

Abstract
Identifying effective small molecules that specifically target the p53 pathway in cancer has been an exciting, though challenging, approach for the development of anti-cancer therapy. We recently identified Inauhzin (INZ) as a novel p53 activator, selectively and efficiently suppressing tumor growth without displaying genotoxicity and with little toxicity to normal cells. In order to reveal the structural features essential for anti-cancer activity of this small molecule, we have synthesized a panel of INZ analogs and evaluated their ability to induce cellular p53 and to inhibit cell growth in cell-based assays. This study as described here leads to the discovery of INZ analog 37 that displays much better potency than INZ in both of p53 activation and cell growth inhibition in several human cancer cell lines including H460 and HCT116+/+ cells. This INZ analog exhibited much less effect on p53-null H1299 cells and HCT116−/− cells, and importantly no toxicity on normal human p53-containing WI-38 cells. Hence, our results not only unveil key chemical features for INZ activity, but also identify the newly synthesized INZ analog 37 as a better small molecule for further development of anti-cancer therapy.
Introduction
The p53 tumor suppressor protein can prevent the formation of tumors through several mechanisms, including the activation of cell-cycle checkpoints to prevent damaged cells from proliferation (cell-cycle arrest and DNA repair), the promotion of senescence (permanent cell-cycle arrest), and/or the triggering of cell death (apoptosis or autophagy) [1], [2]. It can also impede cell migration, metabolism, or angiogenesis, which are needed for cancer cell progression and metastasis [1]. Mutations of the tumor suppressor gene TP53 are detected in ∼50% of all types of human cancers [3], while the functions and stability of the p53 protein are often abrogated via posttranslational mechanisms in the rest of human cancers that contain wild type TP53 [4], [5]. Therefore, the restoration or reactivation of wild-type p53 function can lead to rapid elimination of tumors. As such, compounds that target the p53 pathway have become promising anticancer drug candidates, and several of them have entered clinical trials [4], [6]. For instance, Nutlin-3 and MI-219 can increase p53 level and activity by interfering with the p53-MDM2 binding [7]–[9]. Even though there have been extensive endeavors to find small molecules that target the p53 pathway, none has yet proven to be clinically effective therapeutics due to the inherent undesirable toxicity to normal cells and tissues.
Through our recent efforts in conducting in silico screening and cellular-based assays [10], we discovered Inauhzin (INZ) and its analogs ( Figure 1 ) as a novel class of small molecules that effectively activate p53 and promote p53-dependent apoptosis of human cancer cells without causing apparently genotoxic stress. In addition, INZ stabilized p53 by increasing p53 acetylation and preventing MDM2-mediated ubiquitylation of p53 in cells. Remarkably, INZ inhibited cell proliferation, induced senescence and tumor-specific apoptosis, and repressed the growth of xenograft tumors derived from p53-harboring lung cancer H460 and colon cancer HCT116+/+ cells without causing apparent toxicity to normal tissues.
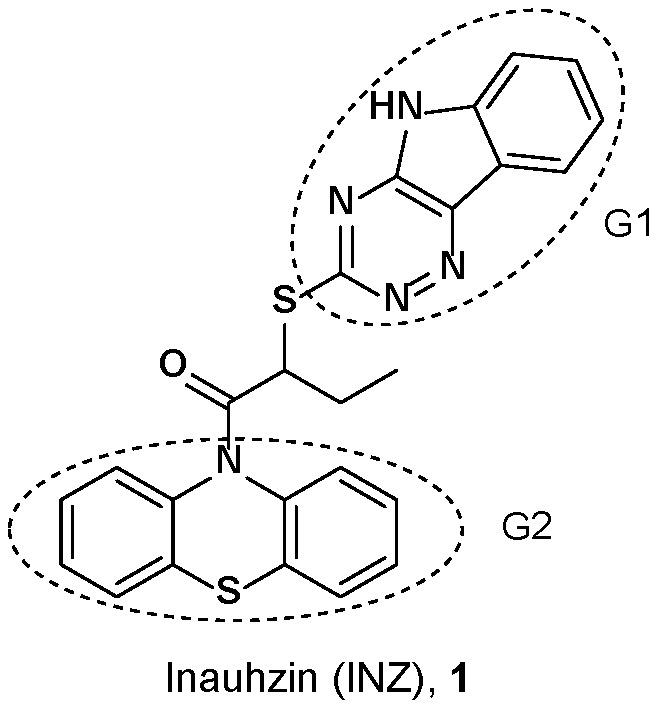
Figure 1. Structure of Inauhzin (INZ).
INZ is an effective anti-cancer agent either alone or in combination with Nutlin treatment [11] or DNA damage agents Cisplatin and Doxorubicin (unpublished). Single treatment with Nutlin-3 is less efficient in inhibiting the growth or promoting apoptosis of some cancer cells, such as HCT116+/+, H460, or A549, in xenograft tumor models even though these cells contain wild type p53. Combination of INZ with Nutlin-3 synergistically promotes apoptosis in HCT116+/+ and H460 cell lines in a p53-dependent fashion. This combination also synergistically activates p53 in xenograft tumors derived from these cancer cells and significantly suppresses their growth.
To further characterize the structural features essential for the activity of this group of small molecules to induce p53 and to suppress cell proliferation, we initiated structure-activity relationship (SAR) analyses of INZ analogs. To this purpose, we synthesized a number of new INZ analogs and also evaluated their capability of p53 induction and cell growth inhibition using cell-based assays. Our study not only reveals critical chemical groups for INZ activity, but also leads to the discovery of INZ derivative 37 that displays better potency in p53 induction and cancer cell growth inhibition than does INZ.
Results and Discussion
Design and Chemical Synthesis
Within its structure, INZ (1) possesses two distinct chemical components: triazino[5,6-b]indol (G1) and phenothiazine (G2) moiety ( Figure 1 ). In our preliminary SAR studies [10], we purchased 46 compounds analogous to INZ with diversities of G1 and G2 and investigated the activity of the compounds in cell-based assays for their ability to induce p53 levels in p53 containing human colon cancer HCT116+/+ cells and/or human lung cancer H460 cells using immunoblotting (IB) ( Figure 2 and Figure 3 ). The results indicated that a unique structure scaffold might be required for the activity of INZ in cells. Removal of the ethyl group at R1 (S1–S3) or modification at both R2 and R3 positions on the indol moiety of INZ (S4) disabled the compound's ability to activate p53 in cells ( Figure 2 ). The R2 position can be modified and substituted without loss of activity by replacing it with some alkyl groups, such as methyl, ethyl and allyl, but not propyl (S5–S8). Both triazino[5,6-b]indol (G1) and phenothiazine (G2) are essential fuctional groups for p53 induction. The analogs containing ethyl group at the R1 position but lacking either functional groups G1 (S9–S10), or G2 (S19–S22) failed to induce p53. Compounds S11–S18, S23–S28, and S29–S34 with different aromatic moieties other than triazino[5,6-b]indol at G1 and/or phenothiazine at G2 had very low or no activity. Overall, the results suggest that a specific chemical structure with the intact triazino[5,6-b]indol-3-ylthio)butanoyl]-10H-phenothiazine might be crucial for p53 activation in cells. Indeed, INZ (1) displayed more potent p53 activation and anticancer inhibition than either of its component fragments, compound 2′ or 3′ ( Figure 4 , and data not shown). It suggests that a synergism is achieved when these two structural units are combined within a single molecule. Therefore, we focused our attention on the structural modifications on the pharmacologically active core: triazino[5,6-b]indol or phenothiazine. Modifications included extension of cabon chain length on R1 (14) ( Figure 5 ), the substitution on the phenothiazine ring (G2) (6–13) ( Figure 5 ) or on the triazino[5,6-b]indol ring (G1) (15–36) ( Figure 5 ).
Figure 2. Cellular Activity of INZ Analogs S1–S34.

(A–B) Initial Inauhzin analogs were purchased and tested the activity on H460 and HCT116+/+ by immunoblotting (IB). Cells were treated with the compounds at 2 µM or 20 µM for 18 hrs and harvested for IB and their p53 induction activity as quantified from IB data as shown in (A–B).
Figure 3. Table of Chemical Structures of Representative Commercial Analogs S1–S34.

Figure 4. Synthesis of INZ Analogs 20–27.

Figure 5. Table of Chemical Structures of INZ Synthetic Analogs 6–36.

The syntheses of these new INZ derivatives are outlined in Figures 4 , 6 , 7 , and 8 .
Figure 6. Synthesis of INZ Analogs 6–19.

Figure 7. Synthesis of INZ Analogs 28–30.

Figure 8. Synthesis of INZ Analogs 31–36.

The synthesis of compounds INZ (1) and 6–19 was outlined as Figure 6 . The 5H-[1, 2, 4] triazino[5,6-b]indole-3-thiol 3 was prepared from the commercial isatin according to the standard procedure [12]. The bromide 5 was synthesized through refluxed thiophenol with the bromobutyryl bromide in toluene. Then the thiol 3 was reacted with bromide 5 in the presence of Et3N and afforded compound 1, and 6–19. Other bases were tested and some byproducts were produced, which gave rise to low yields.
The amide derivatives 20–27 were prepared in one step from INZ (1) in the presence of organic bases as depicted in Figure 4 .
The amine derivative 28 was synthesized from INZ (1) and ethyl bromoacetate in the presence of K2CO3, which was depicted in Figure 7 . Other organic bases, such as Et3N or DIPEA, were tested and the reaction proceeded very slowly with low yields. Compound 28 was hydrolyzed by 1 M NaOH and afforded the acid 29. The alcohol 30 was obtained through reduction of 28 by NaBH4. LiBH4 was tested and several byproducts were produced as revealed by TLC analysis. Figure 8 shows the “click chemistry” for the synthesis of triazol derivatives. Triazols 34–36 were obtained in good yields through the reaction of azide derivative and the propargyl 31 and 32 under the standard conditions [13].
Biological assessments of INZ analogs
The synthetic analogs were then assayed for their potential to induce p53 level and activity in H460 cells and HCT116+/+ cells by IB. Compounds were added into cultured H460 and HCT116+/+ cells at 0.5, 2, 10 µM for 18 hrs and harvested for IB. The p53 activation was assessed by up-regulating the levels of MDM2, p53 and p53 acetylation. The induction level of p53 by each of the tested INZ analogs was normalized against the loading control of GAPDH and compared to the level of p53 in the cells treated with 2 µM INZ ( Figure 9 ). Compounds showing good efficacy in p53 induction were further subjected to a 3-day WST assay to assess their ability to kill cancer cells. INZ was used along with the analogs as a positive control in each assay. The EC50 values for their ability to inhibit cell growth were calculated through serial dilution of their concentrations with the highest concentration at 50 µM in H460 and HCT116+/+ cells or 100 µM in H1299 and HCT116−/− cells. Four-parameter or two-parameter Hill equation was employed to calculate and plot the dose-response curves as shown with some representative compounds in Figure 10 . EC90 values were calculated from the EC50 and Hill slope by a web-based calculator: http://www.graphpad.com/quickcalcs/Ecanything1.cfm.
Figure 9. Cellular Activities of INZ Synthetic Analogs 6–37.

(A–B) Cellular activity of INZ synthetic analogs 6–37 measured using IB that detects p53 levels and activity in H460 and HCT116+/+ cells. Cells were harvested for IB with antibodies as indicated after being treated with each compound for 18 hrs as shown in representative blots from HCT116+/+ cells (A) (number denotes each compound; Inauhzin, INZ), and their p53 induction activity as quantified from IB data as shown in (B).
Figure 10. Cell Growth Inhibition by INZ Synthetic Analogs.

(A) Structure of INZ synthetic analog 37. (B) Representative cell growth inhibition curves of INZ synthetic analogs 8, 30 and 37 in H460, H1299, HCT116+/+ and HCT116−/− cell lines. (C) a EC50 of the selected INZ analogs represent the average of triplicates. The EC50 values were determined by the two-parameter Hill equation where EC50 and the Hill coefficient were allowed to refine while the maximal and minimal values remain fixed. b EC90 values were calculated from the EC50 and Hill slope by a web-based calculator: http://www.graphpad.com/quickcalcs/Ecanything1.cfm. c Not be able to be determined.
Anti-proliferative Effect of Synthetic INZ analogs
In synthetic INZ analogs containing triazino[5,6-b]indol (G1), subtle and major modifications to phenothiazine ring (G2) generally led to less potent molecules. Though subtle changes on the branches of the phenothiazine ring were tolerated (for instance, compounds 6 and 7 with chlorine or methoxy remained active in p53 induction) ( Figure 5 and Figure 9 ), they did not reach 50% p53 induction in H460 cells and HCT116+/+ cells at 2 µM. The removal of any ring of G2, as shown for compound 10–13 ( Figure 5 ), caused loss of activity, and those compounds were essentially inactive ( Figure 9 ). The exception to this trend was substitution of the sulfur atom with methylene (8). 1-acridin-INZ derivative (8) drastically induced p53 at 0.5 µM, whereas compound 9, whose sulfur was substituted with oxygen, was inactive ( Figure 9 ). It should be noted that 1-acridin-INZ (8) also exhibited more than 2 fold higher potency than did INZ in its inhibitory effect on H460 (EC50 = 2.7 µM) ( Figure 10 ) and HCT116+/+ cells (EC50 = 1.3 µM) ( Figure 10 ). The EC90 values of this analog were in the range of 3.5–10 µM, which were 3–10 fold lower than those for INZ.
Compound 14 ( Figure 5 ) with the longer chain containing butyl at R1 position exhibited lower activity for p53 induction, which further indicated that the appropriate length of alkyl chain at R1 position is crucial for the activity of INZ, as INZ activity in p53 activation was reduced or lost when the chain was either longer than 2 carbons (14, Figure 5 ) or removed (S1–S3, Figure 3 ). Compounds 15–19 ( Figure 5 ) were synthesized to determine the effect of different substituents, such as electron-withdrawing group (halogen atoms) and electron donating group (methyl or methoxy), at R3 position of indole ring (G1) on p53 induction. Compounds 16 and 17, which have a chlorine and bromine atom, respectively, exhibited similar activity to that of INZ in HCT116+/+ cells with a dose-dependent induction of p53 acetylation at lysine 382, p53 protein level and the up-regulation of MDM2 level ( Figure 9 ). Compound 18 with a methoxy group displayed a marked decrease in p53 activation. In contrast, the methyl derivative 19 exhibited a significant effect on p53 induction compared to INZ at 0.5 µM. It also inhibited the proliferation of H460 and HCT116+/+ with EC90 values of ∼20–30 µM, which were 1.5 fold lower than that for INZ ( Figure 10C ). These results indicate that the order of influence of these substituents on the antiproliferative activity of INZ is as follows: CH3>Cl>Br>F>OCH3.
The results from our preliminary biological screening of INZ analogs (S5–S8, Figure 3 ) suggested that R2 position could be modified. We conjugated biotin directly to INZ through the formation of the amide bond at the active hydrogen of R2 and gained compound 20 ( Figure 5 ). This biotin-conjugated INZ was initially designed for target identification studies. To our delight, the biotinylated INZ (20) was as effective as INZ in the induction of p53 acetylation and level in both H460 and HCT116+/+ cells [10] ( Figure 9B ). Another biotin-conjugated compound derived from the inactive compound 15 was used as a negative control in the target identification screening (data not shown). In addition to compound 20, some other amide compounds (21–27) ( Figure 5 ) were made through the same procedure. All these compounds with various ketone substitutions on R2 exhibited good activities in p53 induction and cell growth inhibition in comparison with INZ ( Figure 9 and 10 ). Derivatives 20, 21 and 27 showed similar EC90 values of 7.5, 9.6 and 9.0 µM, respectively whereas INZ is about 39.9 µM. Removal (25→32, Figure 5 ) or separation (21→33, Figure 5 ) of the carbonyl group from the indol resulted in a significant decrease in activity ( Figure 9 ). Replacing the ketone with an ester (28) or carboxylic acid (29) resulted in essentially inactive analogs, in striking contrast to its alcohol derivative 30, which was comparable to compound 8 in p53 activation and cell growth inhibition ( Figure 5 , 9 , and 10 ). The EC90 values of compound 30 as tested in H460 and HCT116+/+ cells, respectively, were ∼7.7 µM and 4.6 µM, which was 5 fold lower than that of INZ ( Figure 10 ). Since compounds 8 and 30 displayed more potent activity compared to INZ, we synthesized the analog 37 that contains both substitution of the sulfur atom with methylene on G2 and alcohol substitution on G1. We found that compound 37 was remarkably 10- and 5-fold more active than was INZ in growth inhibition of H460 and HCT116+/+ cells (EC50 = 0.7 µM and 0.5 µM), respectively.
INZ displayed much higher toxicity to p53-containing human cancer cells than to p53-null cancer cells. This was evident in the EC50 and EC90 values, which were 1.5 and 5–7 fold greater in p53-null cells than in p53-containing cell lines, respectively ( Figure 10C ). We further examined the activity of INZ synthetic analogs by conducting in vitro cytotoxicity assays using p53 null lung cancer H1299 cells and colon cancer HCT116−/− cells. Compounds 8, 19, 21, 30 and 37 were demostrated much less effective in H1299 cells and HCT116−/− cells, in contrast to their inhibitory activity on p53-containing cells ( Figure 10C ), as the EC50 values of compounds 8, 30 and 37 on H1299 were 10.4, 16.6 and 11.2 µM, respectivly, which were 3–15 fold higher than those on H460 cells. The EC90 values of compound 8, 30 and 37 on H1299 cells and HCT116−/− cells were greater than 50 µM whereas those on H460 and HCT116+/+ cells were 3.5 and 10, 7.7 and 4.6, and 3.6 and 5.0 µM, respectivly. More remarkably, these synthetic analogs were much less toxic to normal human fiberbrast cell WI-38 ( Figure 10C ), while they were much more potent than was INZ in killing p53-containing cancer cells. For example, the EC50 value of compound 37 for WI-38 was unable to be determined at the highest concentration tested (50 µM) in comparison of its EC50 values of 0.7 and 0.5 µM to p53-containing H460 and HCT116+/+ cells, respectively. Together, these results indicate that these more potent INZ analogs, such as compounds 8, 30 and 37, possess strong p53-dependent cytotoxicity. Among them, compound 37 stands out as the most effective INZ analog from this study.
Conclusion
Our initial studies on the 46 commercial analogs of INZ yielded information on the important functional groups at each of its two scaffolds indentified as triazino[5,6-b]indol ring (G1) and phenothiazine ring (G2). The functional analyses of the commerical and synthetic analogs of INZ for their ability to activate p53 and to inhibit cell growth further as described above validate that each of the functional groups of INZs is critical for p53 activation and inhibition of cancer cell growth ( Figure 11 ). Most modifications to phenothiazine ring G2, such as the branch substitutions (6–7), or replacement with other rings (9–13, S19–S22), led to the decreased activity in p53 induction, with the exception of that the substitution of sulfur in the G2 region by methylene (1→8) showed greater potentcy than compound 1 in both p53 induction and cancer cell inhibition. Analyses of analogs S1–S3, and 14 demonstrate that the ethyl group at R1 is required for the activity of these compounds. The butyl group was tolerated. Modification of R3 position at the region G1 with methyl, but not halide or methoxy substitutions, increased activity in both of the assays (15–19). Most modifications on R2 at the G1 region resulted in the impressive improvement in terms of p53 activation compared to compound 1 (20–27, 30–31). Overall, the best compound from this study was 1-acridin-INZ acohol (37). The potency of this analog, compared to INZ, was improved nearly 5- to 10-fold in cancer growth inhibition. Interestingly and importantly, compounds 8, 30 and 37 were more potent in p53 activation than their parental compound INZ especially with the selective toxicity to p53-containing tumor cells, but not to normal cells.
Figure 11. Structure-Activity Relationships of INZ.

Based on these SAR and cell-based analyses as described here, 1-acridin-INZ acohol (37) represented the most promising candidate for further development and will be selected for further characterization of its biological activity against cancer by using orthotopic lung tumors derived from H460 cells in the near future.
Materials and Methods
Compounds S1–S34
INZ analogs S1–S34 were purchased from Asinex, ChemDiv and ChemBridge. Compounds S1–S5 were described in a preceding paper, re-validated by LC/MS on an Agilent 1200 LC/MS system (Agilent Technology) at the Chemical Genomics Core Facility of Indiana University School of Medicine. The minimum purity of all compounds is higher than 90%.
Cell Culture and Immunoblotting Analysis
Human lung carcinoma H460, non-small-cell lung cancer H1299, human colon cancer HCT116 (HCT116+/+), and human embryonic fibroblast WI-38 were bought from the American Type Culture Collection (ATCC). Human colon cancer HCT116 p53 null cell lines (HCT116−/−) were generously offered by Dr. Bert Vogelstein (Johns Hopkins University) [14]. Those cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U per mL penicillin, and 100 U per mL streptomycin. Compounds were dissolved in DMSO and diluted directly into the medium to the indicated concentrations; 0.1% DMSO was used as a control. After incubation with the compounds for 18 h, cells were harvested and lysed in 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40 supplemented with 1 mM DTT and 0.2 mM PMSF. An equal amount of protein samples (50 µg) was subjected to SDS-PAGE and transferred to a PVDF membrane (PALL Life Science). The membranes with transferred proteins were blocked with 1×TBST containing 5% non-fat, dried milk for 1 h at room temperature, and then incubated with anti-p53-acetylated (lys382, Cell Signaling), anti-p53 (mouse monoclonal, DO-1, Santa Cruz), anti-MDM2 (4B11) [15], or anti-GAPDH antibodies (Sigma) followed by a secondary antibody labeled with horseradish peroxidase (Pierce). The blots were developed by an enhanced chemiluminescence detection kit (Thermo Scientific), and signals were visualized by Omega 12iC Molcular Image System (UltraLUM).
Cell Viability Assay
To assess cell growth, the cell counting kit (Dojindo Molecular Technologies Inc., Gaithersburg, Maryland) was used according to manufacturer's instructions. Cell suspensions were seeded at 3,000 cells per well in 96-well culture plates and incubated overnight at 37°C. Compounds were added into the plates and incubated at 37°C for 72 hrs. Cell growth inhibition was determined by adding WST-8 at a final concentration of 10% to each well, and the absorbance of the samples was measured at 450 nm using a Microplate Reader (Molecular Device, SpecrtraMax M5e). EC50 values were determined by the Hill equation using Igor 4.01 (Lake Oswego, Oregon, USA). EC90 values were calculated from the EC50 and Hill slope by a web-based calculator: http://www.graphpad.com/quickcalcs/Ecanything1.cfm.
General Chemistry
All purchased chemicals were reagent-grade or better. Proton and carbon NMR spectra were recorded on a 500 MHz Bruker Avance II spectrometer. Chemical shifts are reported in δ (parts per million, ppm) with the δ 7.26 signal of CDCl3 (1HNMR), δ 2.50 signal of DMSO-d 6 (1H NMR), or δ 77.2 signal of CDCl3 (13C NMR) as internal standards. All column chromatography was performed using Dynamic Adsorbents 230–400 mesh silica gel (SiO2) with the indicated solvent system unless otherwise noted. TLC analysis was performed using 254 nm glass-backed plates and visualized using UV light (254 nm). HRMS data were obtained at the Mass Spectrometry Facility at IUPUI Chemistry Department on a Waters/Macromass LCT. All the synthetic compounds were analyzed by LC/MS on an Agilent 1200 LC/MS system (Agilent Technology) at the Chemical Genomics Core Facility of Indiana University School of Medicine and the purity was over 95%.
General procedure for synthesis of compounds 1, 6–19
2-((5H-[1,2,4]triazino[5,6-b]indole-3yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one(1, INZ)
2-bromo-1-(10H-phenothiazin-10-yl)butan-1-one (3.675 g, 25 mmol) and 5H-[1,2,4]triazino[5,6-b]indole-3-thiol (2.125 g, 25 mmol) were dissolved in 50 ml anhydrous DMF and cooled to 0°C. 11.1 ml Et3N (250 mmol) was dropped to the above mixture. After stirring for1.5 h, TLC indicated that the reaction was completed and stopped. 300 ml ethyl acetate was added to the reaction mixture. The organic phase was washed by saturated NH4Cl for five times and separated. It was dried by anhydrous Na2SO4 and filtered. The organic phase was concentrated to about 15 ml and the pale solid was formed. The amorphous solid was collected and washed by a few ethyl acetate. 1H NMR (500 MHz, DMSO-d6) δ; 12.63 (br, 1H), 8.34 (d, J = 7.5, 1H), 7.89–7.96 (m, 1H), 7.56–7.73 (m, 4H), 7.37–7.47 (m, 5H), 7.29–7.22 (m, 1H), 5.27 (t, J = 7.0, 1H), 1.86 (br, 1H), 1.74 (br, 1H), 0.85 (br, 3H). 13C NMR (125 MHz, DMSO-d6) δ 170.0, 146.8, 141.7, 140.9, 138.5, 138.3, 131.4, 128.6, 128.2, 128.0, 127.7, 127.4, 123.0, 122.0, 118.0, 113.2, 31.1, 25.9, 11.8. HRMS was calculated for C25H19N5OS2 469.1031 and found 469.1047.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(2-chloro-10H-phenothiazin-10-yl)butan-1-one (6)
Compound 6 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.64 (d, J = 17.5, 1H), 8.33–8.35 (m, 1H), 7.84–7.91 (m, 1H), 7.60–7.34 (m, 4H), 7.39–7.48 (m, 5H), 5.15–5.26 (m, 1H), 1.82 (br, 1H), 1.73 (br, 1H), 0.85 (br, 3H). HRMS calcd for C25H18ClN5OS2 503.0641; found 503.0643.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(2-methoxy-10H-phenothiazin-10-yl)butan-1-one (7)
Compound 7 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.65(d, J = 22.5, 1H), 8.32–8.35 (m, 1H), 7.59–7.73 (m, 1H), 7.36–7.53 (m, 7H), 7.21 (br, 1H), 6.96–7.21 (m, 1H), 5.29 (t, J = 7.0, 1H), 3.51–3.71 (m, 3H), 1.95 (br, 1H), 1.77 (br, 1H), 0.86 (br, 3H). HRMS calcd for C26H21N5O2S2 499.1137; found 499.1144.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(acridin-10(9H)-yl)butan-1-one (8)
Compound 8 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.60 (br, 1H), 8.32 (d, J = 7.5, 1H), 7.68–7.73 (m, 3H) 7.59 (d, J = 8.0, 1H), 7.44–7.47 (m, 1H), 7.17–7.34 (m, 6H), 5.41 (s, 1H), 3.86 (s, 2H), 2.10 (br, 1H), 1.86 (br, 1H), 0.95 (br, 3H). 13C NMR (125 MHz, DMSO-d6) δ 169.9, 166.1, 146.7, 141.8,140.9, 139.2, 135.2, 131.4, 127.8, 126.7, 126.6, 125.5, 122.9, 122.0, 117.9, 113.2, 47.3, 33.4, 26.2, 11.9. HRMS calcd for C26H21N5OS 451.1467; found 451.1474.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenoxazin-10-yl)butan-1-one (9)
Compound 9 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.63 (br, 1H), 8.33 (d, J = 8.0, 1H), 7.70–7.34 (m, 3H), 7.60 (d, J = 8.0, 1H), 7.46 (d, J = 7.5, 1H), 7.19–7.25 (m, 6H), 5.47 (t, J = 7.0, 1H), 1.99–2.03 (m, 1H), 1.81–1.86 (m, 1H), 0.89 (t, J = 7.5, 3H). HRMS calcd for C27H19N5O2S 453.1259; found 453.1270.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(2H-benzo[b][1,4]thiazin-4(3H)-yl)butan-1-one (10)
Compound 10 was synthesized similarly to 1. 1H NMR (500 MHz, CDCl3) δ 10.15 (s, 1H), 8.38 (d, J = 7.5, 1H), 7.60–7.66 (m, 3H), 7.43 (t, J = 7.5, 1H), 7.23 (br, 1H), 7.12–7.14 (m, 2H), 5.30 (br, 1H), 3.30 (br, 3H), 2.14 (d, J = 7.0, 1H), 2.00 (br, 2H), 1.08 (br, 3H). HRMS calcd for C21H19N5OS2 421.1031; found 421.1038.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(3,4-dihydroquinolin-1(2H)-yl)butan-1-one (11)
Compound 11 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.60 (br, 1H), 7.69–7.72 (m, 1H), 7.58 (d, J = 8.0, 1H), 7.45 (t, J = 7.0, 1H), 7.35 (br, 1H), 7.12–7.16 (m, 2H), 7.06 (t, J = 7.5, 1H), 5.28 (t, J = 6.5, 1H), 3.97 (br, 1H), 3.55 (br, 1H), 2.64–2.76 (m, 2H), 2.04 (br, 2H), 1.86 (br, 2H), 0.89 (br, 3H). HRMS calcd for C22H21N5OS 403.1467; found 403.1479.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-thiomorpholinobutan-1-one (12)
Compound 12 was synthesized similarly to 1. 1H NMR (500 MHz, CDCl3) δ 10.88 (br, 1H), 8.37 (d, J = 8.0, 1H), 7.63–7.64 (m, 2H), 7.40–7.43 (m, 1H), 5.35 (t, J = 7.0, 1H), 4.12–4.19 (m, 2H), 4.03–4.05 (m, 1H), 3.86–3.89 (m, 1H), 2.92–2.95 (m, 1H), 2.72–2.77 (m, 1H), 2.60–2.64 (m, 2H), 2.19–2.26 (m, 1H), 2.04–2.10 (m, 1H), 1.13 (t, J = 7.0,3H). HRMS calcd for C17H19N5OS2 373.1031; found 373.1033.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(2,3-dihydro-1H-pyrrolo[3,2-b]pyridine-1-yl)butan-1-one (13)
Compound 13 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.56 (s, 1H), 8.28 (d, J = 8.0, 1H), 8.08 (d, J = 4.5, 1H), 7.67–7.71 (m, 2H), 7.56 (d, J = 8.5, 1H), 7.42 (t, J = 7.5, 1H), 7.00–7.03 (m, 1H), 6.59 (br, 1H), 4.05 (t, J = 8.5, 2H), 3.12 (q, J = 7.5, 2H), 2.10–2.15 (m, 1H), 1.98–2.04 (m, 1H), 1.07 (t, J = 7.5, 3H). HRMS calcd for C20H18N6OS 390.1263; found 390.1274.
2-((5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)hexan-1-one (14)
Compound 14 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.62 (br, 1H), 8.35 (d, J = 7.5, 1H), 7.96 (br, 1H), 7.58–7.74 (m, 4H), 7.37–7.48 (m, 5H), 7.26 (br, 1H), 5.32 (t, J = 7.0, 1H), 1.86 (br, 1H), 1.83 (br, 1H), 1.09–1.24 (m, 4H), 0.73 (br, 3H). HRMS calcd for C27H23N5OS2 497.1344; found 497.1348.
2-((7-fluoro-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one (15)
Compound 15 was synthesized similarly to compound 1. 1H NMR (500 MHz, DMSO-d6) δ 12.70 (br, 1H), 8.17 (d, J = 7.0, 1H), 7.89 (br, 1H), 7.56–7.64 (m, 4H), 7.37–7.43 (m, 4H), 7.22–7.25 (m, 1H), 5.26 (t, J = 7.0, 1H), 1.95 (br, 1H), 1.74 (br, 1H), 0.85 (d, J = 7.0, 3H). HRMS calcd for C25H18FN5OS2 487.0937; found 487.0962.
2-((7-chloro-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one (16)
Compound 16 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.79 (br, 1H), 8.38 (s, 1H), 7.88 (br, 1H), 7.73–7.79 (m, 1H), 7.49–7.63 (m, 3H), 7.37–7.43 (m, 4H), 7.23 (br, 1H), 5.26 (t, J = 7.0, 1H), 1.86 (br, 1H), 1.74 (br, 1H), 0.86 (br, 3H). HRMS calcd for C25H18ClN5OS2 503.0641; found 503.0641.
2-((7-bromo-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one (17)
Compound 17 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.84 (br, 1H), 8.50 (s, 1H), 7.85 (d, J = 8.0, 2H),7.58 (d, J = 8.5, 3H), 7.38–7.41 (m, 4H), 7.23 (br, 1H), 5.26 (s, 1H), 1.86 (br, 1H), 1.73 (br, 1H), 0.85 (br, 3H). HRMS calcd for C25H18BrN5OS2 547.0136; found 547.0136.
2-((7-methoxy-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one (18)
Compound 18 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ; 12.47 (br, 1H), 7.89 (br, 1H), 7.85 (br, 1H), 7.52–7.64 (m, 4H), 7.37–7.43 (m, 3H), 7.32–7.34 (m, 1H), 7.22 (br, 1H), 5.25 (t, J = 7.0, 1H), 1.86 (br, 1H), 1.74 (br, 1H), 0.85 (br, 3H). HRMS calcd for C26H21N5O2S2 499.1137; found 499.1136.
2-((7-methyl-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one (19)
Compound 19 was synthesized similarly to 1. 1H NMR (500 MHz, DMSO-d6) δ 12.50 (br, 1H); 8.13 (s, 1H), 7.87 (br, 1H), 7.49–7.61 (m, 5H), 7.37–7.42 (m, 3H), 7.22 (br, 1H), 5.25 (d, J = 6.5, 1H), 2.52 (s, 3H), 1.90 (br, 1H), 1.74 (br, H), 0.86 (br, 3H). HRMS calcd for C26H21N5OS2 483.1188; found 483.1196.
General procedure for synthesis of compounds 20–27
5-(5-oxo-5-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1,2,4]triazino[5,6-b]indol-5-yl)pentyl)tetrahydro-1H-thieno[2,3-d]imidazole-2(5H)-one (20)
Biotin (100 mg, 0.410 mmol) was placed in 10 ml reaction flask and cooled to 0°C. 2.7 ml SOCl2 was added to the flask and allowed to room temperature. The mixture was stirred for 1 h and excess SOCl2 was evaporated. The residue was co-evaporated with 5 ml anhydrous toluene for three times to give the biotin acid chloride. The crude acid chloride was dissolved in 5 ml anhydrous THF. INZ (65 mg, 0.138 mmol) was dissolved in 3 ml anhydrous THF and injected to the above solution through syringe. The mixture was cooled to 0°C and 100 µl Et3N (0.717 mmol) was dropped to the mixture. The solution was then allowed to room temperature. TLC was used to monitor the reaction. After 11 h, TLC indicated that the reaction was completed. The reaction mixture was diluted with 30 ml ethyl acetate and washed by saturated NaCl for two times. The organic phase was separated and dried by anhydrous Na2SO4. The organic phase was filtered, concentrated in vacuum and was purified by column (DCM/CH3OH, 55∶1). The product was obtained as viscous oil. 1HNMR (500 MHz, CDCl3) δ 8.69 (d, J = 8.5, 1H), 8.40 (d, J = 7.5, 1H), 7.92 (br, 1H), 7.75–7.72 (m, 1H), 7.67 (d, J = 7.0, 1H), 7.59–7.56 (m, 1H), 7.53 (d, J = 3.0, 1H), 7.40 (br, 1H), 7.35–7.29 (m, 3H), 7.18 (br, 1H), 5.60 (d, J = 39.5, 1H), 5.42–5.38 (m, 1H), 5.14 (s, 1H), 4.56–4.53 (m, 1H), 4.41–4.37 (m, 1H), 3.48–3.41 (m, 1H), 3.35–3.26 (m, 1H), 3.25–3.23 (m, 1H), 2.98–2.95 (m, 1H), 2.76 (d, J = 12.5, 1H), 1.90–1.83 (m, 4H), 1.79–1.75 (m, 1H), 1.70 (br, 1H), 1.61–1.58 (m, 2H), 0.98–0.90 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 173.1, 170.0, 167.7,163.7,146.7, 142.4, 139.5, 138.5, 138.3, 132.2, 127.7, 127.5, 127.3, 127.0, 126.8, 125.9, 121.4, 119.6, 117.8, 62.0, 60.4, 55.4, 55.3, 40.6, 39.1, 28.5, 28.4, 26.0, 24.2, 11.7. HRMS calcd for C35H33N7O3S3 695.1807; found 695.1817.
2-((5-benzoyl-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one (21)
Compound 21 was synthesized similarly to 20. 1H NMR (500 MHz, DMSO-d6) δ 8.49 (d, J = 7.5, 1H), 8.23 (d, J = 8.5, 1H), 7.60–7.78 (m, 6H), 7.40–7.55 (m, 4H), 7.17–7.28 (m, 4H), 7.06 (br, 1H), 4.89 (t, J = 6.5, 1H), 1.93 (t, J = 6.5, 1H), 1.57–1.62 (m, 1H), 0.75 (t, J = 7.0, 3H). HRMS calcd for C32H23N5O2S2 573.1293; found 573.1298.
2-((5-nicotinoyl-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)-1-(10H-phenothiazin-10-yl)butan-1-one (22)
Compound 22 was synthesized similarly to 20. 1H NMR (500 MHz, CDCl3) δ 8.97 (s, 1H), 8.91 (d, J = 4.0, 1H), 8.49 (d, J = 8.0, 1H), 8.41 (d, J = 8.5, 1H), 8.03–8.05 (m, 1H), 7.78–7.84 (m, 2H), 7.66 (t, J = 7.5, 1H), 7.58 (d, J = 7.0, 1H), 7.48–7.51 (m, 2H), 7.34 (br, 1H), 7.26–7.28 (m, 3H), 7.12 (br, 1H), 5.08 (t, J = 6.0, 1H), 1.94–1.96 (m, 1H), 1.67–1.70 (m, 1H), 0.83 (t, J = 6.5, 3H).13C NMR (125 MHz, CDCl3) δ 169.9, 167.7, 166.4,153.8, 150.6,146.7, 141.9, 139.3, 138.4,138.2, 137.3, 132.0, 129.6,128.2, 127.7, 127.3,127.2, 127.1, 126.9, 126.7, 126.2, 123.1, 121.9, 119.9, 116.5, 64.4, 25.8, 11.6. HRMS calcd C31H22N6O2S2 574.1246; found 574.1257.
1-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1,2,4]triazino[5,6-b]indol-3-yl)hexan-1-one (23)
Compound 23 was synthesized similarly to 20. 1H NMR (500 MHz, CDCl3) δ 8.70 (d, J = 8.5, 1H), 8.42 (d, J = 8.0, 1H), 7.89 (br, 1H), 7.68–7.76 (m, 2H), 7.51–7.60 (m, 2H), 7.40 (br, 1H), 7.27–7.35 (m, 3H), 7.18 (br, 1H), 5.37 (t, J = 7.0, 1H), 3.26–3.41 (m, 2H), 2.13 (br, 1H), 1.81–1.90 (m, 3H), 1.28–1.46 (m, 4H), 0.98–1.00 (m, 6H). HRMS calcd for C31H29N5O2S2 567.1763; found 567.1763.
1-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1, 2, 4]triazino[5,6-b]indol-5-yl)pent-4-en-1-one (24)
Compound 24 was synthesized similarly to 20. 1H NMR (500 MHz, CDCl3) δ 8.71 (d, J = 8.5, 1H), 8.43 (d, J = 7.5, 1H), 7.89 (br, 1H), 7.73–7.77 (m, 1H), 7.68 (d, J = 6.5, 1H), 7.58–7.61 (m, 1H), 7.51–7.53 (m, 1H), 7.41 (br, 1H), 7.34 (t, J = 7.0, 1H), 7.26–7.28 (m, 2H), 7.18 (br, 1H), 5.90–5.98 (m, 1H), 5.36 (t, J = 7.0, 1H), 5.10–5.20 (m, 2H), 3.48–3.53 (m, 1H), 3.37–3.43 (m, 1H), 2.57–2.62 (m, 2H), 2.12 (br, 1H), 1.89 (br, 1H), 0.98 (br, 3H). 13C NMR (125 MHz, CDCl3) δ 172.5, 169.9, 167.8, 146.7, 142.4, 139.5, 138.6, 138.3, 136.2, 132.2, 128.3, 127.7, 127.4, 127.3, 126.9, 126.8, 125.9, 121.5, 119.6, 117.8, 116.3, 100.0, 45.8, 38.8, 28.2, 26.1, 11.7. HRMS calcd for C30H25N5O2S2 551.1450; found 551.1461.
1-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1, 2, 4]triazino[5,6-b]indol-5-yl)pent-4-yn-1-one (25)
Compound 25 was synthesized similarly to 20. 1H NMR (500 MHz, CDCl3) δ 8.73 (d, J = 8.5, 1H), 8.43 (d, J = 7.5, 1H), 7.86 (d, J = 7.0, 1H), 7.74–7.77 (m, 1H), 7.69 (d, J = 7.0, 1H), 7.59–7.62 (m, 1H), 7.54 (d, J = 7.5, 1H), 7.34–7.40 (m, 2H), 7.24–7.30 (m, 2H), 7.18 (br, 1H), 5.32 (t, J = 7.0, 1H), 3.58–3.62 (m, 1H), 3.45–3.52 (m, 1H), 2.72–2.74 (m, 2H), 2.15 (br, 1H), 1.90 (br, 1H), 1.28 (t, J = 7.5, 1H), 0.98–1.01 (m, 3H). HRMS calcd for C30H23N5O2S2 549.1293; found 549.1294.
5-bromo-1-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1, 2, 4]triazino[5,6-b]indol-5-yl)pentan-1-one (26)
Compound 26 was synthesized similarly to 20. 1H NMR (500 MHz, CDCl3) δ 8.71 (d, J = 8.5, 1H), 8.43 (d, J = 7.5, 1H), 7.92 (br, 1H), 7.76 (t, J = 8.0, 1H), 7.68 (d, J = 7.0, 1H), 7.60 (t, J = 7.5, 1H), 7.52–7.54 (m, 1H), 7.41 (br, 1H), 7.30–7.36 (m, 3H), 7.19 (br, 1H), 5.39 (t, J = 7.0, 1H), 3.31–3.51 (m, 5H), 2.11 (br, 1H), 1.93–1.98 (m, 2H), 1.85–1.91 (m, 4H), 0.98 (br, 3H). HRMS calcd for C29H24BrN5O2S2 617.0555; found 617.0543.
Ethyl 5-oxo-5-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1,2,4]triazino[5,6-b]indol-5-yl)pentanoate (27)
Compound 27 was synthesized similarly to 20. 1H NMR (500 MHz, CD3Cl) δ 8.70 (d, J = 8.5, 1H), 8.42 (d, J = 7.5, 1H), 7.91 (br, 1H), 7.74–7.77 (m, 1H), 7.68 (d, J = 7.5, 1H), 7.58–7.61 (m, 1H), 7.52–7.53 (m, 1H), 7.40 (br, 1H), 7.30–7.35 (m, 3H), 7.17 (br, 1H), 5.39 (t, J = 6.5, 1H), 3.74 (s, 3H), 3.71–3.72 (m, 2H), 3.42–3.52 (m, 2H), 2.42–2.50 (m, 2H), 1.89–2.02 (m, 2H), 0.98 (br, 3H). 13C NMR (125 MHz, CD3Cl) δ 173.3, 172.5, 169.9, 167.7, 146.6, 142.4, 139.4, 138.6, 138.3, 132.2, 128.3, 127.7, 127.5, 127.3, 127.1, 170.0, 126.8, 125.9, 121.4, 119.6, 117.7, 68.0, 51.8, 38.4, 32.9, 26.0, 19.5, 11.7. HRMS calcd for C31H27N5O4S2 597.1504; found 597.1498.
General procedure for synthesis of compounds 28–37
Ethyl 2-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1,2,4]triazino[5,6-b]indol-5-yl)acetate (28)
Compound 1 (0.1407 g, 0.3 mmol) was dissolved in 5 ml anhydrous DMF. 50 mg K2CO3 and ethyl 2-bromoacetate (0.2004 g, 1.2 mmol) were added to the above solution. This reaction was stirred at room temperature. After 6 h, TLC indicated there was no starting material remained and the reaction was stopped. 150 ml ethyl acetate was added to the above mixture. The organic phase was washed by saturated NH4Cl for five times and separated. The organic phase was dried by Na2SO4 and concentrated. The residue was purified by flash column chromatography (hexane/ethyl acetate-2∶1) and viscous oil 28 was obtained. 1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 7.5, 1H), 7.88 (br, 1H), 7.62–7.63 (m, 2H), 7.42–7.45 (m, 2H), 7.29–7.32 (m, 2H), 7.19–7.26 (m, 2H), 7.09 (br, 2H), 5.37 (t, J = 7.0, 1H), 4.97 (s, 2H), 4.19 (q, J = 7.0, 2H), 2.01 (br, 1H), 1.82 (br, 1H), 1.20–1.23 (m, 3H), 0.89–0.91 (m, 3H). HRMS calcd for C29H25N5O3S2 555.1399; found 555.1405.
2-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1,2,4]triazino[5,6-b]indol-5-yl)acetic acid (29)
Compound 28 (161 mg, 0.2901 mmol) was dissolved in 15 ml 1,4-dioxane and 0.58 ml 1 M NaOH was added to the solution. The reaction mixture was stirred at room temperature. After 14 h, TLC indicated that there was no starting material remained and the reaction was stopped. The pH of the reaction was adjusted to 4∼5 using concentrated HAc. The mixture was extracted by ethyl acetate for three times and the organic phase was combined. The organic phase was dried by anhydrous Na2SO4 and concentrated under vacuum. The residue was purified by column (DCM/MeOH-60∶1) and the viscous oil 29 was obtained. 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 8.0, 1H), 7.94 (br, 1H), 7.62–7.68 (m, 2H), 7.35–7.49 (m, 4H), 7.24–7.30 (m, 3H), 7.07 (br, 1H), 5.40 (t, J = 7.0, 1H), 5.01 (s, 2H), 2.03 (br, 1H), 1.84 (br, 1H), 0.92 (br, 3H). HRMS calcd for C27H21N5O3S2 527.1086; found 527.1093.
2-((5(2-hydoxyethyl)-5H-[1,2,4]triazino[5,6-b]indol-5-yl)thiol)-1-(10H-phenothiazin-10-yl)butan-1-one (30)
Compound 28 (95 mg, 0.1712 mmol) was dissolved in 4 ml MeOH/THF (3∶1). The solution was cooled to 0°C. Then NaBH4 (39 mg, 1.027 mmol) was added to the above solution. The reaction mixture was allowed to room temperature. After 6 h, TLC indicated there was no starting material remained and the reaction was stopped. Acetic acid was used to quench the reaction. The mixture was purified by column (hexane/ethyl acetate (5∶1-3∶1-1∶1) and the viscous oil was obtained. 1H NMR (500 MHz, CDCl3) δ 8.44 (d, J = 8.0,1H), 7.99 (br, 1H), 7.72 (t, J = 8.0, 1H), 7.66 (br, 1H), 7.59 (d, J = 8.0, 1H), 7.47–7.51 (m, 2H), 7.40 (br, 1H), 7.27–7.34 (m, 3H), 7.18 (br, 1H), 5.37 (br, 1H), 4.45 (br, 2H), 4.09 (br, 2H), 2.28 (br, 1H), 2.12 (br, 1H), 1.88 (br, 1H), 0.96 (d, J = 7.5, 3H). 13C NMR (125 MHz, CDCl3) δ 170.4, 146.7, 141.6, 138.6, 138.4, 130.9, 127.7, 127.3, 127.2, 126.9, 126.8, 123.1, 122.4, 110.6, 100.0, 60.7, 44.2, 25.9, 11.7. HRMS calcd for C27H23N5O2S2 513.1293; found 513.1303.
1-(10H-phenothiazin-10-yl)-2-((5(prop-2-yn-1-yl)-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thiol)butan-1-one (31)
Compound 31 was synthesized similarly to 28 as amorphous powder. 1H NMR (500 MHz, CDCl3) δ 8.48 (d, J = 7.5 1H), 7.59 (br, 1H), 7.75–7.78 (m, 1H), 7.67–7.70 (m, 2H), 7.51–7.55 (m, 2H), 7.40 (br, 1H), 7.31–7.34 (m, 3H), 7.17 (br, 1H), 5.43 (t, J = 6.5, 1H), 5.08–5.13 (m, 2H), 2.40 (s, 1H), 1.92 (br, 1H), 1.89 (br, 1H), 0.92–1.01 (m, 3H). HRMS calcd for C28H21N5O2S2 507.1188; found 507.1188.
2-((5(but-3-yn-1-yl)-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thiol)-1-(10H-phenothiazin-10-yl)butan-1-one (32)
Compound 32 was synthesized similarly to 28 as amorphous powder. 1H NMR (500 MHz, CDCl3) δ 8.46 (d, J = 7.5, 1H), 7.97 (br, 1H), 7.68–7.74 (m, 2H), 7.48–7.59 (m, 3H), 7.40 (br, 1H), 7.31–7.34 (m, 2H), 7.18 (br, 1H), 5.41 (t, J = 7.0, 1H), 4.45–4.52 (m, 2H), 2.77–2.80 (m, 2H), 2.14 (br, 1H), 1.89 (br, 1H), 0.98 (br, 3H). 13C NMR (125 MHz, CD3Cl) δ 169.4, 165.9, 145.8, 140.9, 140.6, 137.9, 137.8, 130.9, 128.2, 127.8, 127.5, 127.2, 127.0, 123.0, 121.5, 117.3, 111.6, 80.6, 73.3, 25.7, 17.5, 11.3. HRMS calcd for C28H21N5O2S2 507.1188; found 507.1188.
2-((5-(2-oxo-2-phenylethyl)-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thiol)-1-(10H-phenothiazin-10-yl)butan-1-one (33)
Compound 33 was synthesized similarly to 28 as amorphous powder. 1H NMR (500 MHz, CDCl3) δ 8.49 (d, J = 7.5, 1H), 8.05 (d, J = 7.5, 1H), 7.90 (br, 1H), 7.65–7.72 (m, 3H), 7.55–7.58 (m, 2H), 7.49–7.52 (m, 2H), 7.40–7.46 (m, 2H), 7.31–7.39 (m, 1H), 7.21–7.24 (m, 4H), 5.69–5.77 (m, 2H), 5.39 (t, J = 6.5, 1H), 2.05 (br, 1H), 1.87 (d, J = 5.5, 1H), 0.90–0.92 (m, 3H). HRMS calcd for C33H25N5O2S2 587.1450; found 507.1461.
2-((5-((1-phenethyl-1H-1,2,3-triazol-4-yl)methyl)-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thiol)-1-(10H-henothiazin-10-yl)butan-1-one (34)
2-bromoethylbenzene (3.083 g, 0.3 mmol) was dissolved in 17 ml anhydrous DMF. NaN3 (2.1664 g, 33.329 mmol) and 56 mg KI were added to the above solution. The reaction mixture was heated to 90°C for 18 h. TLC indicated that there was no starting material remained. The reaction was stopped and 200 ml DCM was added to the mixture. The organic phase was washed by 50 ml water and dried by anhydrous Na2SO4. The organic phase was concentrated and evaporated under vacuum. The crude azide was obtained and used for the next step directly.
Compound 32 (0.1268 g, 0.25 mmol) and azide (33.4 mg, 0.227 mmol) were dissolved in 3.6 ml t-BuOH/H2O/THF (v/v-1∶1∶1). Sodium ascorbate (98.9 mg, 0.4994 mmol) and CuSO4 (11.3 mg, 0.0454 mmol) in 0.5 ml water were added to the above reaction mixture. The reaction mixture was heated to 55°C. After stirring for 24 h, TLC indicated that there was no starting material remained. The reaction was stopped and cooled to room temperature. 6 ml water was added to the mixture. The solid was collected and washed with a few water. Then, the solid was dissolved in 8 ml acetone and the solution was filtered. The filtrate was evaporated and the residue was dissolved in 3 ml ethyl acetate. The solution was heated and 5 ml hexane was added to the solution. After overnight, gray solid was formed and collected. The amorphous solid 34 was washed by 4 ml hexane and dried. 1H NMR (500 MHz, CDCl3) δ 8.44 (d, J = 8.0, 1H), 8.03 (br, 1H), 7.66–7.77 (m, 3H), 7.48–7.51 (m, 2H), 7.41 (br, 1H), 7.32–7.33 (m, 2H), 7.18 (br, 1H), 7.06–7.13 (m, 4H), 6.93–6.95 (m, 2H), 5.46–5.56 (m, 3H), 4.53 (t, J = 7.5, 2H), 3.14 (t, J = 7.5?, 2H), 2.07 (br, 1H), 1.88 (br, 1H), 0.98 (br, 3H). HRMS calcd for C36H30N8OS2 654.1984; found 654.1991.
2-((5-((1-phenethyl-1H-1,2,3-triazol-4-yl)ethyl)-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thiol)-1-(10H-phenothiazin-10-yl)butan-1-one (35)
Compound 35 was synthesized similarly to 34 as amorphous powder. 1H NMR (500 MHz, CDCl3) δ 8.43 (d, J = 7.5, 1H), 8.00 (br, 1H), 7.64–7.67 (m, 2H), 7.42–7.50 (m, 4H), 7.23–7.33 (m, 6H), 7.18 (br, 1H), 7.00–7.02 (m, 2H), 6.92 (s, 1H), 5.44 (t, J = 6.5, 1H), 4.62 (t, J = 7.0, 2H), 4.48 (t, J = 7.5, 2H), 3.24 (t, J = 7.0, 2H), 3.06 (t, J = 7.5, 2H), 2.04–2.08 (m, 1H), 1.87–1.89 (m, 1H), 0.92–1.01 (m, 3H). HRMS calcd for C37H32N8OS2 668.2140; found 668.2135.
Ethyl 4-(4-(2-(3-((1-oxo-1-(10H-phenothiazin-10-yl)butan-2-yl)thio)-5H-[1,2,4]triazino[5,6-b]indol-5-yl)ethyl)-1H-1,2,3-triazol-1-yl)butanoate (36)
Compound 36 was synthesized similarly to 34 as amorphous powder. 1H NMR (500 MHz, CDCl3) δ 8.41 (d, J = 8.0, 1H), 8.01 (br, 1H), 7.62–7.68 (m, 2H), 7.50 (d, J = 7.5, 1H), 7.40–7.44 (m, 3H), 7.26–7.33 (m, 3H), 7.17 (br, 2H), 5.44 (t, J = 6.5, 1H), 4.66 (t, J = 7.0, 2H), 4.32 (t, J = 7.0, 2H), 4.15 (q, J = 7.0, 2H), 3.29 (t, J = 7.0, 2H), 2.20 (t, J = 7.5, 2H), 2.07–2.11 (m, 2H), 1.87 (br, 1H), 1.77 (br, 1H), 1.28 (t, J = 7.0, 3H), 0.94–1.00 (m, 3H). HRMS calcd for C35H34N8O3S2 678.2195; found 678.2197.
1-(acridin-10(9H)-yl)-2-((5-(2-hydroxyethyl)-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)butan-1-one (37)
Compound 37 was synthesized similarly to 30 as viscous oil. 1H NMR (500 MHz, CDCl3) δ 8.35 (d, J = 7.5, 1H), 7.77 (br, 2H), 7.66–7.70 (m, 1H), 7.55 (d, J = 8.0, 1H), 7.44 (t, J = 8.0, 1H), 7.24–7.29 (m, 4H), 7.16 (br, 2H), 5.47 (br, 1H), 4.33–4.41 (m, 2H), 4.05–4.08 (m, 2H), 3.86 (br, 2H), 2.13–2.19 (m, 1H), 1.98–1.99 (m, 1H), 1.04 (br, 3H). HRMS calcd for C28H25N5O2S 495.1729; found 495.1731.
Acknowledgments
We thank Samy Meroueh for help in initial chemical selection, Sergio C. Chai at St. Jude Children's Research Hospital for help in data analysis of cell growth inhibition, Bert Vogelstein for offering us with cell lines, and all of our colleagues for active discussion and help.
Funding Statement
H.L. was supported by National Institutes of Health-National Cancer Institute grants CA127724, CA095441, CA172468, and CA129828. Q.-Z.Y. was supported by National Institute of Health grant AI065898.
References
- 1. Vousden KH, Prives C (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- 2. Krizhanovsky V, Lowe SW (2009) Stem cells: The promises and perils of p53. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hollstein M, Sidransky D, Vogelstein B, Harris CC (1991) p53 mutations in human cancers. Science 253: 49–53. [DOI] [PubMed] [Google Scholar]
- 4. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP (2009) Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer 9: 862–873. [DOI] [PubMed] [Google Scholar]
- 5. Kruse JP, Gu W (2009) Modes of p53 regulation. Cell 137: 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mandinova A, Lee SW (2011) The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med 3: 64rv61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, et al. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303: 844–848. [DOI] [PubMed] [Google Scholar]
- 8. Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, et al. (2004) Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med 10: 1321–1328. [DOI] [PubMed] [Google Scholar]
- 9. Shangary S, Qin D, McEachern D, Liu M, Miller RS, et al. (2008) Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A 105: 3933–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang Q, Zeng SX, Zhang Y, Ding D, Ye Q, et al. (2012) A small molecule Inauhzin inhibits SIRT1 activity and suppresses tumour growth through activation of p53. EMBO Mol Med 4: 298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Zhang Q, Zeng SX, Mayo LD, Lu H (2012) Inauhzin and Nutlin3 synergistically activate p53 and suppress tumor growth. Cancer Biol Ther 13: 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kgokong JL, Smith PP, Matsabisa GM (2005) 1,2,4-Triazino-[5,6b]indole derivatives: effects of the trifluoromethyl group on in vitro antimalarial activity. Bioorg Med Chem 13: 2935–2942. [DOI] [PubMed] [Google Scholar]
- 13. Vang T, Xie Y, Liu WH, Vidovic D, Liu Y, et al. (2011) Inhibition of lymphoid tyrosine phosphatase by benzofuran salicylic acids. J Med Chem 54: 562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, et al. (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275: 1787–1790. [DOI] [PubMed] [Google Scholar]
- 15. Dai MS, Lu H (2004) Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem 279: 44475–44482. [DOI] [PubMed] [Google Scholar]