

Abstract
Background and Objectives
Application of local anesthetics may lead to nerve damage. Increasing evidence suggests that risk of neurotoxicity is higher in patients with diabetic peripheral neuropathy. Additionally, block duration may be prolonged in neuropathy. We sought to investigate neurotoxicity in vitro and block duration in vivo in a genetic animal model of diabetes mellitus type II.
Methods
In the first experiments, neurons harvested from control Zucker Diabetic Fatty (ZDF) rats were exposed to acute (24 hours) or chronic (72 hours) hyperglycemia, followed by incubation with lidocaine 40 mM (approximately 1%). In a second experiment, neurons harvested from control ZDF rats, or diabetic ZDF rats, were incubated with lidocaine, with or without SB203580, an inhibitor of the p38 Mitogen-Activated Protein Kinase. Finally, we performed sciatic nerve block (lidocaine 2%, 0.2 mL) in control or diabetic ZDF rats, and measured motor and nociceptive block duration.
Results
In vitro, neither acute nor chronic hyperglycemia altered neurotoxic properties of lidocaine. In vitro, incubation of neurons with lidocaine resulted in a slightly decreased survival ratio when neurons were harvested from diabetic (57 ± 19) as compared to control (64 ± 9 %) rats. The addition of SB203580 partly reversed this enhanced neurotoxic effect and raised survival to 71 ± 12 in diabetic and 66 ± 9 % in control rats, respectively. In vivo, even though no difference was detected at baseline testing, motor block was significantly prolonged in diabetic as compared to control rats (137 ± 16 min versus 86 ± 17 min).
Conclusions
In vitro, local anesthetic neurotoxicity was more pronounced on neurons from diabetic animals, but the survival difference was small. In vivo, subclinical neuropathy leads to substantial prolongation of block duration. We conclude that early diabetic neuropathy increases block duration, while the observed increase in toxicity was small.
Introduction
Local anesthetics are widely used to block nerve conduction for surgical anesthesia, or to manage acute and chronic pain. Unfortunately, local tissue damage, in particular neurotoxicity induced by local anesthetics, remains a substantial concern.1 This concern may apply even more to nerves with pre-existing pathology.2 Neuropathy due to diabetes mellitus (DM) may be a relevant risk factor for regional anesthetic procedures.3,4 It is the most common neuropathy worldwide, affecting a large proportion of diabetes patients.5 The relevance of diabetic neuropathy for daily anesthetic practice lies in the fact that because of their frequent comorbidities, diabetic patients are predestined to undergo many types of surgery under regional anesthesia. Results from some experimental trials suggest that diabetic neuropathic nerves are more susceptible to local anesthetic-induced neurotoxicity than healthy control nerves during peripheral nerve blockade,6,7 while another recent trial found no difference in toxicity when effects of spinal anesthesia were examined.8 Epidemiological data suggest that nerve damage following regional anesthesia may be up to 10 times more frequent in diabetic patients as compared to the general patient population.2,9 The effect of isolated hyperglycemia on local anesthetic-induced neurotoxicity has not been investigated.
According to the “double-crush-hypothesis,” pre-existing nerve damage may further pre-dispose nerves to suffer from local anesthetic-induced neurotoxicity.10 One pathogenic pathway activated in both diabetic neuropathy and local anesthetic-induced neuropathy is activation of the p38 Mitogen-Activated Protein Kinase (MAPK).3 Neurotoxicity of the prototype local anesthetic, lidocaine, is mediated by specific activation of the p38 MAPK, while its inhibition significantly attenuates toxicity.11 Similarly, expression of the p38 MAPK has been correlated with the development of diabetic sensorimotor neuropathy in a streptozotocin-induced rat diabetic model, while its inhibition resulted in restoration of normal nerve conduction.12
Further, it has been controversially discussed whether diabetic peripheral neuropathy affects block duration.3 Three studies investigated this in a streptozotocin-induced rat model, with 1 study demonstrating normal,6 and 2 studies showing prolonged block duration.7,8 In diabetic patients, spinal anesthesia has been reported to last longer than in healthy control patients.13
The present study therefore aimed to test in a rat model of Type II diabetes the hypothesis that pre-existing subclinical neuropathy, but not isolated hyperglycemia, would pre-dispose nerves to injury by local anesthetics. Our secondary goals were to determine length of nerve block, and examine potential neuroprotective effects of an inhibitor of the p38 MAPK, SB 203580.
Methods
Experimental procedures were approved by the Animal Care and Use Committee of the Austrian Federal Ministry of Science, Education and Culture (BMWF-66.011/0097-C/GT/2007) and the Harvard Medical Area Standing Committee on Animals (Boston, Massachusetts).
Drugs
For in vitro experiments, the pH of the lidocaine stock solution 1 M was 4.65 (in dimethylsulfoxide). The pH of the final solution added to medium was 7.38 for lidocaine 40 mM (Sigma Aldrich, Vienna, Austria), which corresponds to a concentration of approximately 1%. The concentration of dimethylsulfoxide (Sigma, Vienna) in treated cultures was not significantly higher than in control cultures incubated with vehicle only. The inhibitor of the p38 MAPK, SB203580, was co-incubated with lidocaine in selected cultures at a final concentration of 10 μM. D-Glucose (Sigma, Vienna) was prepared in a 1 M stock solution in distilled water. For experiments, we diluted glucose in Roswell Park Memorial Institute (RPMI)-medium supplemented with B27- and antibiotic additives to achieve a final concentration in medium of 5 mM (control) or 25 mM (hyperglycaemic).14 Incubation of neuronal cell cultures with lidocaine, with or without addition of SB203580 (Sigma, Vienna), was performed for 24 hours. This timeframe was used in previous investigations and, combined with the lidocaine concentration of 40 mM, resulted in approximately 50% of cell death.11,15,16
Animals
We employed adult male Zucker Diabetic Fatty (ZDF) rats, a combined congenital / dietary inbred model of type II diabetes mellitus obtained from Charles River Laboratories (Sulzfeld, Germany or Wilmington, Massachusetts). The main pathogenic mechanism in these animals is a homozygous missense mutation in the leptin receptor (ZDFfa/fa genotype). ZDF rats develop metabolic syndrome, including hyperglycemia.17 Two types of ZDF rats were used: the genotype homozygous for obesity and metabolic syndrome (ZDFfa/fa, “diabetic”), and heterozygous normoglycemic animals (ZDFfa/−, “control”). The progression from pre-diabetic to diabetic state has been described in detail.18 Diabetic rats suffer from obesity, insulin resistance, and overt type II diabetes mellitus starting at 7 to 10 weeks of age. At age 12 to 14 weeks, ZDF rats develop slowing of conduction velocity indicative of diabetic neuropathy.19 Animals were used at 6 weeks of age for cell culture experiments into acute and chronic hyperglycemia, and at 12 weeks of age for in vitro and in vivo experiments into subclinical diabetic neuropathy. Until experiments, diabetic rats received water and irradiated Purina diet #5008 (Charles River Laboratories) ad libitum. This diet has been shown to most reliably promote the progression of metabolic syndrome in ZDF rats, and has been used in previous investigations on diabetic neuropathy in this model, eg,20
Primary sensory neuron culture
Dorsal root ganglion (DRG) cultures were obtained as described previously.21 Neurons were acutely harvested from (ZDFfa/fa) or ZDF control rats, which were euthanized by carbon dioxide narcosis, according to institutional protocol (Animal Care and Use Committee of the Austrian Federal Ministry of Education, Science and Culture, Vienna, Austria). DRGs were de-sheathed and incubated in collagenase 5000 U/mL for 90 minutes at 37°C, followed by 15 minutes in 0.25% trypsin/EDTA. After dissociation in Roswell Park Memorial Institute (RPMI) medium containing 10% horse/5% fetal bovine serum, neurons were plated in RPMI medium supplemented with B27 additives (1:100) and antibiotics (penicillin, 1000 units/mL; streptomycin, 1000 μg/mL; and amphotericin B, 25 μg/mL in 0.85% saline), all purchased from Invitrogen (Carlsbad, California). Neurons were allowed to adhere to the glass floor of dishes coated with poly-D-lysine/laminin for 24 hours. Poly-D-lysine was applied at a concentration of 0.1 mg/mL in distilled H20 and laminin at 7 μg/mL in RPMI solution. Cell cultures were kept at 37°C in a humidified atmosphere containing 5% CO2.
In vitro model of diabetic metabolism
After harvesting ganglions from healthy ZDFfa/− control rats, cell cultures had 24 hours time to stabilize until continued treatment with glucose. Subsequently, we maintained control (5 mM) and high (25 mM) glucose concentrations for 24 or 72 hours to simulate acute or hyperglycemia, or diabetic metabolism as described before.14
Inhibition of p38 MAP kinase
To determine whether blocking of p38 MAPK would increase neuronal cell survival in neurons treated with lidocaine, we co-incubated some cultures with an inhibitor of the p38 MAPK, SB203580, at a concentration of 10μm.11
Quantification of neurotoxicity in vitro
We quantified neurotoxicity using the cytotoxicity detection kit (Roche Diagnostics, Graz, Austria). This assay detects lactate dehydrogenase (LDH) released from injured cells. We used sterile 96-well tissue culture plates and incubated cells as described above with experimental drugs. Cell injury results in release of LDH, which is subsequently present in the culture supernatant. Supernatant is extracted and mixed with reagent containing a tetrazolium salt, which is cleaved by LDH to result in the formation of formazan after incubation at 30 minutes at room temperature under light protection. Detection of water-soluble formazan is done by determining absorption at 492 nm, interpolating values with low and high controls included in the kit. Plausibility control was undertaken to remove extreme outliers. Due to measurement variability, some LDH values resulted in survival values of just above 100%.
Peripheral nerve blockade
In vivo experiments were conducted according to a protocol approved by the Harvard Medical Area Standing Committee on Animals (Boston, Massachusetts). Neurobehavioral investigation was performed as described before by an investigator blinded to experimental group allocation.22-24 We performed in vivo experiments on ZDFfa/fa (“diabetic”) and ZDFfa/− (“control”) rats. All rats were weighed before experiments. We tested nociceptive and sensory function with mechanical pinch with serrated forceps. Tests were performed bilaterally at all measurement time points. In male diabetic ZDF animals, a specialized diet such as applied in our animals results in hyperlipidemia and hyperglycemia by 8 weeks of age, and diabetes by 12 weeks of age (Charles River Laboratories). In functional tests, motor nerve conduction velocity, as a sign of subclinical diabetic neuropathy, is decreased at 12 to 14 weeks of age, indicating first underlying pathological processes.19
Test animals were anesthetized with Sevoflurane (Abbott Laboratories, Abbott Park, Illinois) during all procedures. Sciatic nerves were surgically exposed by lateral incision of the thighs and division of the superficial fascia and muscle as described before.21 A volume of 0.2-mL test drug was injected directly beneath the clear fascia surrounding the nerve, but outside the perineurium, proximal to the sciatic bifurcation. We chose lidocaine 2% (corresponding to approximately 80 mM) as described previously.21 The wound was sutured with 3-0 vicryl suture.
Motor function was assayed before injection, and after 15, 30, 45, 60, 75, 90, 120, and 150 minutes by holding the rat upright with the control hind limb extended so that the distal metatarsus and toes of the target leg supported the animal's weight; the extensor postural thrust was recorded as the force (in grams) applied by each of the 2 hind limbs to a digital platform balance (Ohaus Lopro; Fisher Scientific, Florham Park, New Jersey). The reduction in this force, representing reduced extensor muscle contraction caused by motor block, was calculated as a percentage of the control force (pre-injection control value 90–130 g). The percent reduction in force was assigned a “range” score: 0 = no block (or baseline); 1 = minimal block, force between the pre-injection control value of 100% and 50%; 2 = moderate block, force between 50% of the pre-injection control value and 12 g (approximately 12 g represented the approximate weight of the flaccid limb); 3 = complete block, force 12 g or less.
Nociception was evaluated by the nocifensive withdrawal reflex and vocalization to pinch of a skin fold over the lateral metatarsus (cutaneous pain) with a serrated forceps; the force and duration of this pinch was held as constant as possible. The extent of the nocifensive withdrawal reflex and vocalization were combined on a scale of 0 to 3 for each examination. Grading was as follows: 3 = complete block, no nocifensive reaction or vocalization; 2 = moderate block, vocalization accompanied by slow withdrawal or flexion of the leg; 1 = minimal block, brisk flexion of the leg, with some sideways movement of the body or other escape response and loud vocalization; 0 = baseline with quick nocifensive responses listed above. The contralateral limb was used as control in all experiments. We defined the time to complete absence of block (motor block and nociceptive block score = 0) as block time.
Statistics
We used ANOVA and, if significant, unpaired t-test to compare data between 2 groups. Statistical significance was assumed at P < 0.05. Bonferroni-Holm was used to correct for multiple group comparisons. SPSS 16.0 was used for statistical analyses.
Results
In vitro, acute or chronic hyperglycemia do not increase lidocaine neurotoxicity
The application of control or high concentrations of glucose for 24 hours prior to lidocaine incubation did not alter neurotoxicity. In comparison to control cultures and high glucose alone, incubation with lidocaine alone led to a reduction in survival of about 50% in both control and high-glucose cultures (Table 1). Co-application of SB203580 with lidocaine did not prevent lidocaine-induced neurotoxicity (52 ± 15%, n = 16).
Table 1. Survival in response to lidocaine after acute or chronic hyperglycemia.
| Treatment group | Survival | n | Significance |
|---|---|---|---|
| Acute hyperglycemia (24 hours) | |||
|
| |||
| 1. Control | 105 ± 4 | 9 | S vs. 2 and 4 |
| NS vs. 3 | |||
| 2. Lidocaine | 45 ±15 | 17 | S vs. 1 and 3 |
| NS vs. 4 | |||
| 3. High glucose | 107 ± 6 | 17 | S vs. 2 and 4 |
| NS vs. 1 | |||
| 4. High glucose plus lidocaine | 55 ±13 | 17 | S vs. 1 and 3 |
| NS vs. 2 | |||
| Chronic hyperglycemia (72 hours) | |||
|
| |||
| 1. Control | 103 ± 4 | 9 | S vs. 2 and 4 |
| NS vs. 3 | |||
| 2. Lidocaine (as above) | 45 ± 15 | 17 | S vs. 1 and 3 |
| NS vs. 4 | |||
| 3. High glucose | 102 ±7 | 17 | S vs. 2 and 4 |
| NS vs. 1 | |||
| 4. High glucose plus lidocaine | 53 ± 27 | 16 | S vs. 1 and 3 |
| NS vs. 2 | |||
Legend: S significant NS non-significant.
In both cultures incubated with control medium or high glucose levels for 72 hours, lidocaine incubation led to a reduction in cell survival of approximately 50% (Table 1). Co-application of SB203580 with lidocaine resulted in a cell number of 50 ± 25% (n = 16, non-significant).
In vitro, lidocaine is more neurotoxic in diabetic than in control cells
In neuron cultures from diabetic animals harvested at 12 weeks of age, control cultures had a viability of 117 ± 7 % (n = 18). Cultures from control animals incubated with lidocaine had a cell count of 64 ± 9 % (n = 33, P < 0.001 as compared to controls), whereas neurons from diabetic animals had a cell count of 57 ± 19 % (n = 64, P < 0.001 as compared to controls, P < 0.05 as compared to non-diabetic animals after multiple-group correction, Fig. 1).
Figure 1.
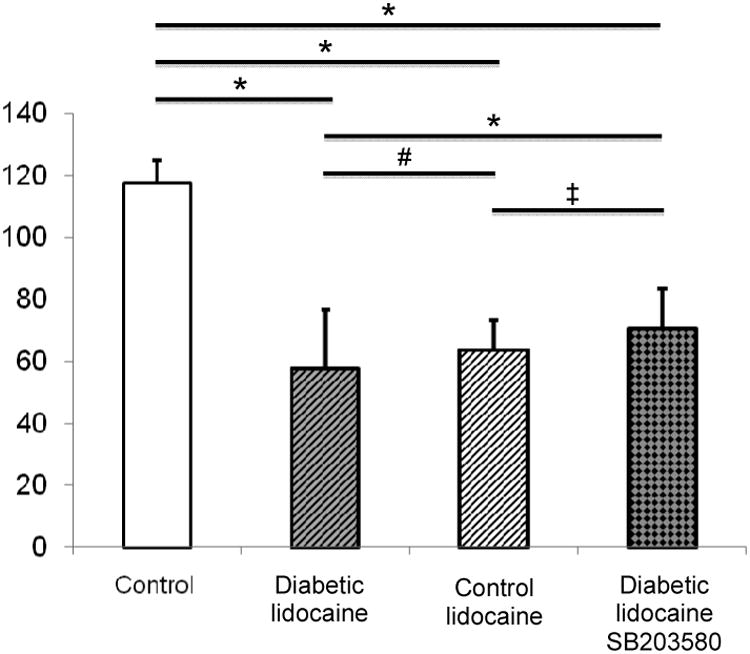
Comparison of viability of dorsal root ganglia primary cultures of diabetic (ZDFfa/fa) and control (ZDFfa/−) animals. Cells were exposed for 24 hours to control medium, lidocaine, or lidocaine and MAP kinase inhibitor SB203580. Data are displayed as mean and standard deviation. Comparisons were made by means of student-t-test with Bonferroni-Holm correction. *delineates p < 0.0001; ‡= p < 0.01; # = p < 0.05.
In vitro, co-incubation of lidocaine with SB203580 reduces neurotoxicity in diabetic neurons
Neurons from diabetic animals incubated with lidocaine had a survival rate of 57 ± 19 % (n = 64), and addition of SB203580 to lidocaine in neurons from diabetic animals resulted in a survival of 71 ± 12 % (n = 66, P < 0.001 to controls, P < 0.001 as compared to lidocaine in diabetic animals). Some non-diabetic cultures were incubated with lidocaine and SB203580, resulting in a survival of 66 ± 9 % (n = 33, non-significant as compared to lidocaine in diabetic animals).
In vivo, subclinical diabetic neuropathy prolongs lidocaine duration of action
At baseline, behavioral tests were not significantly different between diabetic and non-diabetic rats. In detail, motor testing revealed a force of 111 ± 12 g in control and 114 ± 12 g in diabetic animals. Nociceptive testing was 0 in all control, and 0 in all diabetic animals. In diabetic animals, motor block lasted significantly longer than in control animals (137 ± 16 min versus 86 ± 17 min, P < 0.001, Fig. 2A). Response to deep pinch was abolished longer in diabetic than in non-diabetic rats (128 ± 22 min versus 77 ± 10 min, P < 0.001, Fig. 2B). Response to superficial pinch was similarly abolished longer in diabetic than in non-diabetic rats (128 ± 22 min versus 81 ± 8 min, P < 0.001, Fig. 2C). All animals regained the values of the baseline behavioural test within a day. Return to baseline values for neurobehavioral tests was observed in all animals.
Figure 2.


The time course of motor (A), deep sensory (B), and superficial sensory (C) block in control and diabetic animals is displayed during 150 minutes after injection of lidocaine. Pre-defined 4-point scales were chosen with 0 denoting no block, and 3 complete block. Data presented as mean ± standard error, testing was done by means of ANOVA with post hoc Benforoni test. * denotes p < 0.05.
Note: For all nerve conduction tested, a significantly prolonged block was seen in diabetic animals.
Discussion
The main findings of our study are that (A) in vitro, lidocaine neurotoxicity is not altered by acute (24h) or chronic (72h) hyperglycemia; (B) in vitro, despite reaching statistical significance, subclinical diabetic neuropathy does not appear to substantially increase lidocaine neurotoxicity; (C) in vitro, inhibition of p38 MAPK, at least partly, reversed lidocaine neurotoxicity in diabetic neurons; and (D) in vivo, subclinical diabetic neuropathy increases block duration without apparent adverse behavioral effects after the blocks resolve. However, we did not histologically examine these lidocaine-exposed sciatic nerves to address potential disruption of fiber integrity after behavioral tests had returned to baseline. In nerve cell cultures from non-diabetic animals, lidocaine resulted in a reduction in cell survival of about 50%, confirming results from previous investigations and corroborating the experimental in vitro paradigm.11,21
In vitro, acute or chronic hyperglycemia do not increase lidocaine neurotoxicity
We first sought to determine whether acute or prolonged hyperglycemia per se would be sufficient to increase lidocaine neurotoxicity in vitro. Hyperglycemia alone did not enhance lidocaine-induced neurotoxicity. Even though a period of 72 hours has previously been described as sufficient to elicit first general changes indicative of diabetic metabolism in PC12 cell line cultures,14 this approach may not result in long-standing alterations characteristic of severe diabetic neuropathy, which may predispose nerves to injury following exposure to local anesthetics. Using doses of glucose that reflect previous investigations seeking to simulate diabetic conditions in vitro,14 we were unable to detect a negative impact of acute or chronic hyperglycemia on local anesthetic neurotoxicity.
In vitro, lidocaine neurotoxicity is comparable between diabetic and control cells
Secondly, neurons explanted from animals with longer-lasting diabetes (12 weeks) were compared to neurons harvested from non-diabetic animals with regards to sensitivity towards lidocaine neurotoxicity. The slight survival advantage of non-diabetic neurons observed was small, and despite reaching statistical significance, appears not to reflect clinically relevant neurotoxicity. No previous investigations have investigated the potential of neurotoxicity in neurons explanted from a genetically-modified rodent model. The timing of neuron harvesting was chosen to precede development of manifest diabetic neuropathy. It is generally thought that diabetic nerves are more susceptible to exogeneous trauma / ischemia / toxins.6,25,26 However, the results obtained by us are not as pronounced as those reported previously in vivo by others.6,7 Two reasons may account for this discrepancy. First, at the time of harvesting, animals had long-standing hyperglycemia but, presumably only mild, subclinical neuropathy, while animals in other trials6,7 were pronounced neuropathic. Zucker rats reliably develop progressive neuropathy,19 so by choosing a later time point for experiments, we may have found a more pronounced neurotoxic effect. Also, a recent study in Type I diabetic rats subjected to repeated intrathecal injections failed to demonstrate significant neurotoxicity.8 Second, the model employed by us was a model of DM type II, reflecting metabolic syndrome rather than the streptozotocin (STZ) model used in previous investigations.6,7 It has been stated that there is no single best animal model for research of regional anesthesia in diabetic neuropathy.27 We believe the model chosen by us relates reasonably well to the clinical situation, in which the majority of neuropathic patients presenting for surgery are Type II diabetics.
In vitro, co-incubation of lidocaine with SB203580 reduces neurotoxicity in diabetic neurons
Physiologically, the p38 MAPK is a member of the family of mitogen-activated protein kinases, key regulators of eukaryotic cell regulation.28 p38 MAPK is activated following a broad variety of stressors, such as environmental factors (cytotoxic substances, radiation, osmotic stress, heat shock), inflammatory cytokines (eg, Tumor Necrosis Factor α), and growth factors.28 P38 MAPK potently activates transcription factors and other protein kinases.28 Therapeutic inhibition of p38 MAPK activity has been shown to be of potential benefit in, among others, experimental nerve trauma,29 excitotoxicity,30 and growth factor withdrawal.31
In the setting of diabetic neuropathy, activation of p38 MAPK (leading to neuronal degeneration) coincides with the development of functional deficits in a STZ-based rat model, while pharmacologic inhibition of MAPK greatly attenuates functional changes.12 Similarly, p38 MAPK activation occurs when primary sensory neurons are exposed to toxic doses of local anaesthetics,11,21,32 and inhibition of p38 MAPK leads to decreased neurotoxicity in vitro and in vivo.21 We therefore hypothesized that activation of the same neurodegenerative p38 MAPK pathway by both diabetic neuropathy and local anaesthetic aggravates neuronal damage. This should be most prominent when lidocaine is applied, because the latter selectively activates p38 MAPK in neurons.11 We found only minimally increased neurotoxicity when lidocaine was applied to nerves harvested from diabetic animals as compared to control animals. Inhibition of the p38 MAPK, at least in part, abolished this effect in vitro. Inhibition of p38 MAPK reduced the lidocaine-induced neurotoxicity in diabetic neurons, confirming our experimental hypothesis. Effects on healthy neurons were not statistically different. The protective effect of p38 MAPK in healthy neurons was smaller compared to previous investigations.15,16,21 Several factors may account for the unexpected finding that lidocaine neurotoxicity in control cells was unaffected by p38 MAPK inhibition. The model used in this study differs from that used in previous studies as it represents a different genotype of rats (ZDF versus Sprague Dawley rats or PC12 cell line cultures).
In vivo, subclinical diabetic neuropathy prolongs lidocaine duration of action
We next investigated influence of diabetic subclinical neuropathy upon lidocaine block duration. At baseline, we found no difference in sensitivity, supporting our model of subclinical diabetic neuropathy. When lidocaine was applied to diabetic nerves, it led to a longer-lasting sensory and motor block compared to wild-type controls. In a previous study using the STZ diabetes model, Kalichman and Calcutt showed that lidocaine exhibited normal duration of block in diabetic nerves.6 Our results are, however, in concordance with a recent investigation by Kroin et al,7 who demonstrated increased sciatic nerve block duration in STZ diabetic rats, using a slightly longer pre-treatment time.7 In the same STZ experimental model in rats, spinal anesthesia was reported to result in a longer block duration in diabetic animals without a concomitant increase in neurotoxicity.8 In diabetic patients, spinal anesthesia was reported to last longer than in non-diabetic patients.13
The reason for increased block duration may be multifactorial, involving changes in nerve metabolism and physiology, and nerve fiber damage.7 Previous investigations had used open6 or percutaneous7 nerve block. We cannot exclude that the open approach resulted in inflammation around the nerve fascia, but in a recent experimental study, Kroin et al found no difference in block duration whether nerve block was performed open or closed.7
Limitations
Some limitations of the present study should be briefly addressed. First, our in vitro data cannot be directly correlated with clinical practice. As for peripheral nerve blockade (rat sciatic nerve block model), the drugs are applied to the axon rather than the DRG, making dissociated neuronal cultures an imperfect approximation. Moreover, glial cells, usually found in standard DRG cultures, could influence LA-induced neurotoxicity. We used lidocaine as the prototype amide-type local anesthetic. For peripheral nerve block in clinical practice, other drugs, such as mepivacaine (short-acting) or long-acting local anesthetics, are more frequently used. We suggest the ZDF model be used to investigate other local anesthetics in regular clinical use, and to include a group of animals with even longer-standing diabetes to evaluate fully developed diabetic neuropathy as well.
The ZDF rat has been described as a model of type II diabetes mellitus, but features other conditions as well, most notably obesity. In the ZDF model, male rats develop manifest hyperglycemia at 7 weeks of age. At the age of 12 to 14 weeks (the time point used for our experiments), ZDF rats feature an overt neuropathic deficit discernible by electrophysiologic investigation.19 We attest to the fact that we did not confirm these investigations in our test animals, such that presence of subclinical nerve damage was inferred from the literature showing a predictable clinical course of diabetes and complications,17 and a definable window when this damage appears.19 The ZDF model chosen to investigate the effects of long-standing hyperglycemia on regional anesthesia has been widely used in metabolic research. Nevertheless, a recent editorial highlighted the fact that all current diabetes models feature confounding factors.27 In the case of ZDF, it has been argued that it is more a model of metabolic syndrome than exclusively for diabetes, while the same applies for STZ-induced diabetes Type I, which is confounded by hepatic and renal damage, and substantial behavioral changes.27,33 Our model is most likely more representative of clinical reality for anesthesiologists, where patients with neuropathy due to Type II diabetes represent the most controversial patient collective.
Finally, we concluded from behavioral data that no overt neurotoxicity was observed in our model, without performing neurohistopathological investigations once tests had returned to baseline. Future investigations should include histological examination to corroborate our findings. We did not carry out neurobehavioral testing after tests had returned to baseline.
Conclusion
In conclusion, we demonstrate that in vitro, acute, or chronic hyperglycemia per se does not increase lidocaine-induced neurotoxicicity. We observed a small, albeit statistically significant, increase in neurotoxicity when lidocaine was applied to diabetic as compared to non-diabetic neurons, but the difference in our model appears small. The inhibitor of the p38 MAPK, SB203580, can at least partially reverse lidocaine-induced neurotoxicity in diabetic nerves. In vivo, block duration is substantially prolonged by subclinical diabetic neuropathy.
Acknowledgments
Supported by departmental funds and the National Institutes of Health, Bethesda, MD (Research Grant No. GM64051 to P. Gerner)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Auroy Y, Benhamou D, Bouaziz H, et al. Peripheral nerve block: yesterday's facts and tomorrow's challenges. Ann Fr Anesth Reanim. 2006;25:82–83. doi: 10.1016/j.annfar.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Hebl JR, Kopp SL, Schroeder DR, Horlocker TT. Neurologic complications after neuraxial anesthesia or analgesia in patients with preexisting peripheral sensorimotor neuropathy or diabetic polyneuropathy. Anesth Analg. 2006;103:1294–1299. doi: 10.1213/01.ane.0000243384.75713.df. [DOI] [PubMed] [Google Scholar]
- 3.Lirk P, Birmingham B, Hogan Q. Regional anesthesia in patients with preexisting neuropathy. Int Anesthesiol Clin. 2011;49:144–165. doi: 10.1097/AIA.0b013e3182101134. [DOI] [PubMed] [Google Scholar]
- 4.Blumenthal S, Borgeat A, Maurer K, et al. Preexisting subclinical neuropathy as a risk factor for nerve injury after continuous ropivacaine administration through a femoral nerve catheter. Anesthesiology. 2006;105:1053–1056. doi: 10.1097/00000542-200611000-00028. [DOI] [PubMed] [Google Scholar]
- 5.Said G. Diabetic neuropathy--a review. Nat Clin Pract Neurol. 2007;3:331–340. doi: 10.1038/ncpneuro0504. [DOI] [PubMed] [Google Scholar]
- 6.Kalichman MW, Calcutt NA. Local anesthetic-induced conduction block and nerve fiber injury in streptozotocin-diabetic rats. Anesthesiology. 1992;77:941–947. doi: 10.1097/00000542-199211000-00017. [DOI] [PubMed] [Google Scholar]
- 7.Kroin JS, Buvanendran A, Williams DK, et al. Local anesthetic sciatic nerve block and nerve fiber damage in diabetic rats. Reg Anesth Pain Med. 2010;35:343–350. doi: 10.1097/aap.0b013e3181e82df0. [DOI] [PubMed] [Google Scholar]
- 8.Kroin JS, Buvanendran A, Tuman KJ, Kerns JM. Safety of Local Anesthetics Administered Intrathecally in Diabetic Rats. Pain Med. 2012 doi: 10.1111/j.1526-4637.2012.01396.x. [DOI] [PubMed] [Google Scholar]
- 9.Brull R, McCartney CJ, Chan VW, El-Beheiry H. Neurological complications after regional anesthesia: contemporary estimates of risk. Anesth Analg. 2007;104:965–974. doi: 10.1213/01.ane.0000258740.17193.ec. [DOI] [PubMed] [Google Scholar]
- 10.Hebl JR, Horlocker TT, Pritchard DJ. Diffuse brachial plexopathy after interscalene blockade in a patient receiving cisplatin chemotherapy: the pharmacologic double crush syndrome. Anesth Analg. 2001;92:249–251. doi: 10.1097/00000539-200101000-00049. [DOI] [PubMed] [Google Scholar]
- 11.Haller I, Hausott B, Tomaselli B, et al. Neurotoxicity of Lidocaine Involves Specific Activation of the p38 Mitogen-activated Protein Kinase, but Not Extracellular Signal-regulated or c-jun N-Terminal Kinases, and Is Mediated by Arachidonic Acid Metabolites. Anesthesiology. 2006;105:1024–1033. doi: 10.1097/00000542-200611000-00025. [DOI] [PubMed] [Google Scholar]
- 12.Price SA, Agthong S, Middlemas AB, Tomlinson DR. Mitogen-activated protein kinase p38 mediates reduced nerve conduction velocity in experimental diabetic neuropathy: interactions with aldose reductase. Diabetes. 2004;53:1851–1856. doi: 10.2337/diabetes.53.7.1851. [DOI] [PubMed] [Google Scholar]
- 13.Echevarria M, Hachero A, Martinez A, et al. Spinal anaesthesia with 0.5% isobaric bupivacaine in patients with diabetes mellitus: the influence of CSF composition on sensory and motor block. Eur J Anaesthesiol. 2008;25:1014–1019. doi: 10.1017/S0265021508004729. [DOI] [PubMed] [Google Scholar]
- 14.Lelkes E, Unsworth BR, Lelkes PI. Reactive oxygen species, apoptosis and altered NGF-induced signaling in PC12 pheochromocytoma cells cultured in elevated glucose: an in vitro cellular model for diabetic neuropathy. Neurotox Res. 2001;3:189–203. doi: 10.1007/BF03033191. [DOI] [PubMed] [Google Scholar]
- 15.Lirk P, Haller I, Colvin H, et al. In vitro, inhibition of Stress-Activated Protein Kinase pathways protects against bupivacaine- and ropivacaine-induced neurotoxicity. Anesth Analg. 2008;106:1456–1464. doi: 10.1213/ane.0b013e318168514b. [DOI] [PubMed] [Google Scholar]
- 16.Lirk P, Haller I, Colvin HP, et al. In vitro, lidocaine-induced axonal injury is prevented by peripheral inhibition of the p38 mitogen-activated protein kinase, but not by inhibiting caspase activity. Anesth Analg. 2007;105:1657–1664. doi: 10.1213/01.ane.0000286171.78182.e2. table of contents. [DOI] [PubMed] [Google Scholar]
- 17.Chen D, Wang MW. Development and application of rodent models for type 2 diabetes. Diabetes Obes Metab. 2005;7:307–317. doi: 10.1111/j.1463-1326.2004.00392.x. [DOI] [PubMed] [Google Scholar]
- 18.Etgen GJ, Oldham BA. Profiling of Zucker diabetic fatty rats in their progression to the overt diabetic state. Metabolism. 2000;49:684–688. doi: 10.1016/s0026-0495(00)80049-9. [DOI] [PubMed] [Google Scholar]
- 19.Oltman CL, Coppey LJ, Gellett JS, Davidson EP, Lund DD, Yorek MA. Progression of vascular and neural dysfunction in sciatic nerves of Zucker diabetic fatty and Zucker rats. Am J Physiol Endocrinol Metab. 2005;289:E113–122. doi: 10.1152/ajpendo.00594.2004. [DOI] [PubMed] [Google Scholar]
- 20.Brussee V, Guo G, Dong Y, et al. Distal degenerative sensory neuropathy in a long-term type 2 diabetes rat model. Diabetes. 2008(57):1664–1673. doi: 10.2337/db07-1737. [DOI] [PubMed] [Google Scholar]
- 21.Lirk P, Haller I, Myers RR, et al. Mitigation of direct neurotoxic effects of lidocaine and amitriptyline by inhibition of p38 mitogen-activated protein kinase in vitro and in vivo. Anesthesiology. 2006;104:1266–1273. doi: 10.1097/00000542-200606000-00023. [DOI] [PubMed] [Google Scholar]
- 22.Gerner P, Haderer AE, Mujtaba M, et al. Assessment of differential blockade by amitriptyline and its N-methyl derivative in different species by different routes. Anesthesiology. 2003;98:1484–1490. doi: 10.1097/00000542-200306000-00028. [DOI] [PubMed] [Google Scholar]
- 23.Thalhammer JG, Vladimirova M, Bershadsky B, Strichartz GR. Neurologic evaluation of the rat during sciatic nerve block with lidocaine. Anesthesiology. 1995;82:1013–1025. doi: 10.1097/00000542-199504000-00026. [DOI] [PubMed] [Google Scholar]
- 24.Gerner P, Binshtok AM, Wang CF, et al. Capsaicin combined with local anesthetics preferentially prolongs sensory/nociceptive block in rat sciatic nerve. Anesthesiology. 2008;109:872–878. doi: 10.1097/ALN.0b013e31818958f7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zochodne DW, Cheng C, Sun H. Diabetes increases sciatic nerve susceptibility to endothelin-induced ischemia. Diabetes. 1996;45:627–632. doi: 10.2337/diab.45.5.627. [DOI] [PubMed] [Google Scholar]
- 26.Zochodne DW, Cheng C. Diabetic peripheral nerves are susceptible to multifocal ischemic damage from endothelin. Brain Res. 1999;838:11–17. doi: 10.1016/s0006-8993(99)01670-4. [DOI] [PubMed] [Google Scholar]
- 27.Williams BA, Murinson BB, Grable BR, Orebaugh SL. Future considerations for pharmacologic adjuvants in single-injection peripheral nerve blocks for patients with diabetes mellitus. Reg Anesth Pain Med. 2009;34:445–457. doi: 10.1097/AAP.0b013e3181ac9e42. [DOI] [PubMed] [Google Scholar]
- 28.Kyriakis JM, Avruch J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. doi: 10.1152/physrev.2001.81.2.807. [DOI] [PubMed] [Google Scholar]
- 29.Obata K, Yamanaka H, Kobayashi K, et al. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JY, Kim EJ, Kwon KJ, et al. Neuroprotection by fructose-1,6-bisphosphate involves ROS alterations via p38 MAPK/ERK. Brain Res. 2004;1026:295–301. doi: 10.1016/j.brainres.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 31.Horstmann S, Kahle PJ, Borasio GD. Inhibitors of p38 mitogen-activated protein kinase promote neuronal survival in vitro. J Neurosci Res. 1998;52:483–490. doi: 10.1002/(SICI)1097-4547(19980515)52:4<483::AID-JNR12>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 32.Haller I, Lirk P, Keller C, Wang GK, Gerner P, Klimaschewski L. Differential neurotoxicity of tricyclic antidepressants and novel derivatives in vitro in a dorsal root ganglion cell culture model. Eur J Anaesthesiol. 2007;24:702–708. doi: 10.1017/S0265021507000154. [DOI] [PubMed] [Google Scholar]
- 33.Tomlinson KC, Gardiner SM, Hebden RA, Bennett T. Functional consequences of streptozotocin-induced diabetes mellitus, with particular reference to the cardiovascular system. Pharmacol Rev. 1992;44:103–150. [PubMed] [Google Scholar]