

Abstract
The μ-opioid receptor mediates rewarding effects of alcohol and illicit drugs. We hypothesized that altered DNA methylation in the μ-opioid receptor gene (OPRM1) might influence the vulnerability to alcohol dependence (AD). Genomic DNA was extracted from the peripheral blood of 125 European Americans with AD and 69 screened European American controls. Methylation levels of 16 CpGs in the OPRM1 promoter region were examined by bisulfite sequencing analysis. A multivariate analysis of covariance was conducted to analyze AD-associated methylation changes in the OPRM1 promoter region, using days of intoxication in the past 30 days, sex, age, ancestry proportion and childhood adversity (CA) as covariates. Three CpGs (80, 71, and 10 bp upstream of the OPRM1 translation start site) were more highly methylated in AD cases than in controls (CpG-80: P = 0.033; CpG-71: P = 0.004; CpG-10: P = 0.008). Although these sites were not significant after correction for multiple comparisons, the overall methylation level of the 16 CpGs was significantly higher in AD cases (13.6%) than in controls (10.6%) (P = 0.049). Sex and CA did not significantly influence OPRM1 promoter methylation levels. Our findings suggest that OPRM1 promoter hypermethylation may increase the risk for AD and other substance dependence disorders.
Keywords: alcohol dependence (AD), bisulfite sequencing, childhood adversity, comorbid drug dependence, μ-opioid receptor gene (OPRM1), promoter CpG methylation
INTRODUCTION
Alcohol dependence (AD) occurs commonly in the population. The lifetime prevalence of AD in the US population was estimated to be 13.3%.1,2 AD is a heterogeneous and complex disorder, risk for which involves effects of multiple genes and environmental factors.3 Twin and adoption studies have yielded heritability estimates for the disorder of 50–80%.4,5 Despite its high heritability, much of the individual difference in liability to AD is attributable to the effects of environment.
Environmental effects on complex disorders such as AD may be mediated in part by epigenetic mechanisms (for example, DNA methylation and histone modification) that alter gene expression without changing DNA sequence. Epigenetic modification is essential to cellular development and differentiation, and to the adaptation to environmental changes.6,7 Epigenetic modification may also produce long-lasting changes in gene expression with a range of phenotypic outcomes, and could, for example, lead to increased vulnerability to AD and other disorders. A study by Bonsch et al.8 showed a higher peripheral blood DNA methylation level in the α-synuclein gene in AD patients than in healthy controls. Following this finding, studies using peripheral blood (or lymphoblastoid cell lines derived from peripheral blood lymphocytes) showed that several other genes, including the monoamine oxidase A gene,9 the serotonin transporter gene,10 the N-methyl-d-aspartate 2b receptor subunit gene,11 and the proopiomelanocortin gene12 were more highly methylated in patients with AD than in control subjects. Of interest, an inverse correlation between promoter DNA methylation levels and gene expression levels has been observed. For example, Bonsch et al.13 showed that genomic DNA hypermethylation was associated with a lower expression of the DNA methyltransferase gene in patients with AD. Similarly, hypermethylation of the promoter region of the homocysteine-induced endoplasmic reticulum protein gene was associated with downregulation of homocysteine-induced endoplasmic reticulum protein gene expression in patients with AD.14
The μ-opioid receptor gene (OPRM1) is a candidate gene involved in the reward pathway of alcohol or drugs of abuse and may exhibit altered DNA methylation levels in patients with alcohol or drug dependence. Naltrexone, an opioid receptor antagonist, is approved in a number of countries for the treatment of AD.15–17 Variation in OPRM1 moderated naltrexone treatment response in alcoholic individuals in some,18,19 but not all studies.20 Furthermore, while some genetic association studies have demonstrated that OPRM1 variation influences the susceptibility to AD,21,22 others have not.23 As the methylation state of a gene (especially in the promoter region) is a mechanism by which genetic diversity could influence gene expression and function, OPRM1 could, like the above-mentioned genes, also had altered DNA methylation levels in patients with AD. If so, this could reflect a pathophysiological mechanism in which the expression and function of the μ-opioid receptor are altered after chronic alcohol use, leading it to be more or less sensitive to endogenous opioids and opioid antagonists such as naltrexone.
Nielsen et al.24 investigated methylation profiles of 16 CpGs in the OPRM1 promoter region in methadone-maintained former heroin addicts and healthy controls. Different DNA methylation patterns were observed between former heroin addicts and controls. Two CpGs, one 18 bp upstream of the OPRM1 translation start site (TSS) (coded as CpG-18) and another 84 bp downstream of the OPRM1 TSS (coded as CpG + 84), showed increased methylation levels relative to the CpG profiles of controls. Moreover, a recent study also showed hypermethylation of OPRM1 promoter CpGs in blood and sperm from male opioid addicts.25 Although opioid addiction is distinct from AD, a high co-occurrence of AD and opioid dependence has been reported.26,27 Thus, genetic variation or epigenetic modification of OPRM1 may have an overlapping role in both disorders. Considering the previous findings, we hypothesized that hypermethylation of OPRM1 would be observable in subjects with AD. The present study was undertaken to investigate whether there are increased DNA methylation levels in the promoter region of OPRM1 in AD patients. Additionally, the influence of sex, childhood adversity (CA) and comorbid drug dependence on OPRM1 promoter methylation was also analyzed. As brain tissue is not easily accessible, we used genomic DNA from peripheral blood to conduct this study. Support for our hypothesis would be important for the potential utility of the peripheral lymphocyte DNA methylation level as a biomarker for AD.
SUBJECTS AND METHODS
Study subjects
As shown in Table 1, 125 European Americans with AD (81 or 64.8% male) and 69 European American healthy controls (34 or 49.3% male) were recruited from substance abuse treatment centers and through advertisements at the University of Connecticut Health Center (n = 87), Yale University (n = 53), and the Medical University of South Carolina (n = 54). Among the affected subjects, the following drug dependence comorbidities were observed: 45 (36.0%) were diagnosed with cocaine dependence, 14 (11.2%) were diagnosed with opioid dependence, and 31 (24.8%) were diagnosed with marijuana dependence. Cases and controls were 41±13 (mean±s.d.) and 39±15 (mean±s.d.) years old, respectively. Subjects were interviewed using an electronic version of the Semi-Structured Assessment for Drug Dependence and Alcoholism28 to derive lifetime diagnoses for AD according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV). Controls were screened to exclude those with alcohol or drug (for example, cocaine, opioid, marijuana) abuse or dependence. Both case and control subjects were screened to exclude individuals with a psychotic disorder (schizophrenia or bipolar disorder) or a major depressive disorder. Additionally, all participants were asked whether, by age 13, they had witnessed or experienced violent crimes such as shooting or rape or been sexually or physically abused, or by age six, either of their parents had died. Endorsement of any of these adverse childhood experiences was coded as positive for exposure to CA. Forty-six cases (36.8%) and six controls (8.7%) were exposed to CA. Subjects gave informed consent as approved by the institutional review board at each clinical site, and certificates of confidentiality were obtained from the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism.
Table 1.
Characteristics of European American cases and controls
| Cases with AD (n = 125) | Controls (n = 69) | |
|---|---|---|
| AD only, n (%) | 61 (48.8%) | 0 (0%) |
| AD + CD, n (%) | 45 (36.0%) | 0 (0%) |
| AD + OD, n (%) | 14 (11.2%) | 0 (0%) |
| AD + MjD, n (%) | 31 (24.8%) | 0 (0%) |
| Days of intoxication in the past 30 days (mean ± s.d.) | 5 ± 7, t= −4.36, P<0.001 | 1 ± 3 |
| Years of intoxication lifetime (mean ± s.d.) | 12 ± 9, t= −10.06, P<0.001 | 0 ± 1 |
| Sex, male (%) | 81 (64.8%) | 34 (49.3%) |
| Age (meant±s.d.) | 41 ± 13 | 39 ± 15 |
| AD cases with comorbid CD | 45 (36.0%) | 0 (0%) |
| AD cases with comorbid OD | 14 (11.2%) | 0 (0%) |
| AD cases with comorbid MjD | 31 (24.8%) | 0 (0%) |
| Childhood adversity (CA) | 46 (36.8%) | 6 (8.7%) |
Abbreviations: AD, alcohol dependence; CD, cocaine dependence; MjD, marijuana dependence; OD, opioid dependence; AD + CD, AD with comorbid cocaine dependence; AD + OD, AD with comorbid opioid dependence; AD+ MjD, AD with comorbid marijuana dependence.
Genomic DNA extraction and bisulfite modification
Genomic DNA was extracted from peripheral blood. Two hundred nanograms of genomic DNA was treated with the CT Conversion reagent included in the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) according to the manufacturer's protocol. The final bisulfite-treated genomic DNA was eluted from the Zymo-Spin IC Column (Zymo Research) in 20 μl of M-Elution buffer.
Bisulfite DNA sequencing
The PCR primers (F1: 5′-TTTTTTTTTGTTTTAGTTAGG-3′; R1: 5′-CAAATTACCATCTAAATAAA-3′) and nested PCR primers (F2: 5′-TGTAAGAAATAGTAGGAGTTGTGGTAG-3′; R2: 5′-AATAAAACAAATTAACCCAAAAACC-3′) to amplify the promoter region of OPRM1 (harboring 16 CpG sites located at nucleotides −93, −90, −80, −71, −60, −50, and −32, −25, −18, −14, −10, + 12, + 23, + 27, +53 and 84 (the minus symbol identifies the CpG sites located in the upstream of the TSS and the plus symbol identifies the CpG sites located in the downstream of the TSS)) were the same as those described by Nielsen et al.24 The conditions for PCRs and nested PCRs were similar to those described by Nielsen et al.,24 except that we used the PfuUltra II Fusion HS DNA Polymerase (Stratagene, Santa Clara, CA, USA) and a reaction volume of 20 μl. The primer information and CpG locations are illustrated in Figure 1.
Figure 1.
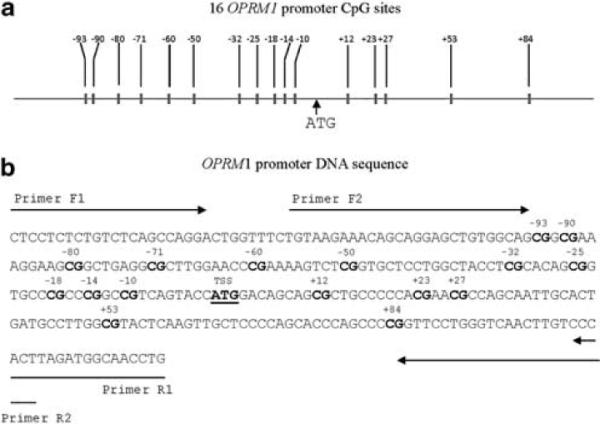
OPRM1 promoter DNA sequence and CpG sites. (a) Locations of 16 OPRM1 promoter CpG sites. (b) OPRM1 promoter CpG sites are numbered as the distance (in bases) upstream (`−') or downstream (`+') of the first base (`A') of the translation start site `ATG'. Sequences of forward and reverse primers (F1, F2, R1 and R2) for polymerase chain reactions are indicated in the figure. A full color version of this figure is available at the Journal of Human Genetics journal online.
Nested PCR products (5 μl) were mixed with 2 μl of ExoSAP-IT (USB Corp., Cleveland, OH, USA) and incubated at 37 °C for 15 min to degrade remaining primers and nucleotides. To inactivate ExoSAP-IT, the mixture was incubated at 80 °C for 15 min. We subjected 3.5 μl of ExoSAP-IT-treated PCR products to direct sequencing using either the nested PCR forward or reverse primers as sequencing primers. Sequence analysis was carried out on a 48-capillary 3730 DNA Analyzer (Applied Biosystems, Carlsbad, CA, USA) at the Yale core facility (The Keck Biotechnology Resource Laboratory, New Haven, CT, USA). The sequencing reaction used fluorescently-labeled dideoxynucleotides included in the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) and the PfuUltra II Fusion HS DNA Polymerase (Stratagene). The sequencing experiment was conducted according to the manufacturer's instructions.
DNA methylation quantification
Sequencing results were in ABI trace file format, which contained information on the peak height of each base. The ABI.pm module of BioPerl version 1.5.229 (http://www.bioperl.org) was used to extract the peak height of cytosine (C) (representing the proportion of methylated base C) and thymine (T) (representing the proportion of unmethylated base C) at each CpG site from the electropherogram file. The percentage of DNA methylation was calculated as the peak height of base C vs the peak height of base C plus the peak height of base T (that is, C/(C+T)%).30 As one OPRM1 promoter CpG site (CpG-50) was also included in the Illumina GoldenGate methylation array (probe ID for CpG-50: cg13887561) (Illumina, San Diego, CA, USA) designed for our recent study31 and 95 samples were determined by both the methods (bisulfate sequencing in this study and methylation array-based assay in our previous study), we were able to validate our bisulfate sequencing-based DNA methylation measurements. At this CpG site (CpG-50), the degree of methylation determined by each method was moderately, but significantly correlated (Pearson's correlation coefficient=0.402, P<0.001), suggesting that the current measurements were accurate.
Statistical and bioinformatics analysis
Methylation levels of 16 CpG sites in the promoter region of OPRM1 were compared between AD cases and healthy controls using the general linear model multivariate analysis of covariance incorporated in the SPSS 17.0 software (SPSS Inc., Chicago, IL, USA). The number of days of intoxication in the past 30 days, sex, age, ancestry proportion, and CA were included as covariates. The European ancestry proportion of each subject was estimated by examining a set of ancestry informative markers as described in our previous study.32 Additionally, the influence of sex, CA and comorbid drug dependence on OPRM1 promoter methylation levels was also analyzed using multivariate analysis of covariance. Bonferroni correction was used to adjust the P-values (the P-value for significance was set at 0.05/16=0.003, for comparison of methylation levels of 16 CpGs between cases and controls).
RESULTS
OPRM1 promoter methylation differences between all AD cases and controls
Three CpG sites (CpG-80, CpG-71 and CpG-10) had significantly higher methylation levels in all AD cases (mean±s.e.m. (%): 27.6±2.0, 24.3±1.7 and 14.2±1.0, respectively) than in healthy controls (mean±s.e.m. (%): 19.9±2.8, 15.7±2.3 and 9.3±1.4, respectively) (CpG-80: P=0.033; CpG-71: P=0.004; CpG-10: P=0.008) (Figure 2). Although the P-values from these three CpGs did not survive multiple testing corrections, the overall methylation level of the 16 OPRM1 CpGs was significantly higher in all AD cases (mean±s.e.m. (%): 13.6±0.9) than in healthy controls (mean±s.e.m. (%): 10.6±1.2) (P=0.049) (Supplementary Table S1).
Figure 2.
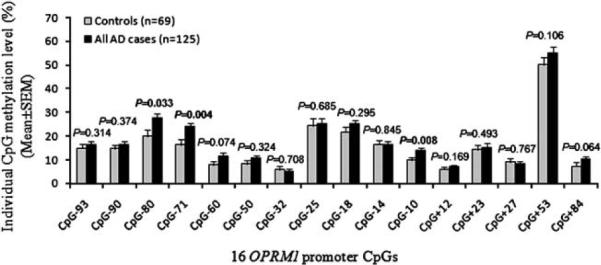
Methylation differences of 16 OPRM1 promoter CpGs between all alcohol dependent cases and healthy controls. The association between alcohol dependence (AD) and OPRM1 promoter CpG methylation was analyzed using the multivariate analysis of covariance with sex, age, ancestry proportion and childhood adversity as covariates. X-axis: 16 OPRM1 promoter CpG sites; Y-axis: Individual CpG methylation level (mean±s.e.m.), and black bars denote all cases with AD (n =125) and gray bars denote control subjects (n = 69).
OPRM1 promoter methylation differences between AD-only cases and controls
When cases with only AD (that is, without comorbid cocaine dependence, opioid dependence or marijuana dependence) were considered, two of the above three CpGs were hypermethylated in cases compared with controls (CpG-71: P=0.005; CpG-10: P=0.013) (Figure 3 and Supplementary Table S2). The P-values did not survive correction for multiple testing.
Figure 3.
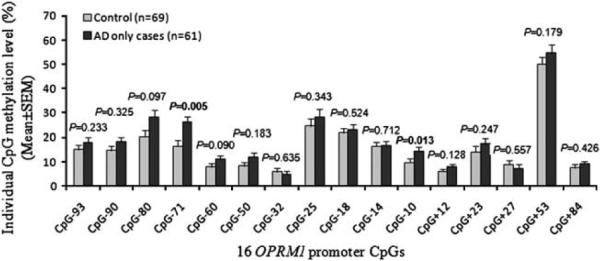
Methylation differences of 16 OPRM1 promoter CpGs between cases with only alcohol dependence (AD) and healthy controls. The association between AD and OPRM1 promoter CpG methylation was analyzed using the multivariate analysis of covariance with sex, age, ancestry proportion and childhood adversity as covariates. X-axis: 16 OPRM1 promoter CpG sites; Y-axis: Individual CpG methylation level (mean±s.e.m.), and black bars denote cases with only AD (n = 61) and gray bars denote control subjects (n = 69).
Association of comorbid drug dependence and OPRM1 promoter methylation
Methylation levels of 16 OPRM1 promoter CpGs were compared between AD cases with comorbid drug dependence and healthy controls. One CpG was hypermethylated in AD cases with comorbid cocaine dependence (CpG-10: P=0.024) (Supplementary Table S3), three CpGs were hypermethylated in AD cases with comorbid opioid dependence (CpG-80: P=0.002; CpG-71: P=0.033; CpG-60: P=0.033) (Supplementary Table S4), and two CpGs were hypermethylated in AD cases with comorbid marijuana dependence (CpG-71: P=0.017; CpG-10: P=0.032) (Supplementary Table S5) compared with healthy controls. The P-values did not survive correction for multiple testing.
Influence of sex and CA on OPRM1 promoter methylation
No significant differences in methylation levels of 16 OPRM1 promoter CpGs were observed between males and females in either cases or controls (Supplementary Figure S1). Although there were significantly more AD cases with CA (36.8%) than healthy controls with CA (8.7%) (χ2=16.50, df=1, P<0.001) (Table 1), CA did not show a striking effect on OPRM1 promoter methylation in either AD cases (except CpG-60, but the P-value (0.046) from CpG-60 did not survive multiple testing corrections) or healthy controls (Supplementary Figure S2).
DISCUSSION
Dependence on alcohol and drugs of abuse has a moderate to high heritability component.33 In addition to variation in base sequence, epigenetic modification of chromosomes may also be associated with substance dependence. The present study demonstrated that there was a significantly higher DNA methylation of the OPRM1 promoter region in AD cases than in healthy controls. In particular, hypermethylation of two closely mapped CpGs (CpG-80 and CpG-71) and one additional CpG (CpG-10) (all in the upstream of the TSS) was observed in AD cases (Figure 2). Moreover, alcohol or drug dependence may influence methylation levels of OPRM1 promoter CpGs in either a common or specific way. Altered methylation of certain OPRM1 promoter CpGs may be associated with a specific substance dependence trait while other OPRM1 promoter CpGs may show changed methylation in subjects with different substance dependence traits. The two CpG sites (CpG-18 and CpG+84) that were found to be hypermethylated in former heroin addicts24 did not show significant methylation differences between AD cases and controls in the present study. Nevertheless, three closely mapped CpGs (CpG-80, CpG-71 and CpG-60) in the OPRM1 promoter were hypermethylated in cases with comorbid AD and opioid dependence compared with controls. Presumably, hypermethylation of these CpGs located in the OPRM1 promoter may block the binding of transcription factors, resulting in lowered transcription of OPRM1and increased risk for substance dependence disorders.
There are two possible interpretations for DNA hypermethylation in subjects with alcohol and/or drug dependence. First, greater DNA methylation may predispose to the development of AD and may have arisen before alcohol consumption and/or drug use. Epigenetic changes (including DNA methylation) accumulate dynamically in the genome during the course of life (especially at specific stages of life).34 As epigenetic modifications, like gene variation, can regulate gene transcription, one possible consequence of such changes could be to increase an individual's vulnerability to alcohol and/or drug dependence. Possible moderators of gene methylation are social bonding and early-life stress—both are known to affect opioid neurochemistry and later behavior.35 However, the present study did not show significant effects of CA on OPRM1 promoter methylation. This may be due to the limited sample size. A second possibility is that the greater DNA methylation is the consequence of long-term heavy drinking. DNA methylation depends upon the availability of a methyl group from S-adenosylmethionine. S-adenosylmethionine is derived from methionine, which is synthesized through the methylation of homocysteine.36 Chronic alcohol intake has been associated with elevated homocysteine plasma concentrations.37 Thus, alcohol drinking or nutrition deficiency may result in altered DNA methylation levels of specific genes such as OPRM1. Additionally, changed DNA methylation levels may result from neuroadaptation to chronic alcohol consumption. Although several research groups have now observed an association between DNA methylation and alcohol and/or drug dependence, differentiating DNA methylation as a predisposing factor from that which is a consequence of alcohol and/or drug use requires further research.
The present study has a number of limitations. First, the sample size was comparatively small. Second, we only included subjects from the European American population. Given the fact that DNA methylation status may vary by populations, subsequent studies should examine methylation patterns in alcoholics from other populations. Third, we considered DNA methylation differences in only one gene. As AD is a complex disorder, DNA methylation levels of a number of other genes participating in either alcohol metabolism or brain reward pathways should be analyzed. Ideally, the association of DNA methylation and AD would be analyzed at a genome-wide level. Finally, we did not examine OPRM1 expression levels in blood samples because they were collected years earlier and were unavailable for RNA extraction. We were not able to directly analyze the correlation between DNA methylation and OPRM1 expression in peripheral blood cells. Most recently, we measured OPRM1 expression levels in postmortem human-brain prefrontal cortex tissues (Brod-mann area 9) in 23 pairs of cases (affected with AD or abuse) and controls of European ancestry (cases and controls were matched by sex, age, postmortem intervals and alcohol consumption amounts, and so on.) using the HumanHT-12 v4 Expression BeadChip assay (Illumina). No significant differences in brain prefrontal cortex OPRM1 expression levels were observed between cases and controls (data not shown). The negative finding may be due to (1) the small sample studied, or (2) tissue-specific methylation/expression of OPRM1. Future studies should also examine AD-associated OPRM1 expression levels in other brain reward regions such as the nucleus accumbens and the ventral tegmental area.
Identification of epigenetic marks (for example, methylated CpG sites in genes) can help to understand both the genetic and epigenetic mechanisms of AD, as well as a variety of other diseases. Importantly, dietary or pharmacological interventions may be used to treat AD and other disorders by modifying epigenetic marks. Additionally, changes in such marks may be useful biomarkers for treatment responses. Current findings suggest that more in-depth research combining genetic and epigenetic approaches could be a powerful means to identify both genetic and environmental risk factors for complex disorders. Importantly, studies that simultaneously examine peripheral and brain gene methylation are crucial for understanding the relevance of this work to behavior and treatment.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by the National Institute of Health (NIH) Grants K99/R00 DA022891 (HZ), R01 DA12690 (JG), R01 AA11330 (JG), R01 DA12849 (JG), R01 AA017535 (JG), R01 DA018432 (HRK) and grants from the VA CT MIRECC Center to JG and the Alcoholic Beverage Medical Research Foundation (ABMRF) to HZ.
Dr Kranzler has received compensation for professional services from the National Institutes of Health (NIAAA and NIDA) and for academic lectures and editorial functions in various scientific venues (including the ACNP). Dr Kranzler has had consulting arrangements with the following pharmaceutical companies: Alkermes, Lilly, Lund-beck, Pfizer and Roche. Dr Anton has had consulting agreements with the following companies: Eli Lilly, GlaxoSmithKline and Alkermes. Drs Kranzler and Anton have also received honoraria from the Alcohol Clinical Trials Initiative (ACTIVE), which Eli Lilly, Schering Plough, Lundbeck, Alkermes, GlaxoSmithKline, Abbott and Johnson & Johnson support. Dr Anton reports research support from Lilly. Dr Gelernter reports that he has received compensation for professional services in the previous 3 years from the following entities: Yale University School of Medicine, Veterans Affairs Healthcare System (VA) and the National Institutes of Health (NIAAA, NIDA, and NIMH), and related to academic lectures and editorial functions in various scientific venues (including the ACNP).
Footnotes
CONFLICT OF INTEREST The remaining authors declare no conflict of interest.
References
- 1.Grant BF. Prevalence and correlates of alcohol use and DSM-IV alcohol dependence in the United States: results of the National Longitudinal Alcohol Epidemiologic Survey. J. Stud. Alcohol. 1997;58:464–473. doi: 10.15288/jsa.1997.58.464. [DOI] [PubMed] [Google Scholar]
- 2.Grant BF, Dawson DA, Stinson FS, Chou SP, Dufour MC, Pickering RP. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend. 2004;74:223–234. doi: 10.1016/j.drugalcdep.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 3.Gelernter J, Kranzler HR. Genetics of alcohol dependence. Hum. Genet. 2009;126:91–99. doi: 10.1007/s00439-009-0701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heath AC, Bucholz KK, Madden PA, Dinwiddie SH, Slutske WS, Bierut LJ, et al. Genetic and environmental contributions to alcohol dependence risk in a national twin sample: consistency of findings in women and men. Psychol. Med. 1997;27:1381–1396. doi: 10.1017/s0033291797005643. [DOI] [PubMed] [Google Scholar]
- 5.Knopik VS, Heath AC, Madden PA, Bucholz KK, Slutske WS, Nelson EC, et al. Genetic effects on alcohol dependence risk: re-evaluating the importance of psychiatric and other heritable risk factors. Psychol. Med. 2004;34:1519–1530. doi: 10.1017/s0033291704002922. [DOI] [PubMed] [Google Scholar]
- 6.Kramer DA. Commentary: Gene-environment interplay in the context of genetics, epigenetics, and gene expression. J. Am. Acad. Child. Adolesc. Psychiatry. 2005;44:19–27. doi: 10.1097/01.chi.0000145804.30112.6b. [DOI] [PubMed] [Google Scholar]
- 7.Frisancho AR. Developmental adaptation: where we go from here. Am. J. Hum. Biol. 2009;21:694–703. doi: 10.1002/ajhb.20891. [DOI] [PubMed] [Google Scholar]
- 8.Bonsch D, Lenz B, Kornhuber J, Bleich SDNA. Hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport. 2005;16:167–170. doi: 10.1097/00001756-200502080-00020. [DOI] [PubMed] [Google Scholar]
- 9.Philibert RA, Gunter TD, Beach SR, Brody GH, Madan A. MAOA methylation is associated with nicotine and alcohol dependence in women. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2008;147B:565–570. doi: 10.1002/ajmg.b.30778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philibert RA, Sandhu H, Hollenbeck N, Gunter T, Adams W, Madan A. The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008;147B:543–549. doi: 10.1002/ajmg.b.30657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biermann T, Reulbach U, Lenz B, Frieling H, Muschler M, Hillemacher T, et al. N-methyl-D-aspartate 2b receptor subtype (NR2B) promoter methylation in patients during alcohol withdrawal. J. Neural. Transm. 2009;116:615–622. doi: 10.1007/s00702-009-0212-2. [DOI] [PubMed] [Google Scholar]
- 12.Muschler MA, Hillemacher T, Kraus C, Kornhuber J, Bleich S, Frieling H. DNA methylation of the POMC gene promoter is associated with craving in alcohol dependence. J. Neural. Transm. 2010;117:513–519. doi: 10.1007/s00702-010-0378-7. [DOI] [PubMed] [Google Scholar]
- 13.Bonsch D, Lenz B, Fiszer R, Frieling H, Kornhuber J, Bleich S. Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. J. Neural. Transm. 2006;113:1299–1304. doi: 10.1007/s00702-005-0413-2. [DOI] [PubMed] [Google Scholar]
- 14.Bleich S, Lenz B, Ziegenbein M, Beutler S, Frieling H, Kornhuber J, et al. Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcohol Clin. Exp. Res. 2006;30:587–591. doi: 10.1111/j.1530-0277.2006.00068.x. [DOI] [PubMed] [Google Scholar]
- 15.Anton RF, O'Malley SS, Ciraulo DA, Cisler RA, Couper D, Donovan DM, et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence: the COMBINE study: a randomized controlled trial. JAMA. 2006;295:2003–2017. doi: 10.1001/jama.295.17.2003. [DOI] [PubMed] [Google Scholar]
- 16.O'Malley SS, Robin RW, Levenson AL, GreyWolf I, Chance LE, Hodgkinson CA, et al. Naltrexone alone and with sertraline for the treatment of alcohol dependence in Alaska natives and non-natives residing in rural settings: a randomized controlled trial. Alcohol Clin. Exp. Res. 2008;32:1271–1283. doi: 10.1111/j.1530-0277.2008.00682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ray LA, Chin PF, Miotto K. Naltrexone for the treatment of alcoholism: clinical findings, mechanisms of action, and pharmacogenetics. CNS Neurol. Disord. Drug Targets. 2010;9:13–22. doi: 10.2174/187152710790966704. [DOI] [PubMed] [Google Scholar]
- 18.Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR, et al. A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology. 2003;28:1546–1552. doi: 10.1038/sj.npp.1300219. [DOI] [PubMed] [Google Scholar]
- 19.Anton RF, Oroszi G, O'Malley S, Couper D, Swift R, Pettinati H, et al. An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch. Gen. Psychiatry. 2008;65:135–144. doi: 10.1001/archpsyc.65.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gelernter J, Gueorguieva R, Kranzler HR, Zhang H, Cramer J, Rosenheck R, et al. Opioid receptor gene (OPRM1, OPRK1, and OPRD1) variants and response to naltrexone treatment for alcohol dependence: results from the VA Cooperative Study. Alcohol Clin. Exp. Res. 2007;31:555–563. doi: 10.1111/j.1530-0277.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Luo X, Kranzler HR, Lappalainen J, Yang BZ, Krupitsky E, et al. Association between two mu-opioid receptor gene (OPRM1) haplotype blocks and drug or alcohol dependence. Hum. Mol. Genet. 2006;15:807–819. doi: 10.1093/hmg/ddl024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van der Zwaluw CS, van den Wildenberg E, Wiers RW, Franke B, Buitelaar J, Scholte RH, et al. Polymorphisms in the mu-opioid receptor gene (OPRM1) and the implications for alcohol dependence in humans. Pharmacogenomics. 2007;8:1427–1436. doi: 10.2217/14622416.8.10.1427. [DOI] [PubMed] [Google Scholar]
- 23.Arias A, Feinn R, Kranzler HR. Association of an Asn40Asp (A118G) polymorphism in the mu-opioid receptor gene with substance dependence: a meta-analysis. Drug Alcohol Depend. 2006;83:262–268. doi: 10.1016/j.drugalcdep.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen DA, Yuferov V, Hamon S, Jackson C, Ho A, Ott J, et al. Increased OPRM1 DNA methylation in lymphocytes of methadone-maintained former heroin addicts. Neuropsychopharmacology. 2009;34:867–873. doi: 10.1038/npp.2008.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chorbov V, Todorov A, Lynskey M, Cicero T. Elevated levels of DNA methylation at the OPRM1 promoter in blood and sperm from male opioid addicts. J. Opioid. Manag. 2011;7:258. doi: 10.5055/jom.2011.0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caetano R, Schafer J. DSM-IV alcohol dependence and drug abuse/dependence in a treatment sample of whites, blacks and Mexican Americans. Drug Alcohol Depend. 1996;43:93–101. doi: 10.1016/s0376-8716(96)01296-3. [DOI] [PubMed] [Google Scholar]
- 27.Ottomanelli G. Methadone patients and alcohol abuse. J. Subst. Abuse Treat. 1999;16:113–121. doi: 10.1016/s0740-5472(98)00030-0. [DOI] [PubMed] [Google Scholar]
- 28.Pierucci-Lagha A, Gelernter J, Feinn R, Cubells JF, Pearson D, Pollastri A, et al. Diagnostic reliability of the Semi-structured Assessment for Drug Dependence and Alcoholism (SSADDA) Drug Alcohol Depend. 2005;80:303–312. doi: 10.1016/j.drugalcdep.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 29.Stajich JE. An Introduction to BioPerl. Methods Mol. Biol. 2007;406:535–548. doi: 10.1007/978-1-59745-535-0_26. [DOI] [PubMed] [Google Scholar]
- 30.Jiang M, Zhang Y, Fei J, Chang X, Fan W, Qian X, et al. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab. Invest. 2010;90:282–290. doi: 10.1038/labinvest.2009.132. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Herman AI, Kranzler HR, Anton RF, Zhao H, Zheng W, et al. Array-based profiling of DNA methylation changes associated with alcohol dependence. Alcohol Clin. Exp. Res. 2012 doi: 10.1111/j.1530-0277.2012.01928.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, Kranzler HR, Weiss RD, Luo X, Brady KT, Anton RF, et al. Pro-opiomelanocortin gene variation related to alcohol or drug dependence: evidence and replications across family- and population-based studies. Biol. Psychiatry. 2009;66:128–136. doi: 10.1016/j.biopsych.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goldman D, Oroszi G, Ducci F. The genetics of addictions: uncovering the genes. Nat. Rev. Genet. 2005;6:521–532. doi: 10.1038/nrg1635. [DOI] [PubMed] [Google Scholar]
- 34.Rutten BP, Mill J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr. Bull. 2009;35:1045–1056. doi: 10.1093/schbul/sbp104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wittmann W, Schunk E, Rosskothen I, Gaburro S, Singewald N, Herzog H, et al. Prodynorphin-derived peptides are critical modulators of anxiety and regulate neurochemistry and corticosterone. Neuropsychopharmacology. 2009;34:775–785. doi: 10.1038/npp.2008.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J. Nutr. 2002;1328(Suppl):2333S–2335S. doi: 10.1093/jn/132.8.2333S. [DOI] [PubMed] [Google Scholar]
- 37.Lutz UC. Alterations in homocysteine metabolism among alcohol dependent patients-clinical, pathobiochemical and genetic aspects. Curr. Drug Abuse Rev. 2008;1:47–55. doi: 10.2174/1874473710801010047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.