

Abstract
Age-related macular degeneration (AMD) is an outer retinal disease that involves aging and immune dysfunction. In the aging retina, microglia aggregate in the outer retina and acquire intracellular autofluorescent lipofuscin deposits. In this study, we investigated whether accumulation of A2E, a key bisretinoid constituent of ocular lipofuscin, alters the physiology of retinal microglia in pathologically relevant ways. Our findings show that sublethal accumulations of intracellular A2E in cultured retinal microglia increased microglial activation and decreased microglial neuroprotection of photoreceptors. Increased A2E accumulation also lowered microglial expression of chemokine receptors and suppressed microglial chemotaxis, suggesting that lipofuscin accumulation may potentiate subretinal microglial accumulation. Significantly, A2E accumulation altered microglial complement regulation by increasing CFB and decreasing CFH expression, favoring increased complement activation and deposition in the outer retina. Taken together, our findings highlight the role of microglia in the local control of complement activation in the retina and present the age-related accumulation of ocular lipofuscin in subretinal microglia as a cellular mechanism capable of driving outer retinal immune dysregulation in AMD pathogenesis.
Keywords: microglia, retina, lipofuscin, A2E, aging, age-related macular degeneration, complement, activation, neuroprotection, photoreceptors, chemokine
1. Introduction
Age-related macular degeneration (AMD), a neurodegenerative retinal disease of the elderly (Jager et al. 2008), is the leading cause of legal blindness in persons 60 years and older in the developed world (Congdon et al. 2004) and represents a major challenge to healthy vision worldwide. Central vision is lost in AMD as a result of neurodegenerative atrophy affecting photoreceptors and retinal pigment epithelial (RPE) cells associated with characteristic extracellular lesions between these cells and their blood supply. Although therapies targeting vascular endothelial growth factor (VEGF) have demonstrated efficacy in limiting the pathological neovascular growth in a subset of cases, comprehensive prevention and treatment measures for AMD remain elusive. In part, a lack of knowledge of AMD pathogenesis have impeded the development of new therapies (Donoso et al. 2010; Zarbin & Rosenfeld 2010).
In AMD, two particular factors, chronic neuroinflammation and aging, appear central in disease risk and progression (Hyman & Neborsky 2002; Donoso et al. 2006; Kanda et al. 2008; Augustin & Kirchhof 2009). For the first, the involvement of chronic neuroinflammation has been underscored by genetic analyses that have implicated individual molecules of the immune system, particularly those in the complement system, as conferring risk for AMD (Swaroop et al. 2009; Donoso et al. 2010; Gehrs et al. 2010). However, how these immune molecules participate in pathogenic cellular mechanisms in AMD is unclear. For the second, advanced age constitutes the largest and most significant risk factor for AMD (Friedman et al. 2004), indicating the importance of senescent effects on relevant cellular mechanisms. Taken together, these findings indicate that aging effects within immune cells that are influential in the outer retina, the tissue locus where AMD develops, are likely to be significant in disease progression.
Microglia, the primary resident immune cell of the retina and a critical regulator in its immune environment (Boehm et al. 2011), have been implicated in the etiology of AMD (Xu et al. 2009; Karlstetter et al. 2010). In the mouse retina, while retinal microglia in young healthy animals are confined to the inner retinal layers and are spatially removed from photoreceptors and retinal pigment epithelial (RPE) cells (Chen et al. 2002), they undergo a remarkable translocation into the outer retina in aged animals to accumulate in the subretinal space (Xu et al. 2008; Damani et al. 2011). Indeed, similar accumulations of subretinal microglia have also been characterized in human AMD (Penfold et al. 2001; Gupta et al. 2003), as well as in AMD-relevant retinopathies in mouse models (Combadiere et al. 2007; Tuo et al. 2007; Luhmann et al. 2009). These findings imply that these accumulations of senescent microglia may dysregulate immune interactions and drive AMD progression (Ma et al. 2009), linking the cellular mechanisms of aging and chronic neuroinflammation in the outer retina.
The aging phenotype of CNS microglia, the mechanisms underlying microglia aging, and microglial contributions to age-related CNS neurodegenerative diseases are topics of current investigation (von Bernhardi et al. 2010). However, these cellular changes may differ between different CNS compartments and the related pathologies induced in each region may vary (Olah et al. 2011). In the mouse retina, the senescent microglial phenotype is characterized not only by their outer retinal translocation but also by the development of intracellular lipofuscin granules whose spectral characteristics are similar to those found in aged RPE cells (Xu et al. 2008). In RPE cells, the age-related accumulation of ocular lipofuscin, and a constituent bisretinoid in particular, A2E (Lamb & Simon 2004), has been related to deleterious effects observed in in vitro studies. These include membrane disruption (Sparrow et al. 1999), lysosomal dysfunction (Holz et al. 1999), loss of antioxidant activity (Shamsi & Boulton 2001), phototoxicity (Schutt et al. 2000; Sparrow et al. 2000; Sparrow & Cai 2001), and immune dysregulation through complement activation (Zhou et al. 2006; Zhou et al. 2009). Whether lipofuscin accumulation in aged subretinal microglia induces parallel alterations that may contribute to age-related retinal pathology has not previously been investigated.
In this study, we address the hypothesis that A2E accumulation in retinal microglia induces significant alterations in microglial physiology that can contribute to immune dysregulation relevant to AMD progression. We have employed an in vitro model of A2E-loaded cultured retinal microglia cells and assessed their activation status, chemotaxis, neuroprotective properties, and immune regulation as a function of their A2E accumulation. Additionally, by using an in vivo cell transplantation model, we assessed the effect of A2E-laden microglia in the subretinal space on complement regulation and photoreceptor apoptosis. These studies enabled us to explore the significance of increasing ocular lipofuscin in retinal microglia and to investigate the mechanisms underlying how aged microglia, altered in their retinal location and physiology in senescence, may contribute to cellular mechanisms driving AMD.
2. Methods
2.1. Experimental animals
Wild type C57BL/6J and heterozygous CX3CR1+/GFP transgenic mice (created by breeding CX3CR1GFP/GFP mice (Jung et al. 2000) to C57BL/6J mice) were used. Animals were purchased from the Jackson Laboratory (Bar Harbor, ME) and from the National Institute of Aging-Aged Rodent Colony (Bethesda, MD). Mice were bred and housed in a National Institutes of Health animal facility in a temperature and light controlled environment with a 12-hour day-light cycle. Animal procedures were conducted according to protocols approved by the local Institutional Animal Care and Use Committee and in concordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
2.2 Human eye tissue
Eyes from two donors with atrophic form of advanced AMD (geographic atrophy) (aged 82 and 85 years) and four donors without history and signs of AMD (aged 45-77 years) were obtained from eye banks through a tissue resource center (National Disease Research Initiative, NDRI, Philadelphia, PA). The AMD diagnoses in these eyes were derived from donor records and from gross examinations of the fundus prior to tissue sectioning. The eyes were fixed in 4% paraformaldehyde, and the maculas dissected. A portion of the macular tissue was processed by cryosectioning (8-10 μm-thick sections). In other macular areas, the retina was dissected free of the choroid-RPE complex and flat-mounted with the photoreceptor layer uppermost. Eye tissue was collected under applicable regulations and guidelines with proper consent, protection of human subjects, and donor confidentiality.
2.2. Synthesis of A2E
A2E was synthesized essentially as described previously (Parish et al. 1998). Briefly, a mixture of all-trans-retinal (100 mM), ethanolamine (50 mM) and acetic acid (50 mM) in methanol was incubated in a sealed vial in the dark at room temperature for 2 days. The reaction mixture was then separated by high-performance liquid chromatography (HPLC) using a reverse phase C18 column (Advantage, 4.6 × 250 mm, 60Å pore size, 5 μm particle diameter; Thomas Instrument Co., Swedesboro, NJ) with a gradient of methanol in water (85-96% methanol + 0.1% TFA, 1 ml/min, Gilson 715 HPLC system). Eluants were monitored at 430 nm and 320 nm; the fractions containing A2E were collected, pooled and dried under nitrogen. The residue was reconstituted in methanol; the identity and purity of the solute was confirmed by mass spectrometry and spectrophotometry. The concentration of A2E was measured using a molar extinction coefficient of 36,900.
2.3. Cell culture
Retinal microglia were isolated from young postnatal day (P) 12 to 15 C57BL/6J and heterozygous CX3CR1+/GFP transgenic mice as previously described (Ma et al. 2009). Briefly, retinal cells were dissociated by digestion in 2 % dispase or papain, followed by trituration, before transfer into 75cm2 flasks containing DMEM/F12 media with 10% fetal bovine serum (FBS) (Gibco, Carlsbad, CA) and 1× MEM non-essential amino acids solution (Sigma, St Louis, MO). The resulting mixed cell cultures that adhered to the bottom of the flasks were allowed to grow to confluence. Microglial cells were detached from the adherent cell layer by shaking, collected, and transferred to a new 75cm2 flask. The culture medium was changed after one day with fresh culture medium containing GM-CSF (1ng/ml, eBioscience, Cat#14-8331, San Diego, CA). When the microglial cells attained 60-70% confluence, they were scraped off and harvested for subsequent experiments.
Cultures containing the photoreceptor cell line 661W (gift from Dr Xuri Li, National Eye Institute) and the RPE cell line, ARPE-19 (gift from Dr Chi-Chao Chan, National Eye Institute) were prepared as previously described (Tuohy et al. 2002; Ding et al. 2009).
2.4. Cellular accumulation of A2E
Retinal microglia were loaded with A2E using the following protocol: A suspension of retinal microglia was seeded into wells of a 6-well plate (1 ml per well, concentration 1×105 cell/ml) and allowed to attach to the bottom over 24 hours. A2E was added to culture media (to a final concentration of 3-24 μM) of cultured microglia for varying durations of 3 to 24 hours to allow cellular uptake of A2E. After A2E loading, the cells were washed twice with fresh A2E-free media, and then either harvested for analysis or allowed to condition 1 ml of DMEM:F12 medium containing 5% heat inactivated FBS for another 24 hours to generate microglia-conditioned media (CM-RMG).
2.5. Subretinal transplantation of microglia
Experimental animals were anesthetized by the intraperitoneal injection of ketamine (90mg/kg) and xylazine (8mg/kg). As previously described (Zhao et al. 2007; Ma et al. 2009), the temporal sclera between the limbus and equator was first exposed and the subretinal space accessed through a small scleral incision using a sharp 31-gauge needle. A suspension containing 1×108/ml of cultured GFP-positive retinal microglia isolated from CX3CR1+/GFP mice and previously loaded with A2E (6 μM for 6 hours) were mixed with growth factor-depletedMatrigel (BD Biosciences, Bedford, MA) at a 1:1 ratio. Non-A2E-loaded microglia were similarly prepared as a control. A 1.5μl volume of the microglia suspension was slowly injected into the subretinal space using a blunt needle attached to a Hamilton micro-syringe. One eye of each experimental animal received subretinal transplantation of A2E-loaded microglia while the contralateral eye received non-loaded control microglia. Animals were euthanized by CO2 inhalation three days after injection and their ocular tissues harvested and analyzed.
2.6. Immunofluorescence labeling of cultured cells and ocular tissues
Retinal and sclerochoroidal flat-mounts were prepared as described previously (Damani et al. 2011). After fixation in 4% paraformaldehyde in phosphate buffered saline (PBS) for 4 hours, flat-mounted tissue was washed three times with PBS for 10 minutes each, transferred into PBS containing 1% Triton-X100 (Sigma, St. Louis, MO) for 1hr at room temperature, and then incubated in blocking buffer (Roche, Indianopolis, IN) for 30 min on a shaker. Sclerochoroidal flat-mounts containing a monolayer of RPE cells uppermost were immunostained with the antibodies to the following antigens: CD11b (AbD Serotec, Oxford, UK, Cat#MCA711, clone#5c6, 1:100) to label microglia cells, phalloidin (Alexa-488 or -633-conjugated, Invitrogen, Carlsbad, CA, #A22284, 1:100) or ZO-1 (Invitrogen, Cat#40-2200, 1:200) to label RPE cells, C3b/iC3b/C3c (mAb 2/11, #HM1078, Hycult, Uden, The Netherlands, 1:200) to reveal complement deposition at the level of the RPE. Retinal flat-mount explants were immunostained with primary antibodies to S-opsin (Cat# AB5405, Millipore, Billerica, MA, 1:300) to reveal photoreceptor outer segments. Slide-mounted sections after immunohistochemical staining were imaged using confocal microscopy (FluoView 1000, Olympus Corp., Tokyo, Japan).
Cultured cells were similarly fixed and permeabilized, and incubated overnight with primary antibodies to the following antigens: retinal microglia were immunolabeled for calcium binding adaptor molecule-1 (Iba1, Wako, Richmond, VA; 1:1500), CFH (Santa Cruz, CA, #sc-17951, 1:200), and CFB (Santa Cruz Biotechnology, Santa Cruz, CA, Cat#sc-67141, 1:200). ARPE-19 cells were immunostained with antibodies to C3b/iC3b/C3c and phalloidin using the same protocol as for RPE cells in sclerochoroidal flat-mounts. Secondary antibodies, conjugated to Alexa-488, Alexa-568, or Alexa-633 (Invitrogen, Carlsbad, CA), were added at a 1:200 dilution and incubated for 1-2 hrs. 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes/invitrogen, Cat# D1306) was used to label cellular nuclei. To label acidic lysosomes of cultured microglia, fluroscently-tagged basic amines (Lysotracker, Cat#MP07525, Invitrogen) was used as described in the manufacturer’s instructions; briefly, 0.5 μM of the tagged amine was added to cultured cells for 1.5 hours at 37°C, washed with PBS, and subsequently fixed in 4% paraformadehyde in PBS before visualization.
2.7. Apoptosis and Cellular Proliferation Assays
Apoptotic cells in culture or in the outer nuclear layer of the retina were labeled using a TUNEL assay according to the manufacturer’s instructions (In Situ Cell Death Detection Kit, TMR Red, Cat#: 12156792001, Roche, Indianopolis, IN). Cell nuclei were also labeled with DAPI, imaged using epifluorescence or confocal microscopy, counted, and the fraction of TUNEL-positive nuclei determined. At least 4 biological replicates were used for all quantifications. Proliferating RPE cells were labeled by bromodeoxyuridine (BrdU) incorporation. BrdU (Sigma, #B5002, 10μg/ml) was added to the cultures for a final concentration of 10 μM and incubated for 2.5 hours. After fixation in 4% paraformaldehyde in PBS, proliferating cells incorporating BrdU were identified by immunohistochemical stainining with an anti-BrdU antibody (G3G4, 1:200, Developmental Studies Hybridoma Bank, Iowa City, IA) and an anti-mouse secondary Alexa-568 antibody (Invitrogen). Proliferation rate was assessed by quantification of BrdU-positive cells in representative 10× fields (6 biological replicates were used for each condition).
2.8. Cell Viability Assay
The reduction of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to purple formazan product by reductase enzymes in living cells was used to assay the viability of cultured cells. To assay the effect of A2E loading on microglial viability, cultured retinal microglia (4×104 cells in a 200μl volume cell suspension) were distributed into each well of a 96-well plate. After one day of culture, microglia were changed into culture media containing different concentrations of A2E (0 μM, 3 μM, 6 μM, 12 μM, 24 μM; 4 biological replicates per concentration) and incubated at 37°C in a 5% CO2 incubator for 6 hours. The conversion of MTT for each condition was quantified according to instructions provided by the assay’s manufacturer (Trevigen, Gaithersburg, MD, Cat#4890-025-K).
2.9. Neuroprotection Assay
661W photoreceptor cells were distributed into 96-well plate (2×104 cells per well in 100μl). The ability of microglia to provide neuroprotection to photoreceptors undergoing oxidative stress was evaluated using the following assay. After one day of culture, conditioned media from microglial cultures were added to 661W cells in the presence of 0.5mM H2O2for 16 hrs. Controls included non-conditioned media (DMEM:F12 medium+5% heat inactivated FBS) in the presence and absence of added 0.5mM H2O2. Each experimental group contained at least 6 replicates.
2.10. Complement activation assays
An in vitro assay was used to assess the ability of microglia-conditioned media to influence complement activation and deposition on ARPE-19 cells. The assay was performed by adding mouse serum to ARPE-19 cells in the presence or absence of conditioned media from cultured microglia. Briefly, 1×105 ARPE-19 cells were added to each chamber of a slide-mounted 4-chambered plate and allowed to grow for 24 hours. Mouse serum was added either alone, with microglial-conditioned media from A2E-loaded microglia, or with microglial-conditioned media from control microglia. Controls included wells in which conditioned microglia medium was added alone without serum. After 3 hours of incubation, ARPE-19 cells were washed twice with PBS, and then fixed with 4% paraformadehyde for 20 minutes. Complement activation was detected by immunohistochemistry for complement breakdown products C3b/iC3b/C3c (mAb 2/11, #HM1078, Hycult Biotech, Uden, The Netherlands). The amount of deposition was quantified as the number of positive pixels located on cellular surfaces per 40× field using image-analysis software (NIH ImageJ, Bethesda, MD) under standardized conditions.
2.11. Immunoblot Analysis
Retinal microglia cells and ARPE-19 cells were suspended in RIPA buffer (R0278, Sigma) containing a protease inhibitor cocktail (Calbiochem, Gibbstown, NJ) and homogenized by ultrasonication. Protein concentrations in the homogenates were measured using the BCA Protein Assay Kit (Pierce, IL, USA). Protein extracts from retinal microglia cells or ARPE-19 cells containing 5-10μg of protein was separated using a 4-12% Bis-Tris Gel (NuPAGE, Invitrogen, USA) and transferred to a nitrocellulose membrane (iBlot Gel Transfer Stacks, Invitrogen). After blocking overnight with a buffer containing 1.5% BSA and 5% blocking reagent (Rodeo Blocker, USB), the blot was incubated with the primary antibodies for CFH (1:1000, Cat#sc-17951), CFB (1:1000, Cat#sc-67141), β-actin (1:3000) (all from Santa Cruz Biotechnology, Santa Cruz, CA,), and C3b/iC3b/C3c (1:1200). Anti-rabbit or rat horseradish peroxidase (HRP) (IVD Research, Carlsbad, CA) or anti-goat HRP (Santa Cruz Biotechnology) were used as secondary antibodies (1:3000, 1 hr). Resulting blots were developed using chemiluminescent detection (Amersham Hyperfilm ECL, GE Healthcare, Piscataway, NJ) and protein expression levels quantitated using image analysis software (ImageQuant, GE Healthcare).
2.12. Quantification of gene expression
Gene expression was quantified in retinal microglia using both RT-PCR and Nanostring nCounter assay. For rt-PCR, total RNA was extracted using the RNeasy Mini kit (Qiagen, Valencia, CA) and reverse-transcribed to cDNA using a RETROscript® Kit (Ambion, Austin, TX) according to the manufacturer’s instructions. cDNA was used for RT-PCR in an amplification reaction volume of 15 μl, comprising of 1 μl of the cDNA, 2 μl of primer mixture, and 7.5 μl of Plus Master Mix (QIAGEN, HotStarTaq Plus) and 4.5 μl H2O. PCR amplification was conducted for 20-35 cycles, and GAPDH was used as an internal control. A list of primers used is provided in Table 1. At least 3 replicates of each experiment were performed.
The NanoString nCounter assay was performed in the NCI DNA Sequencing/Digital Gene Expression Core (National Cancer Institute, NIH, Bethesda, MD) according to the manufacturer’s protocol (NanoString Technologies, Seattle, WA), as described previously (Geiss et al. 2008). In brief, the nCounter CodeSet for these studies contained probe pairs for 116 test and 14 control genes (6 positive control and 8 negative controls). A2E-loaded (6 μM for 6 hrs) microglia and control microglia cells were harvested and their total RNA was extracted with RNeasy Mini kit (QIAGEN, Valencia, CA) and concentrated to 20 ng/μl. Each sample was hybridized in triplicate with 100 ng of total RNA in each reaction. All genes and controls were assayed simultaneously in multiplexed reactions. To account for slight differences in hybridization and purification efficiency, the raw data were normalized to the standard curve generated via the nCounter system spike-in controls present in all reactions and corrected with internal control.
2.13. Statistical analysis
Statistical analyses were performed using statistical software (Graphpad, San Diego, CA, USA). Comparisons between control and A2E-loaded microglial cells were performed using a paired t-test. Conparisons of 3 or more data groups were performed using one-way analyses of variance (ANOVA), and comparisons between pairs of group means performed with the Tukey-Kramer multiple comparison test. In all graphical representations, the error bars indicate standard error of the mean (SEM).
3. Results
3.1. Age-dependent appearance of autofluorescent microglia in the subretinal space
Retinal microglia have been described to undergo age-dependent changes in their distribution, morphology, and behavior (Xu et al. 2008; Damani et al. 2011). In the young adult animal (age 2-6 months), horizontal arrays of regularly spaced and non-overlapping microglia are found in the inner retina, while only the rare, isolated microglial cell can be detected in the outer retina (from the outer nuclear layer to the subretinal space) (Chen et al. 2002; Santos et al. 2008). None of these rare subretinal microglia were found to exhibit significant intracellular autofluorescence (Fig. 1A). In aged mouse retina (age 18-24 months), CD11b-positive microglia accumulate in the subretinal space in increased numbers and demonstrate prominent autofluorescence in their cytoplasm. The emission spectrum of autofluorescence in retinal microglia resembles that for lipofuscin-accumulating RPE cells (Xu et al. 2008) and overlaps with that for the bisretinoid A2E (Sparrow et al. 2010), a well-characterized component of lipofuscin in the fundus (Eldred & Lasky 1993).
Figure 1. Age-dependent accumulation of autofluorescent microglia in the subretinal space of the mouse retina.
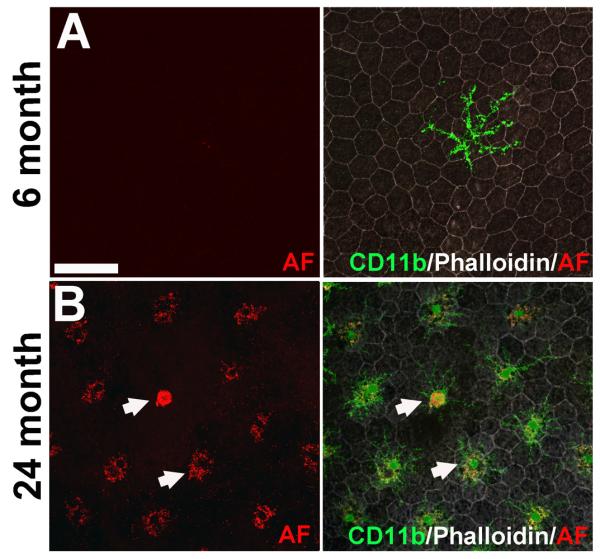
RPE-choroidal whole-mounts were prepared from young (6 months old) and aged mice (24 months old) and immunolabeled with antibodies for CD11b (green) to label subretinal microglia and Alexa 633-conjugated phalloidin (white) to reveal RPE morphology. Cellular autofluorescence (AF) was imaged with excitation illumination at wavelength 568nm and emission was captured in the 570-630nm range. (A) In young adult RPE-choroidal preparations, rare isolated CD11b-positive microglia with clear ramified morphologies (right) without detectable cellular autofluorescence (left) were observed in the subretinal space. (B) In the aged retina, increased numbers of subretinal microglia were observed in the subretinal space (right). Compared to those in the younger retina, these aged microglia had a less ramified morphology with shorter processes, larger somata, and a more prominent punctate pattern of autofluorescence in their cytoplasm (left, arrowhead). Scale bar = 50 μm.
A subset of retinal microglia in human retina was also observed to demonstrate a similar intracellular autofluorescence. In AMD eyes, microglia in the outer retina (Fig. 2A, B) and subretinal space (Fig. 2C-D) demonstrated punctate patterns of cytoplasmic autofluorescence. Microglia located in the inner retina and macrophages in the choroid did not possess intracellular autofluorescence (Fig. 2A). In aged control eyes without AMD, microglia could also be located in the subretinal space, and these also possessed punctate patterns of intracelullar autofluorescence (Fig. 2E) which tended to be lower than that of subretinal microglia in AMD eyes (Fig. 2F). In eyes from younger donors (44 – 55 years), subretinal microglia were rare and non-autofluorescent (data not shown).
Figure 2. Autofluorescent microglia in the subretinal space and outer retina of human retina in aged and AMD eyes.

(A) Retinal cryosection from an eye from a 82 year old AMD donor with advanced atrophic AMD ; section was taken through an area of partial RPE and photoreceptor degeneration. Retinal microglia and choroidal macrophages were immunolabeled with Iba-1 (green) and cellular nuclei with DAPI (blue). Autofluorescent signal (in red) was observed in RPE cells (arrowhead) and also outer retinal microglia cells (inset) but was absent in inner retinal microglia and choroidal macrophages (arrows). (B) High magnification view of outer retinal microglia in (A) showing a punctate intracellular pattern of autofluorescence. (C) Autofluorescent microglia in the subretinal space (arrowhead) in the vicinity of drusen (Dr) in an eye with AMD. (D) Brightfield (left) and fluorescence imaging (right) of an Iba-1 immunopositive microglial cell (green) in the subretinal space (arrowhead) with punctate autofluorescence in the soma (arrowhead). Note that choroidal macrophages (arrows) on the other side of Bruch’s membrane (Br) are devoid of autofluorescence. (E) Confocal image of subretinal microglia in a flat-mounted retina from an aged non-AMD donor (77 years of age) showing punctate intracytoplasmic autofluorescence. (F) Confocal image of subretinal microglia in a flat-mounted retina from an AMD donor with advanced atrophic AMD (85 years of age) demonstrating prominent autofluorescence in cellular somata. Scale bars: in (A) = 500 μm, in (C-F) = 50 μm.
3.2. Accumulation of bisretinoid A2E in cultured retinal microglia
As bisretinoid pigments of lipofuscin, in particular A2E, have previously been associated with physiological changes in RPE cells (Sparrow & Boulton 2005), we hypothesized that A2E accumulation in subretinal microglia in aged animals may underlie age-related autofluorescent changes and exert consequential effects on microglia physiology. We investigated this by creating an in vitro model in which A2E was induced to accumulate in cultured retinal microglia via extracellular uptake. We discovered that when cultured retinal microglia were exposed to extracellular A2E in the culture medium, they accumulated intracellular autofluorescent that was localized to the lysosomal compartment (Fig. 3A, B). As previously described for RPE cells, we found that exposure to culture media containing high A2E concentrations (>6 μM) resulted in decreased microglial cell viability (Fig. 3C), even in the absence of short wavelength irradiation (Sparrow et al. 2002). Prolonged incubations (12-24 hours) at sublethal A2E concentrations (≤6 μM) also induced decreased microglial proliferation (Fig. 3D) and increased apoptotic cell death (Fig. 3E). Although microglia which were loaded with other retinoids (all-trans and 13-cis retinonic acid) also showed increased intracellular autofluorescence localizing to the lysosomal compartment (Supplementary Fig. 1), similar levels of apoptotic cell death was not induced in loaded microglia (Fig 3E).
Figure 3. In vitro accumulation of autofluorescent bis-retinoid A2E in cultured retinal microglial cells.

Primary mouse retinal microglia were cultured in the absence (A) and presence of A2E (6 μM) (B) for 16 hours. The addition of A2E in the culture medium resulted in the accumulation of an autofluorescent signal (red) in the cytoplasm (B, left) of exposed microglial cells that was absent in control cells (A, left). Acidic intracellular organelles such as lysosomes were labeled using a fluorescent acidotropic probe (LysoTracker, green) (A,B, middle). The co-localization of the autofluorescent signal with LysoTracker-positive intracellular organelles (arrowheads, B right) indicated the accumulation of autofluorescent A2E in the lysosomal compartment of microglia. Scale bar = 50 μm. (C) The effect of A2E loading on retinal microglia viability, as measured using a MTT assay, was evaluated following 6-hour incubations in increasing A2E concentrations. Cell viability relative to control (retinal microglia not pre-loaded with A2E) decreased significantly when A2E concentrations exceeded 6 μM. (D) A2E-loading decreased retinal microglial proliferation in culture with increasing duration of A2E (6 μM) incubation as measured by incorporation of BrdU. (E) Microglial apoptosis in response to A2E loading, as evaluated by TUNEL labeling, was also significantly higher after 24 hours incubation compared to 6 hours incubation. Similar exposure of microglia to other retinoids in the visual cycle, all-trans retinal and 13-cis retinal did not produce significant increases of apoptotic rates relative to control (retinal microglia not pre-loaded with A2E). Data comprise of mean ± SEM, n = 3, *indicates comparisons relative to control for which p<0.05, 1-way ANOVA with Dunnett’s multiple comparison test.
3.3. Effect of A2E accumulation on microglial activation
We hypothesized that the effects of sublethal levels of A2E uptake in retinal microglia may induce pathologically significant changes in their physiology, such as an alteration in their activation state. While retinal microglia under standard culture conditions demonstrated a bipolar or ramified morphology, those loaded with A2E transitioned to a more amoeboid morphology typical of activated microglia (Fig 4A). A2E-loaded microglia, relative to control microglia, expressed higher levels of mRNA for genes that constitute a “classically” activated M1 state (Mantovani et al. 2004), such as CD16, CD68, CD86, and CD40 (Fig. 4B). However, mRNA levels for pro-inflammatory cytokines such as TNF-α, IL1β, and IL6 remained stable. On the other hand, genes associated with an “alternatively” activated M2 phenotype, such as CD206, CD200R, and TGF-β, were significantly decreased (Fig. 4C). Together, the effects of A2E accumulation of microglial morphology and gene expression indicated a polarization of microglial activation towards an increased M1/M2 ratio. In comparison, intracellular accumulation of retinoids, all-trans and 13-cis retinonic acid, did not result in similar profile of M1 and M2 markers (Supplementary Fig 2A), indicating a specific effect of A2E on microglial physiology.
Figure 4. Intracellular A2E loading alters the activation state of retinal microglia.

(A) The morphology of retinal microglia cultured under control conditions (top) and after A2E loading (6 μM for 6 hours) were evaluated using brightfield microscopy (left) and Iba1 immunohistochemistry ( right). While cultured retinal microglia under control conditions (top) demonstrated a branched, ramified morphology, those loaded with A2E transitioned into a rounded, more amoeboid morphology typical of activated microglia (bottom). Scale bar = 100 μm. (B) Changes in gene expression in retinal microglia with and without A2E loading (6 μM for 6 hours) were assayed with mRNA expression profiling using a direct, multiplex-capture method (nCounter, Nanostring). The expression of several cell surface markers, cytokines, and enzymes that are indicative of a “classically activated”, proinflammatory (M1) state were found to be predominantly increased (left). CD16, CD68, and CD40 were significantly increased following A2E loading relative to control while the remaining genes assayed remained stable or slightly (and non-significantly) decreased. (C) The expression of genes, such as CD206, CD200R, and TGF-β, which are indicative of an “alternatively activated”, anti-inflammatory (M2) state were predominantly decreased (right). These changes of gene expression profile suggest that A2E accumulation exerted a polarizing influence on retinal microglia, favoring a pro-inflammatory, activated M1 phenotype over a M2 phenotype. (*indicates comparisons relative to control for which p<0.05, unpaired t-test; n = 3 replicates).
3.4. Effect of A2E accumulation on growth factor expression and neuroprotective properties
Microglia have been ascribed both neuroprotective and neurotoxic roles in a variety of contexts, correlating with microglial M1 versus M2 activation states (Czeh et al. 2011). In the retina, microglia-mediated regulation of photoreceptor neuroprotection has been associated with the expression of neurotrophic factors (Harada et al. 2002). To understand the impact of A2E accumulation on microglial neuroprotective functions, we investigated microglial expression of growth factors and assessed their ability of protect photoreceptors in an in vitro neuroprotection assay. We found that A2E accumulation in cultured retinal microglia significantly decreased the mRNA expression of multiple growth factors as assessed by the nCounter assay (Fig. 5A) and by RT-PCR (Fig. 5B), an effect not observed in microglia that were loaded with 13-cis retinal and all-trans retinal (Supplementary Fig. 2B). These decreases correlated to a reduction in the ability of conditioned media from A2E-loaded microglia to rescue 661W photoreceptors undergoing oxidative stress. While the addition of conditioned media from non-A2E-loaded control microglia was able to increase the viability of stressed 661W cells, the neuroprotective effect of conditioned media was abrogated by prior A2E-loading of microglia in a dose-dependent manner (Fig. 5C).
Figure 5. Effects of intracellular A2E on retinal microglial growth factor expression and neuroprotective function.

Changes in gene expression of growth factors following A2E loading (6 μM for 6 hours) were assayed using the nCounter system (A) and also by semi-quantitative RT-PCR (B). mRNA expression of genes for growth factors, brain-derived neurotrophic factor (BDNF), epidermal growth factor (EGF), ciliary neurotrophic factor (CNTF), glial cell-derived neurotrophic factor (GDNF), and nerve growth factor (NGF) were all reduced following A2E loading, relative to unloaded control microglia. (Data comprise the mean ± SEM, n = 3, *indicate comparisons for which p<0.05 relative to control, unpaired t-test.) (C) The ability of microglia to provide neuroprotection to 661W photoreceptor cells was assessed in an in vitro model. Conditioned media (CM-RMG) from microglia that were either (1) cultured under control conditions or (2) pre-loaded with increasing concentrations of A2E were collected and added to cultures of 661W cells exposed to hydrogen peroxide (H202, 0.5mM) for 16 hrs to induce oxidative stress-mediated cell death. An MTT-based assay was used to measure cell viability. CM-RMG from microglia cultured under control conditions (0 μM A2E) increased the proportion of surviving 661W cells significantly. However this protective effect was abrogated for CM-RMG from microglia that had been pre-loaded with A2E of increasing concentrations, indicating that the neuroprotective functions of microglia were reduced by A2E accumulation. (Data are the mean ± SEM, n = 6, *indicates comparisons for which p<0.05, 1-way ANOVA with Tukey-Kramer multiple comparison test.)
3.5. Effect of A2E accumulation on chemokine-mediated microglial migration
The localization and migration of microglia in the CNS are highly influenced by chemokine-based cell-cell interactions that are mediated by the expression of chemokine receptors on microglial cell surfaces (Ransohoff et al. 2007; Biber et al. 2008). In particular, in the retina, communications through the CCL2/CCR2 and CX3CL1/CXCR1 axes have been implicated in localization and mobilization of retinal microglia (Raoul et al. 2010). We hypothesized that the accumulation of autofluorescent compounds in microglia may influence their ability to respond to chemokine-based communications. To address this, we compared the levels of mRNA expression of chemokine receptors before and after accumulation of A2E in cultured retinal microglia by rt-PCR. We found that mRNA levels for CX3CR1 (the receptor for CX3CL1), CCR2 (the receptor for CCL2), CCR1 and CCR5 (receptors for CCL3) were decreased following A2E-loading relative to non-loaded controls (Fig 6A). A2E-loading of microglia also resulted in decreased chemotaxis in response to CCL2 and CCL3 (Fig. 6B) in a dose-dependent manner in a Boyden-chamber assay, indicating that A2E-microglia are less responsive to chemokine signaling, possibly as a result of decreased chemokine receptor expression.
Figure 6. Effects of intracellular A2E on microglial expression of chemokines/chemokine receptors and microglial chemotaxis.

(A) Changes in mRNA expression of chemokine receptors following 6 hours of incubation in 6 μM of A2E were assayed using RT-PCR. Gene expression levels of CX3CR1, CCR2, CCR5, and CCR1 were also lowered relative to non-loaded control microglia. (Data are the mean ± SEM, n = 3, *indicates comparisons for which p<0.05 relative to control, unpaired t-test.) (B) Chemotaxis of cultured retinal microglia with and without A2E loading were also assessed using a Boyden chamber assay using CCL2 (100ng/ml) (upper) and CCL3 (50ng/ml) (lower) as chemoattractants. A2E-loading of microglia induced a dose-dependent decrease in chemotaxis relative to controls (microglia not preloaded with A2E). (Data comprise of mean ± SEM, n = 6, *indicate comparisons for which p<0.05, 1-way ANOVA with Tukey-Kramer multiple comparison test.)
3.6 Effect of A2E accumulation on microglial expression of complement regulatory factors
Recent studies have highlighted the ability of retinal microglia cells to express complement and complement regulatory genes (Luo et al. 2011), indicating that the regulation of complement deposition in the outer retina by microglia may be a relevant factor driving retinal degeneration in AMD (Collier et al. 2011; Rutar et al. 2011). We investigated the possibility that A2E accumulation in retinal microglia may influence their ability to regulate complementactivation. We confirmed by immunohistochemistry (Fig. 7A), RT-PCR (Fig. 7B), and Western blot analyses (Fig 7C) that cultured retinal microglia expressed detectable levels of complement factor B (CFB), a key positive regulator of complement activation in the alternative pathway, as well as complement factor H (CFH), a negative regulator complement activation. We discovered that increasing concentration of A2E exerted reciprocal dose-dependent effects on the mRNA of CFB and CFH, inducing a progressive increase in the levels of CFB mRNA expression and a converse decrease in the levels of CFH expression (Fig. 7B). Analogously, as analyzed by Western blot analysis, increasing durations of A2E exposure that induce increased A2E loading similarly resulted in an elevated expression of CFB and a decreased expression of CFH (Fig. 7C). Exposure of retinal microglia to other retinoids (13-cis retinal and all-trans retinal) did not alter the mRNA expression of CFB and CFH significantly (Supplementary Fig. 2C).
Figure 7. Effects of intracellular A2E on microglial expression of complement regulatory proteins CFB and CFH.

(A) Immunohistochemical staining for CFB (left) and CFH (right) in cultured retinal microglia, demonstrating immunopositivity for both proteins. Scale bar = 50 μm. (B) mRNA expression levels of CFB and CFH were altered in a dose-dependent manner by A2E loading. Representative examples of RT-PCR amplifications of CFB and CFH mRNA in retinal microglia showing upregulation of CFB expression and downregulation of CFH expression as a function of increasing concentration of A2E exposure (upper). Quantification of mRNA levels of CFB and CFH, relative to GADPH expression, demonstrated significant changes from control levels with A2E-loading, which progressed as a function of increasing concentration of A2E. (Data comprise of mean ± SEM, n =4-6, *indicate comparisons relative to control for which p<0.05, 1-way ANOVA with Dunnett’s multiple comparison test.) (C) Corresponding changes in CFB and CFH protein expression as quantitated by Western blotting. A significant increase in CFB expression and a significant decrease in CFH from control levels were observed following A2E loading at 6 μM for 24 hours. (Data comprise of mean ± SEM, n = 3, *indicate comparisons relative to control for which p<0.05, 1-way ANOVA with Dunnett’s multiple comparison test.)
3.7. Microglial regulation of complement activation is altered by A2E accumulation
As CFH and CFB are critical factors in the regulation of complement activation via the alternative pathway, we employed an in vitro model of complement activation to evaluate the ability of retinal microglia to influence complement deposition and the effect of A2E-loading on this function. Using mouse serum as a source of complement, complement deposition on a monolayer of ARPE-19 cells was measured by immunohistochemistry to complement breakdown products, iC3b/C3b/C3c. Conditioned media from cultured retinal microglia were added to the culture system and evaluated for their ability to influence the amount of complement deposition. We found that while cultured media from non-A2E-loaded control microglia were able to strongly inhibit the deposition of iC3b/C3b/C3c, this inhibitory effect was significantly abrogated in those from A2E-loaded microglia (Fig. 8A, B). Quantification of complement activation for the same culture conditions by Western blot analysis of iC3b demonstrated similar effects. These results indicate that retinal microglia are involved in the negative regulation of complement activation, and that this effect is reduced by A2E accumulation as a result of reciprocal changes in expression levels of CFB and CFH that favor complement activation.
Figure 8. Intracellular A2E loading abrogates the ability of microglia to negatively regulate complement activation.

The regulation of complement deposition by retinal microglia was assessed using an in vitro assay. Mouse serum was used as a source of complement. A monolayer of ARPE-19 cells provided a cellular surface for complement deposition. Conditioned media from retinal microglia (CM-RMG) were added to the culture to evaluate their ability to modulate in vitro complement activation and deposition. (A) Representative examples of immunohistochemical staining of iC3b/C3b/C3c deposition (white) on the ARPE-19 cell layer (marked with phalloidin (green) and DAPI (blue)) are shown under different experimental conditions. In column 1, conditioned media from control microglia was added in the absence of serum; minimal to no complement deposition was detected under these conditions. In column 2, serum was added alone, resulting in the prominent induction of complement activation and deposition. In column 3, addition of control (unloaded) microglial conditioned media to serum resulted in a significant suppression of complement deposition. In column 4, the addition of conditioned media from A2E-loaded microglia with serum also resulted in a suppression of complement deposition. However, complement deposition was significantly greater compared to column 3, indicating that an abrogation of negative regulatory effect on complement deposition as a result of A2E loading. (B) Quantification of iC3b/C3b/C3c deposition (in A) by image analysis measuring the intensity of the immunofluorescence of iC3b/C3b/C3c labeling under standard conditions. Asterisks indicate comparisons for which p<0.05 (1-way ANOVA with Dunnett’s multiple comparison test, n=4). (C) Representative Western blots for iC3b/C3b/C3c from whole cell lysates of ARPE-19 cells from the four experimental conditions described in (A), showing similar changes in iC3b deposition (top). Quantitation of the intensity of the iC3b band demonstrates similar comparisons observed in (B). (Data comprise of mean ± SEM, n = 3, *indicates comparisons for which p<0.05, 1-way ANOVA with Dunnett’s multiple comparison test.)
3.8. In vivo effects of A2E-loaded microglia on complement activation and photoreceptor apoptosis
To confirm the regulatory effect of retinal microglia on complement activation, we investigated whether A2E accumulation in microglia in the subretinal space may also influence complement activation in this particular locus in vivo. A2E-loaded retinal microglia were transplanted into the subretinal space in experimental wild type mice; as controls, eyes were either injected with non-A2E-loaded microglia or left uninjected. Ocular tissue were harvested and analyzed 3 days after injection. We found that while subretinal transplantation of non-A2E-loaded microglia induced small amounts of iC3b/C3b/C3c deposition in the subretinal space, the levels of complement deposition were significantly elevated in eyes injected with A2E-loaded microglia (Fig. 9A, B). The increased complement deposition in these eyes also corresponded to a significantly elevated level of photoreceptor apoptosis as measured by TUNEL labeling and increased outer segment loss and disruption (Fig. 10). These results indicate that the accumulation of subretinal A2E-laden microglia can result in the dysregulation of complement regulation in the outer retina, favoring complement activation, leading to increased photoreceptor injury and degeneration.
Figure 9. In vivo effects of subretinal transplantation of A2E-loaded microglia on complement activation.

GFP-positive retinal microglia from CX3CR1+/GFP transgenic mice were cultured in the absence or presence of A2E, then subsequently suspended in diluted Matrigel before injection into the subretinal space of experimental mice (volume of injection = 1.5 μl, density = 1×108 cells/ml). One eye of each experimental animal was injected with microglia previously loaded with A2E; the contralateral control eye was injected with microglia not loaded with A2E. Uninjected eyes from other animals served as additional controls. Animals were euthanized and enucleated 3 days post-injection. Resulting RPE sclerochoroidal flat-mounts were prepared and immunolabeled for the RPE tight-junction protein ZO-1 (red), and for complement activation breakdown products, iC3b/C3b/C3c (white). (A) In uninjected eyes (top row), there was an absence of GFP+ cells, an intact and regular RPE monolayer, and an absence of iC3b/C3b/C3c. In eyes injected with retinal microglia not previously loaded with A2E (middle row), accumulations of GFP+ cells could be located at injection sites. Underlying areas of RPE cells as revealed by ZO-1 labeling showed an irregular mosaic distribution, with low levels of iC3b/C3b/C3c deposited on the apical surfaces of RPE cells. In eyes injected with retinal microglia previously loaded with A2E (6 μM for 6 hours) (bottom row), similar areas demonstrated increased RPE disorganization that were associated with elevated levels of iC3b/C3b/C3c deposition. (B) Quantification of iC3b/C3b/C3c deposition in 20× fields centered on the site of transplanted retinal microglia demonstrated a significant increase in eyes receiving transplantations of A2E-loaded microglia relative to control microglia. (Data comprise of mean ± SEM, n = 3, *indicates comparison for which p<0.05, 1-way ANOVA with Tukey’s multiple comparison test.)
Figure 10. Effects of subretinal transplantated of A2E-loaded microglia on photoreceptor integrity and survival.

Effects of subretinal A2E-loaded microglia on photoreceptors were evaluated 3 days following the transplantation of GFP-positive, A2E-loaded retinal microglia into the subretinal space. Controls included eyes injected with non-A2E-loaded microglia and uninjected eyes. Photoreceptor apoptosis in the areas of cell injection was evaluated in retinal flatmounts by TUNEL-labeling of photoreceptor nuclei in the outer nuclear layer (ONL) by confocal microscopy. The preservation of cone photoreceptor outer segments in the same areas was evaluated by S-opsin immunolabeling. (A) (Top row) For uninjected control eyes, rare or no TUNEL-positive cells were observed in the ONL. S-opsin labeling was prominent in the outer segments which demonstrated a regular arrangement and intact structure. (Middle row) For eyes injected with non-A2E-loaded microglia, a few TUNEL-positive nuclei were detected overlying in injection site. S-opsin-labeled outer segments also appeared sparser than in uninjected controls. (Bottom row) For eyes injected with microglia loaded with A2E, the density of TUNEL-positive nuclei was increased and S-opsin-positive outer segments were decreased in density, appearing shorter and more irregular. (B) Quantification of the number of TUNEL+ positive nuclei per 40× field in the overlying ONL demonstrated a significantly increased rate of photoreceptor apoptosis in eyes containing A2E-loaded subretinal microglia, relative to those containing unloaded microglia or uninjected controls. (C) Quantification of the number of S-opsin-positive outer segments per 40× field demonstrates a significantly decreased number of outer segments in eyes containing A2E-loaded subretinal microglia, relative to those containing unloaded microglia or uninjected controls. (*indicates comparisons for which p<0.05, 1-way ANOVA with Tukey-Kramer multiple comparison test, n= 3 replicates.)
4. Discussion
The translocation of senescent retinal microglia from the inner to the outer retina in aged and AMD eyes in human patients and mouse models highlights a potential cellular mechanism that underlies age-related immune dysregulation that may be relevant in driving AMD progression (Ma et al. 2009; Karlstetter et al. 2010). However, endogenous changes occurring in senescent subretinal microglia that can induce their transition to a pathogenic phenotype are not well understood. Features that are particular to subretinal microglia in the aged retina but which are absent in “resting” parenchymal microglia in young healthy animals may be relevant to this transformation. In the present work, we hypothesize that the accumulation of intracellular lipofuscin and its related compounds in subretinal microglia is one such particular feature with pathogenic consequences. We address this hypothesis by examining the effects of intracellular loading of A2E, a well-characterized bisretinoid component of lipofuscin, on microglial physiology in vitro and the effects of A2E-laden microglia in the retina in vivo.
The sources of lipofuscin accumulating in subretinal microglia are not definitively known. While cellular autophagy, the general process by which many aging cells develop lipofuscin, may be involved, subretinal microglia probably accumulate lipofuscin and its precursors primarily by phagocytosis of material from surrounding photoreceptor outer segments and/or RPE cells. This is suggested by recent in vitro studies in which BV2 microglia following phagocytosis of rod outer segment preparations developed increases in autofluorescence in the lysosomal compartment (Lei et al. 2012). Prolonged residence in the subretinal space appears necessary for lipofuscin accumulation as the few isolated subretinal microglia found in young mouse retina are mostly devoid of autofluorescence. Intracellular lipofuscin content in subretinal microglia have been also observed to increase monotonically with age (Xu et al. 2008), indicating a cumulative effect of continuing lipofuscin acquisition over time. Parenchymal microglia in the inner retina of aged animals also lack strong autofluorescence (Xu et al. 2008; Damani et al. 2011), further supporting an outer retinal source for lipofuscin.
To investigate the functional significance of lipofuscin accumulation in microglia, we employed a model of A2E accumulation that is modeled after in vitro studies in cultured RPE cells (Holz et al. 1999; Sparrow et al. 1999). As was the case for RPE cells, we found that A2E in the culture medum was readily internalized by cultured retinal microglia and accumulated intracellularly in lysosomes. As was also found in RPE cells, high concentrations of A2E in the medium resulted in increased intracellular levels that induced significant cellular toxicity and apoptosis. This induction of cell death may be secondary to the amphiphilic detergent-like nature of A2E that disrupts membrane integrity (Sparrow et al. 1999) and the induction of singlet oxygen production from A2E photoxidation that results in cellular apoptosis (Sparrow et al. 2002). Of greater interest to us however were the effects of sublethal accumulations of A2E on microglial physiology that can model the alterations in viable, lipofuscin-laden microglia cells accumulating in the subretinal space and exerting their effects on outer retinal cells.
Our experiments centered on exploring changes in A2E-fed microglia within the sublethal ranges of A2E incubation. We found that sublethal levels of A2E-loading altered the activation status of retinal microglia as assessed by morphological and molecular means. Cultured retinal microglia following A2E loading decreased in ramification and transitioned to a more amoeboid morphology, indicative of an activated state. Also, molecular markers of classical (M1) activation were predominantly increased while markers of alternative (M2) activation were decreased, indicating an overall M1/M2 polarization. Changes analogous to these A2E-induced changes have been observed in subretinal microglia in aged mice and mouse models of AMD (Combadiere et al. 2007; Xu et al. 2008; Luhmann et al. 2009; Damani et al. 2011); subretinal microglia, relative to microglia in the inner retina, demonstrated decreased ramification and a more rounded cell shape, and were immunopositive for activation markers such as CD68. However, subretinal microglia in aged retina have also been noted to lack MHCII expression (Xu et al. 2008), suggesting that their activation status may differ from that typically induced by activating signals such as lipopolysaccharride (LPS). We found here that A2E-loaded microglia, while expressing increased levels of mRNA for costimulatory molecules such as CD86 and activation markers such as CD68, were not significantly altered in their expression levels of proinflammatory cytokines such as TNFα, IL1β, and IL6. The pathological significance of increasing microglial M1/M2 polarization has previously been related to a predisposition towards proinflammatory responses and away from wound-healing and regenerative activities, a physiological alteration that may promote neuroinflammation and neurodegeneration in CNS diseases (Colton & Wilcock 2010; Czeh et al. 2011). In the retina, altered expression of markers that reflect an increased M1/M2 macrophage polarization was also detected more prominently in eyes with advanced AMD disease compared to age-matched controls (Cao et al. 2011), suggesting that A2E-induced microglial polarization may also confer pathogenic capabilities on subretinal microglia in AMD.
We found that A2E-loading in retinal microglia resulted in a decrease in mRNA expression of multiple growth factors and reduced the ability of microglia to confer neuroprotection on photoreceptors exposed to oxidative stress. The ability of microglia to provide neuroprotection to neurons has been previously documented in the number of injury/disease contexts such as metabolic impairment (Park et al. 2001), nitric oxide-induced injury (Toku et al. 1998), ischemia (Madinier et al. 2009) and excitotoxicity (Vinet et al. 2012). In the retina, microglia have been demonstrated to provide protection to photoreceptors subject to light-induced injury via the production of neurotrophic factors such as NGF, NT-3, CNTF, GDNF (Harada et al. 2002). We observed here a dose-dependent abrogation of the neuroprotective effect of retinal microglia by A2E-loading, indicating that lipofuscin accumulation in aged subretinal microglia may cumulatively reduce supportive microglial functions, such as by the reduced expression of growth factors, leading to an elevated vulnerability of photoreceptors in the aged retina.
We found that A2E-loading in retinal microglia resulted in decreased mRNA expression for a number of chemokine receptors known to be significant in guiding microglial migration. These A2E-loaded microglia were also less able to migrate in response to chemokine stimulation. While the cellular mechanisms driving microglia accumulation in the subretinal space are not fully understood, microglial defects in chemokine signaling, particularly with respect to CX3CR1 and CCR2 axes, have been implicated (Raoul et al. 2010). Transgenic mice deficient in CX3CL1/CX3CR1 and CCL2/CCR2 signaling demonstrate prominent and premature subretinal microglial accumulation relative to age-matched controls, and, perhaps as a result, develop pathological features resembling those found in AMD (Combadiere et al. 2007; Tuo et al. 2007; Luhmann et al. 2009). How can decreased chemokine signaling be related to subretinal microglial accumulation? One mechanism that has been proposed relates subretinal microglial accumulation to the decreased ability of subretinal microglia to exit the subretinal space by translocation across the RPE cell layer to move into the choroid. This mechanism posits that a trans-RPE mode of microglial migration, whose existence has been implicated in several studies (Ng & Streilein 2001; Raoul et al. 2008; Omri et al. 2011), serves to allow the egress of subretinal microglia and which is guided by, and dependent on, chemokine signaling. In the current study, we noted A2E-dependent decreases in microglial expression of chemokine receptor and chemokine-mediated microglia migration. These observations suggest a sequence of events in which an initial entry of microglia to the subretinal space in the aged retina results in microglial accumulation of lipofuscin, decreasing their chemotaxis and retarding their exit from the subretinal space. This prolonged residence time in the subretinal space can further potentiate lipofuscin accumulation, leading to further retention in a positive feedback cycle. In this schema, the lipofuscin-rich environment of the aged outer retina can play a significant role in promoting the retention and accumulation of subretinal microglia.
Our results here demonstrate that cultured retinal microglia are capable of expressing the key complement regulatory proteins, CFH and CFB, and that A2E-loading significantly alters the expression levels of these proteins to favor complement activation in in vitro and in vivo settings. The involvement of the complement system in AMD pathogenesis has been strongly supported by studies demonstrating the presence of complement proteins in drusen (Johnson et al. 2001; Anderson et al. 2002), the hallmark of early AMD, and those that associate AMD prevalence with variants in the genetic sequence of complement genes (Swaroop et al. 2009). While the analysis of the complement composition in drusen have been focused primarily on deposits in the sub-RPE space, presumably from basally-directed secretion from RPE cells (Johnson et al. 2011), the presence of subretinal deposits containing typical drusen-associated components, including complement regulatory proteins, have also been described in AMD eyes (Rudolf et al. 2008). These subretinal deposits, corresponding to clinically-observed reticular pseudodrusen (Zweifel et al. 2010), have been associated with progression to advanced forms of AMD in both the neovascular (Pumariega et al. 2011) and atrophic (Schmitz-Valckenberg et al. 2011) forms. These complement-containing deposits found in both the subretinal and sub-RPE spaces indicate a dysregulation of complement activation in these two outer retinal compartments, and it is yet unclear how similar or distinct their pathogenesis and pathological significances may be. While complement and complement-regulatory proteins may be systemically distributed in the serum are likely to influence complement deposition in the retina (Scholl et al. 2008; Johnson et al. 2011),, there is also evidence for a role for local complement regulation by RPE cells (Ramo et al. 2008; Ma et al. 2010) and also by subretinal microglia.
Recent studies have indicated that retinal microglia can not only influence the inflammatory millieu of the retina by producing cytokines and chemokines but also regulate complement activation by expressing complement molecules and complement regulatory proteins (Luo et al. 2011). In a light-induced injury model in which microglia recruitment to the subretinal space was followed by deposition of C3 in the outer retina, intervention with a serotonin agonist that decreased microglial recruitment also concurrently decreased complement deposition (Collier et al. 2011). Using a similar light-induced injury model, Rutar et al, reported that the recruited retinal microglia in the subretinal space upregulated C3 expression as detected by in situ hybridization and resulted in increased complement activation and deposition in the outer retina, which in turn mediated nearby photoreceptor death (Rutar et al. 2011). Our findings indicate that retinal microglia, by expressing complement regulators such as CFB and CFH, can act as local regulators of complement in the alternative pathway. In addition, the reciprocal effect of A2E-loading on microglial CFB and CFH expression was effective in abrogating the ability of microglia to limit complement activation and deposition. We observed this effect in vitro on a monolayer of ARPE-19 cells (a system that approximates only partially to native RPE cells), but also in vivo on apical RPE cell surfaces when A2E-loaded retinal microglia were transplanted into the subretinal space. These results indicate that altered microglia accumulating in the subretinal space can exert a significant effect on local complement regulation in the outer retina. Our findings posit that A2E-laden subretinal microglia have the effect of shifting the balance towards complement activation in the outer retina, and thereby driving processes of photoreceptor and RPE disruption and degeneration such as those observed in AMD.
The overall complement regulation in the aging outer retina is likely to derive not only from systemic factors but also from microglial- and RPE-derived factors. RPE cells, like retinal microglia, express component regulatory proteins, including CFB and CFH, whose levels of expression are influenced by aging, exposure to inflammatory mediators, and oxidative stress (Chen et al. 2007; Wu et al. 2007; Chen et al. 2008). Like in microglia, A2E-lipofuscin in RPE cells can also play a significant role in regulating complement activation. RPE cells in transgenic abcr4−/− mice, which contain higher levels of A2E compared to wild type animals, have downregulated levels of complement regulatory proteins and increased complement activation (Radu et al. 2011). Sparrow and colleagues have also reported that RPE cells in vitro containing A2E and its oxidation products are capable of inducing complement activation (Zhou et al. 2006; Zhou et al. 2009). How intracellular A2E in RPE cells increase complement activation has been related to altered expression levels of complement regulators (Chen et al. 2008; Radu et al. 2011), and also to direct contact between complement proteins and A2E-associated compounds which have been redistributed outside the RPE cells via cellular fragmentation (Zhou et al. 2006; Zhou et al. 2009). We have related the effect of lipofuscin-A2E on microglial complement regulation to the altered expression of CFH and CFB but we are unable to rule out the influence of other regulatory proteins or the extracellular effects of A2E that may have leached into the extracellular environment. Taken together, the findings here and in other studies (Ma et al. 2009; Wang et al. 2009) indicate that it is likely that aging changes in subretinal microglia and RPE cells may both contribute to complement and immune dysregulation in the aging retina that predisposes towards AMD.
In summary, our findings indicate that age-dependent lipofuscin accumulation in subretinal microglia in the aged retina can exert changes in the physiology of retina microglial that promote their retention in the subretinal space, increase their activation state, decrease their neuroprotective properties, and bias their complement regulatory properties to favoring complement activation. The potential consequence of these microglial changes is the creation of an environment in the outer retina that is more proinflammatory and favorable to complement activation and deposition, which can then drive RPE and photoreceptor injury and apoptosis such as that observed in AMD. As such, these changes may constitute a cellular mechanism underlying AMD pathogenesis in which aging changes and chronic neuroinflammatory events combine to confer increased risks of disease progression. Future studies into the cellular mechanisms regulating lipofuscin uptake and its effect on microglial physiology may present novel targets for AMD intervention and prevention.
Supplementary Material
Supplementary Figure 1. In vitro accumulation of autofluorescence in cultured retinal microglial cells loaded with 13-cis retinal and all-trans retinal. Primary mouse retinal microglia were cultured in the absence (top row) and presence (bottom rows) of retinoids, 13-cis retinal and all-trans retinal (6 μM). Cytoplasmic autofluorescence (green) accumulated in retinoid-exposed microglial cells and remained absent in control cells. The autofluorescent signals in loaded cells colocalized with the lysosomal compartment labeled using LysoTracker (red). Scale bar = 25 μm.
Supplementary Figure 2. Effect of in vitro accumulation of retinoids, 13-cis retinal and all-trans retinal on mRNA gene expression. (A) mRNA expression M1 genes (top) that had been significantly increased following A2E accumulation and M2 genes (bottom) that had been significantly decreased following A2E accumulation were evaluated in retinal microglia following incubation with either 13-cis retinal (hatched bars) and all-trans retinal (black bars) (both 6 μM for 6 hours) by RT-PCR. mRNA levels for each gene are expressed as a percentage of that in unloaded retinal microglia (white bars). Significant changes relative to control are indicated by an asterisk (p<0.05, 1-way ANOVA with Dunnett’s multiple comparison test, n = 4 replicates). No significant increases in M1 genes were noted, while only TGF-β decreased with all-trans retinal loading. CD206 mRNA levels were conversely increased with 13-cis retinal loading. Taken together, loading with either 13-cis retinal or all-trans retinal did not result in an overall increase in M1/M2 gene expression ratio seen with A2E loading. (B) mRNA levels of growth factors, which were decreased with A2E loading, were not significantly altered by either 13-cis retinal or all-trans retinal loading (p>0.05 for all comparisons). (C) mRNA levels of complementary regulatory proteins, CFH and CFB, were not significantly altered by either 13-cis retinal or all-trans retinal loading (p>0.05 for all comparisons).
{kind=link}
Acknowledgements
We are grateful to Dr Maria M. Campos for her assistance with histopathology of aged and AMD eyes. This study was supported by the NEI Intramural by the National Eye Institute Intramural Research Program and a grant from the American Health Assistance Foundation (AHAF). Acknowledgement is made to the donors of ADR, a program of the American Health Assistance Foundation, for support of this research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The data in this manuscript is published or submitted elsewhere, and has been approved by all contributing authors. Procedures involving animals here have been previously approved by institutional review boards.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
All authors indicate that they do not have conflicts of interest to disclose.
REFERENCES
- Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–431. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- Augustin AJ, Kirchhof J. Inflammation and the pathogenesis of age-related macular degeneration. Expert Opin Ther Targets. 2009;13:641–651. doi: 10.1517/14728220902942322. [DOI] [PubMed] [Google Scholar]
- Biber K, Vinet J, Boddeke HW. Neuron-microglia signaling: chemokines as versatile messengers. J Neuroimmunol. 2008;198:69–74. doi: 10.1016/j.jneuroim.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Boehm MR, Oellers P, Thanos S. Inflammation and immunology of the vitreoretinal compartment. Inflamm Allergy Drug Targets. 2011;10:283–309. doi: 10.2174/187152811796117717. [DOI] [PubMed] [Google Scholar]
- Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, Chan CC. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. 2011;61:528–535. doi: 10.1111/j.1440-1827.2011.02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Yang P, Kijlstra A. Distribution, markers, and functions of retinal microglia. Ocul Immunol Inflamm. 2002;10:27–39. doi: 10.1076/ocii.10.1.27.10328. [DOI] [PubMed] [Google Scholar]
- Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res. 2007;84:635–645. doi: 10.1016/j.exer.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Chen M, Muckersie E, Robertson M, Forrester JV, Xu H. Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp Eye Res. 2008;87:543–550. doi: 10.1016/j.exer.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Collier RJ, Wang Y, Smith SS, Martin E, Ornberg R, Rhoades K, Romano C. Complement deposition and microglial activation in the outer retina in light-induced retinopathy: inhibition by a 5-HT1A agonist. Invest Ophthalmol Vis Sci. 2011;52:8108–8116. doi: 10.1167/iovs.10-6418. [DOI] [PubMed] [Google Scholar]
- Colton CA, Wilcock DM. Assessing activation states in microglia. CNS Neurol Disord Drug Targets. 2010;9:174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, Debre P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, Sennlaub F. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon N, O’Colmain B, Klaver CC, Klein R, Munoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- Czeh M, Gressens P, Kaindl AM. The yin and yang of microglia. Dev Neurosci. 2011;33:199–209. doi: 10.1159/000328989. [DOI] [PubMed] [Google Scholar]
- Damani MR, Zhao L, Fontainhas AM, Amaral J, Fariss RN, Wong WT. Age-related alterations in the dynamic behavior of microglia. Aging Cell. 2011;10:263–276. doi: 10.1111/j.1474-9726.2010.00660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding X, Patel M, Shen D, Herzlich AA, Cao X, Villasmil R, Klupsch K, Tuo J, Downward J, Chan CC. Enhanced HtrA2/Omi expression in oxidative injury to retinal pigment epithelial cells and murine models of neurodegeneration. Invest Ophthalmol Vis Sci. 2009;50:4957–4966. doi: 10.1167/iovs.09-3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–152. doi: 10.1016/j.survophthal.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso LA, Vrabec T, Kuivaniemi H. The role of complement Factor H in age-related macular degeneration: a review. Surv Ophthalmol. 2010;55:227–246. doi: 10.1016/j.survophthal.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993;361:724–726. doi: 10.1038/361724a0. [DOI] [PubMed] [Google Scholar]
- Friedman DS, O’Colmain BJ, Munoz B, Tomany SC, McCarty C, de Jong PT, Nemesure B, Mitchell P, Kempen J. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- Gehrs KM, Jackson JR, Brown EN, Allikmets R, Hageman GS. Complement, age-related macular degeneration and a vision of the future. Arch Ophthalmol. 2010;128:349–358. doi: 10.1001/archophthalmol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- Gupta N, Brown KE, Milam AH. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res. 2003;76:463–471. doi: 10.1016/s0014-4835(02)00332-9. [DOI] [PubMed] [Google Scholar]
- Harada T, Harada C, Kohsaka S, Wada E, Yoshida K, Ohno S, Mamada H, Tanaka K, Parada LF, Wada K. Microglia-Muller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. J Neurosci. 2002;22:9228–9236. doi: 10.1523/JNEUROSCI.22-21-09228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz FG, Schutt F, Kopitz J, Eldred GE, Kruse FE, Volcker HE, Cantz M. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 1999;40:737–743. [PubMed] [Google Scholar]
- Hyman L, Neborsky R. Risk factors for age-related macular degeneration: an update. Curr Opin Ophthalmol. 2002;13:171–175. doi: 10.1097/00055735-200206000-00007. [DOI] [PubMed] [Google Scholar]
- Jager RD, Mieler WF, Miller JW. Age-related macular degeneration. N Engl J Med. 2008;358:2606–2617. doi: 10.1056/NEJMra0801537. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Forest DL, Banna CD, Radeke CM, Maloney MA, Hu J, Spencer CN, Walker AM, Tsie MS, Bok D, Radeke MJ, Anderson DH. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108:18277–18282. doi: 10.1073/pnas.1109703108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda A, Abecasis G, Swaroop A. Inflammation in the pathogenesis of age-related macular degeneration. Br J Ophthalmol. 2008;92:448–450. doi: 10.1136/bjo.2007.131581. [DOI] [PubMed] [Google Scholar]
- Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology. 2010;215:685–691. doi: 10.1016/j.imbio.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Lamb LE, Simon JD. A2E: a component of ocular lipofuscin. Photochem Photobiol. 2004;79:127–136. doi: 10.1562/0031-8655(2004)079<0127:aacool>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Lei L, Tzekov R, Tang S, Kaushal S. Accumulation and autofluorescence of phagocytized rod outer segment material in macrophages and microglial cells. Mol Vis. 2012;18:103–113. [PMC free article] [PubMed] [Google Scholar]
- Luhmann UF, Robbie S, Munro PM, Barker SE, Duran Y, Luong V, Fitzke FW, Bainbridge JW, Ali RR, MacLaren RE. The drusenlike phenotype in aging Ccl2-knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Invest Ophthalmol Vis Sci. 2009;50:5934–5943. doi: 10.1167/iovs.09-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Chen M, Xu H. Complement gene expression and regulation in mouse retina and retinal pigment epithelium/choroid. Mol Vis. 2011;17:1588–1597. [PMC free article] [PubMed] [Google Scholar]
- Ma KN, Cashman SM, Sweigard JH, Kumar-Singh R. Decay accelerating factor (CD55)-mediated attenuation of complement: therapeutic implications for age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010;51:6776–6783. doi: 10.1167/iovs.10-5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Zhao L, Fontainhas AM, Fariss RN, Wong WT. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS One. 2009;4:e7945. doi: 10.1371/journal.pone.0007945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madinier A, Bertrand N, Mossiat C, Prigent-Tessier A, Beley A, Marie C, Garnier P. Microglial involvement in neuroplastic changes following focal brain ischemia in rats. PLoS One. 2009;4:e8101. doi: 10.1371/journal.pone.0008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Ng TF, Streilein JW. Light-induced migration of retinal microglia into the subretinal space. Invest Ophthalmol Vis Sci. 2001;42:3301–3310. [PubMed] [Google Scholar]
- Olah M, Biber K, Vinet J, Boddeke HW. Microglia phenotype diversity. CNS Neurol Disord Drug Targets. 2011;10:108–118. doi: 10.2174/187152711794488575. [DOI] [PubMed] [Google Scholar]
- Omri S, Behar-Cohen F, de Kozak Y, Sennlaub F, Verissimo LM, Jonet L, Savoldelli M, Omri B, Crisanti P. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy: role of PKCzeta in the Goto Kakizaki rat model. Am J Pathol. 2011;179:942–953. doi: 10.1016/j.ajpath.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J. Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc Natl Acad Sci U S A. 1998;95:14609–14613. doi: 10.1073/pnas.95.25.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park LC, Zhang H, Gibson GE. Co-culture with astrocytes or microglia protects metabolically impaired neurons. Mech Ageing Dev. 2001;123:21–27. doi: 10.1016/s0047-6374(01)00336-0. [DOI] [PubMed] [Google Scholar]
- Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- Pumariega NM, Smith RT, Sohrab MA, Letien V, Souied EH. A prospective study of reticular macular disease. Ophthalmology. 2011;118:1619–1625. doi: 10.1016/j.ophtha.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu RA, Hu J, Yuan Q, Welch DL, Makshanoff J, Lloyd M, McMullen S, Travis GH, Bok D. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011;286:18593–18601. doi: 10.1074/jbc.M110.191866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramo K, Cashman SM, Kumar-Singh R. Evaluation of adenovirus-delivered human CD59 as a potential therapy for AMD in a model of human membrane attack complex formation on murine RPE. Invest Ophthalmol Vis Sci. 2008;49:4126–4136. doi: 10.1167/iovs.08-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Liu L, Cardona AE. Chemokines and chemokine receptors: multipurpose players in neuroinflammation. Int Rev Neurobiol. 2007;82:187–204. doi: 10.1016/S0074-7742(07)82010-1. [DOI] [PubMed] [Google Scholar]
- Raoul W, Auvynet C, Camelo S, Guillonneau X, Feumi C, Combadiere C, Sennlaub F. CCL2/CCR2 and CX3CL1/CX3CR1 chemokine axes and their possible involvement in age-related macular degeneration. J Neuroinflammation. 2010;7:87. doi: 10.1186/1742-2094-7-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul W, Keller N, Rodero M, Behar-Cohen F, Sennlaub F, Combadiere C. Role of the chemokine receptor CX3CR1 in the mobilization of phagocytic retinal microglial cells. J Neuroimmunol. 2008;198:56–61. doi: 10.1016/j.jneuroim.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Rudolf M, Malek G, Messinger JD, Clark ME, Wang L, Curcio CA. Sub-retinal drusenoid deposits in human retina: organization and composition. Exp Eye Res. 2008;87:402–408. doi: 10.1016/j.exer.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutar M, Natoli R, Kozulin P, Valter K, Gatenby P, Provis JM. Analysis of complement expression in light-induced retinal degeneration: synthesis and deposition of C3 by microglia/macrophages is associated with focal photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2011;52:5347–5358. doi: 10.1167/iovs.10-7119. [DOI] [PubMed] [Google Scholar]
- Santos AM, Calvente R, Tassi M, Carrasco MC, Martin-Oliva D, Marin-Teva JL, Navascues J, Cuadros MA. Embryonic and postnatal development of microglial cells in the mouse retina. J Comp Neurol. 2008;506:224–239. doi: 10.1002/cne.21538. [DOI] [PubMed] [Google Scholar]
- Schmitz-Valckenberg S, Alten F, Steinberg JS, Jaffe GJ, Fleckenstein M, Mukesh BN, Hohman TC, Holz FG. Reticular drusen associated with geographic atrophy in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2011;52:5009–5015. doi: 10.1167/iovs.11-7235. [DOI] [PubMed] [Google Scholar]
- Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, Fritsche LG, Chong NV, Fimmers R, Wienker T, Holz FG, Weber BH, Oppermann M. Systemic complement activation in age-related macular degeneration. PLoS One. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutt F, Davies S, Kopitz J, Holz FG, Boulton ME. Photodamage to human RPE cells by A2-E, a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 2000;41:2303–2308. [PubMed] [Google Scholar]
- Shamsi FA, Boulton M. Inhibition of RPE lysosomal and antioxidant activity by the age pigment lipofuscin. Invest Ophthalmol Vis Sci. 2001;42:3041–3046. [PubMed] [Google Scholar]
- Sparrow JR, Boulton M. RPE lipofuscin and its role in retinal pathobiology. Exp Eye Res. 2005;80:595–606. doi: 10.1016/j.exer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Cai B. Blue light-induced apoptosis of A2E-containing RPE: involvement of caspase-3 and protection by Bcl-2. Invest Ophthalmol Vis Sci. 2001;42:1356–1362. [PubMed] [Google Scholar]
- Sparrow JR, Nakanishi K, Parish CA. The lipofuscin fluorophore A2E mediates blue light-induced damage to retinal pigmented epithelial cells. Invest Ophthalmol Vis Sci. 2000;41:1981–1989. [PubMed] [Google Scholar]
- Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest Ophthalmol Vis Sci. 1999;40:2988–2995. [PubMed] [Google Scholar]
- Sparrow JR, Wu Y, Nagasaki T, Yoon KD, Yamamoto K, Zhou J. Fundus autofluorescence and the bisretinoids of retina. Photochem Photobiol Sci. 2010;9:1480–1489. doi: 10.1039/c0pp00207k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow JR, Zhou J, Ben-Shabat S, Vollmer H, Itagaki Y, Nakanishi K. Involvement of oxidative mechanisms in blue-light-induced damage to A2E-laden RPE. Invest Ophthalmol Vis Sci. 2002;43:1222–1227. [PubMed] [Google Scholar]
- Swaroop A, Chew EY, Rickman CB, Abecasis GR. Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu Rev Genomics Hum Genet. 2009;10:19–43. doi: 10.1146/annurev.genom.9.081307.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toku K, Tanaka J, Yano H, Desaki J, Zhang B, Yang L, Ishihara K, Sakanaka M, Maeda N. Microglial cells prevent nitric oxide-induced neuronal apoptosis in vitro. J Neurosci Res. 1998;53:415–425. doi: 10.1002/(SICI)1097-4547(19980815)53:4<415::AID-JNR3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Tuo J, Bojanowski CM, Zhou M, Shen D, Ross RJ, Rosenberg KI, Cameron DJ, Yin C, Kowalak JA, Zhuang Z, Zhang K, Chan CC. Murine ccl2/cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Invest Ophthalmol Vis Sci. 2007;48:3827–3836. doi: 10.1167/iovs.07-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuohy G, Millington-Ward S, Kenna PF, Humphries P, Farrar GJ. Sensitivity of photoreceptor-derived cell line (661W) to baculoviral p35, Z-VAD.FMK, and Fas-associated death domain. Invest Ophthalmol Vis Sci. 2002;43:3583–3589. [PubMed] [Google Scholar]
- Vinet J, van Weering HR, Heinrich A, Kalin RE, Wegner A, Brouwer N, Heppner FL, van Rooijen N, Boddeke HW, Biber K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J Neuroinflammation. 2012;9:27. doi: 10.1186/1742-2094-9-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bernhardi R, Tichauer JE, Eugenin J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem. 2010;112:1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Ohno-Matsui K, Yoshida T, Shimada N, Ichinose S, Sato T, Mochizuki M, Morita I. Amyloid-beta up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: Another mechanism of complement activation in age-related macular degeneration. J Cell Physiol. 2009;220:119–128. doi: 10.1002/jcp.21742. [DOI] [PubMed] [Google Scholar]
- Wu Z, Lauer TW, Sick A, Hackett SF, Campochiaro PA. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J Biol Chem. 2007;282:22414–22425. doi: 10.1074/jbc.M702321200. [DOI] [PubMed] [Google Scholar]
- Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Xu H, Chen M, Manivannan A, Lois N, Forrester JV. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell. 2008;7:58–68. doi: 10.1111/j.1474-9726.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Zarbin MA, Rosenfeld PJ. Pathway-based therapies for age-related macular degeneration: an integrated survey of emerging treatment alternatives. Retina. 2010;30:1350–1367. doi: 10.1097/IAE.0b013e3181f57e30. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wang Z, Liu Y, Song Y, Li Y, Laties AM, Wen R. Translocation of the retinal pigment epithelium and formation of sub-retinal pigment epithelium deposit induced by subretinal deposit. Mol Vis. 2007;13:873–880. [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103:16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50:1392–1399. doi: 10.1167/iovs.08-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel SA, Spaide RF, Curcio CA, Malek G, Imamura Y. Reticular pseudodrusen are subretinal drusenoid deposits. Ophthalmology. 2010;117:303–312. e301. doi: 10.1016/j.ophtha.2009.07.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. In vitro accumulation of autofluorescence in cultured retinal microglial cells loaded with 13-cis retinal and all-trans retinal. Primary mouse retinal microglia were cultured in the absence (top row) and presence (bottom rows) of retinoids, 13-cis retinal and all-trans retinal (6 μM). Cytoplasmic autofluorescence (green) accumulated in retinoid-exposed microglial cells and remained absent in control cells. The autofluorescent signals in loaded cells colocalized with the lysosomal compartment labeled using LysoTracker (red). Scale bar = 25 μm.
Supplementary Figure 2. Effect of in vitro accumulation of retinoids, 13-cis retinal and all-trans retinal on mRNA gene expression. (A) mRNA expression M1 genes (top) that had been significantly increased following A2E accumulation and M2 genes (bottom) that had been significantly decreased following A2E accumulation were evaluated in retinal microglia following incubation with either 13-cis retinal (hatched bars) and all-trans retinal (black bars) (both 6 μM for 6 hours) by RT-PCR. mRNA levels for each gene are expressed as a percentage of that in unloaded retinal microglia (white bars). Significant changes relative to control are indicated by an asterisk (p<0.05, 1-way ANOVA with Dunnett’s multiple comparison test, n = 4 replicates). No significant increases in M1 genes were noted, while only TGF-β decreased with all-trans retinal loading. CD206 mRNA levels were conversely increased with 13-cis retinal loading. Taken together, loading with either 13-cis retinal or all-trans retinal did not result in an overall increase in M1/M2 gene expression ratio seen with A2E loading. (B) mRNA levels of growth factors, which were decreased with A2E loading, were not significantly altered by either 13-cis retinal or all-trans retinal loading (p>0.05 for all comparisons). (C) mRNA levels of complementary regulatory proteins, CFH and CFB, were not significantly altered by either 13-cis retinal or all-trans retinal loading (p>0.05 for all comparisons).