

Protein drugs, due to their large size and hydrophilic nature, are normally precluded from effective delivery such as cell entry or tissue diffusion. Among the transport barriers, the skin poses as a formidable challenge to proteins due to the impermeable stratum corneum. The existing techniques for percutaneous protein delivery must rely on sophisticated delivery systems, such as the use of complicated nanocarriers or mechanical devices, to overcome the skin barrier for noninvasive delivery. A challenge in manufacturing of such systems is their complicated processes and potential negative impact on protein drug stability. Moreover, the high manufacturing cost of these advanced systems often offsets their remarkable advantages. To circumvent these problems that confront the current methods, we hypothesized the concept of “skin-permeable proteins” which would possess skin penetrating ability, and thereby eliminate a need for a transport vehicle. However, naturally occurring proteins with skin penetrating ability rarely exist. Herein, we present a novel strategy for chemically constructing artificial skin-permeable proteins, featured by a simple conjugation of a protein to a cell-penetrating peptide (CPP), which would display a penetration effect on the stratum corneum barrier, and transport the attached proteins into the skin. Furthermore, the feasibility of application in transcutaneous immunization is demonstrated.
CPPs are known for their versatility in carrying macro- or supra-molecules through the cell membrane barriers that challenge the conventional drug delivery approaches.[1] The CPPs are capable of transporting their cargos, often linked by a covalent bond, into almost all cell types.[2] Among such CPPs, the low molecular weight protamine (LMWP) peptide (VSRRRRRRGGRRRRR), developed in our laboratory by enzymatic digestion of protamine (an FDA approved drug), offers distinct advantages. First, LMWP is as potent as the virus-derived TAT peptide, the most-studied CPP to date, in mediating cellular translocation of the attached cargos.[3] Secondly, unlike other CPPs, the toxicity profile of LMWP has already been thoroughly established. LMWP was shown to be non-immunogenic,[4] and its use in dogs did not elicit acute toxic responses.[5] Lastly, while other CPPs must be chemically synthesized, LMWP can be produced in mass quantities direct from native protamine with limited processing time and cost.[6]
In this investigation, the artificial skin-permeable protein was synthesized by conjugating LMWP to ovalbumin (OVA), a representative antigenic protein, via a cleavable disulfide bond (Scheme 1). The LMWP-OVA conjugates were purified by heparin affinity chromatography, and the final product, generally possessing a 1:1 molar ratio of LMWP:OVA, was verified by MALDI-TOF-MS.
Scheme 1.

Chemical conjugation of LMWP to OVA
As noted, skin keratinocytes are a physical barrier that provides the front line of defense against infection and also poses a challenge to protein delivery. On the other hand, keratinocytes execute a “part-time” antigen-presenting function by secreting immune mediators and transferring antigens to local antigen-presenting cells.[7] LMWP was shown to exhibit an ability to translocate the linked cargos of varying sizes into keratinocytes (Figure 1), demonstrating the potential for percutaneous protein delivery.
Figure 1.

Uptake by human keratinocyte cells of a) rhodamine B; b) OVA; and c) BSA; compared with those of d) LMWP-rhodamine B; e) LMWP-OVA; and f) LMWP-BSA conjugates. Protein cargos were labeled with FITC. (Scale bar = 100 μm)
The plausibility of percutaneous delivery in vivo was examined by topical application of LMWP-linked lysozyme, OVA, or bovine serum albumin (BSA), to represent a broad range of protein sizes. All the LMWP-linked proteins successfully penetrated the stratum corneum and accumulated primarily in the epidermis (Figure 2), whereas the control proteins without LMWP linkage remained on the surface of skin.
Figure 2.

In vivo transcutaneous delivery mediated by LMWP. a), b), and c) represented unmodified free lysozyme, OVA, and BSA, whereas d), e), and f) represented LMWP-linked lysozyme, OVA, and BSA, respectively. Arrows represented the direction of skin penetration. (Scale bar = 200 μm)
The skin penetration mechanism of CPPs is still under debate. However, the interaction between CPP and lipid bilayer is believed to play a major role in the cell penetration process.[8] The skin permeability is governed by the physical state and structural organization of the extracellular lipids.[9] Hence, the skin penetration function of LMWP could account for its interaction with the skin extracellular lipid matrices. Such interaction would lead to disruption of the ordered lipid orientation, thereby creating channels for transducing protein cargos through the stratum corneum.
As a typical example of protein percutaneous delivery, the immunological milieu of the skin is an ideal site for noninvasive vaccine delivery. The epidermis is rich in mature Langerhans cells (LCs), which represent a network of immune cells that underlie 25% of the total surface area in human skin,[10] and thus the epidermis is the target skin layer for transcutaneous immunization (TI). TI can be achieved by topically applying antigens, which, with the aid of a transdermal delivery system, penetrate into skin and subsequently elicit the desired immunity. The network of LCs acts as an immunological line of defense and initiates immune responses by conveying the captured antigens to other cells of the immune system, e.g. lymphocytes, melanocytes and Mercel cells.[11] Therefore, the unique epidermal accumulation of the LMWP-linked proteins offers an ideal situation to alert such antigen-presenting cells.
The constructed skin-permeable antigen of LMWP-OVA was tested for the feasibility of TI on Balb/c mice. Humoral IgG is the primary protection induced by preventive vaccines to neutralize and eliminate of pathogens. Figure 3a revealed that a significant elevation of anti-OVA IgG concentration in the blood was observed following topical application of LMWP-OVA with cholera toxin as adjuvant. The IgG levels in TI groups treated with the high- (TI-H) and medium-dose (TI-M) of antigen displayed no statistical differences (p > 0.05) from those in animals given OVA through the standard intramuscular immunization method (IM group). The control group, receiving topical native OVA, exhibited markedly lower levels of IgG, due to poor percutaneous absorption of unmodified OVA. These findings indicated that the epidermis-accumulated LMWP-OVA was captured by LCs that subsequently migrated to lymphoid tissues and presented the antigens, effectively eliciting robust humoral immune responses. Furthermore, disulfide linkage could be cleaved by the elevated level of glutathione and reductase activity in the cytosol,[12] allowing release of OVA from LMWP, thus retaining a full intrinsic immunogenicity. As evidence, LMWP-OVA in TI method triggered OVA-specific IgG responses comparable to the IM injection of OVA. Since the conjugation of LMWP to OVA might affect its intrinsic antigenic determinants, a cleavable linkage could ease such concern.
Figure 3.
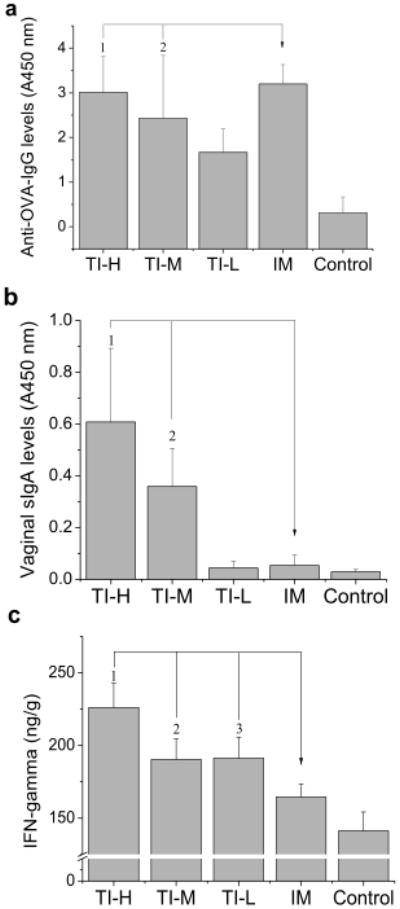
Transcutaneous immunization study. Mice were topically immunized with high- (500 μg; TI-H), medium- (250 μg; TI-M), and low-dose (100 μg; TI-L) of LMWP-OVA. a) High levels of anti-OVA IgG were observed in all TI groups, with no statistically difference between the TI-H (1p = 0.529) or TI-M (2p = 0.150) group and the IM group. Importantly, significant elevation in b) sIgA concentrations (1,2 p < 0.01); and c) IFN-γ levels (1,2,3 p < 0.01) were achieved by TI with LMWP-OVA, compared to that by IM injection of OVA. (n = 10)
TI shows advantages over conventional injection vaccination by offering the opportunity to elicit specific immune responses, such as targeted immunity to the female reproductive tract[13] and cytotoxic T lymphocytes (CTL) effect.[14] Secretory IgA (sIgA) is the predominant humoral defense mechanism at mucosal surface, and it therefore protects the host from initial infections. As shown in Figure 3b, the anti-OVA sIgA levels measured in vaginal secretions were significantly higher in TI-H and TI-M groups than those in the IM group, confirming the promise of TI in achieving local protective immunity against female genital infection. Furthermore, interferon-γ(IFN-γ), the representative cytokine known to enhance the CD8+ CTL-mediated cytotoxicity against infected cells, was also present at a level significantly higher in the TI groups than in the IM group (Figure 3c). Notably, local immune response in skin could also benefit from production of high levels of IFN-γ, due to its effect on promoting CTL recognition of antigen molecules in keratinocytes[15] and subsequently their expedited lysis.[16]
In addition, a primer-booster vaccination conducted by combining the IM injection of OVA with transcutaneous boosters of LMWP-OVA showed the immunity induction comparable to the multi-shot IM standard method (Figure S 1). The self-administrable boosters would eliminate follow-up visits to clinics for a multi-dose protocol. Hence this immunization strategy could improve not only patient compliance but also vaccination coverage in underserved areas with limited medical settings.
In conclusion, this methodology for constructing artificial skin-permeable antigens may offer simple and needle-free vaccination modalities without the need for sophisticated drug carriers or expensive medical devices. Such a method could be beneficial especially to developing countries that struggle to fulfill effective vaccination coverage.
Experimental Section
Detailed experimental procedures are available in Supplementary Information.
Supplementary Material
Footnotes
This study was supported by NIH R01 grant CA114612, and in part by WCU Program funded by the MEST, South Korea (Grant No. R31-2008-000-10103-0). Victor C. Yang is currently a Participating Faculty at the Department of Molecular Medicine and Biopharmaceutical Sciences, Seoul National University, South Korea.
References
- 1.a) Keller S, Sauer I, Strauss H, Gast K, Dathe M, Bienert M. Angew Chem Int Ed. 2005;44:5252–5255. doi: 10.1002/anie.200500519. [DOI] [PubMed] [Google Scholar]; b) Lim Y, Lee E, Lee M. Angew Chem. 2007;119:3545–3548. [Google Scholar]; Angew Chem Int Ed. 2007;46:3475–3478. doi: 10.1002/anie.200604576. [DOI] [PubMed] [Google Scholar]; c) Lim Y, Lee E, Lee M. Angew Chem. 2007;119:9169–9172. [Google Scholar]; Angew Chem Int Ed. 2007;46:9011–9014. doi: 10.1002/anie.200702732. [DOI] [PubMed] [Google Scholar]
- 2.a) Fawell S, Seery J, Daikh Y, Moore C, Chen L, Pepinsky B, Barsoum J. Proc Natl Acad Sci USA. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee S, Liang F, Guo X, Xie L, Cahill S, Blumenstein M, Yang H, Lawrence D, Zhang Z. Angew Chem Int Ed. 2005;44:4242–4244. doi: 10.1002/anie.200462004. [DOI] [PubMed] [Google Scholar]
- 3.a) Park Y, Liang J, Ko K, Kim S, Yang VC. J Gene Med. 2002;5:700–711. doi: 10.1002/jgm.402. [DOI] [PubMed] [Google Scholar]; b) Park Y, Chang L, Liang J, Moon C, Chung C, Yang VC. FASEB J. 2005;19:1555–1557. doi: 10.1096/fj.04-2322fje. [DOI] [PubMed] [Google Scholar]
- 4.Tsui B, Singh V, Liang J, Yang VC. Thromb Res. 2001;101:417–420. doi: 10.1016/s0049-3848(00)00427-8. [DOI] [PubMed] [Google Scholar]
- 5.Lee L, Chang L, Wrobleski S, Wakefield T, Yang VC. AAPS J. 2001;3:24–31. doi: 10.1208/ps030319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang L, Lee H, Yang Z, Yang VC. AAPS J. 2001;3:7–14. [Google Scholar]
- 7.Leggatt G, Dunn L, De Kluyver R, Stewart T, Frazer I. Immunol Cell Biol. 2002;80:415–424. doi: 10.1046/j.1440-1711.2002.01105.x. [DOI] [PubMed] [Google Scholar]
- 8.a) Mishra A, Gordon V, Yang L, Coridan R, Wong G. Angew Chem. 2008;120:3028–3031. doi: 10.1002/anie.200704444. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:2986–2989. doi: 10.1002/anie.200704444. [DOI] [PubMed] [Google Scholar]; b) Joanne P, Galanth C, Goasdoué N, Nicolas P, Sagan S, Lavielle S, Chassaing G, El Amri C, Alves I. Biochim Biophys Acta. 2009;1788:1772–1781. doi: 10.1016/j.bbamem.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Plasencia I, Norlen L, Bagatolli L. Biophys J. 2007;93:3142–3155. doi: 10.1529/biophysj.106.096164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glenn G, Taylor D, Li X, Frankel S, Montemarano A, Alving C. Nat Med. 2000;6:1403–1406. doi: 10.1038/82225. [DOI] [PubMed] [Google Scholar]
- 11.Babiuk S, Baca-Estrada M, Babiuk L, Ewen C, Foldvari M. J Control Release. 2000;66:199–214. doi: 10.1016/s0168-3659(99)00274-6. [DOI] [PubMed] [Google Scholar]
- 12.Saito G, Swanson J, Lee K. Adv Drug Deliv Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 13.Gockel C, Bao S, Beagley K. Mol Immunol. 2000;37:537–544. doi: 10.1016/s0161-5890(00)00074-2. [DOI] [PubMed] [Google Scholar]
- 14.Belyakov I, Hammond S, Ahlers J, Glenn G, Berzofsky J. J Clin Invest. 2004;113:998–1007. doi: 10.1172/JCI20261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niederwieser D, Aubock J, Troppmair J, Herold M, Schuler G, Boeck G, Lotz J, Fritsch P, Huber C. J Immunol. 1988;140:2556–2564. [PubMed] [Google Scholar]
- 16.Symington F, Santos E. J Immunol. 1991;146:2169–2175. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.