

Abstract
Background
An inflammatory component is present in the microenvironment of most neoplastic tissues. Inflammation and elevated C-reactive protein (CRP) are associated with poor prognosis and decreased survival in many types of cancer.
Vitamin C has been suggested as having both a preventative and therapeutic role in a number of pathologies when administered at much higher-than-recommended dietary allowance levels.
Since in vitro studies demonstrated inhibition of pro-inflammatory pathways by millimolar concentrations of vitamin C, we decided to analyze the effects of high dose IVC therapy in suppression of inflammation in cancer patients.
Methods
45 patients with prostate cancer, breast cancer, bladder cancer, pancreatic cancer, lung cancer, thyroid cancer, skin cancer and B-cell lymphoma were treated at the Riordan Clinic by high doses of vitamin C (7.5 g -50 g) after standard treatments by conventional methods.
CRP and tumor markers were measured in serum or heparin-plasma as a routine analysis. In addition, serum samples were collected before and after the IVCs for the cytokine kit tests.
Results
According to our data positive response to treatment, which was demonstrated by measurements of C- reactive protein, was found in 75% of patients and progression of the inflammation in 25% of patients. IVC treatments on all aggressive stage cancer patients showed the poor response of treatment.
There was correlation between tumor markers (PSA, CEA, CA27.29 and CA15-3) and changes in the levels of C-reactive protein.
Our test of the effect of IVC on pro-inflammatory cytokines demonstrated that inflammation cytokines IL-1α, IL-2, IL-8, TNF-α, chemokine eotaxin and CRP were reduced significantly after treatments.
Conclusions
The high dose intravenous ascorbic acid therapy affects C-reactive protein levels and pro-inflammation cytokines in cancer patients. In our study, we found that modulation of inflammation by IVC correlated with decreases in tumor marker levels.
In summary, our data support the hypothesis that high dose intravenous ascorbate treatments may reduce inflammation in cancer patients. Our results suggest that further investigations into the use of IVC to reduce inflammation in diseases where inflammation is relevant are warranted.
Background
Inflammation plays a key role in tumour development, affecting tumour proliferation, angiogenesis, metastasis, and resistance to therapy [1-6]. Key features of cancer-related inflammation (CRI) include leukocyte infiltration, cytokine build-up, tissue remodelling, and angiogenesis. Infiltrating leukocytes such as tumour associated macrophages (TAMs), neutrophils, dendritic cells, and lymphocytes establish an inflammatory microenvironment [7] and are key components in tumours of epithelial origins [8]. These leukocytes secrete pro-inflammatory cytokines such as IL1, IL6, TNFα, TGFβ, FGF, EGF and HGF21, as well as chemokines such as CCL2 and CXCL8 [9]. While immune cells may repress tumour growth in some cases [10-12], there is increasing concern that inflammatory microenvironments caused by infiltrating leukocytes can facilitate cancer development [13-16]. In clinical studies, TAMs are associated with poor prognosis, while the use of anti-inflammatory agents is associated with reduced instances of certain cancers [14,17].
Several studies indicate that inflammation is a marker of high cancer risk and poor treatment outcome [18-22]. In response to systemic inflammation, and in particular in response to elevated IL-6 levels, the liver produces CRP [23], a protein that binds to dead or dying cells to activate the complement system. CRP can be used as a marker of systemic inflammation. It correlates with disease progression and can be used to monitor infection [18,24-27]. For example, subjects with highly elevated CRP concentrations (above 80 mg/L, as compared to values below 5 mg/L) showed 3.5 times the risk for “all-cause” mortality compared to other subjects [28]. There are particularly strong negative correlations between CRP levels and cancer survival [14,24,28-38] in a wide variety of cancer types. For example, cancer patients with highly elevated CRP showed increased mortality by a factor of 28. Thus, CRP concentration data confirm a correlation between cancer progression and inflammation.
Serum CRP concentrations in human subjects are, according to one report, inversely correlated with antioxidant nutrient concentrations [39]. Vitamin C (ascorbate, ascorbic acid, AA) is a water-soluble antioxidant and essential nutrient for immune cells and extracellular matrix production [40,41]. These properties, as well as the correlations between ascorbate depletion in cancer patients and prognosis [42], suggest that vitamin C may have a beneficial effect on inflammation in cancer patients. Experiments by Hartel et al. [43] indicate that 20 mM ascorbate inhibited production of IL-6 and TNF-α in monocytes without affecting IL-1 or IL-8 levels. For lymphocytes, the same ascorbate concentrations inhibited IL-2 production without affecting TNF-α of IFN-γ levels. Ascorbate, at milli molar concentrations, may also inhibit NF-κB activation in endothelial cells [44]. NF-κB is an important transcription factor that mediates changes in gene expression during inflammation. The effect may be concentration dependent, as a different study indicated that low ascorbate doses (0.2 mM) actually enhanced NF-κB in Jurkat T-cells [45]. Other studies show that ascorbate inhibits TNF-α activation of NF-κB in human cell lines in vitro in a concentration dependent fashion, and can also inhibit GM-CSF, IL-3, and IL-5 production [46].
While millimolar ascorbate concentrations are not usually considered ‘physiological’, they can be achieved if the vitamin is administered intravenously at high dose. Intravenous vitamin C therapy has been used in the treatment of cancer [47,48]. Rationales for IVC therapy include preferential toxicity of ascorbate toward cancer cells [49,50], potential benefits of ascorbate for immune cells [48], and ascorbate inhibitory effect on angiogenesis [51,52]. In a study with guinea pigs, tumour growth was significantly reduced in cases where intra-tumour ascorbate concentrations reached the millimolar level [53].
In addition, inflammation and oxidative stress can cause down-regulation of immune system associated with T cell dysfunction, which has been described in cancer, infectious, and autoimmune diseases. In cancer and other inflammatory conditions, the T cell receptor (TCR) zeta chain is cleaved resulting in T-cell and Natural Killer (NK) cell inability to activate [54]. Several studies have shown that chronic inflammation is mandatory for the induction of an immunosuppressive environment, due to the high concentrations of reactive oxygen species formed by myeloid suppressor cells [55,56]. Clinically, this occurrence is associated with poor prognosis [57,58]. As the in vitro tissue cultures have shown a reversal of TCR cleavage by antioxidants [59,60], the result of treatment by ascorbic acid may augment T-cell and NK cell immunity.
Over the course of 35 years, the Riordan Clinic has administered IVC to cancer patients. For some of these subjects, blood chemistry data before, during, and after IVC treatments provide measurements of plasma ascorbate, CRP levels, and the levels of cancer markers such as PSA and CEA. This manuscript details an analysis of these clinical data to determine if a correlation exists between IVC treatments, inflammation (measured by CRP, cytokine levels), and tumour progression (measured by PSA, CEA and other tumour markers).
Methods
Biochemical assays and analysis
CRP concentrations in blood (serum or heparin-plasma) were analysed using a particle-enhanced immune-turbidimetric assay (CRP Ultra WR Reagent kit, Genzyme) according to manufacturer’s instructions on an automated analyser (CobasMIRA, Roche Diagnostics). The upper boundary for the normal range was set to 1.9 mg/L.
Blood concentrations of cytokines INF-γ, IL-1, IL-2, IL-8, TNF-α, and eotaxin were obtained using commercially available cytokine array ELISA kits (Ray Biotech). The chemiluminescent signal was imaged by the BioImage system (Alpha Innotech). The antigen-antibody spots on the image were circled by Spot Denso of FluorChem SP software (Alpha Innotech). Serum samples from non-cancer people were used as normal ranges. The pro-inflammatory markers from eleven patients before and after 6 IVCs were compared.
Serum vitamin C measurements were attained as described elsewhere [61].
Statistical analysis
The data were analysed by Systat software (Systat, Inc) and Kaleidagraph software. Variables were presented as mean values ± SD, or as medians with corresponding 25th percentiles. Association between different factors was assessed using linear models. Statistical significance was accepted if the null hypothesis could be rejected at p ≤ 0.05.
Patients and treatments
The diagnosis of patients as to their malignant disease and status was based on the information provided to Riordan Clinic and sent from pathologist and/or oncologist at the request of the patient.
First, patients are tested for glucose-6-phosphate dehydrogenase deficiency prior to treatment, as this deficiency can cause hemolysis. Patients with G6PDH deficiency were not given IVC. For eligible patients, the Riordan IVC protocol was administered as described elsewhere [48]. Briefly, subjects initially receive of 7.5 to 15 g ascorbate infused by slow drip in saline solution. To ensure that patient has adequate renal function, hydration and urinary voiding capacity, baseline lab tests were performed that include a serum chemistry profile and urinalysis. Provided these first treatments are well tolerated, patients are given the option to continue with 25 to 50 g infusions up to three times per week.
Patients who received IVC treatment signed informed consent. Privacy and patient anonymity guidelines were strictly followed in gathering the data for analysis.
Data were analysed for 45 patients who went through the Riordan IVC protocol: twenty-four with prostate cancer, nine with breast cancer, three each with bladder cancer, pancreatic cancer, and lung cancer, and one each with thyroid cancer, skin cancer and B-cell lymphoma. Several patients had metastasis. Treatment and additional clinical data were obtained from medical records. Many of the patients were treated initially by conventional way, by operation, radiotherapy, and administration of hormones and cytotoxic substances. Characteristics of subjects under analysis are listed in Table 1: the median age of the patients was 68 years, with a range of 47–85 years. Clinical response to IVC treatment was assessed by screening parameters of inflammation, cancer markers, complete blood counts, lipid profiles and nutritional status. The median follow-up time was 7.2 years and ranged from 1 year to 18 years.
Table 1.
Characteristics of subjects under analysis with duration and number of treatments
| Type of cancer | Age | Sex | CRP before | CRP after | Days of treatment | Number of treatments | Conventional treatment |
|---|---|---|---|---|---|---|---|
| Anorectal adenocarcinoma, grade 3, invasive |
60 |
F |
19.6 |
1.9 |
663 |
4 |
low anterior resection, colorectostomy |
| Biliary cancer, cholangiocarcinoma S-III(IV) |
77 |
M |
19.58 |
143.76 |
68 |
33 |
No records (NR) |
| Bladder - transitional cell carcinoma x 2, grade 3 |
66 |
M |
57.5 |
11 |
76 |
7 |
operations, post treatment cycles |
| Bladder, grade 3 |
65 |
M |
6.09 |
0.94 |
167 |
7 |
2 times surgery |
| Breast |
71 |
M |
6.2 |
3.1 |
179 |
2 |
mastectomy, Tomoxifen |
| Breast |
57 |
F |
19.4 |
0.4 |
512 |
47 |
NR |
| Breast |
82 |
F |
22 |
8.9 |
132 |
2 |
radiation, 36 treatments |
| Breast |
66 |
F |
12 |
2.25 |
193 |
3 |
Lamoxifin - 1.5 year |
| Breast |
89 |
F |
4.1 |
8.5 |
454 |
19 |
NR |
| Breast, ductal carcinoma in situ, high nuclear grade |
67 |
F |
21.1 |
5.5 |
555 |
12 |
no surgery |
| Breast carcinoma in situ, Basal cell carcinoma, Lymphoma |
71 |
F |
133.2 |
2.3 |
682 |
|
surgery, mastectomy, chemotherapy |
| Breast, category 5 |
78 |
F |
8.6 |
13.1 |
379 |
20 |
NR |
| Breast, poorly differentiated duct cell carcinoma grade 4, Pancreas, malignant stage III, nonresectable |
78 |
F |
3.7 |
8.9 |
233 |
4 |
no radiation or chemotherapy, mastectomy |
| Breast, infiltrating tubular carcinoma |
77 |
F |
2.7 |
7.9 |
232 |
3 |
no radiation, chemotherapy or mastectomy |
| Breast, stage 1, T1 OMO, grade 2 |
53 |
F |
12 |
3.8 |
44 |
11 |
lumpectomy |
| Breast, Stage IIA; Nottingham grade 2 |
69 |
F |
9.3 |
3 |
121 |
20 |
breast lumpectomy; partial mastectomy; excision (L) breast; |
| Colon, stage IV, liver cancer |
64 |
M |
44 |
3.4 |
232 |
12 |
surgery, chemotherapy, hepatic resection |
| Gastric cancer |
70 |
M |
1.6 |
59.8 |
159 |
15 |
NR |
| Large B cell lymphoma |
25 |
F |
10.5 |
0.3 |
82 |
18 |
NR |
| Lung |
65 |
M |
216 |
35.9 |
174 |
27 |
NR |
| Lung - squamous cell, grade 2 |
80 |
M |
9 |
6.7 |
82 |
20 |
surgery, no chemotherapy |
| Lung, renal |
82 |
F |
19.2 |
59 |
167 |
37 |
NR |
| Lymphoma |
53 |
F |
10.8 |
5.2 |
110 |
5 |
Immune therapy 4 treatments/week every 6 months, Rutuxin |
| Pancreas |
85 |
M |
3.8 |
54.5 |
42 |
11 |
NR |
| Pancreas, breast |
71 |
F |
10.1 |
0.4 |
119 |
44 |
NR |
| Pancreas, liver and bone metastasis |
89 |
M |
2 |
25 |
423 |
|
cholecystojejunostomy and gastrojejunostomy |
| Pros, Gleason score 6-8 |
82 |
M |
3.9 |
8.8 |
279 |
8 |
no surgery or hormonal therapy |
| Prostate |
77 |
M |
26.2 |
0.4 |
261 |
16 |
NR |
| Prostate |
80 |
M |
153.3 |
148.4 |
11 |
5 |
NR |
| Prostate |
93 |
M |
29.2 |
2.1 |
136 |
20 |
NR |
| Prostate, colon cancer |
89 |
M |
43.4 |
31.5 |
113 |
5 |
chemotherapy, colonectomy |
| Prostate, Gleason score 2.5, malignant skin melanoma |
72 |
M |
6 |
0.9 |
140 |
8 |
NR |
| Prostate, Gleason score 6 |
53 |
M |
8.4 |
1.9 |
38 |
4 |
surgery |
| Prostate, Gleason score 6, squamous cell melanoma, bone cancer |
78 |
M |
35.5 |
1.3 |
23 |
4 |
radical prosectomy |
| Prostate, Gleason score 6, stage T2A |
72 |
M |
22.1 |
3.66 |
400 |
|
surgery |
| Prostate, Gleason score 7 |
78 |
M |
16.7 |
4.8 |
401 |
4 |
radiation and hormonal therapy |
| Prostate, pancreas |
74 |
M |
14.9 |
40.7 |
89 |
24 |
NR |
| Prostate, pancreas |
86 |
M |
500 |
54.3 |
542 |
102 |
NR |
| Prostate, stage I |
65 |
M |
2.53 |
8.59 |
82 |
1 |
prostatectomy |
| Prostate, Gleason score 4, increased to 6–9 during 2 years |
81 |
M |
5 |
0.6 |
1449 |
3 |
NR |
| Prostate, Esophageal cancer |
84 |
M |
15.3 |
3.1 |
37 |
6 |
removal of esophagus |
| Prostate, metastatic adenocarcinoma, Gleason score 4 + 4 = 8 |
74 |
M |
25 |
0.7 |
791 |
7 |
radical prostatectomy |
| Rectal adenocarcinoma, invasive through muscle layer |
71 |
M |
2.2 |
7.2 |
137 |
2 |
chemotherapy radiation |
| Renal, Basal cell carcinoma |
82 |
M |
30.5 |
19.5 |
48 |
10 |
operation, 33 treatments by radiation |
| Skin, B-cell Lymphoma, Lung | 83 | M | 8.9 | 1.5 | 754 | 36 | surgery, 40 radiations |
To analyse the effect of IVC on the level of pro-inflammatory cytokines, IVC was given to 11 new coming cancer patients at dose of 15 g, 25 g, 50 g, 50 g, 50 g, and 50 g each time for 6 times. All 11 patients were at relative clinical stable status after the conventional cancer treatments (surgery, chemo, or radiation therapies). Patients had different types of cancers (breast, colon, lung, pancreatic, renal and prostate cancer). For example, all breast-cancer patients in the ascorbate-treated group already had mastectomy and radiotherapy and had been given hormones, but all had relapsed by the time ascorbate supplementation was commenced. Treatment of this group of patients was assigned by IVC 15 g-50 g once a day one time per week. Sera were collected before IVC, before the 6th IVC and after the 6th IVC for the cytokine kit test to see the trend of IVC effects on cytokines. Six patients were tested further for the effect of 50 g IVC infusion on C-reactive protein.
Results
Our database contained forty-five subjects who received IVC therapy (ranging from one to 100 treatments, with a median of nine treatments and an inter-quartile range, IQR, of five to eighteen treatments) and for whom CRP data were available before or after treatment (Table 1). Figure 1 shows CRP values before and after treatment.
Figure 1.

C-reactive protein levels after intravenous vitamin C treatments are compared to CRP levels before treatments. Data points (squares) under the diagonal line demonstrate a positive effect of treatment introducing CRP levels. Data points above the diagonal indicate CRP increases after treatment.
Most of the subjects, 76 ± 13% (95% confidence) showed a reduction in CRP values during IVC therapy. If only subjects who had initially elevated CRP (> 10 mg/L) levels are considered, 86 ± 13% (95% confidence) showed a reduction in CRP values during IVC therapy. This is also indicated by the fact that twenty-eight subjects had elevated CRP prior to therapy, but only 14 had elevated CRP levels after treatment. Among the twenty-eight subjects who started with elevated CRP levels, the median CPR reduction was 80% with an IQR of 39% to 94%.
Example of the changed levels of CRP with indicated periods of IVC treatments by 15 g and 50 g of IVC is shown in Figure 2.
Figure 2.
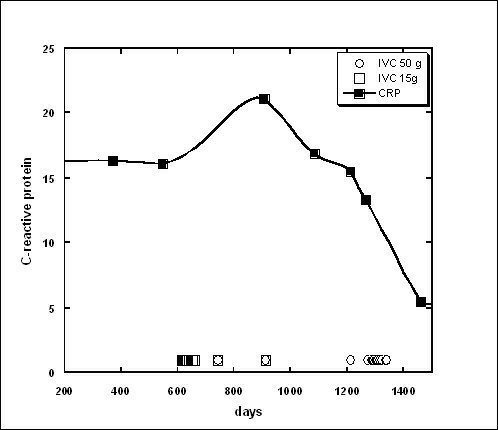
Tracking of CRP concentration (solid squares and curve) over time in a 67 year old subject with ductal carcinoma. The subject was given ten IVC treatments of 15 g (time of treatment indicated by open squares near the x axis) and eleven treatments of 50 g (indicated by open circles near the x axis). Treatments were typically given weekly. CRP concentration decreased from an initial value of 16 mg/L level to a final value of 5.5 mg/L after the last treatment.
Unfortunately, our data covers patients who varied in the number of treatments they received. For patients with prostate cancer, the percentage decrease in CRP concentration after treatment was higher when the patient had more frequent treatments. Median CRP reductions of 95% were attained in subjects who were treated frequently (intervals between IVC infusions being fewer than 10 days) while those treated less frequently. The negative response to IVC treatment with dosages 15 g-50 g was found in patients with gastric cancer (CRP was increased from 1.0 mg/L to 60 mg/L in 160 days, 15 IVC treatments). Progression was measured for three patients with pancreatic cancer. One patient had metastasis in liver and bones, and for this patient CRP was increased from 3.8 mg/L to 54 mg/L in 42 days (13 IVC treatments by dosages 25 g-50 g). For another patient without metastasis, the increase in CRP was from 2 mg/L to 25 mg/L in 400 days. The third patient with pancreatic cancer had 24 treatments during 90 days and CRP was increased from 15 mg/L to 89 mg/L. The patient with cholangiocarcinoma stage IV had 33 IVC treatments in 68 days and CRP was increased from 19 mg/L to 144 mg/L. Negative outcome was found for a patient with lung cancer with metastasis. This patient had 37 IVCs during 170 days and CRP was increased from 19 mg/L to 59 mg/L.
Progression was found for 2 patients with breast cancer (one with infiltrating tubular carcinoma, poorly differentiated duct cell carcinoma with second diagnosis pancreatic cancer in malignant stage 3, non-resectable), and another with breast cancer, category 5. For these patients the increase was mild from 3 mg/L and 8 mg/L to 10 mg/L and 13 mg/L frequently showed median CRP reductions of 64%.
The effects of IVC therapy on PSA tumor marker levels is shown in Figure 3. We had twenty patients for whom PSA levels were assessed before and after IVC therapy. Fifteen of these subjects, 75% with a 95% confidence interval of ± 19%, showed reduced PSA levels during IVC treatment. The examples of the positive response to IVC treatments for patients with prostate cancer are presented in Figures 4 and 5.
Figure 3.

Changes in PSA levels after IVC treatments are compared to PSA levels before treatments. Data points (squares) under the diagonal line demonstrate a positive effect of treatment in reducing PSA levels. Data points above the diagonal line indicate PSA increases after treatment.
Figure 4.

(A, B) – Tracking of PSA levels (circles, squares and curve) over time in two subjects with prostate cancer. Subjects were given IVC treatments at times indicated by the open squares. Subject A: initial Gleason score = 4; treatments typically given monthly at doses of either 25 g (34 treatments) or 50 g (38 treatments); At maximum PSA values (~ 60 ng/mL) treatments were given weekly. PSA levels decreased from initial values of 10–60 ng/mL to final values of 1–2.4 ng/mL. Subject B: initial Gleason score = 6–9; treatments typically given weekly at doses of either 7.5 g (2 treatments) or 25 g (40 treatments); PSA levels decreased from maximum values of 1500 ng/mL to a final value of 7 ng/mL.
Figure 5.

(A, B) - Tracking of PSA levels (solid circles and curve) over time in two subjects with prostate cancer. Subjects were given IVC treatments at times indicated by the open squares. Subject A: initial Gleason score =6; treatments typically given weekly or twice weekly at doses of 25 g; PSA levels decreased from initial values of 60 ng/ml to final values in the normal range. Subject B: initial Gleason score = 6 – 8; treatments typically given weekly or twice weekly at doses of either 25 g (140 treatments) or 50 g (7 treatments); PSA levels rose and fell during treatments, and then experienced a steep increase after treatment was discontinued.
We also had data from nineteen other subjects (mostly breast cancer patients, with a few lung, pancreatic, bladder, and colon cancer patients) for cancer markers CEA, CA27.29, or CA15.3. There was statistically no benefit here, with only 53 ± 22% (95% confidence) of subjects showing reduction in these tumor markers post treatments.
Table 2 summarizes the percentage of patients who saw improvement in various parameters. The fraction of subjects seeing reduced inflammation, as measured by decreases in CRP, was significantly above fifty percent in the prostate cancer group, the breast cancer group, and the entire study group. IVC seemed to have a beneficial effect in reducing PSA levels, but we could not see a significant effect in breast cancer subjects or other cancer markers.
Table 2.
Percentage of patients who saw reduction in CRP or tumour marker levels after IVC therapy “ ± ” indicates 95% confidence intervals, “†” indicates proportions significantly above 50%
| Cancer Type | N | % Improvement | |
|---|---|---|---|
|
CRP Reduction |
|
|
|
| Prostate |
14 |
79 ± 21 |
† |
| Breast |
10 |
80 ± 25 |
† |
| All patients |
45 |
76 ± 13 |
† |
|
Tumour Marker Reduction |
|
|
|
| Prostate |
18 |
77 ± 21 |
† |
| All PSA |
20 |
75 ± 19 |
† |
| Breast |
11 |
73 ± 26 |
|
| All CA/CEA |
19 |
53 ± 22 |
|
| All patients | 40 | 65 ± 15 | † |
In patients for whom both tumor marker and CRP data are available, we found an interesting correlation: patients who had reduced CRP levels tended to also have reduced tumor marker values. This is shown in Figure 6: linear regression shows a positive correlation with r2 = 0.62. Note that we did not observe a correlation between CRP concentrations and tumor marker values pre-treatments (r2 = 0.02)
Figure 6.
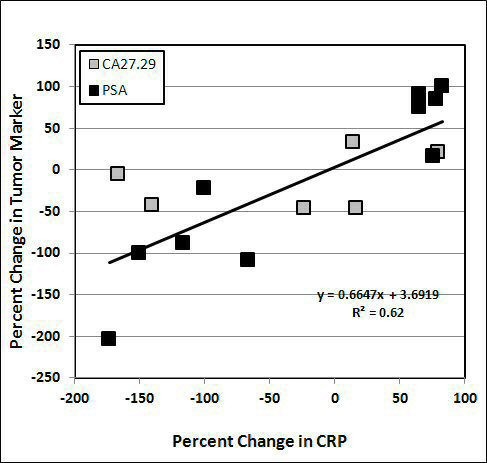
Correlation between changes in CRP levels and changes in tumour markers after IVC therapy. Linear regression indicates a positive correlation (R2 = 0.62) between changes in CRP and changes in the values of tumour markers PSA and CA 27.29. Data excluded several cases of aggressive tumours when the changes in CRP and tumour markers were higher than 300%.
To study the effects of IVC on inflammation in more detail, we measured serum cytokine levels in eleven subjects before and after IVC therapy. These subjects were given six treatments (at doses of 15 g for the first treatment, 25 g for the second treatment, and 50 g for the next four treatments). Plasma ascorbate levels in these subjects were measured to verify that sufficient concentrations were attained for cytokine effects. At the fifty gram treatments, plasma ascorbate concentrations immediately post infusion reached average values of roughly 18 mM. The effects of these treatments on serum concentrations of key pro-inflammatory cytokines are shown in Figure 7. This figure shows two endpoints for each cytokine. The first is the change in cytokine concentration at the point just before the patients are given their sixth IVC dose and after this single 50 g IVC. The second is the change in cytokine concentration (relative to pre-therapy value) at the point just after the patients are given all six IVC dosages. The first endpoint perhaps indicates sustained reductions in cytokine levels due to one treatment while the second indicates an effect of ascorbate after several dosing. After six treatments, patients had noticeably lower levels of IL-2, TNF-α, and eotaxin. All cytokine levels were reduced after the last IVC injection. For six of these subjects, CRP levels were measured before and after the last injection of IVC. In all six cases, CRP concentrations decreased, typically by ten percent, after the infusion.
Figure 7.

Percentage of reduction in serum cytokine levels after IVC therapy. Data present the changes in the level of proinflamatory cytokines after treatment by single dosage of 50 g IVC (pre6/post6) and after 6 treatments by IVC with dosages 15 g-50 g (pre1/post6). In all cancer patients initial level of vitamin C in plasma was low (0.9-1.2 mg/dl) and increased to the levels 200–300 mg/dL after last IVC. In all cases, proinflammatory cytokine levels decreased during IVC treatment.
Discussion
The vitamin C is used in complementary oncology, with intravenous administration being of particular interest since it allows for plasma ascorbate concentrations an order of magnitude beyond those attainable with oral administration [62,63]. Clinical studies suggest that this approach is feasible and potentially beneficial [47,48,64,65]. Several mechanisms of action for ascorbate efficacy against cancer have been proposed over the years, but to our knowledge its potential role in mediating inflammation has not been previously addressed. In the research described in this manuscript, we used CRP as a clinical inflammation marker. Twenty-eight out of forty-five subjects in our study had sharply elevated CRP levels (above 10 mg/L) prior to IVC therapy, suggesting that inflammation is a prevalent problem for cancer patients. This is especially important since other reports indicate that inflammation, particularly elevated CRP, is a marker of a poor prognosis [66]. In 76 ± 13% of our subjects, IVC therapy reduced CRP levels, with improvements being more prevalent (86 ± 13%) in the sub-population with pre-treatment CRP above 10 mg/L. Post-treatment, the number of subjects with these sharply elevated CRP levels was reduced from 28 to 14.
We found that decreases in CRP during treatment correlated with tumour marker decreases. This is consistent with other work [67] demonstrating that plasma CRP levels are well-correlated with serum PSA levels in prostate cancer patients. In the study [67] it was hypothesized that prostatic infection and inflammation may increase serum prostate-specific antigen levels. In addition, authors suggest that plasma CRP measurements may help to differentiate benign conditions from prostate cancer in patients with elevated serum PSA levels. Inflammation might be fundamental in prostate cancer and chronic inflammation may be a legitimate target for prostate cancer chemoprevention and treatment. Inflammatory processes also play a role in the development of histologic benign prostatic hyperplasia [68,69]. Acute and chronic inflammatory infiltrates are routinely found in prostate tissue specimens obtained from men with BPH, and greater levels of inflammation have been observed in larger prostates [70-73].
It is interesting in our study, therefore, that prostate cancer patients showed the most benefit (in terms of reduced PSA and CRP levels) from IVC therapy. A potential effect of IVC in reducing inflammation is also supported by our serum cytokine data, which demonstrate that levels of pro-inflammatory cytokines decrease during IVC therapy. IL-2, TNF-α, and eotaxin appeared to be chronically reduced in patients getting IVC therapy, while all six cytokines studied (IL-1α, IFN-γ, and IL-8, in addition to IL-2, TNF-α, and eotaxin) were acutely reduced after ascorbate infusions of 50 g. Average depression of IL-1 was 20% for six patients and the average decrease for eotaxin was 25%. Interleukin-1 is known to promote inflammatory processes and augment metastasis [74]. It is abundant at tumour sites, where it affects the process of carcinogenesis, tumour growth and invasiveness, and the patterns of tumour-host interactions [75,76]. IL-1 induces uPA expression and NF-kB activation. TNF-alpha, another key inflammatory cytokines, plays a central role in the tumour progression. Constitutive expression of the TNF-alpha from tumour microenvironment is a characteristic of many malignant tumours and its presence is often associated with poor prognosis [77-80]. Eotaxin-1 (CLL11) is a chemo-attractant and lymphocyte activator that has been shown to affect tumour cell growth [81-83]. Chemokine receptor expression in many cancers correlates with poor prognosis, and there is evidence that eotaxin induces angiogenesis and metastasis [84].
In summary, our analysis of data from cancer patients given IVC, along with our tests of cytokine levels, suggest that IVC may reduce inflammation in cancer patients, and that this reduction in inflammation is correlated with reductions in the tumour markers PSA.
The strength of the study is that we used the protocol established in our clinic for the treatment of cancer patients by IVC therapy, lacking at many clinics at least as adjuvant therapy in treatment of malignant disease, and demonstrated the effect of this treatment on the level of inflammation in cancer patients. We analysed the array of patients with various forms of malignant disease, although there were more patients with prostate malignancies than other forms of malignant disease.
The limitations of the study are that the measurements of parameters of inflammation and tumour markers were not very detailed and many patients did not have measurements of the inflammation cytokines during treatment.
Further research in this area and clinical studies of the efficacy of intravenous high dose vitamin C are warranted.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NAM, PRT, AR and JJC analyzed data, interpreted results of analysis and conceptualized the manuscript. All authors read and approved the final manuscript.
Contributor Information
Nina Mikirova, Email: nmikirova@riordanclinic.org.
Joseph Casciari, Email: casciari@sover.net.
Andrea Rogers, Email: arogers@riordanclinic.org.
Paul Taylor, Email: ptaylor@riordanclinic.org.
Acknowledgements
The study was supported by Allan P Markin and Flossie E West Memorial Trust.
References
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- Bartsch H, Nair J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch Surg. 2006;391:499–510. doi: 10.1007/s00423-006-0073-1. [DOI] [PubMed] [Google Scholar]
- de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6:24–37. doi: 10.1038/nrc1782. [DOI] [PubMed] [Google Scholar]
- Hussain SP, Harris CC. Inflammation and cancer: An ancient link with novel potentials. Int J Cancer. 2007;121:2373–2380. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- Lu H, Ouyang W, Huang C. Inflammation, a Key Event in Cancer Development. Mol Cancer Res. 2006;4:221–233. doi: 10.1158/1541-7786.MCR-05-0261. [DOI] [PubMed] [Google Scholar]
- Macarthur M, Hold GL, El-Omar EM. Inflammation and cancer. II. Role of chronic inflammation and cytokine polymorphisms in the pathogenesis of gastrointestinal malignancy. Am J Physiol Gastrointest Liver Physiol. 2004;286:G515–G520. doi: 10.1152/ajpgi.00475.2003. [DOI] [PubMed] [Google Scholar]
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D. et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Eng J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- Dunn G, Bruce A, Ikeda H, Old L, Schreiber R. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- Brigati C, Noonan DM, Albini A, Benelli R. Tumors and inflammatory infiltrates: friends or foes? Clin Exp Metastasis. 2002;19:247–258. doi: 10.1023/A:1015587423262. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res. 2009;15:425–430. doi: 10.1158/1078-0432.CCR-08-0149. [DOI] [PubMed] [Google Scholar]
- Roxburgh CS, McMillan DC. Role of systemic inflammatory response in predicting survival in patients with primary operable cancer. Future Oncol. 2010;6:149–163. doi: 10.2217/fon.09.136. Review. [DOI] [PubMed] [Google Scholar]
- Leek RD, Harris AL. Tumor-associated macrophages in breast cancer. J Mammary Gland Biol Neoplasia. 2002;7:177–189. doi: 10.1023/A:1020304003704. [DOI] [PubMed] [Google Scholar]
- Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor, CSF-1, in mammary gland development and cancer. J Mammary Gland Biol Neoplasia. 2002;7:147–162. doi: 10.1023/A:1020399802795. [DOI] [PubMed] [Google Scholar]
- Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6:130–140. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- McMillan DC. Systemic inflammation, nutritional status and survival in patients with cancer. Curr Opin Clin Nutr Metab Care. 2009;12:223–226. doi: 10.1097/MCO.0b013e32832a7902. Review. [DOI] [PubMed] [Google Scholar]
- Moore MM, Chua W, Charles KA, Clarke SJ. Inflammation and cancer: causes and consequences. Clin Pharmacol Ther. 2010;87:504–508. doi: 10.1038/clpt.2009.254. [DOI] [PubMed] [Google Scholar]
- Wallace JM. Nutritional and botanical modulation of the inflammatory cascade–eicosanoids, cyclooxygenases, and lipoxygenases–as an adjunct in cancer therapy. Integr Cancer Ther. 2002;1:7–37. doi: 10.1177/153473540200100102. [DOI] [PubMed] [Google Scholar]
- Crumley AB, McMillan DC, McKernan M, McDonald AC, Stuart RC. Evaluation of an inflammation-based prognostic score in patients with inoperable gastro-oesophageal cancer. Br J Cancer. 2006;13(94):637–641. doi: 10.1038/sj.bjc.6602998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Murri AM, Bartlett JM, Canney PA, Doughty JC, Wilson C, McMillan DC. Evaluation of an inflammation-based prognostic score (GPS) in patients with metastatic breast cancer. Br J Cancer. 2006;94:227–230. doi: 10.1038/sj.bjc.6602922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfield GM, Pepys MB. C-reactive protein and cardiovascular disease: new insights from an old molecule. QJM. 2003;96:793–807. doi: 10.1093/qjmed/hcg134. [DOI] [PubMed] [Google Scholar]
- Marsik C, Kazemi-Shirazi L, Schickbauer T. C-Reactive Protein and All-Cause Mortality in a Large Hospital-Based Cohort. Clin Chem. 2008;54:343–349. doi: 10.1373/clinchem.2007.091959. [DOI] [PubMed] [Google Scholar]
- Vigushin DM, Pepys MB, Hawkins PN. Metabolic and scintigraphic studies of radioiodinated human C-reactive protein in health and disease. J Clin Invest. 1993;91:1351–1357. doi: 10.1172/JCI116336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt B, Fuhrnrohr B, Muller R, Sheriff A. CRP and the disposal of dying cells: consequences for systemic lupus erythematosus and rheumatoid arthritis. Autoimmunity. 2007;40:295–298. doi: 10.1080/08916930701358925. [DOI] [PubMed] [Google Scholar]
- Mold C, Gewurz H, Du Clos TW. Regulation of complement activation by C-reactive protein. Immunopharmacology. 1999;42:23–30. doi: 10.1016/S0162-3109(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Marsik C, Kazemi-Shirazi L, Schickbauer T. et al. C-reactive protein and all-cause mortality in large hospital-based cohort. Clin Chem. 2008;54:343–349. doi: 10.1373/clinchem.2007.091959. [DOI] [PubMed] [Google Scholar]
- Mahmoud FA, Rivera NI. The role of C-reactive protein as a prognostic indicator in advanced cancer. Curr Oncol Rep. 2002;4:250–255. doi: 10.1007/s11912-002-0023-1. [DOI] [PubMed] [Google Scholar]
- McSorley MA, Alberg AJ, Allen DS, Allen NE, Brinton LA, Dorgan JF. et al. C-reactive protein concentrations and subsequent ovarian cancer risk. Obstet Gynecol. 2007;109:933–941. doi: 10.1097/01.AOG.0000257126.68803.03. [DOI] [PubMed] [Google Scholar]
- Otani T, Iwasaki M, Sasazuki S, Inoue M, Tsugane S. Plasma C-reactive protein and risk of colorectal cancer in a nested case–control study: Japan Public Health Center-based prospective study. Cancer Epidemiol Biomarkers Prev. 2006;15:690–695. doi: 10.1158/1055-9965.EPI-05-0708. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Ikeda Y, Korenaga D, Tanoue K, Hamatake M, Kawasaki K. et al. The impact of preoperative serum C-reactive protein on the prognosis of patients with hepatocellular carcinoma. Cancer. 2005;103:1856–1864. doi: 10.1002/cncr.20976. [DOI] [PubMed] [Google Scholar]
- Reichle A, Grassinger J, Bross K, Wilke J, Suedhoff T, Walter B, Wieland WF, Berand A, Andreesen R. C-reactive Protein in Patients with Metastatic Clear Cell Renal Carcinoma: An Important Biomarker for Tumor-associated Inflammation. Biomarker Insights. 2006;1:87–98. [PMC free article] [PubMed] [Google Scholar]
- Erlinger TP, Platz EA, Rifai N, Helzlsouer KJ. C-reactive protein and the risk of incident colorectal cancer. JAMA. 2004;291:585–590. doi: 10.1001/jama.291.5.585. [DOI] [PubMed] [Google Scholar]
- Shimada H, Nabeya Y, Okazumi S. et al. Elevation of preoperative serum C-reactive protein level is related to poor prognosis in esophageal squamous cell carcinoma. J Surg Oncol. 2003;83:248–252. doi: 10.1002/jso.10275. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Ikeda Y, Korenaga D. et al. The impact of preoperative serum C-reactive protein on the prognosis of patients with hepatocellular carcinoma. Cancer. 2005;103:1856–1864. doi: 10.1002/cncr.20976. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Koga S, Nishikido M. et al. Diagnostic values of acute phase reactants, basic fetoprotein, and immunosuppressive acidic protein for staging and survival in renal cell carcinoma. Urology. 2001;58:161–164. doi: 10.1016/S0090-4295(01)01165-7. [DOI] [PubMed] [Google Scholar]
- Kodama J, Miyagi Y, Seki N. et al. Serum C-reactive protein as a prognostic factor in patients with epithelial ovarian cancer. Eur J Obstet Gynecol Reprod Biol. 1999;82:107–110. doi: 10.1016/S0301-2115(98)00227-9. [DOI] [PubMed] [Google Scholar]
- Ford ES, Liu S, Mannino DM, Giles WH, Smith SJ. C-reactive protein concentration and concentrations of blood vitamins, carotenoids, and selenium among United States adults. Eur J Clin Nutr. 2003;57(9):1157–1163. doi: 10.1038/sj.ejcn.1601667. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JM. Free Radicals in Biology and Medicine. New York: Oxford University Press; 1999. [Google Scholar]
- Calder PC, Albers R, Antoine JM. et al. Inflammatory disease processes and interactions with nutrition. Br J Nutr. 2009;101(Suppl 1):S1–S45. doi: 10.1017/S0007114509377867. [DOI] [PubMed] [Google Scholar]
- Mayland CR, Bennett MI. et al. Vitamin C deficiency in cancer patients. Palliat Med. 2005;19:17–20. doi: 10.1191/0269216305pm970oa. [DOI] [PubMed] [Google Scholar]
- Härtel C, Strunk T, Bucsky P, Schultz C. Effects of vitamin C on intracytoplasmic cytokine production in human whole blood monocytes and lymphocytes. Cytokines. 2004;27(4–5):101–106. doi: 10.1016/j.cyto.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Andrew G, Bowie2, O’Neill LAJ. Vitamin C Inhibits NF-kB Activation by TNF Via the Activation of p38 Mitogen-Activated Protein Kinase. The J Immunol. 2000;165:7180–7188. doi: 10.4049/jimmunol.165.12.7180. [DOI] [PubMed] [Google Scholar]
- Munoz E, Blazquez MV, Ortiz C, Gomez-Diaz C, Navas P. Role of ascorbate in the activation of NF-kB by tumor necrosis factor-a in T-cells. Biochem J. 1997;325:23. doi: 10.1042/bj3250023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie AG, Carcamo JM, Pedraza A, Borquez-Qjeda O, Golde DW. Vitamin C suppresses TNFa-induced NFkB activation by inhibiting IkαB phosphorilation. Biochemistry. 2002;41:12995–13002. doi: 10.1021/bi0263210. [DOI] [PubMed] [Google Scholar]
- Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA. 1976;73:3685–3689. doi: 10.1073/pnas.73.10.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan HD, Casciari JJ, González MJ, Riordan NH, Miranda-Massari JR, Taylor P, Jackson JA. A pilot clinical study of continuous intravenous ascorbate in terminal cancer patients. P R Health Sci J. 2005;24(4):269–276. [PubMed] [Google Scholar]
- Riordan NH, Riordan HRD, Meng X, Li Y, Jackson JA. Intravenous ascorbate as a tumor cytotoxic chemotherapeutic agent. Medical hypothesis. 1995;44:213. doi: 10.1016/0306-9877(95)90137-x. [DOI] [PubMed] [Google Scholar]
- Casciari JJ, Riordan NH. et al. Cytotoxicity of ascorbate, lipoic acid, and other antioxidants in hollow fiber in vitro tumors. Br J Cancer. 2001;84:1544–1550. doi: 10.1054/bjoc.2001.1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikirova NA, Ichim TE, Riordan NH. Anti-angiogenic effect of high doses of ascorbic acid. J Transl Med. 2008;12:6:50. doi: 10.1186/1479-5876-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikirova NA, Casciari JJ, Riordan NH. Ascorbate inhibition of angiogenesis in aortic rings ex vivo and subcutaneous Matrigel plugs in vivo. Journal of Angiogenesis Research. 2010;2:2. doi: 10.1186/2040-2384-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciari JJ, Riordan HD, Mirranda-Massara JR, Gonzalez MJ. Effects of high dose ascorbate administration on L-10 tumor growth in guinea pigs. PR Health Sci. J. 2005;24:145–150. [PubMed] [Google Scholar]
- Dworacki G, Meidenbauer N, Kuss I, Hoffmann TK, Gooding W, Lotze M, Whiteside TL. Decreased zeta chain expression and apoptosis in CD3+ peripheral blood T lymphocytes of patients with melanoma. Clin Cancer Res. 2007;7:947s–957s. [PubMed] [Google Scholar]
- Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182:5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baniyash M. TCR zeta-chain downregulation: curtailing an excessive inflammatory immune response. Nat Rev Immunol. 2004;4(9):675–687. doi: 10.1038/nri1434. [DOI] [PubMed] [Google Scholar]
- Schmielau J, Nalesnik MA, Finn OJ. Suppressed T-cell receptor zeta chain expression and cytokine production in pancreatic cancer patients. Clin cancer research. 2001;17(3 Suppl):933s–939s. [PubMed] [Google Scholar]
- Nakagomi H, Peterson M, Magnusson I, Matsuda M, Mellstedt H, Taupin JL, Vivier E, Anderson P. Kiessling R: Decreased expression of the signal transducing z chains in tumor-infiltrating T-cells and NK cells of patients with colorectal carcinoma. Cancer Res. 1993;53:5610–5612. [PubMed] [Google Scholar]
- Gunji Y, Hori S, Aoe T, Asano T, Ochiai T, Isona K, Saito T. High frequency of cancer patients with abnormal assembly of the T cell receptor-CD3 complex in peripheral blood T lymphocytes. Jpn J Cancer Res. 1994;85:1189–1192. doi: 10.1111/j.1349-7006.1994.tb02927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar MP, Fisher CU, Enyedy EJ, Warke VG, Kumar A, Tsokos GC. Oxidative stress is involved in the heat stress-induced downregulation of TCR zeta chain expression and TCR/CD3-mediated [Ca(2+)](i) response in human T-lymphocytes. Cell Immunol. 2002;215:151–161. doi: 10.1016/S0008-8749(02)00006-0. [DOI] [PubMed] [Google Scholar]
- Duconge J, Miranda-Massari JR, Gonzalez MJ, Jackson JA, Warnock W, Riordan NH. Pharmacokinetics of vitamin C: insights into the oral and intravenous administration of ascorbate. P R Health Sci J. 2008;27(1):7–19. Review. [PubMed] [Google Scholar]
- Duconge J, Miranda-Massari JR, González MJ, Taylor PR, Riordan HD, Riordan NH, Casciari JJ, Alliston K. Vitamin C pharmacokinetics after continuous infusion in a patient with prostate cancer. Ann Pharmacother. 2007;41(6):1082–1083. doi: 10.1345/aph.1H654. [DOI] [PubMed] [Google Scholar]
- Chen Q, Espey MG, Krishna MC. et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102:13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, Rousseau C, Robitaille L, Miller WH. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol. 2008;19(11):1969–1974. doi: 10.1093/annonc/mdn377. Erratum in: Ann Oncol. 2008 19(12):2095. [DOI] [PubMed] [Google Scholar]
- Ohno S, Ohno Y, Suzuki N, Soma G, Inoue M. High-dose vitamin C (ascorbic acid) therapy in the treatment of patients with advanced cancer. Anticancer Res. 2009;29(3):809–815. Review. [PubMed] [Google Scholar]
- St.Sauver JL, Sarma AV, Jacobson DJ, McGree ME, Lieber MM, Girman CJ, Nehra A, Jacobsen SJ. Associations Between C-Reactive Protein and Benign Prostatic Hyperplasia/Lower Urinary Tract Symptom Outcomes in a Population-based Cohort. Am J Epidemiol. 2009;169:1281–1290. doi: 10.1093/aje/kwp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin ATL, Chen KK, Chung HJ, Chang SC. The Significance of Plasma C-reactive Protein in Patients With Elevated Serum Prostate-specific Antigen Levels. Urological Science. 2010;21(2):88–92. doi: 10.1016/S1879-5226(10)60018-8. [DOI] [Google Scholar]
- Eklund CM, Tammela TLJ, Schleutker J, Hurme M. C-reactive protein haplotype is associated with high PSA as a marker of metastatic prostate cancer but not with overall cancer risk. Br J Cancer. 2009;100:1846–1851. doi: 10.1038/sj.bjc.6605081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauver JL, Jacobsen SJ. Inflammatory mechanisms associated with prostatic inflammation and lower urinary tract symptoms. Curr Prostate Rep. 2008;6:67–73. doi: 10.1007/s11918-008-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohnen PW, Drach GW. Patterns of inflammation in prostatic hyperplasia: a histologic and bacteriologic study. J Urol. 1979;121(6):755–760. doi: 10.1016/s0022-5347(17)56980-3. [DOI] [PubMed] [Google Scholar]
- Di Silverio F, Gentile V, De Matteis A. et al. Distribution of inflammation, pre-malignant lesions, incidental carcinoma in histologically confirmed benign prostatic hyperplasia: a retrospective analysis. Eur Urol. 2003;43(2):164–175. doi: 10.1016/S0302-2838(02)00548-1. [DOI] [PubMed] [Google Scholar]
- Gerstenbluth RE, Seftel AD, MacLennan GT. et al. Distribution of chronic prostatitis in radical prostatectomy specimens with up-regulation of bcl-2 in areas of inflammation. J Urol. 2002;167(5):2267–2270. doi: 10.1016/S0022-5347(05)65140-3. [DOI] [PubMed] [Google Scholar]
- Roehrborn C, Kaplan SA, Noble WD. et al. The impact of acute or chronic inflammation in baseline biopsy on the risk of clinical progression of BPH: results of the MTOP study. J Urol. 2005;173:204. doi: 10.1097/01.ju.0000147849.80627.41. [DOI] [PubMed] [Google Scholar]
- Apte RN, Krelin Y, Song X. et al. Effects of microenvironment- and malignant cell-derived interleukin- 1 in carcinogenesis, tumour invasiveness and tumour-host interactions. Eur J Cancer. 2006;42:751–759. doi: 10.1016/j.ejca.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Voronov E, Shouval DS, Krelin Y. et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci USA. 2003;100:2645–2650. doi: 10.1073/pnas.0437939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhou BP. Inflammation. A driving force speeds cancer metastasis. Cell Cycle. 2009;8(20):3267–3273. doi: 10.4161/cc.8.20.9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen EM, Goldberg ID, Liu D. et al. Tumor necrosis factor stimulates epithelial tumor cell motility. Cancer Res. 1991;51:5315–5321. [PubMed] [Google Scholar]
- Arnott CH, Scott KA, Moore RJ. et al. Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene. 2004;23:1902–1910. doi: 10.1038/sj.onc.1207317. [DOI] [PubMed] [Google Scholar]
- Egberts JH, Cloosters V, Noack A. et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008;68:1443–1450. doi: 10.1158/0008-5472.CAN-07-5704. [DOI] [PubMed] [Google Scholar]
- Kulbe H, Thompson R, Wilson JL. et al. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting networkin epithelial ovarian cancer cells. Cancer Res. 2007;67:585–592. doi: 10.1158/0008-5472.CAN-06-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murooka TT, Ward SE, Fish EN. Chemokines and cancer. Cancer Treat Res. 2005;126:15–44. doi: 10.1007/0-387-24361-5_2. [DOI] [PubMed] [Google Scholar]
- Rollins BJ. Inflammatory chemokines in cancer growth and progression. Eur J Cancer. 2006;42:760–767. doi: 10.1016/j.ejca.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Zlotnik A. Chemokines and cancer. Int J Cancer. 2006;119:2026–2029. doi: 10.1002/ijc.22024. [DOI] [PubMed] [Google Scholar]
- Salcedo R, Young HA, Ponce ML. et al. Eotaxin (CCL11) induces in vivo angiogenic responses by human CCR3+ endothelial cells. J Immunol. 2001;166:7571–7578. doi: 10.4049/jimmunol.166.12.7571. [DOI] [PubMed] [Google Scholar]