 120 min, and the Hdac4bko mice were significantly hyperactive compared with Hdac4wt mice (Fig. 2B, inset). To better delineate the alteration in locomotor activity, we analyzed the data in 5 min increments and detected significant differences between Hdac4bko and Hdac4wt mice throughout this time period (Fig. 2B).
To assess whether the loss of Hdac4 impacts anxiety-related behavior, we tested mice in the elevated plus maze and the open-field test, behavioral tasks that incorporate aspects of human anxiety and have predictive validity for anxiolytic drugs (Holmes, 2001; Crawley, 2008). In the elevated plus maze, the Hdac4bko mice spent significantly more time in the center and the open arms and significantly less time in the closed arms compared with Hdac4wt mice, suggesting a decrease in anxiety-like behavior (Fig. 2C). To further examine whether the loss of Hdac4 decreases anxiety-related behavior, we used the open-field test. In the open-field test, the Hdac4bko mice spent significantly more time in the complete center of the field and significantly less time in the periphery compared with Hdac4wt animals, consistent with a decrease in anxiety-related behavior (Fig. 2D).
Since previous data have suggested that HDAC inhibition augments learning and memory, we tested the Hdac4bko mice in context-dependent fear conditioning. In this behavioral test, mice learn to associate a context (the environment) with an aversive footshock. When animals are reexposed to the same environment (context) 24 h later, they generate a fear response in which they “freeze” (Phillips and LeDoux, 1992). Baseline freezing between Hdac4bko and Hdac4wt mice was indistinguishable (data not shown). Ninety minutes after the training session, we tested the animals for STM formation and found that Hdac4bko mice displayed indistinguishable freezing behavior to Hdac4wt mice (Fig. 2E). In contrast, 24 h after the training when the Hdac4bko mice were examined for LTM, they showed significantly less freezing in response to the context compared with Hdac4wt mice, suggestive of a deficit in learning and memory (Fig. 2E). The decrease in freezing of the Hdac4bko mice was not due to differences in pain sensitivity to footshock between the Hdac4bko and Hdac4wt mice (data not shown). These data suggest that the behavioral impairments during fear conditioning observed with the Hdac4bko mice may be due to deficits in associative learning. To evaluate this possibility, we used the Morris water maze paradigm to test hippocampal-dependent learning and memory, independent of locomotor activity (Vorhees and Williams, 2006). During the training period (four trials per day for 10 d), each animal was placed in the pool and allowed to swim ad libitum for up to 1 min or until the platform was located. The Hdac4bko mice were able to locate the platform as rapidly as Hdac4wt animals and showed no significant differences in swim velocity, indicating that the basic neurological functions needed for swimming and sight were normal (Fig. 2G,H). This was particularly important as previous work has shown that HDAC4 is essential for survival of retinal neurons (Chen and Cepko, 2009). A probe test was performed on day 11 in which the platform was removed and mice were allowed to swim ad libitum for 1 min to quantify the amount of time spent in each quadrant. We found that Hdac4wt mice spent significantly more time in the target quadrant where the submerged platform had been located, while in contrast the Hdac4bko mice did not prefer the target quadrant (Fig. 2F), suggesting that Hdac4bko mice have significant impairments in spatial learning and memory.
Hdac5−/− KO mice display no overt behavioral phenotypes
The constitutive Hdac5−/− knock-out mice had been previously generated (Chang et al., 2004). We investigated whether the loss of Hdac5, another key member of the class II HDAC family, produced similar behavioral phenotypes as those observed in the HDAC4bko mice. We found that the Hdac5−/− mice were indistinguishable from wild-type littermate controls, referred to as Hdac5wt, in motor coordination as assessed by the rotarod test (Fig. 3A). The Hdac5−/− and Hdac5wt mice displayed similar total locomotor activity for 120 min (Fig. 3B, inset), as well as when data was analyzed in 5 min increments (Fig. 3B). The Hdac5−/− mice also exhibited normal anxiety-related behaviors as assessed in the elevated plus maze (Fig. 3C) and open-field test (Fig. 3D) relative to Hdac5wt mice. To examine the involvement of HDAC5 in learning and memory, we tested the mice in the fear conditioning behavioral task. We found that the Hdac5−/− had similar freezing behavior in context-dependent fear conditioning to Hdac5wt mice (Fig. 3E). In addition, the Hdac5−/− had indistinguishable levels of baseline freezing and pain sensitivity to footshock compared with Hdac5wt mice (data not shown). These data show that, in striking contrast to the loss of Hdac4, loss of Hdac5 does not impact context-dependent fear conditioning, underscoring the specificity of our observations.
Figure 3.
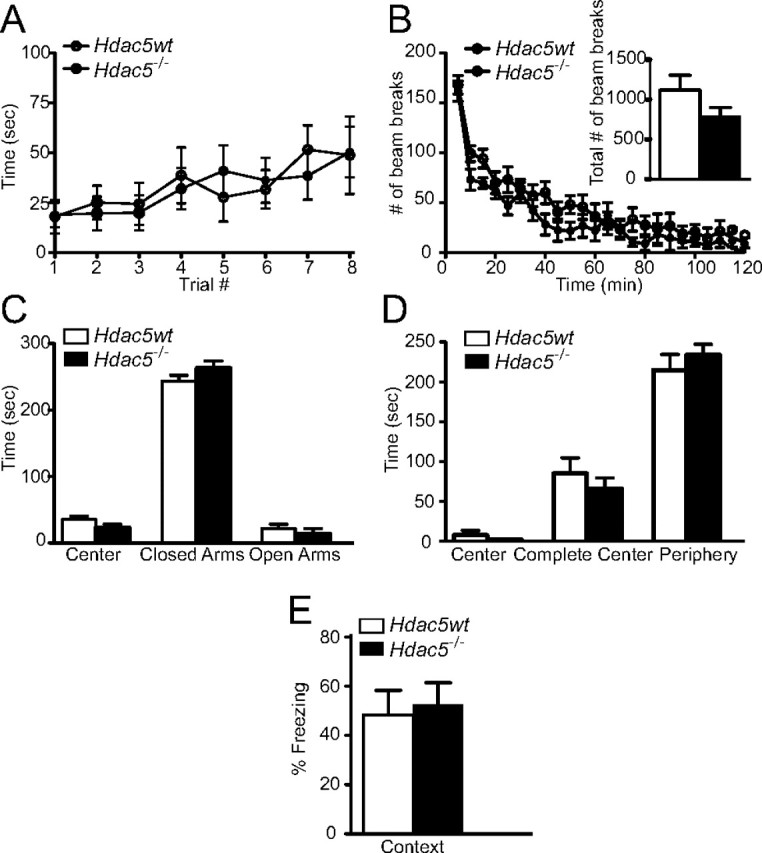
Hdac5−/− mice have normal behavior including memory formation. A, Hdac5−/− mice have normal motor coordination, staying on the rotarod as well as littermate Hdac5wt. The Hdac5−/− mice were indistinguishable from Hdac5wt mice in motor coordination as assessed by the rotarod test, as repeated measures with mixed-model analysis revealed no significant group effect (F(1,6) = 0.06; value of p = 0.8109). B, The Hdac5−/− mice were examined for alterations in total locomotor activity for 120 min, and using Student's t test analysis, there was no significant difference between Hdac5−/− and Hdac5wt mice. We also assessed ambulation in 5 min increments and, based on repeated measures with mixed-model analysis, found no significant group effect between Hdac5−/− and Hdac5wt mice (F(1,6) = 5.87; value of p = 0.0517), while there is a significant time effect (F(23,138) = 38.20; value of p < 0.0001). C, In the elevated plus maze test, Hdac5−/− mice show no significant difference in time spent in the center of the maze, closed arms, or open arms compared with Hdac5wt mice. D, In the open-field test, the Hdac5−/− mice spent a similar amount of time in the complete center and periphery compared with Hdac5wt mice. E, Hdac5−/− mice show normal context-dependent fear conditioning. The time spent freezing 24 h after training in a one-trial fear-conditioning paradigm was assessed. Results indicate no difference in fear memory in the Hdac5−/− mice compared with Hdac5wt mice (Hdac5−/−, n = 7; Hdac5wt, n = 8).
Impaired long-term synaptic plasticity in Hdac4bko mice
Long-term changes in synaptic plasticity are widely believed to be the cellular basis for learning and memory. Therefore, we examined whether the impaired learning and memory behaviors seen after the loss of Hdac4 were associated with changes in LTP by performing field recordings on hippocampal slices prepared from Hdac4bko and Hdac4wt mice. LTP was induced in the CA1 of the hippocampus by three trains of theta burst stimulation on the Schaffer collateral pathway. The Hdac4bko and Hdac4wt mice developed an immediate increase in the field EPSP (fEPSP) slope (*p < 0.05), although the magnitude of enhancement of LTP was markedly reduced in Hdac4bko mice, suggesting a role for HDAC4 in the modulation of long-term synaptic plasticity in the hippocampus (Fig. 4A). We investigated whether the decreased LTP observed in Hdac4bko mice might be associated with alterations in basal synaptic properties. Input–output curves, generated by plotting fEPSP slopes against fiber volley amplitudes, were unchanged in Hdac4bko mice compared with Hdac4wt mice, indicating that basal synaptic transmission is largely unaltered in Hdac4bko mice (Fig. 4B). However, PPF, a short-term form of synaptic plasticity associated with the probability of neurotransmitter release, was decreased in Hdac4bko mice over a range of interevent intervals (20, 30, and 50 ms) consistent with an increase in presynaptic release probability, suggesting that loss of Hdac4 leads to alterations in presynaptic function (Fig. 4C). Together, these results indicate that the deficit seen in LTP after loss of Hdac4 is presumably independent of the increase in release probability as we could not detect a corresponding increase in input–output curves.
Figure 4.

Impaired hippocampal plasticity in Hdac4bko mice. A, Hdac4bko mice display impaired LTP at Schaffer collateral/CA1 pyramidal cell synapses compared with Hdac4wt animals as assessed by a two-way ANOVA with repeated measures (*p < 0.05; Hdac4wt, n = 7; Hdac4bko, n = 8). B, The averaged fEPSP plotted against presynaptic volley amplitude shows no significant difference in input–output function of CA1-fEPSPs in Hdac4bko mice compared with Hdac4wt controls (Hdac4wt, n = 7; Hdac4bko, n = 8). Input–output slopes were fit by linear regression and between group slopes were subsequently compared using unpaired t tests. C, The averaged paired-pulse facilitation for Hdac4wt and Hdac4bko slices at various intervals at SC-CA1 synapses. Using Student's t test, significant decreases in Hdac4bko mice were observed at the first three interstimulus interval data (20, 30, and 50 ms) (*p < 0.05; Hdac4wt, n = 7; Hdac4bko, n = 8).
Loss of Hdac4 or Hdac5 does not impact basal synaptic transmission
To further evaluate whether the LTP impairments observed with the Hdac4bko mice may be linked to differences in basal synaptic properties, we investigated whether loss of Hdac4 impacts unitary synaptic properties using whole-cell voltage-clamp recordings in primary hippocampal cultures. Knockdown of Hdac4 expression was achieved by infecting homozygous Hdac4loxP/loxP hippocampal cultures with a lentivirus expressing Cre recombinase at 4 DIV and then recording on 14–17 DIV. The successful knockdown of Hdac4 in culture following lentiviral-Cre expression was confirmed by quantitative PCR (data not shown). The knockdown of Hdac4 did not impact either the frequencies or amplitudes of mEPSCs, indicating lack of alterations in parameters such as excitatory synapse numbers, spontaneous release probability, as well as postsynaptic AMPA receptor numbers (Fig. 5A–C). We also investigated whether loss of Hdac5 impacts unitary synaptic properties in primary hippocampal cultures. We did not detect any alterations in mEPSC properties of Hdac5−/− compared with Hdac5wt hippocampal neurons (Fig. 5D–F). Together, these results support the premise that HDAC4 or HDAC5 are not required for maintenance of unitary properties of synaptic transmission in the hippocampus.
Figure 5.

Deletion of Hdac4 or Hdac5 in hippocampal neurons in culture does not perturb basal synaptic transmission. A, Representative recordings of miniature excitatory events recorded in 1 μm tetrodotoxin and 50 μm picrotoxin from homozygous Hdac4loxP/loxP neurons infected with either high-titer lentivirus expressing GFP and CRE-GFP. B, C, Bar graph depicts that mEPSC frequency and amplitudes remain unchanged upon Hdac4 knockdown. The numbers on the bars indicate the number of experiments. D, Representative recordings of miniature excitatory events recorded from Hdac5−/− and Hdac5wt neurons. E, F, Bar graphs reveal no changes in mEPSC frequency or amplitudes upon Hdac5 deletion. All the recordings were made on 14–17 DIV hippocampal neuronal cultures. Error bars indicate ±SEM.
Discussion
Our results provide the first genetic evidence that an individual class IIa HDAC, namely HDAC4, is specifically required for learning and memory and synaptic plasticity in mice. Although these findings appear to contrast with previous work showing that HDAC inhibition is a negative regulator of these processes (Korzus et al., 2004; Fischer et al., 2007; Guan et al., 2009), HDAC inhibition has largely been achieved with sodium butyrate, valproate, and suberoylanilide hydroxamic acid (SAHA), agents that preferentially target class I, but not class IIa or IIb, HDACs (Kilgore et al., 2010). Furthermore, our results demonstrate unique and functionally non-redundant roles for HDAC4 and HDAC5 in learning and memory as well as synaptic plasticity, suggesting that broad-scale manipulation of class I or class II HDACs may have differing effects on these processes.
The positive role for HDAC4 in hippocampal-dependent learning and memory, as well as well as LTP, agrees with recent work linking a haploinsufficiency of HDAC4 with brachydactyly mental retardation syndrome (Williams et al., 2010). Our findings imply that inhibition of HDAC4 activity may exert detrimental effects on learning and memory, as well related synaptic processes. Our initial assumption was that HDAC4 involvement in transcriptional control mediated the learning and memory and synaptic plasticity deficits. However, the use of microarray analysis to identify genes differentially regulated in the hippocampus by the loss of Hdac4 did not yield significant changes in gene expression (data not shown). Moreover, our findings that LTP is impaired immediately after its induction, at a time point typically insensitive to transcriptional inhibitors, in the Hdac4bko mice further suggests that these deficits are not due to acute activity-dependent regulation of transcriptional processes. The lack of gene expression changes by our microarray analysis suggests that the loss of Hdac4 is not altering the expression of a signaling molecule that inhibits early-phase LTP but rather that HDAC4 may play a role in nontranscriptional processes involved in LTP. Future studies will be necessary to better delineate the involvement of HDAC4 in these processes to determine whether HDAC4's activity-dependent shuttling, cytoplasmic functioning, or possibly deacetylation of endogenous proteins other than histones are involved.
Depending on the phosphorylation state and cellular location, class IIa HDACs associate with myocyte enhancer factor 2 (MEF2) proteins or the chaperone protein 14-3-3 (Haberland et al., 2009). Previously, we analyzed the role of MEF2C in neurons by generating a brain-specific deletion of MEF2C and showed that MEF2C plays a role in hippocampal-dependent learning and memory by suppressing the number of excitatory synapses and thus regulating basal and evoked synaptic transmission (Barbosa et al., 2008). Therefore, in future work, it will be critical to address whether HDAC4-dependent regulation of learning and memory requires MEF2C function.
Behavioral analysis of brain-specific Hdac4bko mice also revealed impairments in motor coordination, hyperactivity, and decreased anxiety-like behaviors. HDAC4 has been associated with muscle and bone development (Miska et al., 1999, 2001; Lu et al., 2000; McKinsey et al., 2000; Vega et al., 2004; Arnold et al., 2007) through specific effects mediated by its deactylase domain (Rajan et al., 2009). The loss of Hdac4 selectively in the brain resulted in decreased performance on the rotarod test, suggesting impairments in motor coordination. Together, these findings highlight the critical role of HDAC4 in neuromuscular development and function. In contrast, the finding that loss of Hdac4 in mice results in a decrease in anxiety-related behavior reveals an unexpected association between HDAC inhibition and anxiety. Chronic infusion of SAHA, a class I HDAC inhibitor, into the basolateral amygdala of adult mice has been shown to heighten anxiety-related behavior (Adachi et al., 2009). However, it has also been suggested that a putative link between HDACs and anxiety may encompass processes related to early life events. Early life experiences can impact adult behavior and have been suggested to involve epigenetic mechanisms. Administration of trichostatin A (TSA), a broad-acting HDAC inhibitor, into adult rodents reverses anxiety-related behavioral alterations that were the result of deficient maternal care (Weaver et al., 2004, 2006). These data support the hypothesis that the beneficial effects of trichostatin A on anxiety-related behavior may be due to the selective reduction of HDAC4. The ability of HDAC4 to negatively regulate anxiety-related behavior, as well as the demonstration it does not adversely impact synaptic transmission, may make it an attractive potential target for anxiety disorders.
We found that the global loss of Hdac5 does not impact any of the examined behavioral measures, including learning and memory. HDAC5 has been previously implicated in reward-related behaviors in that it is downregulated by chronic cocaine or stress, and loss of Hdac5 in mice results in hypersensitivity to chronic cocaine and stress (Renthal et al., 2007). Together, these findings suggest that HDAC5 does not play a significant role in hippocampal-dependent and other associated behaviors under normal conditions but may be a crucial factor under certain disease states such as addiction or chronic stress.
Whole-cell voltage-clamp recordings from hippocampal neurons showed that loss of Hdac4 or Hdac5 does not impact unitary synaptic transmission. Recently, our group has demonstrated that the pan HDAC inhibitor drug, TSA, triggers a decrease in mEPSC frequency in mature hippocampal neurons without altering synapse numbers, via a mechanism dependent on new gene expression (Nelson et al., 2006). A subsequent study showed that deletion of Hdac2, but not Hdac1, in mature hippocampal neurons results in a decrease in the frequency of mEPSCs with no change in synapse number, similar to the deficits in synaptic transmission observed with TSA (Akhtar et al., 2009). In these studies, the lack of an effect of HDAC1 on synaptic transmission in mature hippocampal cultures may not be that surprising given the low level of Hdac1 expression in postmitotic hippocampal neurons. In contrast, Hdac4 and Hdac5 are expressed at high levels in postmitotic hippocampal neurons similar to expression of Hdac2 (Broide et al., 2007). Yet the loss of Hdac4 or Hdac5 did not impact unitary synaptic transmission, suggesting specificity for individual HDACs in these processes. Together with this earlier work, our findings suggest that class I HDACs that are expressed in postmitotic neurons play critical roles in regulating unitary properties of excitatory neurotransmission as well as plasticity, while class II HDACs, which shuttle between the nucleus and cytoplasm in an activity-dependent manner, are not involved in unitary properties of synaptic function. Future work examining other individual HDACs in mature neurons will be critical to test the premise that class I and II HDACs are specialized with respect to their roles in basal neurotransmission.
HDAC inhibition has been linked to beneficial effects in learning and memory as well as synaptic plasticity for a range of neurological and psychiatric disorders. Our finding that HDAC4 acts as a positive regulator of learning and memory as well as synaptic plasticity highlights the complex roles HDACs play in the CNS and underscores the critical need for understanding the role of individual members of this gene family in the brain. Moreover, since transport of HDAC4 from the nucleus to the cytoplasm is regulated by calcium signaling (Backs et al., 2008), and alterations in calcium-dependent pathways have been linked to a wide variety of brain disorders with learning and memory deficits (Bezprozvanny and Hayden, 2004), it will be interesting to examine HDAC4 expression and activity in these pathological conditions. Given the recent efforts to develop HDAC inhibitors for a variety of disorders, the data from the Hdac4 and Hdac5 mutant mice reveal specific roles in neuronal plasticity deviating from the role of class I HDACs in these processes and emphasize the need for caution inferred by broad-scale pharmacological inhibition.
Footnotes
This work was supported by National Institutes of Health Grants MH081060 (L.M.M.) and MH066198 (E.T.K.) and The Robert A. Welch Foundation Grant I-0025 (E.N.O.).
The authors declare no competing financial interests.
References
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci. 2009;29:4218–4227. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar MW, Raingo J, Nelson ED, Montgomery RL, Olson EN, Kavalali ET, Monteggia LM. Histone deacetylases 1 and 2 form a developmental switch that controls excitatory synapse maturation and function. J Neurosci. 2009;29:8288–8297. doi: 10.1523/JNEUROSCI.0097-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell. 2007;12:377–389. doi: 10.1016/j.devcel.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Backs J, Backs T, Bezprozvannaya S, McKinsey TA, Olson EN. Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol. 2008;28:3437–3445. doi: 10.1128/MCB.01611-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa AC, Kim MS, Ertunc M, Adachi M, Nelson ED, McAnally J, Richardson JA, Kavalali ET, Monteggia LM, Bassel-Duby R, Olson EN. MEF2C, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc Natl Acad Sci U S A. 2008;105:9391–9396. doi: 10.1073/pnas.0802679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezprozvanny I, Hayden MR. Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun. 2004;322:1310–1317. doi: 10.1016/j.bbrc.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1–11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping strategies for mutant mice. Neuron. 2008;57:809–818. doi: 10.1016/j.neuron.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Dittgen T, Nimmerjahn A, Komai S, Licznerski P, Waters J, Margrie TW, Helmchen F, Denk W, Brecht M, Osten P. Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc Natl Acad Sci U S A. 2004;101:18206–18211. doi: 10.1073/pnas.0407976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 2006;59:468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes A. Targeted gene mutation approaches to the study of anxiety-like behavior in mice. Neurosci Biobehav Rev. 2001;25:261–273. doi: 10.1016/s0149-7634(01)00012-4. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Langley B, Lubin FD, Renthal W, Wood MA, Yasui DH, Kumar A, Nestler EJ, Akbarian S, Beckel-Mitchener AC. Epigenetics in the nervous system. J Neurosci. 2008;28:11753–11759. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Klingauf J, Tsien RW. Activity-dependent regulation of synaptic clustering in a hippocampal culture system. Proc Natl Acad Sci U S A. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279:40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Nef S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25:3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 1999;18:5099–5107. doi: 10.1093/emboj/18.18.5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Langley E, Wolf D, Karlsson C, Pines J, Kouzarides T. Differential localization of HDAC4 orchestrates muscle differentiation. Nucleic Acids Res. 2001;29:3439–3447. doi: 10.1093/nar/29.16.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16:710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Invest. 2007;117:2459–2467. doi: 10.1172/JCI31960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan I, Savelieva KV, Ye GL, Wang CY, Malbari MM, Friddle C, Lanthorn TH, Zhang W. Loss of the putative catalytic domain of HDAC4 leads to reduced thermal nociception and seizures while allowing normal bone development. PLoS One. 2009;4:e6612. doi: 10.1371/journal.pone.0006612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Shelton JM, Lee MH, Richardson JA, Patel SB. Microsomal triglyceride transfer protein expression during mouse development. J Lipid Res. 2000;41:532–537. [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell. 2004;119:555–566. doi: 10.1016/j.cell.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Vorhees CV, Williams MT. Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc. 2006;1:848–858. doi: 10.1038/nprot.2006.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver IC, Cervoni N, Champagne FA, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–854. doi: 10.1038/nn1276. [DOI] [PubMed] [Google Scholar]
- Weaver IC, Meaney MJ, Szyf M. Maternal care effects on the hippocampal transcriptome and anxiety-mediated behaviors in the offspring that are reversible in adulthood. Proc Natl Acad Sci U S A. 2006;103:3480–3485. doi: 10.1073/pnas.0507526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, Zondag S, Toriello HV, Magenis RE, Elsea SH. Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010;87:219–228. doi: 10.1016/j.ajhg.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]