

Abstract
A deimmunized bispecific anti-cancer agent was constructed to simultaneously target both the overexpressed EGF receptor on carcinomas and urokinase receptor (uPAR), that is found on the endothelial cells of the neovasculature within tumors. Flow cytometry assays were performed confirming that several different carcinoma lines overexpressed EGFR. Specifically, UMSCC-11B and NA, two head and neck squamous cell carcinomas were highly positive. These head and neck lines, as well as several other carcinoma lines, were then tested in leucine incorporation assays to determine the efficacy of dEGFATFKDEL. Human vein endothelial primary cells, which are largely EGFR negative but uPAR positive, were also tested to determine whether the ATF portion of the molecule that binds uPAR was effective. Both the carcinoma lines and the endothelial cells were inhibited at sub-nanomolar concentrations by dEGFATFKDEL. Furthermore, mouse studies were performed to determine whether this bispecific-targeted toxin was effective at inhibiting tumor growth in vivo. UMSCC-11B tumors were treated with either dEGFATFKDEL, irrelevant control CD19KDEL, or left untreated. The tumors receiving dEGFATFKDEL were significantly inhibited whereas the negative control and untreated tumors progressed. In a separate in vivo study involving another carcinoma line, MDA-MB-231, the effectiveness of dEGFATFKDEL was confirmed. No toxicity was seen at the doses used in either of these mouse studies. This bispecific agent is potently effective in a mouse model of head and neck squamous cell carcinoma.
Keywords: EGFR, uPAR, ATF, head and neck cancer, breast cancer, xenograft model, pseudomonas exotoxin, targeted toxins
Introduction
Head and Neck squamous cell carcinoma (HNSCC) is the sixth most common worldwide form of cancer (1). While many new therapeutics have been developed over the past 20 years to treat HNSCC, survival rates remain virtually unchanged. A major contributing problem to this in HNSCC and other carcinomas is chemo-resistance (1–4). Therefore, new drugs and new drug combinations are urgently needed to overcome the problem of chemoresistance.
Targeting over-expressed tumor markers is a common strategy in HNSCC. Perhaps the most well known of these over-expressed markers is epithelial growth factor receptor or EGFR (5–6). EGFR activates cellular pathways responsible for cancer proliferation, invasion, metastasis, angiogenesis, and resistance to apoptotic signals (7). Thus, new drugs are currently under development to target EGFR in many carcinomas, including HNSCC (8–10).
Urokinase-type plasminogen activator receptor (uPAR) is expressed in a number of solid tumors such as HNSCC. Importantly, uPAR is also expressed on tumor associated stromal cells particularly on the cells that make up the endothelial neovasculature. uPAR normally functions by catalytically converting its ligand pro-uPA into active uPA which causes proteolytic degradation of a number extracellular matrix proteins (11–12). However, uPAR overexpression in cancer corresponds with poor prognosis because of its pro-invasive, proliferative, and metastatic functions. Thus, uPAR has been an attractive target for anti-cancer therapies (13–15).
Targeted toxins (TT) are a type of biological drug consisting of a ligand that specifically recognizes a receptor expressed on cancer cells fused to a catalytic protein toxin that are extremely potent. The activity of the TT is dependent on the ligand binding its receptor and becoming internalized. Following internalization the toxin inhibits protein translation within the target cell causing apoptosis (16).
Recently we reported the activity of a deimmunized bispecific TT, dEGFATFKDEL, in glioblastoma (17–18). This bispecific fusion protein is made up of human EGF and the amino terminal fragment (ATF) of uPA linked to a deimmunized truncated form of Pseudomonas exotoxin A (PE38). This enables the simultaneous targeting of both the overexpressed EGFR on tumor cells and the uPAR on the tumors endothelial neovasculature via enzymatic ADP ribosylation of Elongation Factor-2 (19). Thus, targeted tumor cells die and the tumor neovasculature is also destroyed thereby starving the tumor. Importantly, this toxin is deimmunized which significantly reduces its ability to elicit neutralizing antibodies (17–18). Here we studied the efficacy of dEGFATFKDEL for the first time in an intratumoral therapy model of human HNSCC.
Methods
Construction and Purification of dEGFATFKDEL
For this study, dEGFATFKDEL was constructed and purified as described previously (17). Briefly, synthesis of dEGFATFKDEL was accomplished by fusion of the genes encoding human EGF and the amino terminal fragment (ATF) from uPA. These were then genetically linked to a deimmunized, truncated pseudomonas exotoxin 38. This fusion gene product was then spliced into the Novagen pET28c bacterial expression vector and transfected into competent cells. The bacteria were grown up and protein expression induced using isopropyl-b-D-thiogalactopyranoside (FisherBiotech). Inclusion bodies were isolated and the protein refolded, dialyzed, and purified over a fast protein liquid chromatography ion exchange column (Q sepharose Fast Flow, Sigma) as well as a size exclusion column (Superdex 200, Pharmacia). The resulting column fractions of the protein peak were pooled and purity was determined by SDS-PAGE stained with Commasie Brilliant Blue.
Cell Lines
The squamous cell carcinoma line UMSCC-11B was derived from a larynx tumor following chemotherapy treatment at the University of Michigan (20). Another squamous cell carcinoma, NA-SCC, was isolated from a tongue tumor (21). Dr. Frank Ondrey (University of Minnesota) obtained these lines from their originator Dr. Thomas E. Carey (Department of Otolaryngology-Head and Neck Surgery, University of Michigan) and supplied us with the cells. STR testing was done at John Hopkins University's Fragment Analysis Facility to authenticate the UMSCC-11B cell line. MDA-MB-231 cells were originally obtained from pleural effusion of stage III breast carcinoma patients. These cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Raji cells are a B cell line derived from a Burkitt's Lymphoma and were also obtained from ATCC. The colorectal carcinoma line, Caco-2 was also obtained from ATCC. Human umbilical vein endothelial cells (HUVEC) were obtained from Lonza (Lonza Group Ltd).
Flow cytometry
Flow cytometry was performed to determine the level of EGF and uPAR expression on the surface of the cell lines. A fluorescence-activated cell sorting (FACS) Caliber was used for all flow experiments at the University of Minnesota's Flow Cytometry Core Facility. Antibodies were labeled with fluorescein isothiocyanate (FITC) and data analysis was performed using FLOWJO. The percentage of positive cells was determined by gating control cells that were not incubated with antibody.
Bioassays
Protein synthesis assays measuring [3H]leucine incorporation were used to determine the effect of dEGFATFKDEL on the cell lines. Proliferation assays were also performed on occasion using [3H]thymidine incorporation. These assays have been described previously (17–18, 22). Briefly, cells are plated in 96-well flat-bottomed plates and allowed to adhere overnight in a 37°C and 5% CO2 incubator. The targeted toxins were added in triplicate at 10-fold serial dilutions and incubated for 48 hours. Wells are then pulsed with either [3H]leucine (protein synthesis assay) or [3H]thymidine (proliferation assay) with 1 μCi per well and allowed to incubate for another 24 hours. Plates are then frozen to detach the cells, harvested onto glass fiber filters, washed, dried, and counted using standard scintillation methods. [3H]leucine assays were done using Leucine-free medium. Data are reported as the percentage of control counts.
In vivo studies
The in vivo studies were done on male nu/nu mice purchased from the National Cancer Institute, Frederick Cancer Research and Development Center, Animal Production Area, and housed in microisolator cages in the pathogen-free AAALAC accredited facility under the care of the Department of Research Animal Resources at the University of Minnesota. All animal research protocols had been approved by the University of Minnesota Institutional Animal Care and Use Committee. For the flank tumor studies, UMSCC-11B and MDA-MB-231 cells were stably transfected with a vector containing a firefly luceriferase (luc) gene and a blastocidin resistant gene. This allowed the tumors to be imaged in real time to track the growth or regression of the tumors. For the UMSCC-11B/luc study, four million tumor cells were injected subcutaneously in the left flanks of the mice (5 mice/group). The mice were treated with 2ug's of dEGFATFKDEL intratumorally MTWTh starting on day 7 post-inoculation and ending on day 35. For the MDA-MB-231/luc study, 3 million cells were injected subcutaneously into the left flanks of the mice. Tumors were injected intratumorally with 3ug of dEGFATFKDEL starting on day 25 and 8 courses of treatment were given where one week of injections (MWF) constitute one course. Tumors for both studies were measured using digital calipers and the volume was calculated as the product of the width, length, and height. The mice were weighed regularly in order to monitor for treatment-related toxicities that typically cause a drop in body weight. Mice were imaged once a week in real time using Xenogen Ivis imaging system (Xenogen Corporation, Hopkington MA). Imaging was done as described previously (18, 22). For the imaging, mice were anesthetized using isoflurane gas and then injected with 100ul of a 30 mg/ml D-luciferin solution (Gold Biotechnology, St. Louis MO) 10 minutes prior to imaging. Mouse images represent a 5 minute exposure time and the units of luminescence is expressed as photons/s/cm2/sr. Prism 4 (Graphpad Software, San Diego CA) was used for all statistical analysis.
Results
Flow Cytometry Expression Analysis
In order to determine the level of expression of EGFR and uPAR present on the surface of the head and neck carcinoma cell line, UMSCC-11B, flow cytometry was performed. As shown in figure 1, UMSCC-11B cells are 100% EGFR positive, while only 1.51% uPAR positive. The cells were also probed with the FITC labeled negative control anti-CD19 antibody, HD37, which showed no reactivity against the carcinomas, but did against the B cell lymphoma line Raji. Table 1 shows another head and neck carcinoma, NA-SCC, also highly expresses EGFR at 99%. The breast carcinoma line MDA-MB-231 and colon carcinoma line Caco-2 also highly express EGFR at 91.2% and 98.8% respectively. Human umbilical vein endothelial cells, or HUVECs, were 61% uPAR positive, but had minimal EGFR expression (3.7%). Thus, the head and neck carcinoma lines (UMSCC-11B and NA), as well as the breast carcinoma line (MDA-MB-231) are excellent targets for an EGFR targeted therapy. The primary Human umbilical vein endothelial cells (HUVEC's) show that human endothelial cells, which are part of the neovasculature, are uPAR positive and will serve as targets for our bispecific drug.
Figure 1.

Flow cytometry dot plot of the head and neck carcinoma line UMSCC-11B probed with either FITC conjugated anti-EGFR antibody or anti-uPAR antibody. Cells were also probed with the negative control HD37-FITC (an anti-CD19 antibody).
Table 1.
Expression levels of EGFR, uPAR, and negative control CD19 was determined by flow cytometry. UMSCC-11B, NA-SCC, MDA-MB-231, and Caco-2 all highly express EGFR, but have negligible uPAR expression. HUVEC contained high levels of uPAR receptor as expected. The negative control antibody, HD-37FITC, bound very highly to Raji cells, a CD19 positive B-cell lymphoma line.
| EGFR | uPAR | CD19 | |
|---|---|---|---|
| UMSCC-11B | 100.0 | 1.5 | 0.7 |
| NA-SCC | 99.0 | 1.3 | 0.4 |
| MDA-MB-231 | 91.2 | 1.2 | 1.3 |
| HUVEC | 3.7 | 61.2 | 2.8 |
| Raji | 5.6 | 7.7 | 95.8 |
| Caco-2 | 98.8 | .4 | .8 |
In vitro Assays
Based on the results of the flow cytometry studies, [3H]leucine incorporation assays were performed to determine the level of selective protein synthesis inhibition of dEGFATFKDEL on these cell lines. As shown in figure 2a and b, dEGFATFKDEL selectively inhibited both of the head and neck cell lines, UMSCC-11B, and NA-SCC, with IC50 values at sub-nanomolar concentrations (1.38e−4nM and 4.37e−5nM respectively). MDA-MB-231, a breast carcinoma line, had a similar IC50 value of 5.01e−4nM as seen in figure 2c. Figure 2d shows a Leucine assay graph of a colorectal carcinoma cell line, Caco-2. While dEGFATFKDEL was not quite as potent (IC50=2.19e−3nM) against Caco-2, it was still selectively active. The HUVEC's were tested in figure 2e to determine whether cells that were uPAR positive, but EGFR negative could still be killed by dEGFATFKDEL. Indeed our bivalent dEGFATFKDEL has an IC50 concentration of 0.03nM and can therefore bind uPAR-expressing cells, internalize, and inhibit protein synthesis. Thus, both the EGF and ATF portions of our molecule are highly active. Raji cells (a B-cell lymphoma line) were tested to determine whether dEGFATFKDEL could still inhibit protein synthesis even though the line is EGFR and uPAR negative. Figure 2f shows that while a targeted toxin that binds CD22 and CD19 (d2219ARLKDEL) can inhibit protein synthesis, dEGFATFKDEL cannot. Thus, dEGFATFKDEL is a highly specific and bifunctional drug.
Figure 2.

The activity of dEGFATFKDEL against the carcinoma lines was determined by Leucine and Thymidine incorporation assays. UMSCC-11B, NA-SCC, MDA-MB-231, and Caco-2 Leucine incorporation assays showed that dEGFATFKDEL was selectively active while the negative control in each case had no effect (a–d). HUVEC primary cells were also tested and were selectively inhibited by dEGFATFKDEL as well. However, dEGFATFKDEL had no effect on the EGF and uPAR negative B cell lymphoma line Raji, while a positive control targeted toxin specific against CD22 and CD19 had activity (f).
In vivo Studies
To determine the activity of dEGFATFKDEL in vivo, a flank xenograft tumor model was used. For the study, UMSCC-11B cells that had been stably transfected with a gene expressing firefly luciferase (luc) were injected into the left flanks of nude mice. UMSCC-11B/luc tumors were treated intratumorally for 4 weeks starting on day 7 with dEGFATFKDEL. Figure 3a shows a clear inhibition of the tumor growth in treated verse untreated animals. There were 5 mice per group and the results are statistically significant. To show the selective efficacy of dEGFATFKDEL, a negative control group was treated with CD19KDEL (a targeted toxin specific for CD19). Even though the mice were treated aggressively with these targeted toxins, there were no signs of toxicity at the concentrations used based on the unchanging average mouse weights shown in figure 3b. Figure 4 shows luminescent images of representative mice from the 3 groups. The images of the dEGFATFKDEL treated group show a regression over time of the luminescent signal indicating a corresponding tumor regression. Unlike dEGFATFKDEL, CD19KDEL was not effective at causing tumor regression as the CD19KDEL treated mice showed no decrease in luciferase activity.
Figure 3.

Mouse tumor model of UMSCC-11B show dEGFATFKDEL is effective at inhibiting tumor growth. As seen in (a) tumor growth was inhibited in dEGFATFKDEL treated mice as compared with the no treatment mice. Student t test analysis was performed for dEGFATFKDEL treated tumors compared with untreated tumors on each day. The tumor size between groups became significantly different on day 23 (p<0.05). Body weight was measured in (b) and shows that treated mice did not have any significant weight loss due to drug toxicity throughout the study.
Figure 4.
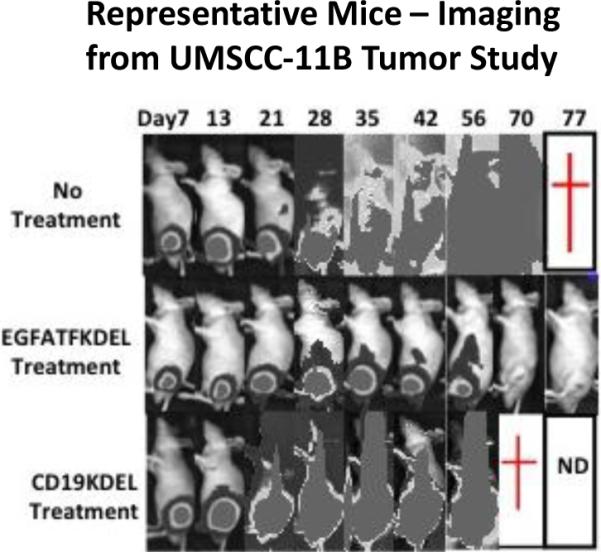
Real time images of representative mice from the UMSCC-11B study are shown. Mice treated with dEGFATFKDEL exhibited tumor reduction over time shown by the corresponding decrease in luminescence. Control treated and untreated mice, however, had continually increasing luminescent levels.
To confirm the effectiveness of dEGFATFKDEL in another carcinoma, MDA-MBA-231/luc tumors were treated intratumorally for 8 weeks. MDA-MB-231/luc tumors grow slower than UMSCC-11B/luc tumors, thus a modified does schedule of 3ug every MWF was used instead of the more aggressive MTWTh regimen. As seen in figure 5, untreated tumors grew normally, while dEGFATFKDEL treated tumors were completely inhibited. No drug toxicity was seen in the treated mice in the study either (data not shown). These results show that dEGFATFKDEL is effective at inhibiting tumor growth and causing tumor regression in both head and neck carcinoma as well as in a second model of carcinoma.
Figure 5.
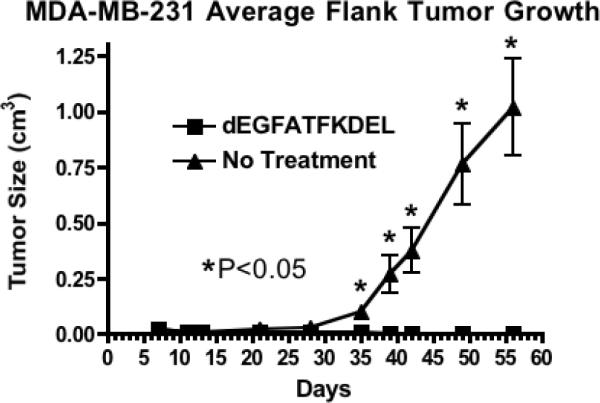
A second mouse carcinoma model was used to confirm the results from the UMSCC-11B study. MDA-MB-231 tumors treated with dEGFATFKDEL were inhibited, while untreated tumors grew normally over time. Student t tests were performed on every day. The dEGFATFKDEL treated tumors were statistically different (p<0.05) from the untreated controls on day 35, and on every subsequent day.
Discussion
This is the first report of a bispecific targeted toxin that simultaneously targets EGFR and the neovasculature in carcinomas. Tumors were chosen that were EGFR positive and uPAR negative. The anti-EGFR moiety of the hybrid protein bound EGFR on the tumors and the anti-uPAR moiety bound on endothelial cells as evidenced by the ability of the drug to kill HUVEC cells. These studies show that dEGFATFKDEL selectively inhibited protein synthesis in both head and neck and breast carcinoma tumors in vitro and caused regression of tumors in vivo. While many therapies have been developed to target either the tumor or the neovasculature, this one simultaneously targets both.
Targeted toxins (TT) are a type of biological drug consisting of ligands fused to a catalytic protein toxin that specifically bind receptors on cancer cells, internalize, and inhibit protein translation thereby causing apoptosis in the target cells. Historically, one main issue has curtailed the use of targeted toxins in the clinic. The development of antibodies against the toxin portion of the fusion protein has limited the efficacy of targeted toxins because multiple treatments are needed to penetrate solid tumors and cause regression (23). Pastan and Onda originally showed that mutations made in the immunogenic regions of PE38 allow for repeated treatments using dEGFATFKDEL without anti-toxin antibodies developing (24). We used these mutations to develop dEGFATFKDEL and showed that mice immunized with dEGFATFKDEL had a greatly reduced capacity for anti-toxin antibody response as compared to unmodified parental EGFATFKDEL (18). Indeed, these mutations have allowed us to create several non-immunogenic targeted toxins that are effective against a range of cancers (18, 25–27). Furthermore, TT act synergistically when added as an adjunct to classical chemotherapy and are more effective in combination than using either therapy separately (28–29).
Targeting the EGF receptor has shown to be effective in cancer therapy because it is over expressed on many human carcinomas. Several studies using targeted toxins specific for EGFR have been undertaken with promising results (30–32). Furthermore, the Food and Drug Administration (FDA) have approved no less than five EGFR inhibitors for the treatment of various types of cancer. They include Cetuximab (head and neck and colorectal cancer), gefitinib (non-small cell lung cancer), erlotinib (non-small cell lung and pancreatic cancer), panitumumab (colorectal cancer), and lapatinib (breast cancer) (10, 33). While these drugs have been an important step forward in the treatment of these cancers, advances have been incremental and there is still much room for improvement.
Angiogenesis is a critical step in tumorigenesis in which new blood vessels are formed. Without a sufficient blood supply, tumor growth is limited and the tumor regresses. Therefore, many anti-angiogenic agents, such as Thalidomide, Bevacizumab, sunitinib, among many others have been developed and approved by the FDA for the treatment of many types of cancer (34). Since uPAR plays a role in neoangiogenesis within tumors, and is also involved in tumor proliferation, tissue invasion, and metastasis, it has become an important target (13–15,35). Thus, we designed dEGFATFKDEL to kill uPAR positive cells in the tumor microenvironment. Interestingly, dEGFATFKDEL is a more potent inhibitor of protein synthesis than the equimolar combination of the two separate TT EGFKDEL and ATFKDEL (18). One explanation could be that the affinity of the bispecific may be greater than the monospecifics or that the bispecific somehow stabilizes the binding of the scFv's to their specific receptors. In vivo, the bispecific would also have two different targets in the tumor microenvironment it could bind, thus providing an increased opportunity for the necessary binding and internalization to take place while monospecifics may be able to diffuse out of the tumor more readily. While the mouse model used here does not provide in vivo proof of vascular effects, the in vitro studies clearly show that dEGFATFKDEL is a potent inhibitor of uPAR positive endothelial cells as shown by the HUVEC assays.
Development of bispecific-targeted therapeutics for cancer treatment is on the rise. Many of these function by either retargeting effector molecules or effector cells (36). One of these is a bispecific antibody developed recently that focuses on inhibiting angiogenesis by targeting PDGFR and VEGFR-A. This bispecific was effective at inhibiting A673 rhabdomyoscarcoma tumors (a typical anti-angiogenesis model) in vivo (37). However, only targeting the angiogenesis pathway has only been mildly effective in the clinic (38). Furthermore, the immune system in many cancer patients is compromised either by the cancer itself or by many chemotherapy treatments (39–40). Bispecific TT, however, do not require immune effector cells to mediate cell death. The catalytic protein toxin enables these molecules to specifically target the tumor cells and tumor microenvironment.
Here we show for the first time that dEGFATFKDEL targets both EGFR positive tumor cells as well as the uPAR positive neovasculature cells in head and neck carcinoma and confirmed its activity in a second carcinoma model as well. This two-pronged approach enables dEGFATFKDEL to potently inhibit protein synthesis in vitro and tumor growth and progression in vivo. Its efficacy, potency, selectivity, and mechanism of action give it an advantage over other biological drugs. Thus, dEGFATFKDEL warrants further study and characterization for the clinical treatment of carcinoma.
Acknowledgments
Financial Support: This work was supported in part by the US Public Health Service Grants R01-CA36725, RO1 HL077923 awarded by the NCI and the NIAID, DHHS, the Randy Shaver Foundation, the Lion's Children's Cancer Fund, and the William Lawrence and Blanche Hughes Fund.
Abbreviations
- HNSCC
- EGFR
- uPAR
- ATF
- TT
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
References
- 1.Bianchini C, Ciorba A, Pelucchi S, Piva R, Pastore A. Targeted therapy in head and neck cancer. Tumori. 2011 Mar-Apr;97(2):137–41. doi: 10.1177/030089161109700201. [DOI] [PubMed] [Google Scholar]
- 2.Yadav A, Kumar B, Teknos TN, Kumar P. Sorafenib enhances the anti-tumor effects of chemo-radiation treatment by down-regulating ERCC-1 and XRCC-1 DNA repair proteins. Mol Cancer Ther. 2011 Jul;10(7):1241–51. doi: 10.1158/1535-7163.MCT-11-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitra D, Malkoski SP, Wang XJ. Cancer Stem Cells in Head and Neck Cancer. Cancers. 2011;3:415–427. doi: 10.3390/cancers3010415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of Resistance to Systemic Therapy in Patients with Breast Cancer. Adv Exp Med Biol. 2007;608:1–22. doi: 10.1007/978-0-387-74039-3_1. [DOI] [PubMed] [Google Scholar]
- 5.Atalay G, Cardoso F, Awada A, Piccart MJ. Novel therapeutic strategies targeting the epidermal growth factor receptor (EGFR) family and its downstream effectors in breast cancer. Annals of Oncology. 2003;14:1346–1363. doi: 10.1093/annonc/mdg365. [DOI] [PubMed] [Google Scholar]
- 6.Uribe P, Gonzalez S. Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: Molecular bases for EGFR-targeted therapy. Pathol Res Pract. 2011 Jun 15;207(6):337–42. doi: 10.1016/j.prp.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 8.Machiels JP, Schmitz S. Molecular-targeted therapy of head and neck squamous cell carcinoma: beyond cetuximab-based therapy. Curr Opin Oncol. 2011 May;23(3):241–8. doi: 10.1097/CCO.0b013e328344f581. [DOI] [PubMed] [Google Scholar]
- 9.Del Campo JM, Hitt R, Sebastian P, Carracedo C, Lokanatha D, Bourhis J, Temam S, et al. Effects of lapatinib monotherapy: results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer. 2011 Aug 23;105(5):618–27. doi: 10.1038/bjc.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Modjtahedi H, Essapen S. Epidermal growth factor receptor inhibitors in cancer treatment: advances, challenges and opportunities. Anticancer Drugs. 2009 Nov;20(10):851–5. doi: 10.1097/CAD.0b013e3283330590. [DOI] [PubMed] [Google Scholar]
- 11.Prager GW, Breuss JM, Steurer S, Mihaly J, Binder BR. Vascular endothelial growth factor (VEGF) induces rapid prourokinase (pro-uPA) activation on the surface of endothelial cells. Blood. 2004;103:955–962. doi: 10.1182/blood-2003-07-2214. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Lin L, Huai Q, Huang M. Challenges for drug discovery - a case study of urokinase receptor inhibition. Comb Chem High Throughput Screen. 2009 Dec;12(10):961–7. doi: 10.2174/138620709789824727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobsen B, Ploug M. The Urokinase Receptor and its Structural Homologue C4.4A in Human Cancer: Expression, Prognosis and Pharmacological Inhibition. Curr Med Chem. 2008;15(25):2559–73. doi: 10.2174/092986708785909012. [DOI] [PubMed] [Google Scholar]
- 14.Reuning U, Sperl S, Kopitz C, Kessler H, Krüger A, Schmitt M, Magdolen V. Urokinase-type plasminogen activator (uPA) and its receptor (uPAR): development of antagonists of uPA/uPAR interaction and their effects in vitro and in vivo. Curr Pharm Des. 2003;9(19):1529–43. doi: 10.2174/1381612033454612. [DOI] [PubMed] [Google Scholar]
- 15.Ulisse S, Baldini E, Sorrenti S, D'Armiento M. The urokinase plasminogen activator system: a target for anti-cancer therapy. Curr Cancer Drug Targets. 2009 Feb;9(1):32–71. doi: 10.2174/156800909787314002. [DOI] [PubMed] [Google Scholar]
- 16.Fitzgerald D, Pastan I. Targeted toxin therapy for the treatment of 672 cancer. J Natl Cancer Inst. 1989;81:1455–63. doi: 10.1093/jnci/81.19.1455. [DOI] [PubMed] [Google Scholar]
- 17.Tsai AK, Oh S, Chen H, Shu Y, Ohlfest JR, Vallera DA. A novel bispecific ligand-directed toxin designed to simultaneously target EGFR on human glioblastoma cells and uPAR on tumor neovasculature. J Neurooncol. 2011;103:255–66. doi: 10.1007/s11060-010-0392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh S, Tsai AK, Ohlfest JR, Panoskaltsis-Mortari A, Vallera DA. Evaluation of a bispecific biological drug designed to simultaneously target glioblastoma and its neovasculature in the brain. J Neurosurg. 2011 Jun;114(6):1662–71. doi: 10.3171/2010.11.JNS101214. [DOI] [PubMed] [Google Scholar]
- 19.Castro MG, Candolfi M, Kroeger K, King GD, Curtin JF, Yagiz K, Mineharu Y. Gene therapy and targeted toxins for glioma. Curr Gene Ther. 2011 Jun;11(3):155–80. doi: 10.2174/156652311795684722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Worsham MJ, Chen KM, Meduri V, Nygren AO, Errami A, Schouten JP, et al. Epigenetic events of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2006;132:668–77. doi: 10.1001/archotol.132.6.668. [DOI] [PubMed] [Google Scholar]
- 21.Abu-Ali S, Fotovati A, Shirasuna K. Tyrosine-kinase inhibition results in EGFR clustering at focal adhesions and consequent exocytosis in uPAR down-regulated cells of head and neck cancers. Mol Cancer. 2008;7:47. doi: 10.1186/1476-4598-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stish BJ, Oh S, Chen H, Dudek AZ, Kratzke RA, Vallera DA. Design and modification of EGF4KDEL 7mut, a novel bispecific ligand-directed toxin, with decreased immunogenicity and potent anti-mesothelioma activity. Br J Cancer. 2009;101:1114–23. doi: 10.1038/sj.bjc.6605297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJ, Wilson WH, Pastan I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009 Jun 20;27(18):2983–90. doi: 10.1200/JCO.2008.20.2630. Epub 2009 May 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B-cell epitopes. Proc Natl Acad Sci USA. 2008;105:11311–6. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stish BJ, Oh S, Chen H, Dudek AZ, Kratzke RA, Vallera DA. Design and modification of EGF4KDEL 7mut, a novel bispecific ligand-directed toxin, with decreased immunogenicity and potent anti-mesothelioma activity. Br J Cancer. 2009;101:1114–23. doi: 10.1038/sj.bjc.6605297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vallera DA, Oh S, Chen H, Shu Y, Frankel AE. Bioengineering a unique deimmunized bispecific targeted toxin that simultaneously recognizes human CD22 and CD19 receptors in a mouse model of B-cell metastases. Mol Cancer Ther. 2010;9:1872–83. doi: 10.1158/1535-7163.MCT-10-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waldron NN, Kaufman DS, Oh S, Inde Z, Hexum MK, Ohlfest JR, Vallera DA. Targeting tumor-initiating cancer cells with dCD133KDEL shows impressive tumor reductions in a xenotransplant model of human head and neck cancer. Mol Cancer Ther. 2011 Oct;10(10):1829–38. doi: 10.1158/1535-7163.MCT-11-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. Anti–Mesothelin Immunotoxin SS1P in Combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 2007 Dec 1;13(23):7166–71. doi: 10.1158/1078-0432.CCR-07-1592. [DOI] [PubMed] [Google Scholar]
- 29.Pearson JW, Sivam G, Manger R, Wiltrout RH, Morgan AC, Jr, Longo DL. Enhanced therapeutic efficacy of an immunotoxin in combination with chemotherapy against an intraperitoneal human tumor xenograft in athymic mice. Cancer Res. 1989 Sep 15;49(18):4990–5. [PubMed] [Google Scholar]
- 30.Engebraaten O, Hjortland GO, Juell S, Hirschberg H, Fodstad O. Intratumoral immunotoxin treatment of human malignant brain tumors in immunodeficient animals. Int J Cancer. 2002;97:846–852. doi: 10.1002/ijc.10137. [DOI] [PubMed] [Google Scholar]
- 31.Liu TF, Hall PD, Cohen KA, Willingham MC, Cai J, Thorburn A, et al. Interstitial diphtheria toxin-epidermal growth factor fusion protein therapy produces regressions of subcutaneous human glioblastoma multiforme tumors in athymic nude mice. Clin Cancer Res. 2005;11:329–334. [PubMed] [Google Scholar]
- 32.Liu TF, Tatter SB, Willingham MC, Yang M, Hu JJ, Frankel AE. Growth factor receptor expression varies among high grade gliomas and normal brain: epidermal growth factor receptor has excellent properties for interstitial fusion protein therapy. Mol Cancer Ther. 2003;2:783–787. [PubMed] [Google Scholar]
- 33.Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008 Mar 13;358(11):1160–74. doi: 10.1056/NEJMra0707704. [DOI] [PubMed] [Google Scholar]
- 34.Fan F, Schimming A, Jaeger D, Podar K. Targeting the tumor microenvironment: focus on angiogenesis. J Oncol. 2012;2012:281261. doi: 10.1155/2012/281261. Epub 2011 Aug 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Q, Xu Q, Dong X, Cao L, Huang X, Hu Q, Hua ZC. A hybrid protein comprising ATF domain of pro-UK and VAS, an angiogenesis inhibitor, is a potent candidate for targeted cancer therapy. Int J Cancer. 2008 Aug 15;123(4):942–50. doi: 10.1002/ijc.23537. [DOI] [PubMed] [Google Scholar]
- 36.Hollander N. Bispecific antibodies for cancer therapy. Immunotherapy. 2009 Mar;1(2):211–22. doi: 10.2217/1750743X.1.2.211. [DOI] [PubMed] [Google Scholar]
- 37.Mabry R, Gilbertson DG, Frank A, Vu T, Ardourel D, Ostrander C, Stevens B, Julien S, et al. A dual-targeting PDGFRbeta/VEGF-A molecule assembled from stable antibody fragments demonstrates anti-angiogenic activity in vitro and in vivo. MAbs. 2010 Jan-Feb;2(1):20–34. doi: 10.4161/mabs.2.1.10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerbel RS. Reappraising antiangiogenic therapy for breast cancer. Breast. 2011 Oct;20(Suppl 3):S56–60. doi: 10.1016/S0960-9776(11)70295-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rasmussen L, Arvin A. Chemotherapy-induced immunosuppression. Environ Health Perspect. 1982 Feb;43:21–5. doi: 10.1289/ehp.824321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salavoura K, Kolialexi A, Tsangaris G, Mavrou A. Development of cancer in patients with primary immunodeficiencies. Anticancer Res. 2008 Mar-Apr;28(2B):1263–9. [PubMed] [Google Scholar]