

Abstract
Objective:
This study aimed to evaluate the antidiarrheal efficacy and pharmacological properties of ethyl 2-(4-oxo-3-o-tolyl-3,4-dihydroquinazolin-2-ylthio)acetate (DQA) as an inhibitor of cystic fibrosis transmembrane conductance regulator protein (CFTR) both in vitro and in vivo.
Materials and Methods:
The effects of DQA on CFTR function and cell viability were investigated in Fisher rat thyroid (FRT) cells expressing human CFTR and human intestinal epithelial T84 cells by short-circuit current measurements and MTT assays, respectively. In vivo antidiarrheal efficacy of DQA was evaluated in a closed loop model of cholera in mice.
Results:
In permeabilized FRT cells, apical chloride current induced by CFTR agonists (10 μM forskolin, 100 μM CPT-cAMP, and 20 μM apigenin) was inhibited by DQA with IC50 ~ 20 μM and complete inhibition at 200 μM. The inhibitory effect was reversible and not associated with cytotoxicity to FRT cells (5–500 μM DQA for 24 h). Likewise, DQA effectively inhibited both forskolin and cholera toxin-induced transepithelial chloride secretion in T84 cells. In mice, intraluminal injection of 100 μM DQA reduced cholera toxin (1 μg/closed loop)-induced intestinal fluid secretion by 85% without affecting intestinal fluid absorption.
Conclusions:
DQA represents a new class of small molecule CFTR inhibitor with potential application in treatment of cholera.
KEY WORDS: CFTR, CFTR inhibitor, chloride secretion, cholera, secretory diarrhea
Introduction
Diarrhea is a leading cause of death worldwide with developing countries being the most affected.[1] Diarrhea caused by cholera is associated with high mortality and morbidity especially in South Asia and Africa.[2] The causative agent of cholera, Vibrio cholerae, induces diarrhea through production of the virulent cholera toxin,[3] which binds to its ganglioside receptor before being internalized into the enterocyte, where the toxin stimulates cAMP-mediated transepithelial Cl- secretion, which in turn provides an osmotic impetus for secretion of Na+ and water through paracellular routes into intestinal lumen.[4] This intestinal fluid loss can be so deleterious as to leading to dehydration and hypovolumic shock in the patient unless adequate fluid replacement therapy is provided.[5]
At present, the mainstay treatment of cholera is rehydration therapy and the use of antibiotic drugs. However, application of these two approaches is made difficult by the lack of effect of rehydration on the course of diarrhea and by the increase in prevalence of antibiotic resistant V. cholerae strains.[5,6] Thus, there is a need for a more specific and effective treatment of cholera.
One pharmacological approach, which holds great promise in the treatment of cholera, is inhibition of cAMP-activated intestinal chloride secretion.[7] There are a number of transport proteins functioning to facilitate cAMP-activated Cl- secretion. Cl- ions are transported into intestinal cells via Na+-K+-2Cl- transporters located at the basolateral membrane of intestinal cells and exit into the intestinal lumen via a cystic fibrosis transmembrane conductance regulator (CFTR), which is a cAMP-activated Cl- channel located at apical membrane.[8,9] Furthermore, activities of cAMP-activated K+ channels and Na+-K+ ATPases located at the basolateral membrane are necessary for maintaining sustainability of this process.[8] Among these transport proteins, CFTR is the most favorable target for antidiarrheal drug development due to its intestinal lumen-facing localization.[8,10,11]
Previous studies using high-throughput screening revealed ethyl 2-(4-oxo-3-o-tolyl-3,4-dihydroquinazolin-2-ylthio)acetate (DQA) [Figure 1] as a CFTR inhibitor with potency of 20 μM as determined by halide influx assays in Fisher rat thyroid (FRT) cells transfected with human CFTR and a halide-sensitive yellow fluorescent protein.[12] The present study aimed to evaluate basic pharmacological properties of this compound using both electrophysiological and biochemical methods. Furthermore, potential antidiarrheal application of DQA was evaluated in vivo. Evidence provided shows that DQA has antidiarrheal efficacy in both human intestinal cells and a mouse model of cholera.
Figure 1.
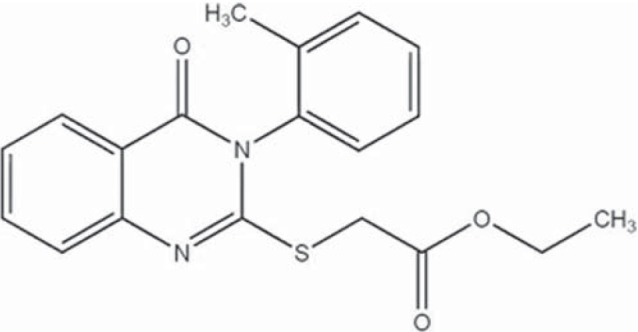
Structure of ethyl 2-(4-oxo-3-o-tolyl-3,4-dihydroquinazolin- 2-ylthio) acetate (DQA)
Materials and Methods
Cell Lines and Chemicals
FRT cells expressing human CFTR were generated as previously described.[13] A human colonic carcinoma T84 cell line was purchased from the American Type Culture Collection (VA, USA). CFTRinh -172 was purchased from Calbiochem (San Diego, CA, USA); cholera toxin from List Biological Laboratories Inc. (CA, USA); trypsin-EDTA, fetal bovine serum, penicillin, and streptomycin from HyClone (UT, USA); carbachol, amiloride, and amphotericin B from Sigma-Aldrich (MO, USA); and DQA from ChemDiv (CA, USA).
Cell Culture
FRT cells were cultured in a Coon's F12 medium, supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. T84 cells were cultured in a 1:1 mixture of Dulbecco's modified Eagle's medium and a nutrient mixture of F-12 Ham supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin. For apical chloride current and short-circuit current measurements, both FRT and T84 cells were sub-cultured in 0.05% trypsin-EDTA and seeded on Snapwell filters (1 cm2 surface area; Corning-Corstar Corp., Corning, NY, USA) at a density of 5 × 105 cells/filter. Cells were kept in a humidified incubator under 95% O2 and 5% CO2 at 37°C with a fresh medium being replaced every 24 h. FRT and T84 cells were grown on Snapwell filters for 7 and 14 days, respectively, allowing the formation of epithelial monolayer with an electrical resistance of more than 1,000 Ω/cm2.
Short-circuit Current and Apical Cl- Current Measurement
Snapwell filters containing FRT and T84 cells were mounted in an Ussing chamber system. For apical chloride current measurements in FRT cells, the hemichamber on basolateral side was filled with Ringer's solution containing (in mM) 130 NaCl, 2.7 KCl, 1.5 KH2PO4, 1 CaCl2, 0.5 MgCl2, 10 Na-HEPES, and 10 glucose (pH 7.3). In order to create a chloride gradient, 65 mM NaCl and 65 mM Na-gluconate were used in place of 130 mM NaCl with a concentration of CaCl2 being increased to 2 mM in the apical hemichamber solution. In addition, the basolateral membrane was permeabilized with 250 μg/ml amphotericin B for 20 min prior to measurement. For short-circuit current measurements in T84 cells, both hemichambers were filled with Kreb's solution containing (in mM) 120 NaCl, 25 NaHCO3, 3.3 KH2PO4, 0.8 K2HPO4, 0.8 MgCl21.2 CaCl2, and 10 glucose (pH 7.3). Both hemichambers were continuously bubbled with 100% O2 (for FRT cells) or 5% CO2 in 95% O2 (for T84 cells) and maintained at 37°C during experimental period. Apical chloride and short-circuit currents were continuously recorded by a DVC-1000 voltage clamp (World Precision Instruments, Sarasota, FL, USA) with Ag/AgCl electrode and 1 M KCl agar bridge.
Intestinal Fluid Secretion and Absorption Measurements in Mice
Mice (30–35 g, ICR strain; The National laboratory Animal Center, Bangkok, Thailand) were fasted for 24 h before experiment. Mice were anesthetized by an intraperitonial injection of sodium thiopenthal (50 mg/kg). Body temperature was maintained between 36°C and 37°C using an electrical lamp. After making a small vertical incision in the abdomen, the small intestine was exteriorized and the mid-jejunum was isolated by suture to create a closed loop (2–3 cm in length). Loops were injected with 100 μl of phosphate-buffered saline (PBS) or PBS containing cholera toxin (1 μg/loop) with or without DQA (100 μM). Abdominal incisions were closed with suture and mice were allowed to recover from anesthesia. After 4 h, mice were reanesthetized and intestinal loops were removed and, length and weight of the intestinal loops were then measured to calculate weight/length ratio. For intestinal absorption studies, loops were injected with 200 μl of PBS with or without DQA (100 μM), incisions sutured and mice left to recover. After 30 min, mice were reanesthetized, and intestinal loops were removed to measure loop weight/length ratio. All animal protocols were approved by the Laboratory Animal Ethical Committee of Mahidol University, Thailand.
Cell Viability Assay
FRT cell viability after exposure to DQA was determined using 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.[14] FRT cells were seeded in 96-well plate at a density of 2 × 104 cells/well, grown for 24 h in a humidified incubator under 5% CO2/95% O2 at 37°C, followed by incubation for a further 24 h in serum-free culture medium containing vehicle or various concentrations of DQA. For MTT assay, medium was removed and MTT (10% v/v) was added into each well and the plate was incubated for 4 h. Then, an aliquot of 100% Dimethyl sulfoxide (DMSO) was added, and cells were incubated for another 3 h with shaking. Cell viability was determined from measurement of absorbance at 590 nm (BMG LABTECH spectrophotometer, Victoria, Australia).
Statistical Analysis
Results of all experiments are presented as mean ± SE. Statistical difference between control and treatment groups was determined using Student's t test with P value < 0.05 considered as statistically significant.
Results
Potency of DQA in Inhibiting CFTR-mediated Apical Chloride Conductance in FRT Cells
The effect of DQA on CFTR function in FRT cells stably expressing human wild-type CFTR was determined by measuring apical chloride current induced by various CFTR agonists, namely, forskolin (FSK), an adenylate cyclase activator, CPT-cAMP, a cell-permeable cAMP, and apigenin, a flavone-type direct CFTR-activator. Basolateral membrane permeabilization together with the presence of apical-directed chloride gradient allows direct measurements of the Cl- transport function of CFTR, which is located at the apical side of FRT cells. DQA inhibited in a dose-response manner CFTR-mediated apical chloride current activated by all agonists with an IC50 of ~ 20 μM [Figure 2a–c], with near complete inhibition at 200 μM DQA. Control experiments (without DQA) showed that the stimulated apical chloride current was stably sustained during the entire course of experiments [Figure 2a–c, insets].
Figure 2.
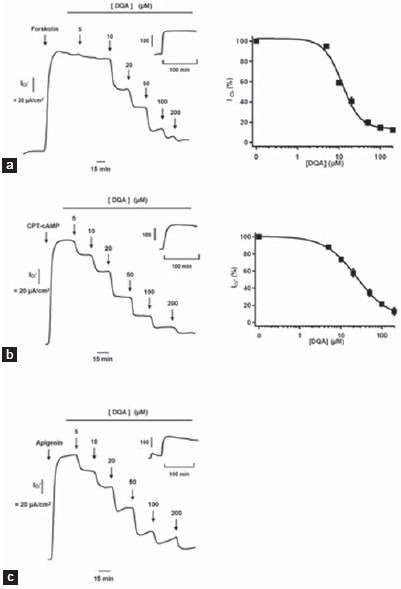
Inhibitory effect in FRT cells of DQA on apical chloride current induced by CFTR agonists, Forskolin (10 μM) (a), CPT-cAMP (100 μM) (b), and apigenin (20 μM (c). (Left) Representative current tracing and (Right) summary of the dose-response analysis (n = 3–5). Insets in the representative tracing show a time tracing of apical chloride current of control (no DQA)
Reversibility of Inhibitory Effect and Cytotoxicity of DQA
A prerequisite characteristic of a potential drug is that it acts reversibly. Recovery of forskolin-activated apical chloride current of FRT cells following removal of DQA (20 μM) treatment was about 90% [Figure 3a]. Exposure of FRT cells to 5–500 μM DQA for 24 h produced no changes in cell viability as determined by MTT assay compared with control [Figure 3b].
Figure 3.
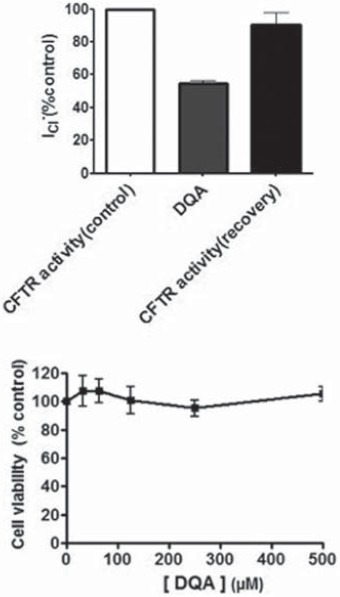
Reversibility of DQA inhibition of CFTR function and DQA cytotoxicity in FRT cells. (a) Reversibility of DQA's effects determined by apical Cl- current measurements. Data are expressed as mean of percent control ± S.E.; n = 3; P < 0.05. (b) Viability of FRT cells exposed to DQA. Data are expressed as mean of percent cell viability compared with vehicle-treated group ± S.E.; n = 3
Inhibition by DQA of cAMP-activated Chloride Secretion Across Human Intestinal Epithelial Cells
CFTR mediates chloride secretion induced by secretagogues, which elevate intracellular cAMP in intestinal cells.[8] The potency of DQA in inhibiting cAMP-activated chloride secretion in intact human intestinal tissue was evaluated using a monolayer of human intestinal T84 cells as a model. Following pretreatment with amiloride (10 μM), an inhibitor of epithelial sodium channels, forskolin (10 μM) stimulated cAMP-mediated chloride secretion that was dose-dependently inhibited by DQA with the IC50 of 10 μM and almost complete inhibition was seen at 100 μM [Figure 4]. Control experiments (without treatment with DQA) showed that an increase in cAMP-activated Cl- current was stable over the period of measurement [Figure 4, inset].
Figure 4.
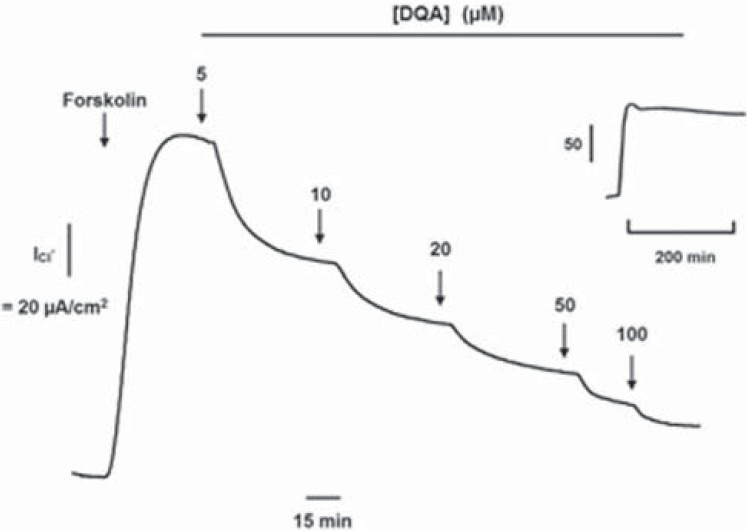
Cumulative inhibitory effect of DQA on cAMP-activated chloride secretion in human intestinal T84 cells. Recording of the current is a representative of three similar sets of measurements. Inset shows a time tracing of forskolin-activated short-circuit current of control (no DQA)
In vitro and in vivo Antidiarrheal Efficacy of DQA
Transepithelial transport of the intestine in either absorptive or secretory directions requires functional cooperation between transport proteins distributed in both luminal and basolateral membranes of enterocytes.[7] The cholera toxin stimulates transepithelial chloride secretion by increasing intracellular cAMP level.[15] The antidiarrheal efficacy of DQA was assessed in both in vitro and in vivo models of cholera toxin-induced intestinal chloride/fluid secretion. In vitro study using short-circuit current analysis of human intestinal T84 cells showed that DQA (10 μM) completely abolished transepithelial chloride secretion induced by cholera toxin (1 μg/ml) [Figure 5a]. The increase in cholera toxin-induced chloride current was steady during the entire period of experiments in control cells [Figure 5a, inset].
Figure 5.
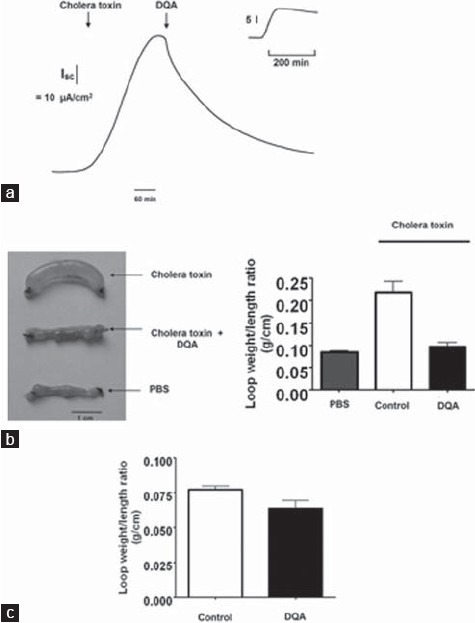
Antidiarrheal efficacy of DQA in human intestinal T84 cells and mouse closed-loop model of cholera. (a) Inhibitory effect of DQA on cholera toxin-induced active chloride secretion across T84 cell monolayer. Inset shows a time tracing of cholera toxin-induced short-circuit current of control (no DQA). (b) In vivo efficacy of DQA in murine closed-loop model of cholera toxin-induced intestinal fl uid secretion. (Left) Photograph of isolated mouse midjejunal loops at 4 h after luminal injection of cholera toxin (1 μg), cholera toxin (1 μg) plus DQA and saline control. (Right) Summary of loop weight/length ratios. Data are expressed as mean ± S.E.; n = 5; P < 0.05. (c) Effect of DQA on intestinal fluid absorption in murine closed-loop model. Data shown are mean ± S.E.; n = 3
For in vivo study, antidiarrheal efficacy of DQA was evaluated using a mouse closed-loop model of cholera. Intestinal fluid secretion was quantified from the weight/length ratio of intestinal loops injected with PBS containing cholera toxin (1 μg/loop). Although complete inhibition by 10 μM DQA of cholera-toxin-induced chloride secretion was obtained in T84 cells, a higher dose of DQA (100 μM) was used to ensure availability of the compound to intestine over the 4-h period of experiment. Simultaneous intraluminal injections of DQA reduced cholera toxin-induced intestinal fluid secretion by 85% [Figure 5b]. The weight/length ratio of intestinal loop treated with saline, cholera toxin, and cholera toxin plus DQA was 0.08 ± 0.01, 0.21 ± 0.07, and 0.10 ± 0.02 g/cm, respectively. DQA had no effect on intestinal fluid absorption as determined from weight/length ratio measurements of intestinal closed-loops after a 30-min period of saline injection [Figure 5c].
Discussion
The purpose of the present study was to characterize pharmacological properties of DQA, a dihydroquinazolin CFTR inhibitor, and to evaluate its potential as an antidiarrheal agent. Its effect on inhibiting apical chloride current stimulated by agonists (forskolin, CPT-cAMP, and apigenin) in FRT cells expressing human wild-type CFTR was reversible and not associated with cytotoxicity (even when exposed to 500 μM DQA for 24 h). These findings are of particular importance as acute diarrhea normally can last up to a week and a permanent inhibitory effect on CFTR could raise the possibility of inducing cystic fibrosis-like pathology.[16] Moreover, DQA was effective in inhibiting transepithelial chloride secretion activated by both the forskolin and cholera toxin in monolayer of human intestinal epithelial T84 cells. Of particular relevance to its antidiarrheal utility, DQA reduced cholera toxin-induced intestinal fluid secretion by 85% without affecting intestinal fluid absorption in a murine closed-loop model.
The IC50 of DQA (10 μM) in inhibiting CFTR of intact human intestinal T84 cells was half of that for FRT cells carrying human wild-type CFTR, a finding in contrast to that found using CFTRinh -172 in which the IC50 value is 10-fold lower for permeabilized FRT than intact T84 cells.[17] This latter phenomenon may be attributed to the intracellular negative membrane potential of T84 cells, which would limit the accumulation of negatively charged CFTRinh -172 (at physiologic pH of 7.4).[17] The cause of higher potency of DQA in T84 cells remains unclear, but it may be related to differences in membrane permeability and/or in amounts of CFTR present in the two cell types.
Surprisingly, 10 μM DQA caused nearly complete inhibition of cholera toxin-induced chloride secretion (using 1 μg/ml toxin). We speculate that, in addition to its inhibitory effect on CFTR, DQA may have other beneficial pleiotropic effects. In vivo studies showed that administration of DQA remarkably reduced intestinal fluid secretion with no effect on intestinal fluid absorption. It is worth noting that cAMP-activated chloride secretion is not only involved in the pathophysiology of enterotoxin-induced secretory diarrhea, but is also involved in other infections and inflammation-related diarrhea,[4,8,18] as cAMP is an intermediate in the process of the release of several endogenous mediators, namely adenosine, from immune cells.[4,19]
In summary, DQA is a novel quinazolin CFTR inhibitor with favorable pharmacological properties, such as reversibility, lack of cytotoxicity and in vivo efficacy. It provides a lead compound for future development of potent antidiarrheal agents based on this class of compounds.
Acknowledgments
This work was supported by the Thailand Research Fund, Office of the Higher Education Commission and Mahidol University (grant MRG5380125), Mahidol University research grant, Faculty of Science, Mahidol University, and the Office of the Higher Education Commission and Mahidol University under the National Research Universities Initiative (NRU). Authors thank Professor Prapon Wilairat for critical reading of the manuscript.
Jenvit Patanayindee and Chatchai Muanprasat have contributed equally to this work.
Footnotes
Source of Support: Thailand Research Fund, Office of the Higher Education Commission and Mahidol University (grant MRG5380125), Mahidol University research grant.
Conflict of Interest: None declared.
References
- 1.Kosek M, Bern C, Guerrant RL. The global burden of diarrhoeal disease, as estimated from studies published between 1992 and 2000. Bull World Health Organ. 2003;81:197–204. [PMC free article] [PubMed] [Google Scholar]
- 2.Boschi-Pinto C, Velebit L, Shibuya K. Estimating child mortality due to diarrhoea in developing countries. Bull World Health Organ. 2008;86:710–7. doi: 10.2471/BLT.07.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanchez J, Holmgren J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell Mol Life Sci. 2008;65:1347–60. doi: 10.1007/s00018-008-7496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest. 2003;111:931–43. doi: 10.1172/JCI18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sack DA, Sack RB, Nair GB, Siddique AK. Cholera. Lancet. 2004;363:223–33. doi: 10.1016/s0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekhar MR, Krishna BV, Patil AB. Changing characteristics of Vibrio cholerae: Emergence of multidrug resistance and non-O1, non-O139 serogroups. Southeast Asian J Trop Med Public Health. 2008;39:1092–7. [PubMed] [Google Scholar]
- 7.Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: Molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–72. doi: 10.1146/annurev.physiol.62.1.535. [DOI] [PubMed] [Google Scholar]
- 8.Thiagarajah JR, Verkman AS. CFTR pharmacology and its role in intestinal fluid secretion. Curr Opin Pharmacol. 2003;3:594–9. doi: 10.1016/j.coph.2003.06.012. [DOI] [PubMed] [Google Scholar]
- 9.Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79(Suppl 1):S23–45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- 10.Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–8. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–71. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: Mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–37. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galietta LV, Jayaraman S, Verkman AS. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol. 2001;281:C1734–42. doi: 10.1152/ajpcell.2001.281.5.C1734. [DOI] [PubMed] [Google Scholar]
- 14.Wongsamitkul N, Sirianant L, Muanprasat C, Chatsudthipong V. A plant-derived hydrolysable tannin inhibits CFTR chloride channel: A potential treatment of diarrhea. Pharm Res. 2010;27:490–7. doi: 10.1007/s11095-009-0040-y. [DOI] [PubMed] [Google Scholar]
- 15.Field M, Fromm D, al-Awqati Q, Greenough WB., 3rd Effect of cholera enterotoxin on ion transport across isolated ileal mucosa. J Clin Invest. 1972;51:796–804. doi: 10.1172/JCI106874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79(Suppl 1):S215–55. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- 17.Thiagarajah JR, Broadbent T, Hsieh E, Verkman AS. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology. 2004;126:511–9. doi: 10.1053/j.gastro.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Lohi H, Mäkelä S, Pulkkinen K, Höglund P, Karjalainen-Lindsberg ML, Puolakkainen P, et al. Upregulation of CFTR expression but not SLC26A3 and SLC9A3 in ulcerative colitis. Am J Physiol Gastrointest Liver Physiol. 2002;283:G567–75. doi: 10.1152/ajpgi.00356.2001. [DOI] [PubMed] [Google Scholar]
- 19.Strohmeier GR, Reppert SM, Lencer WI, Madara JL. The A2b adenosine receptor mediates cAMP responses to adenosine receptor agonists in human intestinal epithelia. J Biol Chem. 1995;270:2387–94. doi: 10.1074/jbc.270.5.2387. [DOI] [PubMed] [Google Scholar]