

Abstract
Although tricyclic antidepressants rapidly activate monoaminergic neurotransmission, these drugs must be administered chronically to alleviate symptoms of depression. This observation suggests that molecular mechanisms downstream of monoamine receptor activation, which include the induction of gene transcription, underlie chronic antidepressant-induced changes in behavior. Here we show that methyl-CpG-binding protein 2 (MeCP2) regulates behavioral responses to chronic antidepressant treatment. Imipramine administration induces phosphorylation of MeCP2 at Ser421 (pMeCP2) selectively in the nucleus accumbens and the lateral habenula, two brain regions important for depressive-like behaviors. To test the role of pMeCP2 in depressive-like behaviors, we used male mice that bear a germ-line mutation knocked into the X-linked Mecp2 locus that changes Ser421 to a nonphosphorylatable Ala residue (S421A). MeCP2 S421A knock-in (KI) mice showed increased immobility in forced-swim and tail-suspension tests compared with their wild-type (WT) littermates. However, immobility of both MeCP2 WT and KI mice in forced swim was reduced by acute administration of imipramine, demonstrating that loss of pMeCP2 does not impair acute pharmacological sensitivity to this drug. After chronic social defeat stress, chronic administration of imipramine significantly improved social interaction in the MeCP2 WT mice. In contrast, the MeCP2 KI mice did not respond to chronic imipramine administration. These data suggest novel roles for pMeCP2 in the sensitivity to stressful stimuli and demonstrate that pMeCP2 is required for the effects of chronic imipramine on depressive-like behaviors induced by chronic social defeat stress.
Introduction
Activation of monoamine receptors is essential to the mechanism by which tricyclic antidepressants and selective-serotonin reuptake inhibitors (SSRIs) alleviate symptoms of depression (Manji et al., 2001). However, despite the fact that these drugs rapidly increase extracellular levels of monoamine neurotransmitters, including serotonin (5-HT) and norepinephrine, these antidepressants must be administered for several weeks or months before they produce alterations in depression-like behavior (Krishnan and Nestler, 2008).
In addition to the acute activation of monoaminergic neurotransmission, antidepressant drugs drive long-lasting changes in neuronal gene expression. Transcriptional changes contribute to chronic antidepressant action by altering the physiology of neurons within circuits that underlie depressive-like behaviors (Thome et al., 2000; Berton et al., 2006; Tsankova et al., 2006). Chromatin regulatory proteins have been of particular interest in this process because of the potential for epigenetic modifications of histone proteins and genomic DNA to confer very lasting changes on gene transcription that correlate with persistent changes in depressive-like behaviors (Tsankova et al., 2007; Covington et al., 2009; Wilkinson et al., 2009; Jiang et al., 2010). Both histone modifying enzymes and proteins that regulate DNA methylation can be targets of regulation by environmental exposures or antidepressant drugs that affect depressive-like behaviors. Specifically, expression of mRNA encoding the histone deacetylase HDAC5 is reduced in the nucleus accumbens (NAc) after chronic social defeat stress, whereas expression of the de novo DNA methyltransferase Dnmt3a is enhanced (Renthal et al., 2007; LaPlant et al., 2010). In support of a functional role for these expression changes in depressive-like behaviors, both Hdac5 knock-out mice and mice overexpressing Dnmt3a in the NAc show enhanced social avoidance after defeat (Renthal et al., 2007; LaPlant et al., 2010).
We have shown that the methyl-CpG-binding protein-2 (MeCP2) is a target of signaling cascades activated by monoamine neurotransmitters. Specifically, we find that either dopamine (DA) or 5-HT receptor activation is sufficient to induce the phosphorylation of MeCP2 at Ser421 (pMeCP2) in specific populations of neurons in the NAc (Deng et al., 2010; Hutchinson et al., 2012). Because the SSRI antidepressant citalopram is among the drugs that induce pMeCP2 in vivo (Hutchinson et al., 2012), we considered the possibility that phosphorylation of MeCP2 may contribute to both 5-HT-regulated depressive-like behaviors and the behavioral response to antidepressant treatment. Here we show that, like citalopram, the tricyclic antidepressant imipramine also induced pMeCP2 in brain regions relevant to depressive-like behaviors. Mice bearing a genetic knock-in (KI) mutation that eliminates this phosphorylation site in MeCP2 (Cohen et al., 2011) show enhanced sensitivity to environmental stressors and fail to respond to chronic imipramine treatment after chronic social defeat stress. These data for the first time implicate MeCP2 in regulation of depressive-like behaviors.
Materials and Methods
Animals.
Adult (8–10 weeks old) male C57BL/6 mice (The Jackson Laboratory), retired CD1 breeders (The Jackson Laboratory), and MeCP2 S421A wild-type (WT) and KI mice (Cohen et al., 2011) were used in these studies. MeCP2 WT and KI littermates were generated from heterozygous breedings in which Mecp2S421A/+ females were crossed to C57BL/6 males. Because Mecp2 is on the X chromosome, male Mecp2+/Y WT and Mecp2S421A/Y KI littermates were used for experiments. Animals were weaned at 21–30 d of age. They were housed in groups of three to five for tail-suspension and forced-swim studies but were singly housed for sucrose preference and chronic social defeat stress. All animals were given access to standard laboratory chow and water ad libitum and were housed in a humidity- and temperature-controlled room on a 14/10 h light/dark cycle. All experiments were conducted with an approved protocol from the Duke University Institutional Animal Care and Use Committee in accordance with guidelines from the National Institutes of Health for the Care and Use of Laboratory Animals.
Immunofluorescent staining of brain sections.
Unless otherwise indicated, 2 h after vehicle or drug administration, mice were perfused transcardially with 4% paraformaldehyde in 0.1 m PBS. Brains were postfixed in 4% paraformaldehyde/PBS overnight and then sunk into 20% (w/v) sucrose/PBS overnight. Coronal sections (40 μm) were cut on a freezing microtome, and brain regions were identified by anatomical landmarks. One section from each brain region of interest was selected for each mouse based on anatomical structures to represent the closest approximation of identical sections between individual mice. To minimize technical variations in immunostaining across genotypes, sections from multiple individual mice were first photographed for visual identification. The sections were then pooled and incubated with antibodies in a single chamber, and finally they were separated after processing for image analysis (Deng et al., 2010). For immunostaining, tissue sections were permeabilized with 1% (v/v) Triton X-100 for 1 h and then blocked with 16% (v/v) goat serum in PBS. Sections were incubated with the following primary antibodies overnight at 4°C: rabbit anti-phospho-Ser421 MeCP2 at 1:15,000 (Deng et al., 2010) and mouse anti-glutamic acid decarboxylase 67 (GAD67) at 1:500 (MAB5406; Millipore Bioscience Research Reagents). After three washes in PBS, sections were incubated with the following species-specific fluorescent-conjugated secondary antibodies for 1 h at room temperature: goat anti-mouse antibodies conjugated to Cy3 at 1:500 (Jackson ImmunoResearch) and Alexa Fluor 488 goat anti-rabbit antibodies at 1:500 (Invitrogen). Sections were washed in PBS, nuclei were labeled with Hoechst dye (Sigma) to facilitate anatomical localization of brain structures, and sections were mounted, coverslipped, and analyzed as described below.
Image analyses.
For quantitative immunofluorescence, images were captured on a Leica DMI4000 inverted fluorescence microscope using a Cascade 512B camera. Digital images were quantified using MetaMorph 7 Image Analysis software (Molecular Devices). To minimize variation between samples, images were captured with a uniform exposure time within a single experiment, and immunofluorescence was quantified across a constant-sized region from a single field of each section. We used the Count Nuclei module in MetaMorph 7.0 to first count the total number of cells per section that were positive for pMeCP2 immunoreactivity, and then we determined the integrated immunofluorescence intensity of all the pMeCP2-positive nuclei in each image. We defined nuclei as objects of 3–8 μm in diameter. For each experiment, an investigator blind to genotype and treatment condition chose a random section from which to set a single threshold value of fluorescence intensity above background to score objects of this size as positive for pMeCP2 expression. The threshold value was adjusted until the output of detected nuclei by the program most closely matched the distribution of positive nuclei seen by eye. This threshold value was then held constant for every image within the experimental set. The first result of this analysis is a count of the number of cells that are pMeCP2 positive in each image. The Count Nuclei module then creates a mask that covers the area of all of the nuclei identified as positive on each section and quantifies the total immunofluorescence intensity under this area (the “integrated intensity”) for each image. Thus, our analysis allows us to tell how many cells have induced expression of pMeCP2 in each image as well as to evaluate the magnitude of pMeCP2 induction in these nuclei.
Tail suspension.
Thirty minutes before the start of the test, MeCP2 KI mice and their WT littermates were injected with saline (Veh) or imipramine (20 mg/kg). The tail-suspension test was conducted in a Med Associates apparatus in which mice were hung by their tails for 6 min. The body weight of the mouse was used as a control to determine the magnitude of its struggling activity, and the duration of immobility and struggling behavior was determined using Med Associates software (Fukui et al., 2007).
Forced swim.
In the forced-swim test (Porsolt et al., 1977), mice were injected with Veh or imipramine (10 or 20 mg/kg, i.p., as indicated) 30 min before testing. The mice were placed into a beaker (15 cm in diameter) of water held at 25°C with a depth of 15 cm. The test was videotaped for 6 min from the side of the beaker and scored subsequently for struggling behavior. Immobility time refers to the time that mice spent floating or engaged in minimal activity to stay afloat. Subtle movements of the feet, tail, or head required to maintain the eyes, nose, and ears above the surface of the water were excluded as immobility. The videotapes were scored using Noldus Observer 5 (Noldus Information Technology) by an observer blind to genotype and treatment condition. In addition to Mecp2 Ser421Ala WT mice (n = 24), C57BL/6 mice (n = 11) were included in the WT group equally distributed among the treatment groups. Importantly, we determined that there was no significant genotype difference between the immobility times of Mecp2 WT mice and the C57BL/6 mice in this assay (F(1,35) = 0.42, p < 0.52).
Sucrose preference test.
Mice were housed individually for 7 d before and throughout the study. All studies were conducted in the home cage. Water bottles were removed 2.5 h before the beginning of the dark cycle. The mice were supplied with two bottles containing water 1.5 h after the start of the dark cycle. The mice were allowed to drink for 1 h, and then the original water bottle was returned. This protocol was performed for several days until stable water consumption was reached. Once consistent water drinking was achieved, the mice were tested in a two-bottle choice test over consecutive days. Water was paired with 0.1, 0.25, 0.5, 1, and 2% sucrose. The total volume of liquid consumed from each bottle was measured each day, and the preference for sucrose was determined by dividing the volume of sucrose consumed by total liquid consumption.
Chronic social defeat stress.
Chronic social defeat stress was performed using a protocol similar to previously described methods for mice (Berton et al., 2006; Tsankova et al., 2006; Krishnan et al., 2007; Golden et al., 2011). KI mice and their WT littermates were exposed to a different CD1 aggressor mouse 10 min each day for 10 consecutive days in the aggressor's home cage. After 10 min of physical contact, the test mouse was returned to its home cage. Bedding from the aggressor's cage was placed in the test mouse's cage to allow for sensory contact during the following 24 h. Control mice were singly housed but were not exposed to CD1 aggressors. Twenty-four hours after the last defeat session, defeated mice and controls were tested for social interaction. Mice were placed in a new arena with a small animal cage at one end, and their movement was tracked for 2.5 min in the absence of a CD1 aggressor, followed by 2.5 min in the presence of the caged aggressor. The 5 min interaction test was filmed, and time spent in the interaction zone and corners was manually scored by an observer blind to genotype and treatment using Noldus Observer 5 (Noldus Information Technology). For 4 weeks after the first interaction test, defeated mice in the chronic treatment group received daily intraperitoneal injections of imipramine (20 mg/kg) in the home cage. Mice in the acute treatment group received Veh for 27 d and one dose of imipramine (20 mg/kg) on the last day of injections. All control mice received Veh injections for 28 d. The day after the last injection, the mice were retested for social interaction as described previously.
Statistical analyses.
Statistical analyses were performed using SPSS version 11.0 statistical software (SPSS). The data are depicted as means and SEM. Data were analyzed by univariate, two-way, or repeated-measures ANOVA as indicated. Bonferroni's corrected pairwise comparisons were used as the post hoc tests. In all cases, p < 0.05 was considered statistically significant.
Results
Imipramine induces pMeCP2 in GABAergic interneurons of the NAc
Previously, we showed that administration of the SSRI citalopram (10 mg/kg, i.p.) is sufficient to induce pMeCP2 in specific neuron populations of the NAc (Hutchinson et al., 2012). To determine whether the induction of pMeCP2 is common to other classes of antidepressant drugs, we asked whether imipramine, a tricyclic antidepressant that inhibits the serotonin (SERT) and norepinephrine (NET) transporters, is also sufficient to induce pMeCP2 in the NAc. C57BL/6 mice were administered either Veh or imipramine (20 mg/kg), and the mice were perfused at 0, 0.5, 2, 4, or 24 h after injection to assess pMeCP2 immunoreactivity in the NAc. For pMeCP2 immunoreactivity in mice treated with imipramine, an ANOVA indicated a significant effect of time (F(4,22) = 7.928, p < 0.001), and Bonferroni's comparisons showed that pMeCP2 intensity in the 2 h group was significantly higher than baseline levels (p < 0.003) (Fig. 1a). Imipramine stimulated a robust but transient induction of pMeCP2 in a small population of neurons in the NAc (Fig. 1b). We showed previously that amphetamine (AMPH), as well as drugs that selectively activate DA or 5-HT signaling in the NAc, induce pMeCP2 in a specific population of GABAergic interneurons that express the marker proteins GAD67 and parvalbumin (Deng et al., 2010; Hutchinson et al., 2012). To determine whether imipramine induces pMeCP2 in this cell type as well, we colabeled NAc sections from imipramine-treated mice with antibodies against both pMeCP2 and GAD67. Similar to our previous findings with AMPH, the selective DA transporter blocker GBR12909 vanoxerine, and the 5-HT3 receptor agonist quipazine, we found that imipramine induced pMeCP2 selectively in GAD67-positive GABAergic interneurons of the NAc (Fig. 1c). Together with our previous findings, these data indicate that at least two classes of antidepressants that modulate monoamine neurotransmission, SSRIs and tricyclic antidepressant drugs, can induce pMeCP2 in the NAc.
Figure 1.
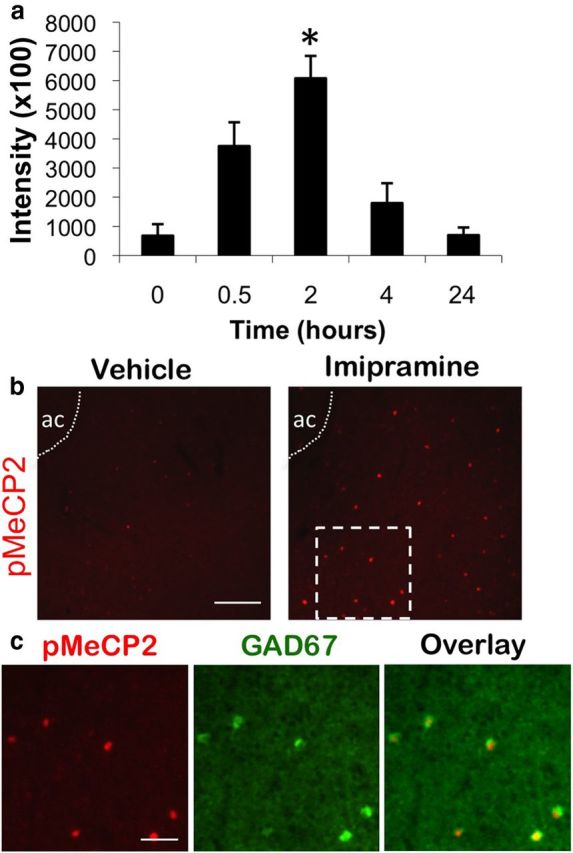
Imipramine induces pMeCP2 in the GABAergic interneurons of the NAc. a, C57BL/6 mice were injected with imipramine (20 mg/kg, i.p.) in the home cage and then perfused at 0, 0.5, 2, 4, or 24 h after injection. pMeCP2 immunoreactivity in the NAc was quantified and averaged across mice at each time point. b, pMeCP2 induction after vehicle or 20 mg/kg imipramine (2 h after injection). Dashed white box was used as the inset image to examine pMeCP2 and GAD67 overlay in c. ac, Anterior commissure. c, pMeCP2 (red), GAD67 (green), and overlay immunostaining in the NAc 2 h after treatment with 20 mg/kg imipramine. White arrows indicate neurons coimmunolabeled with antibodies for pMeCP2 and GAD67. Scale bars, 20 μm. n = 4–5 mice per group. For a, *p < 0.05.
MeCP2 KI mice show enhanced sensitivity to environmental stressors
To investigate a role for pMeCP2 in depressive-like behaviors and antidepressant action, we used a strain of mice that bear a germ-line mutation knocked into the Mecp2 locus that changes Ser421 to a nonphosphorylatable Ala residue (Cohen et al., 2011). The expression levels and chromatin binding patterns of MeCP2 are unchanged in the KI mice compared with their WT littermates. Furthermore, our previous behavioral profiling of this strain revealed no differences between MeCP2 WT and KI littermates in motor function, social interaction in a sociability test, or anxiety-like behaviors (Cohen et al., 2011).
We examined the behavior of MeCP2 Ser421Ala KI mice and their WT littermates in a series of behavioral tasks that model depressive-like behaviors. First, we subjected MeCP2 KI and WT mice to the tail-suspension test, in which the time to cessation of struggling is used as a measure of depression-like behavior (Steru et al., 1985). Repeated-measures ANOVA for time spent immobile showed a significant within-subjects effect of time (F(5,55) = 13.424, p < 0.001), indicating that the amount of time immobile increased over the duration of the test. Furthermore, there was also a significant time × genotype interaction (F(5,55) = 2.788, p < 0.026). Although both genotypes spent similar amounts of time immobile during the first 2 min, the MeCP2 KI mice spent significantly more time immobile during minutes 3 and 4 compared with their WT littermates (p < 0.041 and p < 0.010) (Fig. 2a).
Figure 2.
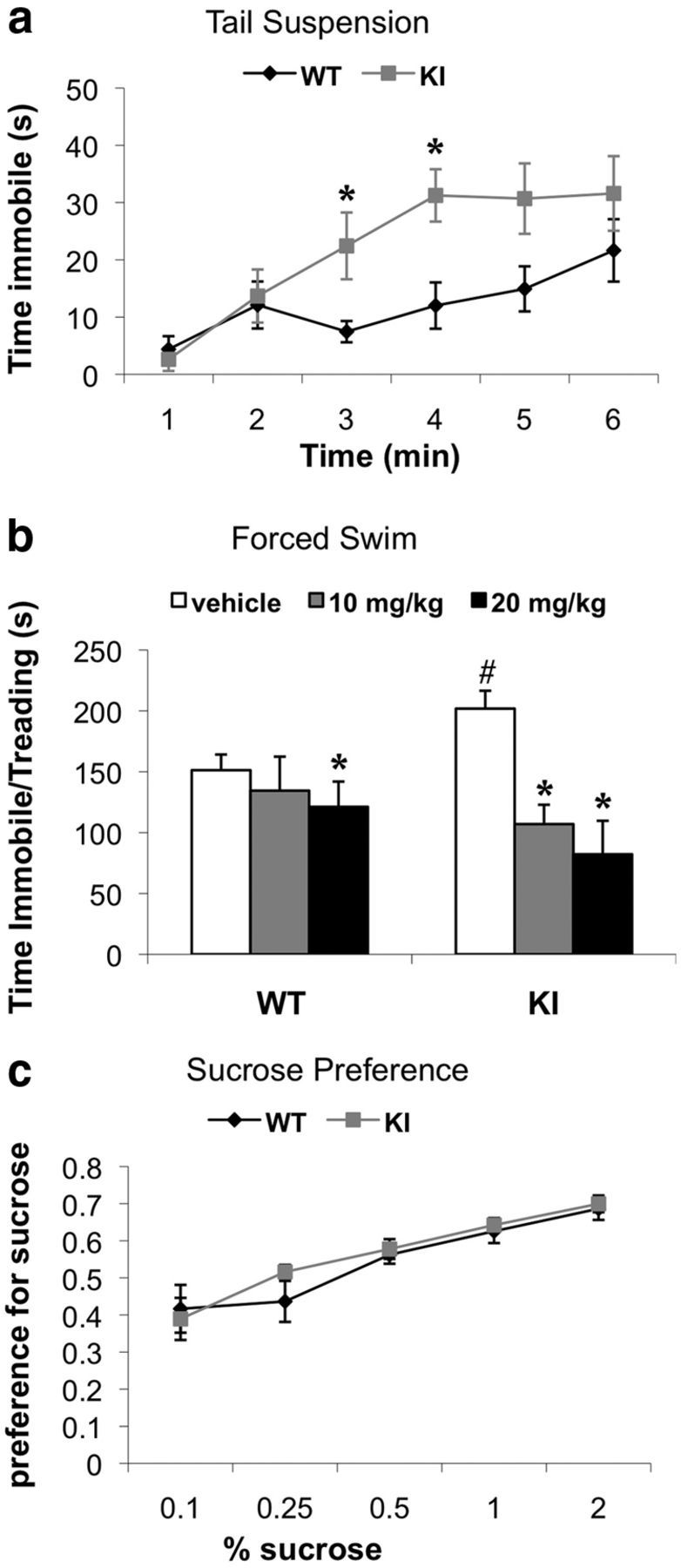
MeCP2 S421A KI mice exhibit increased sensitivity to environmental stressors. a, MeCP2 KI mice and their WT littermates were injected with Veh 30 min before the tail-suspension test, and the amount of time immobile each minute of the 6 min test was recorded. b, MeCP2 KI mice and their WT littermates were injected with either Veh or imipramine (10 or 20 mg/kg) 30 min before the forced swim. Total time immobile and treading over the 6 min test was recorded. c, KI mice and WT littermates were tested for their preference for increasing concentrations of sucrose in a two-bottle choice test. n = 6–7 mice per group for a, n = 4–14 mice per group for b, and n = 7–10 mice per group for c. For a, *p < 0.05 compared with WT at each time point. For b, *p < 0.05 Veh compared with imipramine at 10 or 20 mg/kg within each genotype, and #p < 0.05 WT Veh compared with KI Veh.
Similar to the tail-suspension test, increased immobility/treading time in the forced-swim test is also used as an index of depressive-like behavior in rodents (Porsolt et al., 1977). Importantly, in addition to screening for depressive-like phenotypes, the forced-swim test is widely used to quantify behavioral response to acute antidepressant treatment. The MeCP2 KI and WT mice were injected with either Veh or imipramine (10 or 20 mg/kg) 30 min before the forced-swim test. For the duration of time spent immobile and treading, a two-way ANOVA detected main effects of treatment (F(2,58) = 17.312, p < 0.001) and a significant treatment × genotype interaction (F(2,58) = 3.646, p < 0.032). We observed that the MeCP2 KI mice treated with Veh displayed significant increases in time spent immobile and treading compared with WT controls (p < 0.025) (Fig. 2b). Bonferroni's-corrected pairwise comparisons revealed that the time spent immobile and treading for the KI mice was significantly decreased by 10 and 20 mg/kg imipramine (p < 0.001), and the time spent immobile and treading for the WT littermates was reduced for the 20 mg/kg dose (p < 0.007) (Fig. 2b). These data demonstrate that both MeCP2 WT and KI mice show a behavioral response to acute imipramine treatment.
In addition to sensitivity to stressors, another common depressive-like behavior is anhedonia, which can be characterized as diminished interests or pleasure in otherwise rewarding stimuli. To determine whether the KI mice exhibit this feature of depressive-like behavior, we performed a sucrose preference test, which has been used to model anhedonia in rodents (Barrot et al., 2002; Fukui et al., 2007). The MeCP2 KI mice and their WT littermates were tested for their preference to drink water versus water with increasing percentages of sucrose (0.1, 0.25, 0.5, 1, or 2% sucrose). For sucrose, repeated-measures ANOVA showed significant main effects of concentration but failed to show effects of genotype (F(1,15) = 0.398, p < 0.54). Thus, both genotypes showed a stronger preference for sucrose as the concentration of sucrose increased, and there was no difference between the genotypes in the preference for sucrose at any of the concentrations tested (Fig. 2c). This evidence that the MeCP2 KI mice do not display anhedonia in this task suggests that the enhanced immobility of the MeCP2 KI mice in the tail-suspension and forced-swim tests reflects a selective increased sensitivity to environmental stressors rather than a general increase in depressive-like behaviors in this strain.
MeCP2 KI mice fail to respond to chronic imipramine after chronic social defeat stress
Because MeCP2 is a transcriptional regulator that is a target of phosphorylation by antidepressant-induced signaling cascades, we hypothesize that pMeCP2 contributes to the transcriptional plasticity that underlies behavioral adaptations to chronic antidepressant treatment. To test this hypothesis, we examined the behavior of MeCP2 WT and KI mice in a chronic social defeat stress paradigm. In this paradigm, repeated social defeat interactions have been shown to induce depressive-like symptoms (e.g., social avoidance, anhedonia) that are alleviated by chronic but not acute antidepressant treatment (Kudryavtseva et al., 1991; Golden et al., 2011). Singly housed mice were exposed to a different CD1 aggressor for 10 consecutive days and then tested for social interaction as described previously. The effects of defeat were monitored in a social interaction assay by assessing time spent in the interaction zone or the corners of a social interaction arena after a CD1 aggressor was placed in an enclosed, perforated chamber at one end of the arena (Fig. 3a). Decreased time spent in the interaction zone as well as increased time spent in the corners of the arena has been used to indicate defeat.
Figure 3.

MeCP2 S421A KI mice fail to show behavioral response to chronic imipramine treatment after chronic social defeat stress. a, Schematic representation of the chamber used for the social interaction test with dimensions. The interaction zone is defined as the region directly surrounding the chamber that is either empty (first 2.5 min of test) or contains a CD1 mouse (last 2.5 min of test). Chamber, interaction zone, and corners dimensions shown in inches. b, c, After 10 consecutive days of social defeat, KI and WT mice were tested for social interaction. The total time spent in the interaction zone (b) and the total time spent in the corners (c) in the presence of a CD1 aggressor. d, e, After 10 d of social defeat, KI and WT mice were treated acutely or chronically with imipramine for 28 d. On the day after the last injection, the mice were tested once again for social interaction. The total time in the interaction zone (d) and the total time in the corners (e) in the presence of a CD1 aggressor. n = 10 (WT) or 10 (KI) control and 17 (WT) or 22 (KI) defeated mice per group for b and c, and n = 8–13 mice per group for d and e. For b and c, *p < 0.001 defeated compared with control. For d and e, *p < 0.001 compared with control within each genotype, and #p < 0.001 WT chronic compared with WT acute.
A two-way ANOVA applied to total time in the interaction zone revealed an overall main effect of treatment (F(1,58) = 19.59, p < 0.001). However, there was no effect of genotype (F(1,58) = 0.22, p < 0.64), and the genotype × treatment interaction was not significant (F(1,58) = 0.62, p < 0.43). The defeated mice spent significantly less time in the interaction zone compared with non-defeated controls (p < 0.0001) (Fig. 3b). Similarly, for total time spent in the corners, a two-way ANOVA showed an overall effect of treatment (F(1,58) = 19.59, p < 0.001) but failed to show an effect of genotype (F(1,58) = 0.3, p < 0.64), and the genotype × treatment interaction (F(1,58) = 0.36, p < 0.43) was not significant. The defeated mice spent more time in the corners compared with non-defeated controls (p < 0.0001) (Fig. 3c). Together, these results demonstrate that the MeCP2 KI mice and their WT littermates show comparable levels of social avoidance after 10 d of social defeat.
After defeating the mice and testing for social avoidance, we then treated the mice acutely (27 d of Veh injection with one injection of imipramine on day 28) or chronically (28 d of imipramine injections) with imipramine (20 mg/kg) in their home cages. A control group of mice that did not undergo defeat was administered 28 d of Veh. After the last injection, the behavior of all mice was assessed in a second social interaction test. For time in the interaction zone, ANOVA revealed an effect of treatment for both MeCP2 WT (F(2,26) = 9.18, p < 0.001) and MeCP2 KI (F(2,31) = 7.18, p < 0.003) mice. For both genotypes, defeated mice that had been treated acutely with imipramine exhibited a significant reduction in time spent in the interaction zone compared with controls that had not been defeated (p < 0.001 for WT control vs WT acute, p < 0.007 for KI control vs KI acute) (Fig. 3d). Thus, consistent with previous results, we find that a single dose of imipramine is not sufficient to rescue social interaction after this paradigm of chronic social defeat. Defeated WT mice that had been treated chronically with imipramine exhibited a significant increase in the time spent in the interaction zone compared with the acute treatment group (p < 0.023), demonstrating that chronic imipramine treatment significantly improves social interaction after defeat. In contrast, defeated KI mice that had been treated chronically with imipramine failed to show increased time spent in the interaction zone compared with the acute treatment group (p < 1.00). Similar effects of genotype and treatment were observed when we analyzed time spent in the corners (Fig. 3e). ANOVA revealed an effect of treatment for both MeCP2 WT (F(2,26) = 10.87, p < 0.0004) and MeCP2 KI (F(2,31) = 9.17, p < 0.0007) mice. These data showed that defeated mice of both genotypes treated with acute imipramine spent significantly more time in the corners compared with controls that had not been defeated (WT p < 0.0003 acute vs control, KI p < 0.0043 acute vs. control). Chronic imipramine treatment resulted in a significant decrease in time spent in the corners for MeCP2 WT mice compared with the acute treatment group (p < 0.03), again demonstrating the efficacy of this treatment. In contrast, defeated MeCP2 KI mice treated with chronic imipramine did not show a difference in time spent in the corners compared with the KI acute treatment group (p < 0.96). Together, these data suggest that phosphorylation of MeCP2 at Ser421 is required for the ability of chronic imipramine to rescue social interaction after chronic defeat.
Chronic but not acute imipramine induces pMeCP2 in the lateral habenula
Our observations that the MeCP2 KI mice respond to acute imipramine in the forced swim but fail to show a behavioral response to chronic imipramine in the chronic social defeat stress paradigm suggest that pMeCP2 may be involved in the downstream molecular and cellular plasticities that underlie behavioral responses to chronic antidepressant treatment. To gain insight into the brain regions in which pMeCP2 may contribute to these behavioral responses, we compared the pattern of pMeCP2 after Veh, acute, or chronic imipramine treatment after chronic social defeat stress. C57BL/6 mice were subjected to chronic social defeat stress and then treated acutely (27 d of Veh injection with one injection of imipramine on day 28) or chronically (28 d of imipramine injections) with imipramine (20 mg/kg) in their home cages. As in the behavioral studies described above, a control group of mice that were not defeated were delivered daily injections of Veh for this 28 d period. Brain sections from regions relevant to depressive-like behaviors were analyzed by quantitative immunostaining with an antibody specific for pMeCP2 2 h after the final injection. We found that both acute and chronic imipramine treatment led to similar levels and patterns of pMeCP2 induction in the NAc compared with Veh treatment (Fig. 4a). Relatively high levels of pMeCP2 were found even under Veh control conditions in many regions of the cortex, and, similar to what we reported previously for other drugs that activate 5-HT neurotransmission (Hutchinson et al., 2012), we observed no obvious change in pMeCP2 immunostaining in the cingulate cortex or the prelimbic cortex after either acute or chronic imipramine treatment. Furthermore, neither acute nor chronic imipramine treatment was sufficient to induce pMeCP2 in the dorsal hippocampus, basal lateral amygdala, dorsal striatum, or the ventral tegmental area (VTA) (Fig. 3a and data not shown).
Figure 4.

pMeCP2 in the LHb is induced by chronic but not acute imipramine treatment after chronic social defeat stress. a, C57BL/6 mice were subjected to 10 consecutive days of social defeat stress and then given acute imipramine (27 d of Veh injections followed by imipramine injection on day 28) or chronic imipramine (28 d). The control group was not subjected to defeat but received 28 d of Veh. pMeCP2 immunoreactivity was analyzed in the NAc, prelimbic cortex (PLC), cingulate cortex (Cing. cortex), hippocampus (Hipp.), and LHb 2 h after the final injection. Overlay of pMeCP2 (red) in the LHb and nuclear Hoechst stain (blue) is shown to highlight the subpopulation of pMeCP2-positive cells. Dotted lines indicate the borders of the LHb and the medial habenula (MHb). b, Inset image of the dashed white box in a of pMeCP2 immunoreactivity in the LHb. c, Quantification of pMeCP2 immunoreactivity in the LHb after Veh or acute or chronic imipramine treatment. n = 8–9 mice per group. Scale bars: a, 10 μm; b, 40 μm. For c, *p < 0.05 compared with Veh.
In addition to these brain regions, we also examined pMeCP2 induction in the lateral habenula (LHb) because this structure has been implicated in modulating the behavioral responses to pain, stress, anxiety, sleep, and reward (Morris et al., 1999; Ullsperger and von Cramon, 2003; Lecourtier and Kelly, 2007; Hikosaka, 2010). We found little pMeCP2 in the LHb after Veh treatment and no significant induction after acute imipramine treatment (Fig. 4a,b). In contrast, chronic imipramine treatment produced a significant induction of pMeCP2 in the LHb that was localized to a subset of cells in the medial part of the LHb (Fig. 4a,b). For pMeCP2 immunoreactivity in the LHb, an ANOVA indicated a significant effect of treatment (F(2,23) = 4.699, p < 0.021), and Bonferroni's comparisons showed that pMeCP2 levels were significantly higher in the chronic treatment group compared with Veh control (p < 0.022) (Fig. 4c). Therefore, chronic but not acute imipramine treatment is sufficient to induce pMeCP2 in the LHb. These findings raise the possibility that pMeCP2 induction in the LHb may contribute to the cellular plasticities that underlie the recovery of social interaction induced by chronic imipramine treatment after chronic social defeat.
Discussion
This study is the first to causally implicate MeCP2 in depressive-like behaviors and antidepressant effects. By analyzing the distribution of imipramine-induced pMeCP2 in the brain, we have not only gained insight into how MeCP2 may affect brain function but also advanced understanding of the neural circuit basis of depressive-like behaviors.
MeCP2 is required for aspects of neuronal synapse development and function that underlie a broad range of behaviors (Amir et al., 1999; Deng et al., 2010; Skene et al., 2010; Guy et al., 2011). As a methyl-DNA binding protein, MeCP2 is thought to affect cellular physiology by modulating gene transcription (Guy et al., 2011). However, identifying specific gene targets of MeCP2 has been challenging. MeCP2 is globally bound across the genome in a pattern that mirrors the distribution of methylated CpG dinucleotides, and loss of MeCP2 results in subtle upregulation and downregulation of many mRNAs (Chahrour et al., 2008; Skene et al., 2010). Ser421 phosphorylated MeCP2 shows a similar genomic distribution, raising the possibility that this posttranslational modification may be permissive for changes in transcriptional regulation rather than instructive for the transcription of specific genes (Cohen et al., 2011). In this context, it is notable that we find pMeCP2 restricted to specific populations of neurons within neural circuits that underlie selected behavioral responses (Deng et al., 2010; Hutchinson et al., 2012) (Fig. 4a). These data suggest that local neural circuit-induced Ser421 phosphorylation of MeCP2 may provide a mechanism to confer specificity on the regulation of this chromatin regulator in vivo.
MeCP2 Ser421Ala KI mice show a mild behavioral phenotype compared with mice bearing null or severely hypomorphic Mecp2 alleles (Guy et al., 2011; Goffin et al., 2012). The MeCP2 KI mice perform no differently from their WT littermates on measures of motor function, sociability, spatial learning and memory, or anxiety (Cohen et al., 2011). However, the MeCP2 KI mice displayed enhanced sensitivity to environmental stress in the forced-swim and tail-suspension tests. Interestingly, sucrose preference did not differ between MeCP2 WT mice and their KI littermates, suggesting that valuation of natural rewards is not impaired in the KI mice. These data are interesting in light of our previous observation that the MeCP2 KI mice show impaired performance in two behavioral tasks (novel object recognition and novel social interactions) that depend on novelty (Cohen et al., 2011). Although novel objects and social interactions are thought to confer reward in these tasks, novelty also induces stress in rodents, for example, as measured by the novelty-suppressed feeding task (Dulawa and Hen, 2005). It is possible that disruption of the balance between novelty-induced stress and reward may underlie the impaired performance of the MeCP2 KI mice in novelty-dependent learning tasks.
Our observation that the MeCP2 KI mice are resistant to the effect of imipramine after chronic social defeat reveals a novel requirement for pMeCP2 in chronic antidepressant actions. The fact that the MeCP2 KI mice respond to acute imipramine in the forced-swim test indicates that the molecular pathways that mediate the pharmacological response to this drug are intact in the absence of pMeCP2. Because signaling cascades activated downstream of 5-HT receptors induce the phosphorylation of MeCP2 in brain regions relevant to the emotional actions of these drugs, we hypothesize that the loss of pMeCP2 disrupts the imipramine-induced transcriptional responses that support the plasticity of these neural circuits induced after chronic imipramine administration. However, we cannot rule out the possibility that the constitutive Ser421Ala Mecp2 mutation alters brain development in ways that selectively impair sensitivity to chronic but not acute imipramine. Future studies that restrict the spatial and temporal expression of pMeCP2 to specific regions of the adult brain will be required to directly test this hypothesis.
We showed previously pMeCP2 induction in the NAc after in vivo administration of a number of drugs, including the SSRI citalopram, which selectively activate DA and 5-HT transmission (Deng et al., 2010; Hutchinson et al., 2012). Imipramine, a tricyclic compound, was first identified as an effective antidepressant >50 years ago and is Food and Drug Administration-approved to treat depression (Kuhn, 1958; Richelson, 2003). Imipramine has multiple targets, but its strongest affinity is for the SERT and NET (Richelson, 2003). Because selective inhibition of the NET is not sufficient to induce pMeCP2 in the NAc (Hutchinson et al., 2012), we suspect that imipramine-dependent increases in 5-HT neurotransmission induce pMeCP2. However, activation of pMeCP2 in neurons is not restricted to stimuli that activate G-protein-coupled receptors, and pMeCP2 has been shown to be mediated by intracellular calcium signaling pathways and calcium–calmodulin kinases after membrane depolarization or synaptic NMDA receptor activation (Zhou et al., 2006). These data are interesting because they raise the question of whether antidepressants that act through 5-HT-independent mechanisms also induce pMeCP2. For example, both the noncompetitive NMDA receptor antagonist ketamine and the muscarinic ACh receptor agonist scopolamine have antidepressant effects in animal models of depression, as well as clinical studies (Berman et al., 2000; Furey and Drevets, 2006; Autry et al., 2011; Li et al., 2011b). These antidepressants are of particular clinical interest because ketamine and scopolamine more rapidly alleviate depression-like symptoms within hours to days of treatment (Zarate et al., 2006; Sanacora et al., 2008; Autry et al., 2011). Whether these drugs require pMeCP2 for their behavioral effects remains to be determined.
Our data complement a body of evidence implicating transcriptional changes in the NAc as both positive and negative regulators of depressive-like behaviors (Nestler and Carlezon, 2006). For example, expression of ΔFosB, a Fos family transcription factor implicated in behavioral responses to both reward and stress, is induced after chronic social defeat stress in the NAc (Perrotti et al., 2004). This appears to be an adaptive response that limits depressive-like stress-induced behaviors because increases in ΔFosB expression in the NAc are both sufficient and required for resilience to social defeat (Vialou et al., 2010). In contrast, decreases in Hdac5 expression and increases in Dnmt3 expression, which are seen in the NAc after social defeat, appear to be positively associated with depressive-like behaviors (Renthal et al., 2007; LaPlant et al., 2010). Our observation that imipramine induces pMeCP2 in neurons of the NAc and that this phosphorylation is required for chronic antidepressant action in the chronic social defeat stress paradigm suggests that pMeCP2 acts to induce adaptive neural responses in the NAc that ameliorate the behavioral consequences of stress. Interestingly, we showed previously that MeCP2 also acts in the NAc to limit the rewarding properties of the psychostimulant AMPH (Deng et al., 2010). Whether Ser421 phosphorylation of MeCP2 is required for the effects of MeCP2 on reward remains unknown, but these data raise the question of how a single transcriptional regulator can oppose both reward and stress. It is notable that the induction of pMeCP2 after both AMPH and imipramine administration is restricted to a specific population of GAD67 and parvalbumin-positive fast-spiking GABAergic interneurons (FSIs) within the NAc (Fig. 1c) (Deng et al., 2010; Hutchinson et al., 2012). The function of FSIs in the behavioral output of NAc circuits is poorly understood. However, FSIs exert robust feedforward inhibition over both direct and indirect pathway medium spiny neurons (Koós and Tepper, 1999; Gittis et al., 2010). Given the evidence that selective activation of the direct and indirect pathways induce distinct behavioral states (Lobo et al., 2010), we speculate that FSIs in the NAc may be able to modulate both reward-related and depressive-like behaviors depending on the circuit context in which they are activated.
Finally, we find that chronic but not acute imipramine induces pMeCP2 in the LHb, a brain region shown to regulate stress and reward (Hikosaka, 2010; Bromberg-Martin and Hikosaka, 2011; Li et al., 2011a). The LHb serves as a connection between the forebrain and midbrain structures that regulate emotional behaviors, and importantly LHb neurons modulate the activity of both DA and 5-HT neurons (Hikosaka, 2010). In response to stress, aversive stimuli, or smaller-than-expected rewards, glutamatergic neurons in the LHb inhibit DA neurons in the VTA via the activation of GABAergic neurons in the rostromedial tegmental nucleus that project to VTA (Ji and Shepard, 2007; Matsumoto and Hikosaka, 2009; Hong et al., 2011). Dopaminergic hypoactivity after repeated exposure to aversive or stressful stimuli then results in reduced motor activity and decreased motivation, two features of depressive-like behaviors. Neurons in the LHb also project to serotonergic neurons in the dorsal raphe nucleus and the medial raphe nucleus. Interestingly, activation of the LHb has been shown to both inhibit and facilitate 5-HT release (Kalén et al., 1985; Nishikawa and Scatton, 1985); therefore, activation of the LHb may have differential effects on stress responses depending on the specific consequences for 5-HT transmission. Interestingly, our data (Fig. 4a) suggest that pMeCP2 is induced in a subpopulation of neurons on the medial side of the LHb. Determining the specific projections of these pMeCP2-expressing LHb neurons has the potential to enhance understanding of the midbrain circuits that modulate depressive-like behaviors.
Footnotes
This work was supported by National Institutes of Health (NIH) Predoctoral National Research Service Award F31MH090754 (A.N.H.) and NIH Grants R01DA022202 and R21MH096310 (A.E.W.). We thank Dr. Michael E. Greenberg for the generous gift of the Mecp2 Ser421Ala knock-in mice, Dr. William Wetsel and Dr. Ramona Rodriguiz for assistance with the behavioral analyses, Dr. Herb Covington for advice on experimental design and critical reading of this manuscript, and Young May Cha for assistance with the statistical analyses.
The authors declare no competing financial interests.
References
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bromberg-Martin ES, Hikosaka O. Lateral habenula neurons signal errors in the prediction of reward information. Nat Neurosci. 2011;14:1209–1216. doi: 10.1038/nn.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, Wetsel WC, West AE, Greenberg ME. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington HE, 3rd, Maze I, LaPlant QC, Vialou VF, Ohnishi YN, Berton O, Fass DM, Renthal W, Rush AJ, 3rd, Wu EY, Ghose S, Krishnan V, Russo SJ, Tamminga C, Haggarty SJ, Nestler EJ. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, West AE. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci. 2010;13:1128–1136. doi: 10.1038/nn.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulawa SC, Hen R. Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci Biobehav Rev. 2005;29:771–783. doi: 10.1016/j.neubiorev.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Fukui M, Rodriguiz RM, Zhou J, Jiang SX, Phillips LE, Caron MG, Wetsel WC. Vmat2 heterozygous mutant mice display a depressive-like phenotype. J Neurosci. 2007;27:10520–10529. doi: 10.1523/JNEUROSCI.4388-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–1129. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Nelson AB, Thwin MT, Palop JJ, Kreitzer AC. Distinct roles of GABAergic interneurons in the regulation of striatal output pathways. J Neurosci. 2010;30:2223–2234. doi: 10.1523/JNEUROSCI.4870-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D, Allen M, Zhang L, Amorim M, Wang IT, Reyes AR, Mercado-Berton A, Ong C, Cohen S, Hu L, Blendy JA, Carlson GC, Siegel SJ, Greenberg ME, Zhou Z. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat Neurosci. 2012;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden SA, Covington HE, 3rd, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nat Protoc. 2011;6:1183–1191. doi: 10.1038/nprot.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- Hikosaka O. The habenula: from stress evasion to value-based decision-making. Nat Rev Neurosci. 2010;11:503–513. doi: 10.1038/nrn2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Jhou TC, Smith M, Saleem KS, Hikosaka O. Negative reward signals from the lateral habenula to dopamine neurons are mediated by rostromedial tegmental nucleus in primates. J Neurosci. 2011;31:11457–11471. doi: 10.1523/JNEUROSCI.1384-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson AN, Deng JV, Aryal DK, Wetsel WC, West AE. Differential regulation of MeCP2 phosphorylation in the CNS by dopamine and serotonin. Neuropsychopharmacology. 2012;37:321–337. doi: 10.1038/npp.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Shepard PD. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABAA receptor-mediated mechanism. J Neurosci. 2007;27:6923–6930. doi: 10.1523/JNEUROSCI.0958-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Jakovcevski M, Bharadwaj R, Connor C, Schroeder FA, Lin CL, Straubhaar J, Martin G, Akbarian S. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci. 2010;30:7152–7167. doi: 10.1523/JNEUROSCI.1314-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalén P, Karlson M, Wiklund L. Possible excitatory amino acid afferents to nucleus raphe dorsalis of the rat investigated with retrograde wheat germ agglutinin and d-[3H]aspartate tracing. Brain Res. 1985;360:285–297. doi: 10.1016/0006-8993(85)91244-2. [DOI] [PubMed] [Google Scholar]
- Koós T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryavtseva NN, Bakshtanovskaya IV, Koryakina LA. Social model of depression in mice of C57BL/6J strain. Pharmacol Biochem Behav. 1991;38:315–320. doi: 10.1016/0091-3057(91)90284-9. [DOI] [PubMed] [Google Scholar]
- Kuhn R. The treatment of depressive states with G 22355 (impramine hydrochloride) Am J Psychiatry. 1958;115:459–464. doi: 10.1176/ajp.115.5.459. [DOI] [PubMed] [Google Scholar]
- LaPlant Q, Vialou V, Covington HE, 3rd, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iñiguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolaños CA, Kabbaj M, Xiao G, Neve RL, Hurd YL, Oosting RS, Fan G, Morrison JH, Nestler EJ. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecourtier L, Kelly PH. A conductor hidden in the orchestra? Role of the habenular complex in monoamine transmission and cognition. Neurosci Biobehav Rev. 2007;31:658–672. doi: 10.1016/j.neubiorev.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011a;470:535–539. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, Li XY, Aghajanian G, Duman RS. Glutamate N-methyl-d-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011b;69:754–761. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Hikosaka O. Representation of negative motivational value in the primate lateral habenula. Nat Neurosci. 2009;12:77–84. doi: 10.1038/nn.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JS, Smith KA, Cowen PJ, Friston KJ, Dolan RJ. Covariation of activity in habenula and dorsal raphe nuclei following tryptophan depletion. Neuroimage. 1999;10:163–172. doi: 10.1006/nimg.1999.0455. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Scatton B. Inhibitory influence of GABA on central serotonergic transmission. Involvement of the habenulo-raphe pathways in the GABAergic inhibition of ascending cerebral serotonergic neurons. Brain Res. 1985;331:81–90. doi: 10.1016/0006-8993(85)90717-6. [DOI] [PubMed] [Google Scholar]
- Perrotti LI, Hadeishi Y, Ulery PG, Barrot M, Monteggia L, Duman RS, Nestler EJ. Induction of deltaFosB in reward-related brain structures after chronic stress. J Neurosci. 2004;24:10594–10602. doi: 10.1523/JNEUROSCI.2542-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Richelson E. Interactions of antidepressants with neurotransmitter transporters and receptors and their clinical relevance. J Clin Psychiatry. 2003;64(Suppl 13):5–12. [PubMed] [Google Scholar]
- Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–437. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, Storm D, Duman RS. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Ullsperger M, von Cramon DY. Error monitoring using external feedback: specific roles of the habenular complex, the reward system, and the cingulate motor area revealed by functional magnetic resonance imaging. J Neurosci. 2003;23:4308–4314. doi: 10.1523/JNEUROSCI.23-10-04308.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vialou V, Maze I, Renthal W, LaPlant QC, Watts EL, Mouzon E, Ghose S, Tamminga CA, Nestler EJ. Serum response factor promotes resilience to chronic social stress through the induction of ΔFosB. J Neurosci. 2010;30:14585–14592. doi: 10.1523/JNEUROSCI.2496-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MB, Xiao G, Kumar A, LaPlant Q, Renthal W, Sikder D, Kodadek TJ, Nestler EJ. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29:7820–7832. doi: 10.1523/JNEUROSCI.0932-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK. A randomized trial of an N-methyl-d-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]