

Abstract
Diabetes mellitus encompasses two distinct disease processes: autoimmune Type 1 (T1D) and nonimmune Type 2 (T2D) diabetes. Despite the disparate aetiologies, the disease phenotype of hyperglycemia and the associated complications are similar. In this paper, we discuss the role of the CD39-adenosinergic axis in the pathogenesis of both T1D and T2D, with particular emphasis on the role of CD39 and CD73.
1. Introduction
Extracellular nucleotides, such as adenosine triphosphate (ATP), are important signalling molecules involved in many biological processes. Under basal conditions extracellular concentrations of ATP are maintained at low levels. Endogenous regulation of ATP concentration is mediated by ectoenzymes: the family of ectonucleotidases (E-NTPDases) and ecto-5′-nucelotidase (CD73; E.C. 3.1.3.5) located on the cell surface. Four plasma membrane-bound E-NTPDaess have been cloned: NTPDase1 (CD39; E.C.3.6.1.5), NTPDase2, NTPDase3, and NTPDase8 [1], each with distinct localization and biological properties. NTPDase1 hydrolyzes ATP and adenosine diphosphate (ADP) equally well; NTPDase2 preferentially hydrolyzes ADP; NTPDase3; NTPDase8 have intermediate hydrolysis profiles [2]. The hydrolysis of ATP and ADP generates adenosine monophosphate (AMP), which is then hydrolysed by CD73 to adenosine. CD39 is the rate-limiting enzyme [3] in this cascade and thus is the prime regulator of nucleotide and adenosine concentrations within the microenvironment.
Both CD39 and CD73 expressions are dynamic and change under pathophysiological conditions. Hypoxia upregulates both ectoenzymes—CD39 through Sp1-dependent pathways [4] and CD73 through binding of HIF-1 [5]. Further, within the CD73 gene, promoter region is a cAMP response element (CRE) which regulates transcription through cAMP-dependent CRE-binding protein (CREB). Activation of adenosine receptors increases cAMP and CREB suggesting that the enzymatic product of CD73 (adenosine) may transcriptionally regulate its expression (reviewed in [6]). Finally, the glucocorticoid dexamethasone increases AMP hydrolysis and CD73 expression which is mitigated by protein kinase C (PKC) inhibition [7]. PKC has been shown to activate the transcription of specific genes concluding CD73 [8].
Like ATP, adenosine is constitutively expressed at low levels with a dramatic increase during metabolic stress such as hypoxia and ischemia consequent to ATP hydrolysis. Adenosine is a biologically active molecule that signals through four G-protein-coupled receptors denoted A1, A2A, A2B, and A3. Activation of A1 and A3 inhibits adenylyl cyclase activity through coupling to Gi resulting in a decrease in intracellular cyclic AMP (cAMP), whereas A2A and A2B subtypes are coupled to Gs or Go to stimulate adenylyl cyclase and lead to an increase of cAMP. A change in cAMP concentrations induces downstream signalling by phosphorylating key enzymes. Furthermore, the A2BR is also coupled to Gq/11 stimulating phospholipase C (PLC) reviewed in [9] and the A3R signals via PLC-β2/β3 [10]. Adenosine can also activate phosphoinositide 3-kinase (PI3K), mitogen-activated protein kinases (MAPKs) and extracellular receptor signal-induced kinase (ERK). Additional effector mechanisms include activation of Akt to inhibit apoptosis by A3R, A1R activation which promotes the influx of Ca2+ and efflux of K+ and the activation of the arrestin pathway by adenosine receptors (reviewed in [9]). The adenosine receptors are ubiquitously distributed in the body and the overriding effect of adenosine receptor activation in any one cell is dependent on the repertoire of receptors expressed. The main biological role of adenosine is to maintain vascular and immune homeostasis.
Diabetes, a disorder of glucose homeostasis, is an increasingly prevalent disease worldwide. Two distinct subtypes are recognised: autoimmune diabetes (Type 1 diabetes, T1D) typically afflicting the young and associated with destruction of β-cells and nonimmune diabetes (Type 2 diabetes, T2D) typically arising in those of older age, obese, and with the metabolic syndrome. Although the pathogenesis of the two disorders is distinct, central to both is that of pancreatic β-cell failure and hypoinsulinemia. Features unique to T2D include peripheral insulin resistance and failure of the incretin effect. Despite the disparate aetiologies, the sequelae hyperglycemia and its associated complications are common to both disorders. In this paper, the role of purinergic signalling via the CD39-adenosinergic axis will be discussed in the context of the pathogenesis of T1D and T2D.
2. Pancreatic Expression of Ectoenzymes
Insulin synthesis and secretion is tightly regulated in order to maintain stable blood glucose levels. When blood glucose levels increase, insulin secretion is augmented; conversely in the face of hypoglycaemia, insulin secretion is negligible. Hyperglycemia increases the metabolic demand of β-cells causing a rise in intracellular ATP concentrations [11] and ATP is released with insulin [12] reaching concentrations of 25 μm at the cell surface. Extracellular nucleotides activate P2 receptors—inotropic P2X and metabotropic P2Y. A number of P2 receptors have been implicated in nucleotide-mediated regulation of insulin including P2Y1, P2Y6, P2Y13, P2X3, and amongst others [12–15].
Ectonucleotidase expression has been defined within the mouse, rat, and human endocrine pancreas [16, 17]. Using immunohistochemical and enzyme histochemical techniques, NTPDase1/CD39 was expressed in all blood vessels and acinar tissue; NTPDase2 was localised in capillaries and in connective tissue surrounding islets and acini and NTPDase3 was expressed exclusively in Langerhan islet cells. NTPDase8 was not detected. Inhibition of NTPDase3 activity was shown to facilitate insulin release from rat β-cells. Similarly inhibition of ectonucleotidases with ARL 67156, an inhibitor of NTPDase1 and 3, augmented insulin secretion from human pancreas [13]. Intriguingly CD73 was expressed exclusively in rat islet cells [16] but not in human or mouse [16] (and Chia et al., submitted manuscript). Notably both CD39 and CD73 are secreted from acinar tissue together with ATP directly into the fluid controlling pancreatic exocrine function (reviewed in [18]).
3. Autoimmune Type 1 Diabetes (T1D)
In T1D islet destruction secondary to the autoimmune infiltration of CD4+ T cells and macrophages results in the loss of insulin secretory capacity of β-cells. Treatment with multiple daily injections of insulin slows but does not prevent the development of complications. Transplantation of the whole pancreas or islets is a potential cure for the disease; however, there remains the risk of recurrent disease culminating in graft failure.
3.1. Mouse Models of T-Cell-Mediated Diabetes
The nonobese diabetic (NOD) mouse is the prototypical mouse model for T1D and shares a number of clinical, serological, and immunological features with the human condition. NOD mice spontaneously develop diabetes at ~25 weeks of age after progressing through a prediabetic stage correlating with increasing insulitis.
T-cell-mediated diabetes can also be induced chemically using multiple low dose streptozotocin (MLDS). Streptozotocin is a glucosamine-nitrosourea compound that enters the pancreatic β-cell through the specific glucose transporter 2 (GLUT2) expressed on its surface. Administered in high dose (250 mg/kg) streptozotocin is cytotoxic causing islet death. The onset of diabetes is immediate and there is an absolute lack of insulin. However, streptozotocin administered in low dose (50 mg/kg for 5 days) results in repetitive low-grade β-cell damage, which incites a local inflammatory response comprised principally of CD4+ T cells that is maximal at 12–14 days [19]. The delay in the onset of hyperglycemia suggests immune-mediated damage to β-cells, rather than direct toxicity predominates. Further T cell depleted [20] or deficient mice [21] are resistant to MLDS-induced diabetes.
3.2. Role of E-NTPDase1/CD39 in T-Cell-Mediated Diabetes
CD4+ regulatory T cells are integral to the maintenance of immune homeostasis and abnormalities in number and or function results in autoimmune disease. Indeed low numbers of resting regulatory T cells have been reported in NOD mice [22] and human patients with T1D [23]. CD39 is expressed on both murine [3, 24] and human [25, 26] CD4+ regulatory T cells and is essential for the full suppressive activity of these cells in mice. Further, mice deficient in CD39 (CD39KO) develop an immune diathesis and spontaneous autoimmune alopecia [27]. As anticipated, these mice are highly susceptible to MLDS-induced diabetes with a rapid rate of onset of diabetes (within 10 days) and 100% incidence. Insulitis and reduction in insulin staining was evident at the onset of diabetes. When reconstituted with wild-type bone marrow comprising functional regulatory T cells, the kinetics and incidence are reduced to that of wild-type mice with the development of diabetes at day 42 and overall diabetes incidence of 57% (Chia et al., submitted manuscript). CD39KO mice also have evidence of hepatic insulin resistance [28], which will be discussed in detail below.
Mice have been genetically engineered to overexpress CD39 [29]. CD39 colocalises to β-cells without perturbing glucose homeostasis [30] and these mice are resistant to MLDS-induced diabetes: minimal insulitis was evident and diabetes occurred in only 14% of animals. This robust protection persisted even following reconstitution with bone marrow from immunodeficient CD39KO mice (Chia et al., submitted manuscript) which may reflect enhanced cell regenerative capacity due to increased pancreatic NTPDase activity. Recent work by Andersson et al. [31] and Annes et al. [32] indicate a role for adenosine signalling in cell specific regeneration. In a zebrafish model, the nonselective agonist NECA did not alter protection against cell death but promoted cell regeneration by increasing the proportion of new cells that proliferate through A2A-dependent mechanisms. Interestingly, NECA did not significantly increase the number of cells in normal development. Further in mice treated with streptozotocin at 150 mg/kg for 2 days, BGL were 30% lower in mice concurrently treated with NECA and cell mass was 8 times larger.
3.3. Role of Ecto-5′-nucelotidase/CD73 in T-Cell-Mediated Diabetes
Although CD73 is not expressed in mouse or human islets [16], it is widely expressed on leukocytes and plays an essential role in leukocyte trafficking. Further, like CD39, CD73 plays an integral role in providing immune competence. CD73 is expressed on CD4+ regulatory T cells in mice [3, 33], but interestingly is not expressed by human CD4+ regulatory T cells [26]. CD73KO mice have been generated [34] and extensively characterised. Markedly, reduced CD73 enzymatic activity [34] results in reduced levels of adenosine [35]. The biological relevance of CD73 had become evident from a number of small animal models: CD73 activity attenuates hypoxia-induced vascular leakage FMLP (formyl-Met-Leu-Phe-OH)-stimulated neutrophil adhesion to endothelial cells and neutrophil accumulation in tissues [34, 36, 37]. CD73KO mice have a proinflammatory phenotype with increased VCAM-1 expression on endothelial cells and heightened susceptibility to vascular inflammation and neointima formation [35]. These effects are a consequence of the loss of both enzymatic and nonenzymatic functions of CD73 [38]. Contrary to these reports, we have shown that CD73KO mice are protected in a model of renal ischemia-reperfusion injury [39–41]. Similarly, CD73KO mice are resistant to MLDS-induced diabetes (Figure 1), presumably a consequence of impaired leukocyte trafficking. In alloxan-induced diabetes in rats, a model which produces a pattern of T1D, both platelet-associated CD39 and CD73 activities are increased [42].
Figure 1.
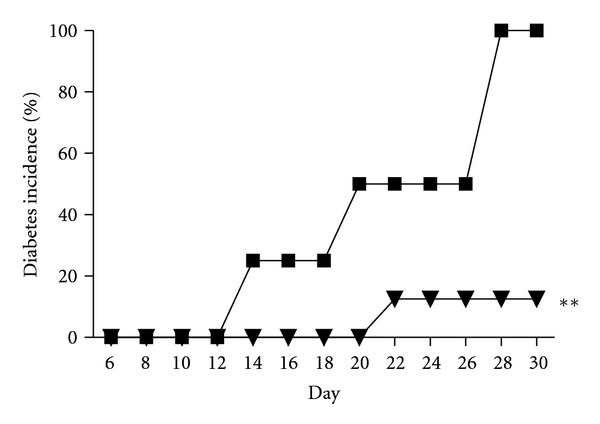
Mice deficient in CD73 are resistant to MLDS-induced diabetes. Diabetes incidence in C57BL/6 wild-type (WT) mice (black squares, n = 4) and CD73KO (black triangles, n = 8) mice following MLDS. **P < 0.01 versus WT mice.
3.4. Adenosine Signalling in T-Cell-Mediated Diabetes
Adenosine signalling has emerged as a regulator of glucose homeostasis through modulating insulin and glucagon release. All four adenosine receptors are expressed in whole pancreas of CD-1 mice [43]; in isolated islets A1, A2A, and A2B receptors are expressed at the mRNA level (Chia et al., submitted manuscript). The A1 and A2A receptors have also been identified on α-cells [44]. Following MLDS, A1 receptor expression is downregulated, A2A expression is unchanged, and A2B receptor expression is augmented (Chia et al., submitted manuscript).
Basal levels of adenosine in isolated islets are in the micromolar range [45], which is sufficient to stimulate glucagon release [46] and inhibit insulin release [47] via the A1 receptor. Thus the peri-islet adenosine concentration is inversely related to extracellular glucose concentrations and may act as a paracrine or autocrine signal [45]. Using the β-cell line INS-1 cells in vitro, treatment with the nonspecific agonist NECA or A1, A2A, and A3 agonists reduced insulin secretion in a dose dependent manner. The effect of NECA was completely antagonised by A2B receptor inhibition [48].
In two mouse models of diabetes (cyclophosphamide treated NOD and MLDS), A1 receptor agonism mitigated diabetes but was less efficacious than the nonspecific agonist NECA [43]. In our hands, antagonism or agonism of the A1 receptor did not influence the rate of diabetes in C57BL/6 wild-type (WT) mice (Chia et al., submitted manuscript).
The A2A receptor is widely expressed on both tissues and circulating cells. Mice lacking the A2A receptor (A2ARKO) are highly susceptible to MLDS-induced diabetes with rapid onset (within 10 days) and 100% diabetes incidence. Like CD39KO mice, the A2ARKO mice are immunocompromised and have hyperproliferative T cells [3]. To delineate the site-specific importance of the A2A receptor, a series of adoptive transfer experiments were performed. Deletion of the A2A receptor either on the tissues or the circulating cells increased the susceptibility of these mice to the effects of MLDS (Chia et al., submitted manuscript). NECA ameliorated diabetes in A2ARKO mice and treatment with an A2AR agonist had no effect in wild-type mice following MLDS [43].
The prevention of MLDS-induced diabetes in CD-1 mice by NECA was reversed by pretreatment with a selective A2B receptor inhibitor [43]. Similarly, we have identified a role for the A2B receptor particularly in the early response to MLDS. The rise in blood glucose following MLDS in C57BL/6 wild-type mice was quicker, reaching hyperglycemia by 8–10 days, although the overall rate of diabetes was unchanged (Chia et al., submitted manuscript).
The protection conferred by CD39 overexpression was mitigated by deletion of the A2A receptor or by pharmacological inhibition of the A2B receptor. Complete blockade of both receptors did not further exaggerate the diabetic phenotype (Chia et al., submitted manuscript). Involvement of more than one adenosine receptor parallels the effects of adenosine in renal IRI, where A2A receptor signaling predominates on circulating CD4+ T cells [49] and macrophages [50], while A2B receptor signaling within the renal parenchyma is also important [51]. Intriguingly, CD73KO mice coadministered with an A2B receptor inhibitor became susceptible to the effects of MLDS, with an onset of diabetes at day 10 and a diabetes incidence of 66% (Figure 2).
Figure 2.
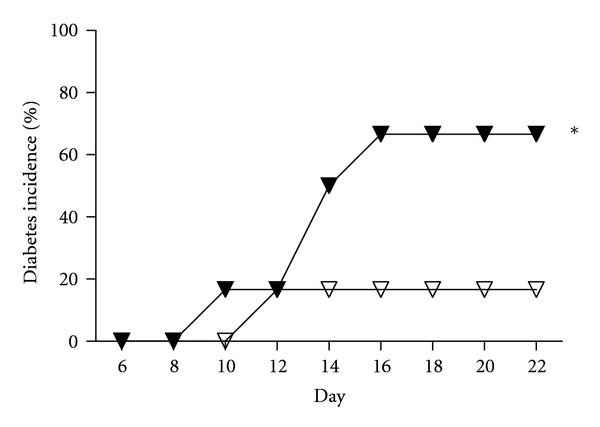
Inhibition of A2B receptor in CD73KO mice increases susceptibility to MLDS-induced diabetes. Diabetes incidence of CD73KO mice treated with either saline (open triangles, n = 6) or the A2BR inhibitor (dose: 0.5 μg/g body weight (BW), twice daily) (black triangles, n = 6). *P < 0.05 versus saline-treated mice.
4. Nonimmune Type 2 Diabetes (T2D)
Insulin resistance characterises T2D, however, β-cell dysfunction must coexist for hyperglycemia to occur. Indeed it is progressive β-cell dysfunction that underpins the progression from normoglycemia to impaired glucose tolerance to overt diabetes.
4.1. Role of CD39 and CD73 in T2D
Mice deficient in CD39 demonstrate impaired glucose tolerance following oral glucose tolerance testing a consequence of hepatic insulin resistance rather than peripheral muscle resistance. There was an associated increased level of hepatocyte c-Jun NH2-terminal kinase (c-JNK) in response to extracellular nucleotides and aberrant insulin receptor substrate (IRS)—2 phosphorylation in the liver of these mice [28]. There was no abnormality in glucose handling following an intraperitoneal glucose load in mice overexpressing CD39 [30] nor intriguingly in CD73KO mice (Figure 3). In human T2D, CD39 expression has been determined in peripheral blood mononuclear cells. Poor glycemic control was associate with proportions of CD39+ cells particularly within the CD19+ subset. Further an increase in CD39 enzymatic activity was observed in this patient cohort [52]. Further, platelet-associated CD39 enzymatic activity was increased in patients with T2D, hypertension, and coexisting T2D and hypertension. Platelet-associated CD73 enzymatic activity was only increased in patients with hypertension or coexisting hypertension and T2D and not T2D alone [53]. CD39 expression also influences the susceptibility to diabetes-induced renal disease in both mice [54] and humans [55]. In African Americans, a common ENTPD1 (CD39) two-single nucleotide polymorphism haplotype was associated with an increased risk for end stage renal disease secondary to T2D.
Figure 3.
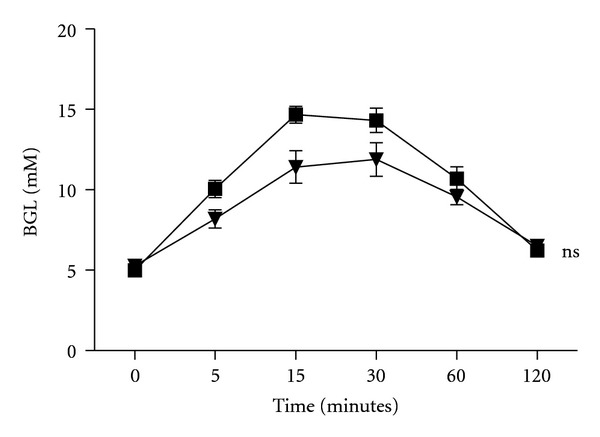
Normal glucose handling in CD73KO mice. Blood glucose levels following 1 mg/g BW of intraperitoneal glucose. WT mice (black squares, n = 6); CD73KO mice (black triangles, n = 8); ns—not significant versus WT mice.
4.2. Adenosine Signalling in T2D
All adenosine receptors are expressed at the mRNA level in skeletal muscle of mice [56] and the role of adenosine receptor blockade in reversing insulin resistance in skeletal muscle from diabetic rats has been realised for some time [57, 58]. In keeping with this treatment of wild-type C57BL/6 mice with NECA promoted impaired glucose tolerance by inhibiting glucose disposal [59]. Although initially thought to be mediated by the A1 receptor, studies with A1RKO [56] and A2RKO [59] mice show that these receptors have a minimal effect on skeletal muscle uptake of glucose. Rather it appears that activation of A2B receptor promotes peripheral insulin resistance and blockade of the receptor in diabetic KKAY mice enhances glucose disposal into skeletal muscle and adipose tissue as well as reducing hepatic glucose production [59]. Further, in Goto-Kakizaki rats, which resemble T2D, insulin levels were increased temporarily following A2B receptor inhibition, although without effecting blood glucose level [48]. There may however be a role for A1 receptor activation through the suppression of lipolysis and free fatty acid levels (FFA) [60] both of which are involved in the pathogenesis of T2D. Indeed, mice overexpressing the A1 receptor in diet-induced insulin resistant mice have lower FFA levels and insulin resistance compared to controls [61]. The effect of the null mutation of A1R on glucose homeostasis following a high fat diet is controversial: Faulhaber-Walter et al. [62] demonstrated decreased glucose tolerance with increased BGL and insulin levels in A1RKO mice (C57BL/6 and Swiss compared to controls) as early as 5 weeks following a high fat diet. Yang et al. [63], however, reported A1RKO mice (C57BL/6) clear blood glucose more efficiently, however, following a high fat diet both WT and A1RKO mice develop glucose intolerance.
4.3. Adenosine and the Incretin Effect
The incretin hormones glucagon-like peptides-1 (GLP-1) and glucagon intestinal peptide (GIP) are released from the gastrointestinal tract in response to food and promote insulin secretion in a glucose concentration-dependent manner in β-cells and inhibit glucagon secretion. The incretins are rapidly metabolised by dipeptidyl peptidase-4 (DPP-4) and drugs that inhibit this enzyme are very effective in the treatment of T2D. DDP-4, also known as CD26 or adenosine deaminase (ADA), enzymatically and irreversibly converts adenosine to inosine. ADA activity has been found in most organs but is notably high in adipose tissue, liver, skeletal muscle and heart. An increase in ADA activity has been reported in patients with T2D and a relationship with insulin resistance has been postulated [64]. High ADA activity is associated with low adenosine levels; however a direct relationship between adenosine and the incretin effect in T2D has not yet been defined.
5. Concluding Remarks
The CD39-adenosinergic axis is involved in the pathophysiology of pancreatic dysfunction and thus drug development targeting different components of the pathway may be of relevance in the treatment of both type 1 and type 2 diabetes. There remain a number of unanswered questions including the source of CD73 enzymatic activity given the lack of expression within the pancreas; the mechanisms behind protection observed with CD73 deletion in MLDS- induced diabetes and the role of purinergic signalling in the incretin effect, which is of particular importance in the pathogenesis of T2D.
Acknowledgments
The authors would like to thank the BioResources Centre (St. Vincent's Hospital, Melbourne, Victoria, Australia) for all aspects of mouse care. This work is supported by Grants from the St. Vincent's Hospital Research Endowment Fund (J. S. J. Chia) and JDRF ITP 4-2006-1025 (K. M. Dwyer).
References
- 1.Robson SC, Sévigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance. Purinergic Signalling. 2006;2(2):409–430. doi: 10.1007/s11302-006-9003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kukulski F, Levesque SA, Lavoie EG, et al. Comparative hydrolysis of P2 receptor agonists by NTPDases 1, 2, 3 and 8. Purinergic Signal. 2005;1(2):193–204. doi: 10.1007/s11302-005-6217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. Journal of Experimental Medicine. 2007;204(6):1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eltzschig HK, Kö hler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. 2009;113(1):224–232. doi: 10.1182/blood-2008-06-165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Synnestvedt K, Furuta GT, Comerford KM, et al. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. Journal of Clinical Investigation. 2002;110(7):993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5'-nucleotidase (CD73) Purinergic Signalling. 2006;2(2):351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bavaresco L, Bernardi A, Braganhol E, Wink MR, Battastini AMO. Dexamethasone inhibits proliferation and stimulates ecto-5'-nucleotidase/CD73 activity in C6 rat glioma cell line. Journal of Neuro-Oncology. 2007;84(1):1–8. doi: 10.1007/s11060-007-9342-2. [DOI] [PubMed] [Google Scholar]
- 8.Node K, Kitakaze M, Minamino T, et al. Activation of ecto-5'-nucleotidase by protein kinase C and its role in ischaemic tolerance in the canine heart. British Journal of Pharmacology. 1997;120(2):273–281. doi: 10.1038/sj.bjp.0700890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobson KA. Introduction to adenosine receptors as therapeutic targets. Handbook of Experimental Pharmacology. 2009;193:1–24. doi: 10.1007/978-3-540-89615-9_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng J, Wang R, Zambraski E, Wu D, Jacobson KA, Liang BT. Protective roles of adenosine A1, A2A, and A3 receptors in skeletal muscle ischemia and reperfusion injury. American Journal of Physiology. 2007;293(6):H3685–H3691. doi: 10.1152/ajpheart.00819.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newgard CB, Denis McGarry J. Metabolic coupling factors in pancreatic β-cell signal transduction. Annual Review of Biochemistry. 1995;64:689–719. doi: 10.1146/annurev.bi.64.070195.003353. [DOI] [PubMed] [Google Scholar]
- 12.Richards-Williams C, Contreras JL, Berecek KH, Schwiebert EM. Extracellular ATP and zinc are co-secreted with insulin and activate multiple P2X purinergic receptor channels expressed by islet beta-cells to potentiate insulin secretion. Purinergic Signalling. 2008;4(4):393–405. doi: 10.1007/s11302-008-9126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacques-Silva MC, Correa-Medina M, Cabrera O, et al. ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic β cell. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(14):6465–6470. doi: 10.1073/pnas.0908935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Léon C, Freund M, Latchoumanin O, et al. The P2Y1 receptor is involved in the maintenance of glucose homeostasis and in insulin secretion in mice. Purinergic Signalling. 2005;1(2):145–151. doi: 10.1007/s11302-005-6209-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohtani M, Suzuki JI, Jacobson KA, Oka T. Evidence for the possible involvement of the P2Y6 receptor in Ca2+ mobilization and insulin secretion in mouse pancreatic islets. Purinergic Signalling. 2008;4(4):365–375. doi: 10.1007/s11302-008-9122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavoie EG, Fausther M, Kauffenstein G, et al. Identification of the ectonucleotidases expressed in mouse, rat, and human Langerhans islets: potential role of NTPDase3 in insulin secretion. American Journal of Physiology. 2010;299(4):E647–E656. doi: 10.1152/ajpendo.00126.2010. [DOI] [PubMed] [Google Scholar]
- 17.Künzli BM, Berberat PO, Giese T, et al. Upregulation of CD39/NTPDases and P2 receptors in human pancreatic disease. American Journal of Physiology. 2007;292(1):G223–G230. doi: 10.1152/ajpgi.00259.2006. [DOI] [PubMed] [Google Scholar]
- 18.Novak I. Purinergic receptors in the endocrine and exocrine pancreas. Purinergic Signalling. 2008;4(3):237–253. doi: 10.1007/s11302-007-9087-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McEvoy RC, Andersson J, Sandler S, Hellerstrom C. Multiple low-dose streptozotocin-induced diabetes in the mouse. Evidence for stimulation of a cytotoxic cellular immune response against an insulin-producing beta cell line. Journal of Clinical Investigation. 1984;74(3):715–722. doi: 10.1172/JCI111487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin M, Yin N, Murphy B, et al. Immune cell-derived C3 is required for autoimmune diabetes induced by multiple low doses of streptozotocin. Diabetes. 2010;59(9):2247–2252. doi: 10.2337/db10-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamura M, Nagafuchi S, Yamaguchi K, Takaki R. The role of thymic immunity and insulitis in the development of streptozocin-induced diabetes in mice. Diabetes. 1984;33(9):894–900. doi: 10.2337/diab.33.9.894. [DOI] [PubMed] [Google Scholar]
- 22.Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4+CD25+ treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity. 2002;16(2):183–191. doi: 10.1016/s1074-7613(02)00279-0. [DOI] [PubMed] [Google Scholar]
- 23.Kukreja A, Cost G, Marker J, et al. Multiple immuno-regulatory defects in type-1 diabetes. Journal of Clinical Investigation. 2002;109(1):131–140. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borsellino G, Kleinewietfeld M, Di Mitri D, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110(4):1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- 25.Dwyer KM, Deaglio S, Crikis S, et al. Salutary roles of CD39 in transplantation. Transplantation Reviews. 2007;21(1):54–63. [Google Scholar]
- 26.Dwyer KM, Hanidziar D, Putheti P, et al. Expression of CD39 by human peripheral blood CD4+CD25+ T cells denotes a regulatory memory phenotype. American Journal of Transplantation. 2010;10(11):2410–2420. doi: 10.1111/j.1600-6143.2010.03291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dwyer KM, Deaglio S, Gao W, Friedman D, Strom TB, Robson SC. CD39 and control of cellular immune responses. Purinergic Signalling. 2007;3(1-2):171–180. doi: 10.1007/s11302-006-9050-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Enjyoji K, Kotani K, Thukral C, et al. Deletion of Cd39/Entpd1 results in hepatic insulin resistance. Diabetes. 2008;57(9):2311–2320. doi: 10.2337/db07-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dwyer KM, Robson SC, Nandurkar HH, et al. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. Journal of Clinical Investigation. 2004;113(10):1440–1446. doi: 10.1172/JCI19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dwyer KM, Mysore TB, Crikis S, et al. The transgenic expression of human CD39 on murine islets inhibits clotting of human blood. Transplantation. 2006;82(3):428–432. doi: 10.1097/01.tp.0000229023.38873.c0. [DOI] [PubMed] [Google Scholar]
- 31.Andersson O, Adams BA, Yoo D, et al. Adenosine signaling promotes regeneration of pancreatic beta cells in vivo. Cell Metabolism. 2012;15(6):885–894. doi: 10.1016/j.cmet.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Annes JP, Ryu JH, Lam K, et al. Adenosine kinase inhibition selectively promotes rodent and porcine islet beta-cell replication. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(10):3915–3920. doi: 10.1073/pnas.1201149109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5'-adenosine monophosphate to adenosine. Journal of Immunology. 2006;177(10):6780–6786. doi: 10.4049/jimmunol.177.10.6780. [DOI] [PubMed] [Google Scholar]
- 34.Thompson LF, Eltzschig HK, Ibla JC, et al. Crucial role for ecto-5'-nucleotidase (CD73) in vascular leakage during hypoxia. Journal of Experimental Medicine. 2004;200(11):1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zernecke A, Bidzhekov K, Özüyaman B, et al. CD73/Ecto-5'-nucleotidase protects against vascular inflammation and neointima formation. Circulation. 2006;113(17):2120–2127. doi: 10.1161/CIRCULATIONAHA.105.595249. [DOI] [PubMed] [Google Scholar]
- 36.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111(4):2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eltzschig HK, Thompson LF, Karhausen J, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104(13):3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 38.Takedachi M, Qu D, Ebisuno Y, et al. CD73-generated adenosine restricts lymphocyte migration into draining lymph nodes. Journal of Immunology. 2008;180(9):6288–6296. doi: 10.4049/jimmunol.180.9.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu B, Rajakumar SV, Robson SC, et al. The impact of purinergic signaling on renal ischemia-reperfusion injury. Transplantation. 2008;86(12):1707–1712. doi: 10.1097/TP.0b013e31819022bc. [DOI] [PubMed] [Google Scholar]
- 40.Rajakumar S, Dwyer K. Ischaemia reperfusion injury in kidney transplantation. In: Randhawa G, editor. Organ Donation and Transplantation—Public Policy and Clinical Perspectives. Vienna, Austria: InTech; 2012. pp. 173–190. [Google Scholar]
- 41.Rajakumar SV, Lu B, Crikis S, et al. Deficiency or inhibition of CD73 protects in mild kidney ischemia-reperfusion injury. Transplantation. 2010;90(12):1260–1264. doi: 10.1097/TP.0b013e3182003d9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lunkes GIL, Lunkes DS, Morsch VM, et al. NTPDase and 5'-nucleotidase activities in rats with alloxan-induced diabetes. Diabetes Research and Clinical Practice. 2004;65(1):1–6. doi: 10.1016/j.diabres.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 43.Németh ZH, Bleich D, Csóka B, et al. Adenosine receptor activation ameliorates type 1 diabetes. The FASEB Journal. 2007;21(10):2379–2388. doi: 10.1096/fj.07-8213com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tudurí E, Filiputti E, Carneiro EM, Quesada I. Inhibition of Ca2+ signaling and glucagon secretion in mouse pancreatic α-cells by extracellular ATP and purinergic receptors. American Journal of Physiology. 2008;294(5):E952–E960. doi: 10.1152/ajpendo.00641.2007. [DOI] [PubMed] [Google Scholar]
- 45.Yang GK, Squires PE, Tian F, Kieffer TJ, Kwok YN, Dale N. Glucose decreases extracellular adenosine levels in isolated mouse and rat pancreatic islets. Islets. 2012;4(1):64–70. doi: 10.4161/isl.19037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chapal J, Loubatieres-Mariani MM, Petit P, Roye M. Evidence for an A2-subtype adenosine receptor on pancreatic glucagon secreting cells. British Journal of Pharmacology. 1985;86(3):565–569. doi: 10.1111/j.1476-5381.1985.tb08932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verspohl EJ, Johannwille B, Waheed A, Neye H. Effect of purinergic agonists and antagonists on insulin secretion from INS-1 cells (insulinoma cell line) and rat pancreatic islets. Canadian Journal of Physiology and Pharmacology. 2002;80(6):562–568. doi: 10.1139/y02-079. [DOI] [PubMed] [Google Scholar]
- 48.Rüsing D, Müller CE, Verspohl EJ. The impact of adenosine and A2B receptors on glucose homoeostasis. Journal of Pharmacy and Pharmacology. 2006;58(12):1639–1645. doi: 10.1211/jpp.58.12.0011. [DOI] [PubMed] [Google Scholar]
- 49.Day YJ, Huang L, Ye H, Li L, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: the role of CD4+ T cells and IFN-γ. Journal of Immunology. 2006;176(5):3108–3114. doi: 10.4049/jimmunol.176.5.3108. [DOI] [PubMed] [Google Scholar]
- 50.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. American Journal of Physiology. 2005;288(4):F722–F731. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 51.Grenz A, Zhang H, Hermes M, et al. Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. The FASEB Journal. 2007;21(11):2863–2873. doi: 10.1096/fj.06-7947com. [DOI] [PubMed] [Google Scholar]
- 52.García-Hernández MH, Portales-Cervantes L, Cortez-Espinosa N, et al. Expression and function of P2X7 receptor and CD39/Entpd1 in patients with type 2 diabetes and their association with biochemical parameters. Cellular Immunology. 2011;269(2):135–143. doi: 10.1016/j.cellimm.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 53.Lunkes GI, Lunkes D, Stefanello F, et al. Enzymes that hydrolyze adenine nucleotides in diabetes and associated pathologies. Thrombosis Research. 2003;109(4):189–194. doi: 10.1016/s0049-3848(03)00178-6. [DOI] [PubMed] [Google Scholar]
- 54.Friedman DJ, Rennke HG, Csizmadia E, Enjyoji K, Robson SC. The vascular ectonucleotidase ENTPD1 is a novel renoprotective factor in diabetic nephropathy. Diabetes. 2007;56(9):2371–2379. doi: 10.2337/db06-1593. [DOI] [PubMed] [Google Scholar]
- 55.Friedman DJ, Talbert ME, Bowden DW, et al. Functional ENTPD1 polymorphisms in african americans with diabetes and end-stage rnal disease. Diabetes. 2009;58(4):999–1006. doi: 10.2337/db08-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johansson SM, Salehi A, Sandström ME, et al. A1 receptor deficiency causes increased insulin and glucagon secretion in mice. Biochemical Pharmacology. 2007;74(11):1628–1635. doi: 10.1016/j.bcp.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 57.Budohoski L, Challiss RAJ, Cooney GJ. Reversal of dietary-induced insulin resistance in muscle of the rat by adenosine deaminase and an adenosine-receptor antagonist. Biochemical Journal. 1984;224(1):327–330. doi: 10.1042/bj2240327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Challis RAJ, Budohoski L, McManus B, Newsholme EA. Effects of an adenosine-receptor antagonist on insulin-resistance in soleus muscle from obese Zucker rats. Biochemical Journal. 1984;221(3):915–917. doi: 10.1042/bj2210915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Figler RA, Wang G, Srinivasan S, et al. Links between Insulin resistance, adenosine A2B receptors, and inflammatory markers in mice and humans. Diabetes. 2011;60(2):669–679. doi: 10.2337/db10-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Large V, Arner P. Regulation of lipolysis in humans. Pathophysiological modulation in obesity diabetes, and hyperlipidaemia. Diabetes and Metabolism. 1998;24(5):409–418. [PubMed] [Google Scholar]
- 61.Dong Q, Ginsberg HN, Erlanger BF. Overexpression of the A1 adenosine receptor in adipose tissue protects mice from obesity-related insulin resistance. Diabetes, Obesity and Metabolism. 2001;3(5):360–366. doi: 10.1046/j.1463-1326.2001.00158.x. [DOI] [PubMed] [Google Scholar]
- 62.Faulhaber-Walter R, Jou W, Mizel D, et al. Impaired glucose tolerance in the absence of adenosine A1 receptor signaling. Diabetes. 2011;60(10):2578–2587. doi: 10.2337/db11-0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang GK, Fredholm BB, Kieffer TJ, Kwok YN. Improved blood glucose disposal and altered insulin secretion patterns in adenosine A1 receptor knockout mice. American Journal of Physiology. 2012;303(2):180–190. doi: 10.1152/ajpendo.00050.2012. [DOI] [PubMed] [Google Scholar]
- 64.Lee JG, Kang DG, Yu JR, et al. Changes in adenosine deaminase activity in patients with type 2 diabetes mellitus and effect of DPP-4 inhibitor treatment on ADA activity. Diabetes and Metabolism Journal. 2011;35(2):149–158. doi: 10.4093/dmj.2011.35.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]