

Abstract
Objective:
The aim of this study was to investigate the effects of pravastatin on the pharmacokinetics of nimodipine in rats.
Materials and Methods:
The effect of pravastatin on P-glycoprotein (P-gp) and cytochrome P450 (CYP) 3A4 activity was evaluated. Nimodipine was administered to rats intravenously (3 mg/kg) and orally (12 mg/kg) with pravastatin (0.3 and 1 mg/kg).
Results:
Pravastatin inhibited CYP3A4 enzyme activity in a concentration-dependent manner with a 50% inhibition concentration (IC50) of 14 µM. Compared with the oral control group, the area under the plasma concentration-time curve (AUC0-∞) of nimodipine was increased significantly. Consequently, the absolute bioavailability (AB) of nimodipine with pravastatin (1 mg/kg) was 31.1%, which was significantly enhanced compared with the oral control group. Moreover, the relative bioavailability (RB) of nimodipine was 1.12- to 1.31-fold greater than that of the control group.
Conclusions:
The enhanced oral bioavailability of nimodipine might be mainly due to inhibition of the CYP3A-mediated metabolism of nimodipine in the small intestine and/or in the liver and due to reduction of the total body clearance rather than both to inhibition of the P-gp efflux transporter in the small intestine and reduction of renal elimination of nimodipine by pravastatin. The increase in the oral bioavailability of nimodipine with pravastatin should be taken into consideration of potential drug interactions between nimodipine and pravastatin.
KEY WORDS: Bioavailability, CYP3A4, nimodipine, P-gp, pharmacokinetics, pravastatin
Introduction
Nimodipine is a dihydropyridine calcium channel blocker that selectively dilates cerebral arteries and increases cerebral blood flow in animals and humans.[1] Its major therapeutic indication is the prevention and treatment of delayed ischemic neurological disorders that often occur in patients with subarachnoid hemorrhages.[2,3] Nimodipine is rapidly absorbed after oral administration and is widely distributed throughout the body. Orally administered nimodipine is subject to extensive first-pass metabolism from the portal circulation, resulting in low systemic bioavailability.[4,5] Usually only the parent compound is active and most of the metabolic steps involve reactions catalyzed by cytochrome P450 (CYP) enzymes.
The reduced bioavailability of nimodipine after oral administration might be due not only to the metabolizing enzyme CYP3A4, but also due to the P-glycoprotein (P-gp) efflux transporter in the small intestine. Saeki et al. reported that nimodipine is a substrate for P-gp and Wacher et al. reported that nimodipine is both a CYP3A4 and P-gp substrate.[6,7]
Pravastatin, HMG-CoA reductase inhibitors (statins) widely used in the management of hypercholesterolemia.[8] Pravastatin is rapidly but incompletely absorbed from the gastrointestinal tract and undergoes extensive first-pass metabolism in the liver, its primary site of action.[9] Cytochrome P450 (CYP) 3A is mainly responsible for 3’’-hydroxy pravastatin formation.[10]
There is no clinically important pharmacokinetic interaction of pravastatin with a number of common CYP3A inhibitors. Itraconazole, diltiazem, and grapefruit juice have all no statistically significant effect on pharmacokinetics of pravastatin.[11–13] The contribution of CYP-dependent biotransformation to pravastatin elimination is minor. Pharmacokinetics interaction of pravastatin with other drugs are rare compared with those of other statins, this may be due to the dual routes of elimination and low plasma protein binding of pravastatin. Pravastatin seems to be more favorable in the management of hypercholesterolemia compared with the other statins.
We attempted to evaluate P-gp activity using the rhodamine-123 retention assay in P-gp overexpressing MCF-7/ADR cells, and assessed CYP3A4 activity.
Antihypertensive agents are commonly coadministered with cholesterol-lowering agents in clinics. Therefore, orally administered pravastatin would affect the pharmacokinetics of nimodipine because it is a substrate of CYP3A4.
However, little information is available regarding the effects of pravastatin on the pharmacokinetics of nimodipine. Therefore, the aim of this study was to examine the effects of pravastatin on CYP3A4 and P-gp activity and the pharmacokinetics of nimodipine in rats after oral and intravenous (i.v.) administration with pravastatin.
Materials and Methods
Chemicals and Apparatus
Nimodipine, nitrendipine (internal standard), and pravastatin were purchased from the Sigma Chemical Co. (St. Louis, MO, USA). Ethyl acetate and methanol were purchased from Merck Co. (Darmstadt, Germany). All other chemicals were reagent grade and were used without further purification. The apparatuses used included High-Performance Liquid Chromatograph (HPLC) (Model LC-10A, Shimadzu Co., Kyoto, Japan), a syringe pump (Model341B, Sage Co., Kyoto, Japan), a vortex mixer (Scientific Industries, Seoul, Korea), and a centrifuge (Abbott Co., TX, USA).
Animal Experiments
Male Sprague-Dawley rats (weighing 270–300 g) were purchased from the Dae Han Laboratory Animal Research Co. (Choongbuk, Korea), and were given access to a commercial rat chow diet (No. 322-7-1, Superfeed Co., Gangwon, Korea) and tap water. The animals were housed, two per cage, and maintained at 22°C ± 2°C and 50–60% relative humidity, under a 12:12 h light-dark cycle. The experiments were initiated after acclimation under these conditions for at least 1 week. The Animal Care Committee of Chosun University (Gwangju, Korea) approved the design and conduction of this study. The rats were fasted for at least 24 h prior to the experiments. The left femoral artery and vein were cannulated using polyethylene tubing (SP45, i.d. 0.58 mm, o.d. 0.96 mm; Natsume Seisakusho Co. Ltd., Tokyo, Japan) for blood sampling and i.v. injection, respectively.
Drug Administration
The rats were divided into six groups (n = 6, each): An oral control group (12 mg/kg of nimodipine dissolved in distilled water, 3.0 mL/kg) with or without 0.3 and 1 mg/kg of pravastatin (mixed in distilled water, 3.0 mL/kg), and an i.v. control group (3 mg/kg of nimodipine, dissolved in 0.9% NaCl solution, 1.5 mL/ kg) with or without 0.3 and 1 mg/ kg of pravastatin (mixed in distilled water, 3.0 mL/kg). Oral nimodipine was administered intragastrically using a feeding tube, and pravastatin was administered in the same manner 30 min prior to the oral administration of nimodipine. Nimodipine for i.v. administration was injected through the femoral vein within 0.5 min. A 0.4-mL aliquot of blood was collected into heparinized tubes from the femoral artery at 0, 0.25, 0.5, 1, 2, 3, 4, 8, 12, and 24 h after nimodipine administration. The blood samples were centrifuged at 13,000 rpm for 5 min and the plasma samples were stored at -40°C until HPLC analysis.
HPLC Assay
The plasma nimodipine concentration was determined by an HPLC assay using a modification of the method reported by Qian et al.[14]
The mobile phase consisted of methanol:water (65:35, v/v). The mobile phase was filtered by passing through a 0.45-μm pore size membrane filter. The retention times at a flow rate of 1.0 mL/min were as follows: Internal standard, 7.6 min; nimodipine, 9.1 min. Linear regression analysis using a least-square fit was performed. A calibration curve was obtained from the standard samples at the following concentrations: 10, 20, 50, 100, 500, and 1000 ng/mL. The following regression equation was obtained: y = 206.0 × + 18.1 (r = 0.999).
The detection limit of nimodipine in rat plasma was 5 ng/ mL. The coefficient of variation was below 4.9%.
CYP3A4 Inhibition Assay
The assay of inhibition on human CYP3A4 enzyme activity was performed in a multiwell plate using CYP inhibition assay kit (GENTEST, Woburn, MA, USA) as described previously.[15] Metabolite concentrations were measured by spectrofluorometer (Molecular Device, Sunnyvale, CA, USA) at an excitation wavelength of 409 nm and an emission wavelength of 530 nm. Positive control (1 μM ketoconazole for CYP3A4) was run on the same plate and produced 99% inhibition. All experiments were done in duplicate, and the results were expressed as the percent of inhibition.
Rhodamine-123 Retention Assay
The procedures used for the Rho-123 retention assay were similar to a reported method.[16] After incubation of the cells with 20 μM rhodamine-123 with pravastatin (10, 30, and 100 μM) for 90 min, the medium was completely removed. Rhodamine-123 fluorescence in the cell lysates was measured using excitation and emission wavelengths of 480 and 540 nm, respectively. Fluorescence values were normalized to the total protein content of each sample and are presented as the ratio to control.
Pharmacokinetic Analysis
The plasma concentration data were analyzed using a noncompartmental method from WinNonlin software version 4.1 (Pharsight Co., Mountain View, CA, USA). The elimination rate constant (Kel) was calculated by the log-linear regression of nimodipine concentration data during the elimination phase, and the terminal half-life (t½) was calculated by 0.693/Kel. The peak concentration (Cmax) and time to reach the peak concentration (Tmax) of nimodipine in the plasma were obtained by visual inspection of the data from the concentration-time curve. The area under the plasma concentration-time curve (AUC0-t) from time zero to the time of the last measured concentration (Clast) was calculated using the linear trapezoidal rule. The AUC zero to infinity (AUC0-∞) was obtained by adding AUC0-t and the extrapolated area was determined by Clast/Kel. The total body clearance for i.v. route (CLt) was calculated from D/AUC, where D is the dose of nimodipine. The absolute bioavailability (AB) of nimodipine was calculated by AUCoral/AUCi.v. × Dosei.v./Doseoral × 100, and the relative bioavailability (RB) was calculated by (AUCwith pravastatin/AUCcontrol) × 100.
Statistical Analysis
All data were presented as means with standard deviation. An analysis of variance (ANOVA) with Scheffe's test was used to determine any significant differences between the control groups and coadministration or pretreatment groups. P value < 0.05 was considered significant.
Results
Inhibitory Effect of Pravastatin on CYP3A4 Activity
The inhibitory effect of pravastatin on CYP3A4 activity is shown in Figure 1. Pravastatin inhibited CYP3A4 enzyme activity and the 50% inhibition concentration (IC50) value of pravastatin on CYP3A4 activity was 14 μM.
Figure 1.
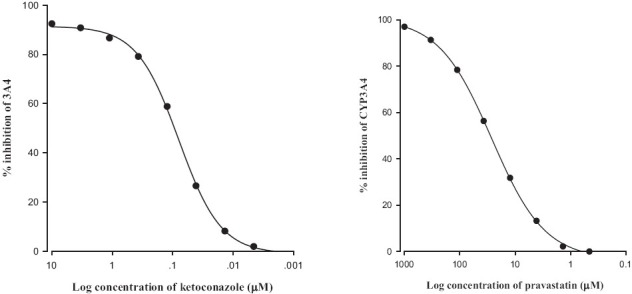
Inhibitory effect of ketokonazole (a) and pravastatin (b) on CYP3A4 activity. All experiments were done in duplicate, and the results were expressed as the percent of inhibition
Rhodamine-123 Retention Assay
Accumulation of rhodamine-123, a P-gp substrate, was not raised in MCF-7/ADR cells overexpressing P-gp compared with that in MCF-7 cells lacking P-gp, as shown in Figure 2. The concurrent use of pravastatin did not enhance the cellular uptake of rhodamine-123 in a concentration-dependent manner ranging from 10 to 100 μM. This result suggests that pravastatin did not significantly inhibit P-gp activity.
Figure 2.
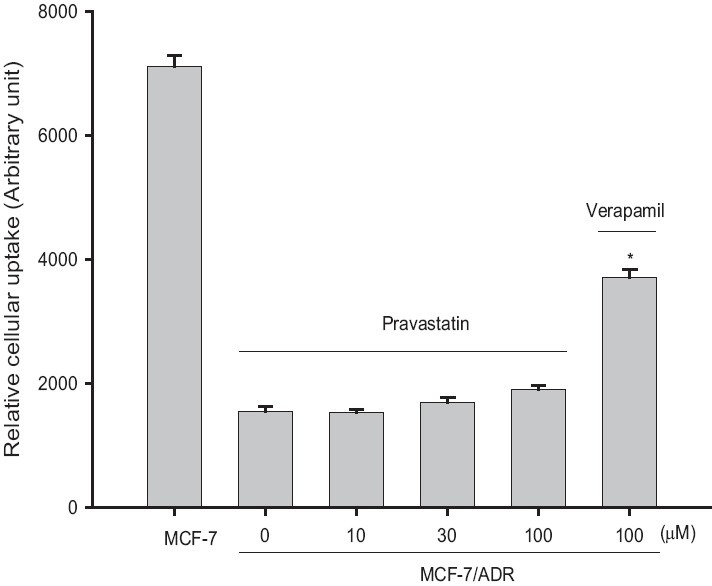
Effects of pravastatin on the cellular accumulation of rhodamine-123 in MCF-7 and MCF-7/ADR cells. Data represents mean ± SD of six separate samples. (Signifi cant at *P < 0.05 difference compared with positive control, verapamil)
Effect of Pravastatin on The Pharmacokinetics of Oral Nimodipine
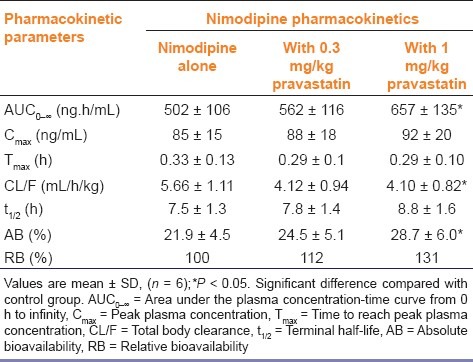
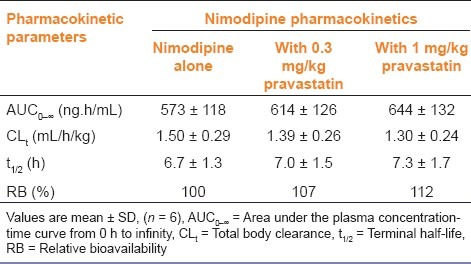
Mean arterial plasma concentration-time profiles of nimodipine following oral administration of nimodipine (12 mg/ kg) to rats with pravastatin (0.3 and 1 mg/kg) are shown in Figure 3. The corresponding pharmacokinetic parameters are shown in Table 1. Compared with the control group, pravastatin significantly (P < 0.05 for 1 mg/kg) increased (30.9%) the area under the plasma concentration-time curve (AUC0-∞) and significantly (P < 0.05) decreased total body clearance of nimodipine. The AB of nimodipine with pravastatin (1 mg/ kg) was 31.1%, which was significantly enhanced (1 mg/kg, P < 0.05) compared with the oral control group, and the RB of nimodipine was increased by 1.12- to 1.31-fold.
Figure 3.
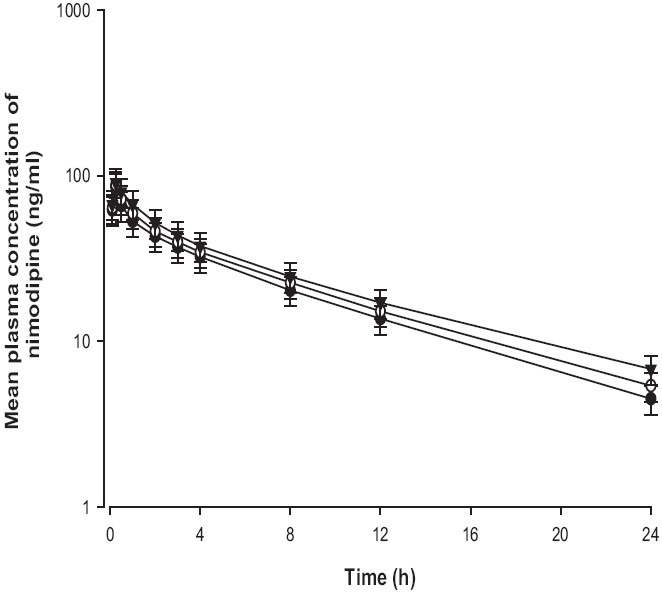
Mean plasma concentration-time profi les of nimodipine after oral administration of nimodipine (12 mg/kg) without (●) or with 0.3 mg/kg (○) and 1 mg/kg (▼) of pravastatin to rats. Bars represent the standard deviation (n = 6)
Table 1.
Pharmacokinetic parameters of nimodipine after oral administration of nimodipine (12 mg/kg) to rats in the presence or absence of pravastatin (0.3 and 1 mg/kg)
Effect of Pravastatin on The Pharmacokinetics of i.v. Nimodipine
Mean arterial plasma concentration-time profiles of nimodipine following an i.v. administration of nimodipine (3 mg/kg) to rats with pravastatin (0.3 and 1 mg/kg) are shown in Figure 4, the corresponding pharmacokinetic parameters are shown in Table 2.
Figure 4.
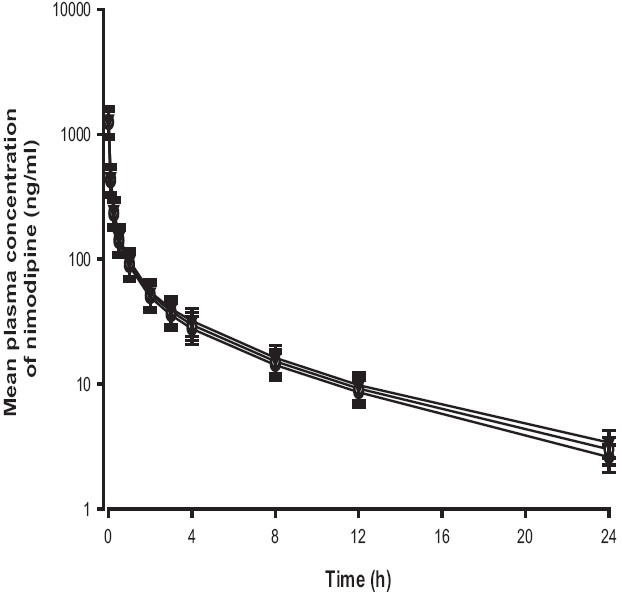
Mean plasma concentration-time profiles of nimodipine after intravenous administration of nimodipine (3 mg/kg) without (●) or with 0.3 mg/kg (○) and 1 mg/kg (▼) of pravastatin to rats. Bars represent the standard deviation (n = 6)
Table 2.
Pharmacokinetic parameters of nimodipine after its intravenous administration of nimodipine (3 mg/kg) to rats in the presence or absence of pravastatin (0.3 and 1 mg/kg)
The AUC of nimodipine was increased, but was not statistically significant compared with that in the control. The t½ of nimodipine was also prolonged, but this increase was not significant. The pharmacokinetics of i.v. nimodipine was not affected by the concurrent use of pravastatin in contrast to those of oral nimodipine.
Discussion
Total CYP P450 content increased slightly proceeding from the duodenum to the jejunum and then decreased sharply to the ileum.[17] Using in situ hybridization with a probe specific for CYP3A4, McKinnon confirmed CYP3A expression throughout the entire small intestine, with highest levels in the proximal regions.[18] The most abundant CYP isoenzyme in the intestine is 3A4.[19]
Based on their broad overlap in substrate specificities as well as their colocalization in the small intestine, the primary site of absorption for orally administered drugs, CYP3A4 and P-gp have been recognized as a concerted barrier to drug absorption.[20,21] The prescription of more than one drug as a combination therapy is increasingly common in current medical practice. Cholesterol-lowering agents such as HMG-CoA reductase inhibitors could be coadministered with calcium channel blockers for the treatment of hypertension.[22]
In the present study, cell-based P-gp activity tests using rhodamine-123 showed that pravastatin (10–100 μM) did not inhibit P-gp activity, but pravastatin significantly inhibited CYP3A4 activity [Figures 1 and 2]. These results are consistent with a report showing that simvastatin effectively inhibited CYP3A4 activity.[23,24] Therefore, pravastatin, an inhibitor of CYP3A4 may significantly impact the bioavailability of nimodipine, a substrat of CYP3A4. As CYP3A4 expressed in rat is corresponding and similar to function of CYP3A4 in human.[25] Therefore, the pharmacokinetic characteristics of nimodipine were evaluated with pravastatin in rats.
This study evaluated the influence of pravastatin, an HMG-CoA reductase inhibitor, on the pharmacokinetics of nimodipine in rats in order to assess the potential drug interactions between pravastatin and nimodipine.
As shown in Table 1, pravastatin significantly (1 mg/kg, P < 0.05) increased area under the plasma concentration-time curve (AUC0-∞) of nimodipine. Pravastatin also significantly increased the AB of nimodipine by 31.1% compared with the oral control group, and the RB of nimodipine was increased by 1.12- to 1.31-fold. These results were consistent with reports that simvastatin significantly increased the AUC and Cmax of verapamil in rats,[26] and atorvastatin also significantly increased the bioavailability of diltiazem in rats.[27] However, these results are not consistent with reports by Yang et al. showing that pravastatin did not significantly increase the AUC and Cmax of losartan in rats.[28] These suggested that the extraction ratio of nimodipine across the rat intestinal tissue was significantly reduced by CYP3A subfamily. The pharmacokinetic profiles of i.v. nimodipine were also evaluated with pravastatin [Table 2]. The AUC and Cmax of nimodipine were not significantly increased by pravastatin, suggesting that the presence of pravastatin did not affect renal elimination in rats. These results were similar to reports by Chung et al. showing that lovastatin did not affect pharmacokinetic parameters of i.v. nicardipine in rats.[29] The pharmacokinetics of i.v. nimodipine was not affected by the concurrent use of pravastatin in contrast to those of oral administration of nimodipine.
Pravastatin significantly enhanced the oral bioavailability of nimodipine. The increase in the oral bioavailability of nimodipine might be mainly attributed to reduced first-pass metabolism of nimodipine via the inhibition of the CYP3A subfamily in the small intestine and/or in the liver rather than both to inhibition of P-gp in the intestine and to reduction of renal elimination of nimodipine by pravastatin. Concomitant use of pravastatin and nimodipine will require close monitoring of potential drug interaction for safe therapy of cardiovascular diseases. Furthermore, the clinical importance of these findings should be investigated in clinical trials.
Footnotes
Source of Support: Nil.
Conflict of Interest: No.
References
- 1.Kazda S, Garthoff B, Krause HP, Schlossmann K. Cerebrovascular effects of the calcium antagonistic dihydropyridine derivative nimodipine in animal experiments. Arzneimittelforschung. 1982;32:331–8. [PubMed] [Google Scholar]
- 2.Scholz H. Pharmacological aspects of calcium channel blockers. Cardiovasc Drugs Ther. 1997;10(Suppl 3):S869–72. doi: 10.1007/BF00051613. [DOI] [PubMed] [Google Scholar]
- 3.Epstein M, Loutzenhister RD. Effects of calcium antagonists on renal hemodynamics. Am J Kidney Dis. 1990;16:10–4. [PubMed] [Google Scholar]
- 4.Maruhn D, Siefert HM, Weber H, Ramsch K, Suwelack D. Pharmacokinetics of nimodipine. II. Communication: Absorption, concentration in plasma and excretion after single administration of [14C] nimodipine in rat, dog and monkey. Arzneimittelforschung. 1985;35:1781–6. [PubMed] [Google Scholar]
- 5.Suwelack D, Weber H, Maruhn D. Pharmacokinetics of nimodipine. II. Communication: Absorption, concentration in plasma and excretion after single administration of [14C] nimodipine in rat, dog and monkey. Arzneimittelforschung. 1985;35:1787–94. [PubMed] [Google Scholar]
- 6.Saeki T, Ueda K, Tanigawara Y, Hori R, Komano T. P-glycoprotein-mediated transcellular transport of MDR-reversing agents. FEBS Lett. 1993;324:99–102. doi: 10.1016/0014-5793(93)81540-g. [DOI] [PubMed] [Google Scholar]
- 7.Wacher VJ, Silverman JA, Zhang Y, Benet LZ. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci. 1998;87:1322–30. doi: 10.1021/js980082d. [DOI] [PubMed] [Google Scholar]
- 8.Hatanaka T. Clinical pharmacokinetics of pravastatin: Mechanisms of pharmacokinetic events. Clin Pharmacokinet. 2000;39:397–412. doi: 10.2165/00003088-200039060-00002. [DOI] [PubMed] [Google Scholar]
- 9.Quion JA, Jones PH. Clinical pharmacokinetics of pravastatin. Clin Pharmacokinet. 1994;27:94–103. doi: 10.2165/00003088-199427020-00002. [DOI] [PubMed] [Google Scholar]
- 10.Jacobsen W, Kirchner G, Hallensleben K, Mancinelli L, Deters M, Hackbarth I, et al. Small intestinal metabolism of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor lovastatin and comparison with pravastatin. J Pharmacol Exp Ther. 1999;291:131–9. [PubMed] [Google Scholar]
- 11.Neuvonen PJ, Kantola T, Kivistö KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther. 1998;63:332–41. doi: 10.1016/S0009-9236(98)90165-5. [DOI] [PubMed] [Google Scholar]
- 12.Azie NE, Brater DC, Becker PA, Jones DR, Hall SD. The interaction of diltiazem with lovastatin and pravastatin. Clin Pharmacol Ther. 1998;64:369–77. doi: 10.1016/S0009-9236(98)90067-4. [DOI] [PubMed] [Google Scholar]
- 13.Fukazawa I, Uchida N, Uchida E, Yasuhara H. Effects of grapefruit juice on pharmacokinetics of atorvastatin and pravastatin in Japanese. Br J Clin Pharmacol. 2004;57:448–55. doi: 10.1046/j.1365-2125.2003.02030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qian M, Gallo JM. High-perfomance liquid chromatographic determinination of the calcium channel blocker nimodipine in monkey plasma. J Chromatogr. 1992;578:316–20. doi: 10.1016/0378-4347(92)80432-p. [DOI] [PubMed] [Google Scholar]
- 15.Crespi CL, Miller VP, Penman BW. Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem. 1997;248:188–90. doi: 10.1006/abio.1997.2145. [DOI] [PubMed] [Google Scholar]
- 16.Han CY, Cho KB, Choi HS, Han HK, Kang KW. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis. 2008;29:1837–44. doi: 10.1093/carcin/bgn092. [DOI] [PubMed] [Google Scholar]
- 17.Zhang QY, Dunbar D, Ostrowska A, Zeisloft S, Yang J, Kaminsky L. Characterization of human small intestinal cytochromes P-450. Drug Metab Dispos. 1999;27:804–9. [PubMed] [Google Scholar]
- 18.McKinnonand RA, McManus ME. Localization of cytochromes P450 in human tissues: Implications for chemical toxicity. Pathology. 1996;28:148–55. doi: 10.1080/00313029600169783. [DOI] [PubMed] [Google Scholar]
- 19.Tubic-Grozdanis M, Hilfinger JM, Amidon GL, Kim JS, Kijek P, Staubach P, et al. Pharmacokinetics of the CYP 3A substrate simvastatin following administration of delayed versus immediate release oral dosage forms. Pharm Res. 2008;25:1591–600. doi: 10.1007/s11095-007-9519-6. [DOI] [PubMed] [Google Scholar]
- 20.Cummins CL, Jacobsen W, Benet LZ. Unmasking the dynamic interplay between intestinal P-glycoprotein and CYP3A4. J Pharmacol Exp Ther. 2002;300:1036–45. doi: 10.1124/jpet.300.3.1036. [DOI] [PubMed] [Google Scholar]
- 21.Wacher VJ, Salphati L, Benet LZ. Active secretion and enterocytic drug metabolism barriers to drug absorption. Adv Drug Deliv Rev. 2001;46:89–102. doi: 10.1016/s0169-409x(00)00126-5. [DOI] [PubMed] [Google Scholar]
- 22.Mason RP. A rationale for combined therapy with a calcium channel blocker and a statin: Evaluation of basic and clinical evidence. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:89–501. doi: 10.2174/156800605774962112. [DOI] [PubMed] [Google Scholar]
- 23.FDA Guidance for industry. In vivo drug metabolism/drug interaction studies-study design, data analysis, and recommendations for dosing and labeling. [Last accessed on 1999 Nov 24]. Available from: http://www.fda.gov/cder/guidance/index.htm .
- 24.Bogman K, Peyer AK, Torok M, Kusters E, Drewe J. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol. 2001;132:1183–92. doi: 10.1038/sj.bjp.0703920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly PA, Wang H, Napoli KL, Kahan BD, Strobel HW. Metabolism of cyclosporine by cytochromes P450 3A9 and 3A4. Eur J Drug Metab Pharmacokinet. 1999;24:321–8. doi: 10.1007/BF03190040. [DOI] [PubMed] [Google Scholar]
- 26.Choi DH, Li C, Choi JS. Effects of simvastatin on the pharmacokinetics of verapamil and its main metabolite, norverapamil, in rats. Eur J Drug Metab Pharmacokinet. 2009;34:163–8. doi: 10.1007/BF03191168. [DOI] [PubMed] [Google Scholar]
- 27.Hong SP, Chang KS, Choi DH, Choi JS. Effects of atorvastatin on the pharmacokinetics of diltiazem and its metabolite, desacetyldiltiazem, in rats. Arch Pharm Res. 2007;30:90–5. doi: 10.1007/BF02977783. [DOI] [PubMed] [Google Scholar]
- 28.Yang SH, Choi JS, Choi DH. Effects of HMG-CoA reductase inhibitors on the pharmacokinetics of losartan and its main metabolite EXP-3174 in rats: Possible role of CYP3A4 and P-gp inhibition by HMG-CoA reductase inhibitors. Pharmacology. 2011;88:1–9. doi: 10.1159/000328773. [DOI] [PubMed] [Google Scholar]
- 29.Chung JW, Yang SH, Choi JS. Effects of lovastatin on the pharmacokinetics of nicardipine in rats. Biopharm Drug Dispos. 2010;31:436–41. doi: 10.1002/bdd.721. [DOI] [PubMed] [Google Scholar]