

Abstract
This paper describes a comparative study on the performances of ethosomes and solid lipid nanoparticle as delivery systems for acyclovir. Ethosomes were spontaneously produced by dissolution of phosphatidylcholine and acyclovir in ethanol followed by addition of an aqueous buffer while solid lipid nanoparticle were produced by homogenization and ultrasonication. Both colloidal systems were morphologically characterized by cryo-transmission electron microscopy. The encapsulation efficiency was 94.2±2.8% for ethosomes and 53.2±0.2% for solid lipid nanoparticle. Concerning Z potential, both formulations are close to neutrality. The diffusion coefficients of the drug from ethosomes and solid lipid nanoparticle, determined by a Franz cell method, were 9.4 and 1.2-fold lower as compared to the free acyclovir in solution, thus evidencing the ability of both colloidal systems in enhancing the diffusion of the drug. The antiviral activity against HSV-1 of both systems was tested by plaque reduction assay in monolayer cultures of Vero cells. Data showed that no significant differences in the antiviral activity were observed by acyclovir in the free or loaded forms. Taken together these results, colloidal systems could be interesting to mediate the penetration of acyclovir within Vero cells.
Keywords: Acyclovir, antiviral activity, ethosomes, HSV-1, SLN
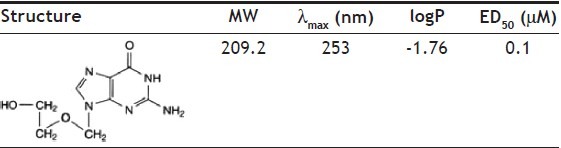
Acyclovir (ACY), {9-[(2-hydroxyethoxy)methyl]guanine} (Table 1), is a synthetic purine nucleoside analogue derived from guanine considered the safest and most efficacious drug able to act against herpes simplex viruses (HSV-1 and HSV- 2)[1–3]. The action mechanism of ACY is related to the inhibition of DNA replication of herpes virus. The antiviral drug ACY is widely used topically especially for treatment of Herpes labialis[4–6]. To this aim, colloidal systems, such as ethosomes and solid lipid nanoparticle (SLN), can be considered drug delivery vehicles playing a major role in the transport and targeting of active agents.
TABLE 1.
CHEMICAL STRUCTURE AND SOME PHYSICOCHEMICAL CHARACTERISTICS OF ACYCLOVIR
Ethosomes are multilamellar vesicles composed of phospholipids, ethanol and water. These lipid vesicular systems embody ethanol in relatively high concentrations[7]. Ethosomes have been demonstrated to be more efficient than liposomes or hydro-alcoholic solutions to deliver topical agent to the skin, probably in reason of their ability in enhancing permeability after their fusion with skin lipids[8–13]. For instance, ACY-containing ethosomes improved clinical efficacy of the drug as compared to that of Zovirax cream (Glaxo Wellcome plc) in the treatment of recurrent herpes labialis[14].
Solid lipid nanoparticles (SLN) are nanoparticles based on solid lipids (i.e. triglycerides) joining the advantages of colloidal lipid emulsions with those of solid matrix particles[15–17]. The solid matrix of SLN should be able to protect chemically labile agents from degradation and to modulate drug release profiles. Moreover SLN can be employed to increase the bioavailability of drugs by improving their diffusion through biological membranes and by protecting them against metabolizing enzymes[18]. SLN offer a number of potential advantages as delivery systems, such as better availability for poorly water-insoluble molecules, the use of physiological lipids and a wide application range (dermal, per os, intravenous)[19].
Aims of this study were (a) the production and characterization of two different colloidal systems, such as ethosomes and SLN, as vehicles for acyclovir, (b) the determination of the drug encapsulation and its release kinetics from both ethosomal and SLN dispersions and (c) the evaluation of the in vitro antiviral activity of drug-containing ethosomes and SLN on Vero cells as compared to that of the sole acyclovir.
Although ACY is slightly soluble in water (1.3 mg/ml at 25°); very slightly soluble in ethanol (0.2 mg/ml) and soluble in dilute aqueous solutions of alkali hydroxides and mineral acids; ethosomes were spontaneously produced by dissolution of phosphatidylcholine (PC) and ACY in ethanol followed by slow addition of an aqueous buffer under continuous stirring at room temperature. To prepare ethosomes, PC (50 mg/ml) and ACY (50 mM) were dissolved in 2 ml of ethanol, then 8 ml of isotonic Palitzsch buffer (IPB) (5 mM Na2B4O7, 180 mM H3BO3, 18 mM NaCl) were slowly added to the alcoholic solution, at 30°, stirring at 700 rpm for 5 min. During preparation, dispersions displayed initial optical transparency due to the high ethanol concentration able to maintain PC in solution. By adding increasing concentrations of aqueous buffer, PC molecules reorganize themselves resulting in a turbid ethosomal suspension. After production, ethosomes were extruded once through two stacked 400 nm pore size filters and three-fold through two stacked 200 nm pore size membranes (Nucleopore Corp., Pleasanton, CA) in order to size the vesicles[20].
The separation of the free drug from the ACY contained within ethosomes was obtained by a gel filtration on Sepharose 4B column (Pharmacia, Uppsala, Sweden) (1.5 cm diameter, 50 cm length) pre-equilibrated and eluted with borate buffer. ACY content was determined by RP-HPLC analysis using a Vydac C18 column (25×0.46 cm) stainless steel packed with 5 mm particles eluted at room temperature with a mobile phase consisting of 10% methanol and 90% NaH2PO4 buffer 0.02 M pH 3 at 0.8 ml/min. The assay method for the determination of acyclovir was validated according to the United States Pharmacopeia (USP). Under these conditions acyclovir showed a limit of quantitation of 10 ng/ml and a retention time of 4 min. The calibration curve was linear in the concentration range 10-5000 ng/ml, R=0.9968.
Tristearine SLN was produced by homogenization and ultrasonication as previously described[21,22]. Briefly, pure tristearine at a concentration of 5% w/w with respect to the total weight of dispersions, was fused and then dispersed in aqueous poloxamer 188 solution (2.5% w/w) at 13,500 rpm, 70° for 1 min, using a high-speed stirrer (Ultra Turrax T25, IKA, Germany). The obtained emulsion was subjected to ultrasonication (Microson™, Ultrasonic cell Disruptor) at 6.75 kHz for 15 min and then cooled down to room temperature. In the case of drug-containing dispersions, 3 mg of the drug were added to the molten lipids and dissolved before adding to the aqueous solution.
The use of pure tristearine give rise to the production of stable and homogenous dispersions, free from aggregates.
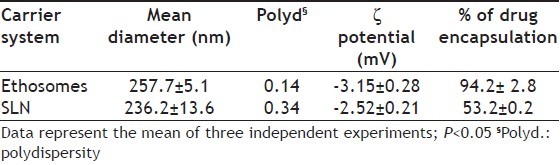
Table 2 summarizes the results concerning size, zeta potential and percentage of drug encapsulation of both ethosomes and SLN encapsulating acyclovir. From the analysis of these data it can be achieved that both colloidal systems show a similar mean size, being 257.7±5.1 nm (P.I. 0.14) for ethosomes and 236.2±13.6 nm (P.I. 0.34) for SLN, and are quite neutral in terms of ionic charge. In fact, as expected by the use of uncharged excipients for the production of both SLN and ethosomes, the zeta potential values of both formulations are around -3 mV. Considering that a value of 25 mV (positive or negative) is taken as the arbitrary value that indicates charged surfaces, the obtained SLN and ethosomes can be considered neutral. In addition, SLN dispersions maintained their dimensions almost unchanged for more than 6 months (data not shown). The best results in terms of percentage of encapsulation were obtained by ethosomes as compared to SLN (94.2% vs 53.2%).
TABLE 2.
AVERAGE DIAMETER, ζ POTENTIAL AND PERCENTAGE OF DRUG ENCAPSULATION OF THE PRODUCED CARRIER SYSTEMS
Cryo-transmission electron microscopy (Cryo-TEM) analyses have been conducted in order to shed light on the internal structure of the dispersed particles in both ethosome and SLN dispersions. Cryo-TEM analyses were performed using a Zeiss EM922 transmission electron microscope for imaging. Images were recorded digitally by a CCD camera (Ultrascan 1000, Gatan) using a image processing system (GMS 1.4 software, Gatan).
Fig. 1 reports cryo-TEM images of colloidal dispersion containing ACY, namely ethosomes (panel A) and SLN (panel B). The electron microscopic analysis demonstrates that ethosomal suspension is mainly characterized by the presence of unilamellar vesicles with few multi-oligo-lamellar vesicles. However, the suspension shows a low poly-dispersion with an average diameter reflecting the pore size of the membrane used for the extrusion.
Fig. 1.
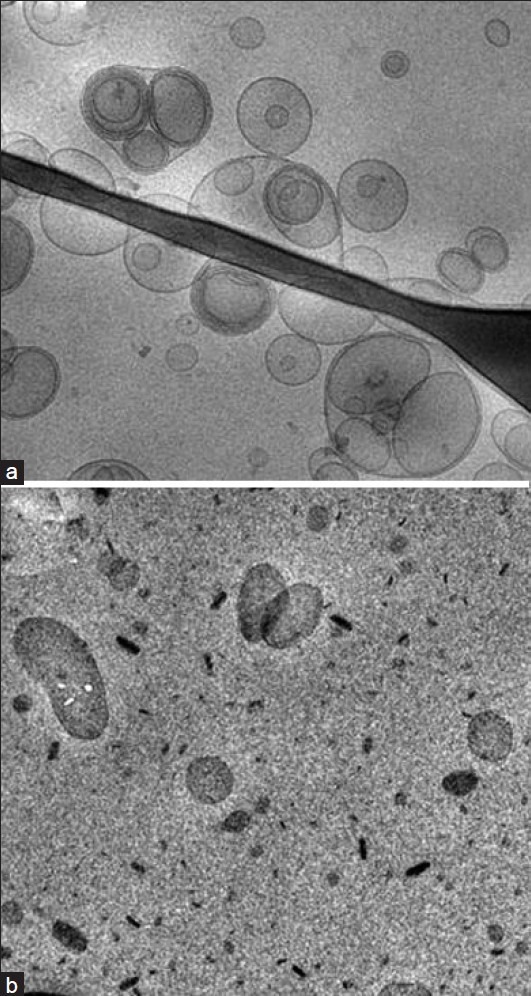
Cryo-TEM photographs of ACY-containing ethosomes (PanelA) and SLN (Panel B) studied in the present paper. Bar represents 180 nm
Concerning SLN, the three dimensional particles are projected in a two dimensional way. In panel B one can observe elongated circular platelet-like crystalline particles and dark, “needle” like structures edge-on viewed. The calculated thickness of nanoparticles was 4.7 nm. It addition, the presence of the drug does not affect colloidal system aspect (data not shown).
In order to investigate the diffusion of ACY from both the ethosomes and SLN an in vitro test based on a Franz cell was employed[23,24]. Particularly, the use of diffusion cell with synthetic membranes allows to determine the drug diffusion characteristic from ethosome or SLN and thus it can be used as a quality control procedure to assure batch-to-batch uniformity[25]. As in vitro system able to mimic the human skin barrier properties, in the present study we utilized a multilaminated membrane system consisting of a hydrophilic membrane of cellulose acetate and nitrate esters (0.2 μm pore size, Schleicher and Schuell, Germany) sandwiched between two lipophilic polydimethylsiloxane (silicone) membranes (SilasticR 500.3, 250 μm thickness, Dow Corning Corporation, Midland, USA)[24–26]. The experiments were carried on using a standard glass Franz cell with 1 cm diameter orifice (0.78 cm2 area).
As receptor phase phosphate buffer 0.1 M (pH 7.4) was used. The receptor phase was always degassed before use to avoid air bubbles formation and poured in the cell body to overflowing. To study the drug release rate, 1 ml of drug solution or drug containing-colloidal suspension was placed into the donor cell compartment and tamped down on the membrane, previously moistened with the receptor phase. The upper part of the chamber was sealed to avoid evaporation. The receptor phase was stirred by means of a constantly spinning bar magnet and thermostated at 37°. At predetermined time intervals comprised between 1 and 8 h, 0.15 ml of receptor phase were withdrawn and the drug concentration in the receptor phase was measured by HPLC.
The amount of drug released per unit area (mg/cm2) was plotted against the square root of time. The cumulative amount of drug released was linear and proportional to the time both for ethosomes and SLN. The slope, which represents the release rate, steady-state flux, was calculated by linear regression. In order to compare formulations loaded with different concentrations of drug or to compare different types of formulations containing the same type of drug (i.e. SLN and NLC), the observed flux of the drug through the membrane has to be normalized[23,26]. In this view, the release rate coefficients were expressed both as experimentally observed fluxes (Jo) and as normalized fluxes (Jn) (Jn=Jo/C, where C is the drug concentration expressed in mg/ml). Correlation coefficients of the regression line were always higher than 0.979 (Table 3).
TABLE 3.
IN VITRO DIFFUSION COEFFICIENTS AND IN VITRO EFFECT ON PLAQUE REDUCTION OF ACYCLOVIR
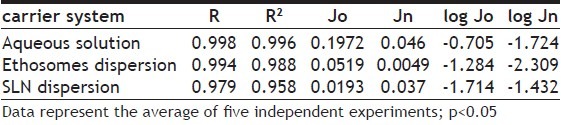
The calculated diffusion coefficients for ACY incorporated within ethosomes and SLN are reported in Table 3 and fig. 2. The diffusion coefficients of ACY from ethosomes and SLN (Jn values) are 9.4 and 1.2-fold lower (P<0.05), respectively, as compared to the free ACY in solution thus evidencing the ability of both colloidal systems in controlling the diffusion of the drug. However, as expected, ethosomes showed a higher effect with respect to SLN probably due to the presence of ethanol in the aqueous compartment of the ethosomes that favored the encapsulation of ACY and controlled its flux through the in vitro system mimicking the skin barrier.
Fig. 2.
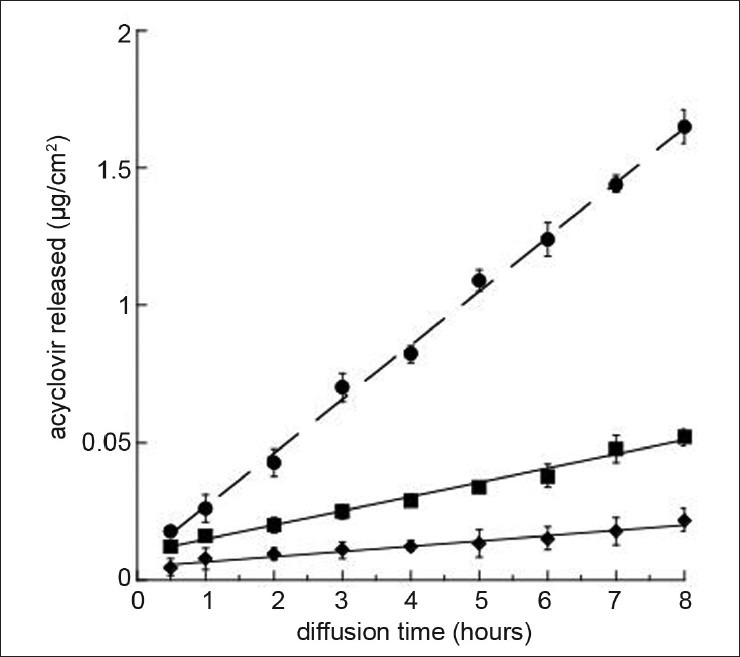
In vitro release kinetics of ACY from solutions (circle), ethosomes (square) and SLN (diamond).
Experiments were performed by a cellulose ester membrane with 0.2 mm pore size and phosphate buffer pH 7.4 as receptor phase. The reported results represent the mean values±SD of six independent experiments
The mechanism of release kinetics was evaluated by fitting the permeation data to the zero-order, first-order and Higuchi diffusion models. All permeation profiles fit the Higuchi diffusion model and a linear relationship was found between the amount of drug released and the square root of time. It could be concluded that the colloidal particles acted as reservoir systems for continuous delivery of the encapsulated drug. However, it has to be taken in mind that several factors may affect transdermal flux, such as drug affinity to colloidal particles and drug solubility within stratum corneum lipids.
The antiviral activity against HSV-1 of drug-containing ethosomes was tested by plaque reduction assay in monolayer cultures of Vero cells (African Green monkey kidney) according to the standard method[27]. Several authors indicated Vero cells as a suitable host for HSV- 1[27–28]. 5×105 cells/well were seeded in 12-well plates and incubated at 37° and 5% CO2. When the cells reached 95% of confluence, they were infected with 100 pfu of HSV-1. After incubation for 1 h at 37° to allow viral adsorption, the plates were washed and the medium replaced with maintenance medium containing different concentrations of empty or drug-containing ethosomes or SLN (i.e. 50, 33, 25, 16.6, 8.3, 5 mM). After 48 h incubation, the medium was removed and the number of plaques was counted by using a light microscope. The antiviral activity was evaluated as plaque reduction with respect to control cells[28,29].
Fig. 3 reports the antiviral activity of ACY after 48 h from infection with HSV-1 MOI 0.1 (panel A) or HSV-1 MOI 1 (panel B). Particularly, after infection Vero cells were incubated in the presence of serial dilutions of ACY in the free form or loaded within ethosomes or SLN. By the analysis of data reported in fig. 3, it is evident that, in general, no significant differences in the antiviral activity were observed by ACY in the free or loaded forms. Although especially at lower ACY concentrations the encapsulation within carrier systems seems to reduce the in vitro antiviral activity of the drug either at MOI 0.1 or MOI 1. Possibly, the high lipophilicity of SLN with respect to ethosomes seems to be the more critical parameter influencing the antiviral activity when higher viral infections are used (i.e. MOI 1). It should be supposed that ethosomes and SLN could be useful in transporting ACY within Vero cells. For appropriate comparison, the activity of both empty colloidal systems was evaluated. As expected, the plaque reduction assay revealed the absence of activity of empty colloidal systems in infected Vero cells (data not shown).
Fig. 3.

Antiviral activity of ACY after 48 h from infection.
Vero cells were infected with HSV-1 MOI 0.1 (panel A) or HSV-1 MOI 1 (panel B) and incubated in the presence of serial dilutions of ACY as free solution (black column), ethosome dispersion (gray column) and SLN suspension (white column). Data represent the mean of three replicates of three independent experiments
By the analysis of the obtained results, it is evident that both SLN and ethosomes are able to encapsulate quite high amount of ACY and, as determined by Franz cell method, to increase the diffusion of the drug as compared to the ACY solution. Thus, it should be supposed that the transport of the ACY by colloidal systems into the stratum corneum bypasses the main barrier to drug permeation, improving skin delivery. However, the antiviral activity against HSV-1 of both formulations tested by plaque reduction assay on Vero cells showed no significant differences as compared to that observed by ACY in solution. Taking into account these considerations, it should be concluded that colloidal systems could be interesting to mediate the penetration of ACY within cells and thus to propose both ethosomes and SLN as possible means for topical administration of anti-herpetic molecules.
ACKNOWLEDGEMENTS
Authors are grateful to Dr. P. Marconi for helpful discussion. Authors also thank Italian Ministry of University and Research (PRIN2006), Fondazione Cassa di Risparmio di Cento and Emilia-Romagna Region (Italy) for financial support.
Footnotes
Cortesi, et al.: Colloidal Dispersions to Deliver Acyclovir
REFERENCES
- 1.Richards DM, Carmine AA, Brogden RN, Heel RC, Speight TM, Avery GS. Acyclovir. A review of its pharmacodynamic properties and therapeutic efficacy. Drugs. 1983;26:378–438. doi: 10.2165/00003495-198326050-00002. [DOI] [PubMed] [Google Scholar]
- 2.Whitley RJ. Herpes simplex virus infection. Semin Pediatr Infect Dis. 2002;13:6–11. doi: 10.1053/spid.2002.29752. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30:115–33. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Blum MR, Liao SH, Miranad PD. Overview of acyclovir pharmacokinetic disposition in adults and children. Am J Med. 1982;73:186–92. doi: 10.1016/0002-9343(82)90088-2. [DOI] [PubMed] [Google Scholar]
- 5.Modiano P, Salloum E, Gillet-Terver MN, Barbaud A, Georges JC, Thouvenot D, et al. Acyclovir-resistant chronic cutaneous herpes simplex in Wiskott-Aldrich syndrom. Br J Dermatol. 1995;133:475–8. doi: 10.1111/j.1365-2133.1995.tb02682.x. [DOI] [PubMed] [Google Scholar]
- 6.Kaufman HE. Treatment of viral diseases of the cornea and external eye. Prog Retin Eye Res. 2000;19:69–85. doi: 10.1016/s1350-9462(99)00004-x. [DOI] [PubMed] [Google Scholar]
- 7.Touitou E, Dayan N, Bergelson L, Godin B, Eliaz M. Ethosomes-novel vesicular carriers for enhanced delivery: Characterization and skin penetration properties. J Control Release. 2000;65:403–18. doi: 10.1016/s0168-3659(99)00222-9. [DOI] [PubMed] [Google Scholar]
- 8.Touitou E, Godin B, Dayan N, Weiss C, Piliponsky A, Levi-Schaffer F. Intracellular delivery mediated by an ethosomal carrier. Biomaterials. 2001;22:3053–9. doi: 10.1016/s0142-9612(01)00052-7. [DOI] [PubMed] [Google Scholar]
- 9.Touitou E. Drug delivery across the skin. Exp Opin Biol Ther. 2002;2:723–33. doi: 10.1517/14712598.2.7.723. [DOI] [PubMed] [Google Scholar]
- 10.Touitou E, Godin B. Dermal drug delivery with ethosomes: Therapeutic potential. Therapy. 2007;4:465–72. [Google Scholar]
- 11.Godin B, Touitou E. Ethosomes: New prospects in transdermal delivery. Crit Rev Ther Drug Carrier Syst. 2003;20:63–72. doi: 10.1615/critrevtherdrugcarriersyst.v20.i1.20. [DOI] [PubMed] [Google Scholar]
- 12.Jain S, Tiwary AK, Sapra B, Jain NK. Formulation and evaluation of ethosomes for transdermal delivery of lamivudine. AAPS PharmSciTech. 2007;8:E111. doi: 10.1208/pt0804111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bendas ER, Tadros MI. Enhanced transdermal delivery of salbutamol sulfate via ethosomes. AAPS PharmSciTech. 2007;8:E107. doi: 10.1208/pt0804107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horwitz E, Pisanty S, Czerninski R, Helser M, Eliav E, Touitou E. A clinical evaluation of a novel liposomal carrier for acyclovir in the topical treatment of recurrent herpes labialis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;87:700–5. doi: 10.1016/s1079-2104(99)70164-2. [DOI] [PubMed] [Google Scholar]
- 15.Muller RH, Mader K, Gohla S. Solid lipid nanoparticles (SLN) for controlled delivery-A review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–77. doi: 10.1016/s0939-6411(00)00087-4. [DOI] [PubMed] [Google Scholar]
- 16.Bunjes H, Drechsler M, Koch MH, Westesen K. Incorporation of the model drug ubidecarenone into solid lipid nanoparticles. Pharm Res. 2001;18:287–93. doi: 10.1023/a:1011042627714. [DOI] [PubMed] [Google Scholar]
- 17.Westesen K, Bunjes H, Koch MH. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J Control Release. 1997;48:223–33. [Google Scholar]
- 18.Westesen K, Siekmann B. Biodegradable colloidal drug carrier systems based on solid lipids. In: Benita S, editor. Microencapsulation. New York: Marcel Dekker; 1996. pp. 213–58. [Google Scholar]
- 19.Lippacher A, Muller RH, Mader K. Preparation of semisolid drug carriers for topical application based on solid lipid nanoparticles. Int J Pharm. 2001;214:9–12. doi: 10.1016/s0378-5173(00)00623-2. [DOI] [PubMed] [Google Scholar]
- 20.Mayer LD, Hope MJ, Cullis PR. Vesicles of variable size produced by a rapid extrusion procedure. Biochim Biophys Acta. 1986;858:161–8. doi: 10.1016/0005-2736(86)90302-0. [DOI] [PubMed] [Google Scholar]
- 21.Esposito E, Fantin M, Marti M, Drechsler M, Paccamiccio L, Mariani P, et al. Solid lipid nanoparticles as delivery systems for bromocriptine. Pharm Res. 2008;25:1521–30. doi: 10.1007/s11095-007-9514-y. [DOI] [PubMed] [Google Scholar]
- 22.Mehnert W, Mader K. Solid lipid nanoparticles: Production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–96. doi: 10.1016/s0169-409x(01)00105-3. [DOI] [PubMed] [Google Scholar]
- 23.Nastruzzi C, Esposito E, Pastesini C, Gambari R, Menegatti E. Comparative study on the release kinetics of methyl nicotinate from topic formulations. Int J Pharm. 1993;90:43–50. [Google Scholar]
- 24.Esposito E, Menegatti E, Cortesi R. Ethosomes and liposomes as topical vehicles for azelaic acid: A preformulatory study. J Cosmet Sci. 2004;55:253–64. [PubMed] [Google Scholar]
- 25.Shah PV, Elkins JS, Hanus J, Noorizadeh C, Skelly JP. In vitro release of hydrocortisone from topical preparations and automated procedure. Pharm Res. 1991;8:55–9. doi: 10.1023/a:1015826205930. [DOI] [PubMed] [Google Scholar]
- 26.Nastruzzi C, Esposito E, Pastesini C, Gambari R, Menegatti E. Comparative study on the release kinetics of methyl-nicotinate from topic formulations. Int J Pharm. 1993;90:43–50. [Google Scholar]
- 27.Hill EL, Ellis MN, Nguyen DP. Antiviral and antiparasitic susceptibility testing. In: Balows A, Hausler WJ Jr, Herrmann KL, Isenberg HD, Shadomy HJ, editors. Manual of Clinical Microbiology. 5th ed. Washinton DC: American Society for Microbiology; 1991. pp. 1184–91. [Google Scholar]
- 28.Pope LE, Marcelletti JF, Katz LR, Lin JY, Katz DH, Parish ML, et al. The anti-herpes simplex virus activity of n-docosanol includes inhibition of the viral entry process. Antiviral Res. 1998;40:85–94. doi: 10.1016/s0166-3542(98)00048-5. [DOI] [PubMed] [Google Scholar]
- 29.Walro DG, Rosenthal KS. The antiviral xanthate compound D609 inhibits herpes simplex virus type 1 replication and protein phosphorylation. Antiviral Res. 1997;36:63–72. doi: 10.1016/s0166-3542(97)00040-5. [DOI] [PubMed] [Google Scholar]