

Abstract
Approximately one third of all deaths are attributed to cardiovascular disease (CVD), making it the biggest killer worldwide. Despite a number of therapeutic options available, the burden of CVD morbidity continues to grow indicating the need for continued research to address this unmet need. In this respect, investigation of the mechanisms underlying the protection that premenopausal females enjoy from cardiovascular-related disease and mortality is of interest. In this review, we discuss the essential role that rodent animal models play in enabling this field of research. In particular, we focus our discussion on models of hypertension and atherosclerosis.
Keywords: SHR, Dahl, deoxycorticosterone, angiotensin, gender, rat, mouse, apolipoprotein, low-density lipoprotein, blood pressure, oestrogen, testosterone
Introduction
Cardiovascular disease (CVD) now stands as the major cause of mortality worldwide. According to the World Health Organization, an estimated 17.1 million people died from CVD in 2004, and recent estimates suggest that in the west, a third of all deaths are a direct consequence of CVD (http://www.BHF.org.uk). The scale of this worldwide epidemic underlies the urgency for identifying novel potential targets and therapeutics.
In this respect, perhaps one of the most striking characteristics of this epidemic is that in the main, irrespective of country, the incidence of most forms of CVD is lower in premenopausal women than in age-matched men (Barrett-Connor, 1997; Lerner and Kannel, 1986; Danaei et al., 2011), a pattern that is rapidly lost and, at worst, completely reversed post-menopause (Kannel et al., 1976). This fact has led scientists to believe that sex hormones play an important role in determining susceptibility to CVD. In particular, there is a wealth of evidence suggesting that female sex hormones exert beneficial effects and that the loss of these at menopause underlies the increase in incidence of CVD post-menopause. Fortunately, these prominent sex differences are similarly evident in animal models. For instance, spontaneously hypertensive rats (SHR), the most commonly used animal model of hypertension (Doggrell and Brown, 1998; Pinto et al., 1998), show age-dependent increases in blood pressure (BP) that is greater in males compared with age-matched females. This example demonstrates that animal models of disease can faithfully replicate the sex bias evident in the clinical situation and support its utilization for elucidation of mechanisms and therapeutics of hypertension. However, the similarity between humans and animal disease models is not always evident. A good exemplar of this is mouse models of atherosclerosis where, in contrast to humans, a greater plaque burden is evident in females compared with age-matched males.
The primary aim of this review is to highlight the sexual dimorphism evident in animal models of CVD. In particular, we limit our attention to hypertension and atherosclerosis in rodents and discuss the limitations of some of the techniques used. In addition, we discuss the role that sex hormones play in any diversity evident. Despite their importance, we do not extend our review further to wider models of CVD such as ischaemic disease where sex differences have also been identified. We refer the readers to some excellent reviews that cover these aspects (Murphy and Steenbergen, 2007; Vaccarino et al., 2011).
I. Hypertension
Prior to menopause, BP is lower in women than in men resulting in a lower incidence overall of hypertension in women. In the National Health and Nutrition Examination Survey III of 9901 adults in the USA, women had lower systolic BP than men, as measured by automated clinic brachial artery BP measurement, across the ages of 18–59. However, with increasing age, this sex difference was lost (Burt et al., 1995). Such differences in BP are also evident in 24 h ambulatory BP measures (Wiinberg et al., 1995). Importantly, 24 h ambulatory BP monitoring (ABPM) is becoming increasingly accepted as the most accurate method for determination of BP and diagnosis of hypertension due to avoidance of the white coat effect (Mancia et al., 2007). Even in a small cohort of healthy, young and normotensive volunteers in our own clinical study, rested clinic systolic BP was 25 mmHg lower in women than in men (Kapil et al., 2010). These sex differences have variously been attributed to the predominantly beneficial or detrimental activity of the sex hormones oestrogen (Ashraf and Vongpatanasin, 2006) or testosterone (Kaushik et al., 2010) respectively.
Despite these findings in humans, studies in commonly used healthy laboratory animals tend to demonstrate no differences between males and females in resting conscious BP measurement, using tail-cuff (Ashton and Balment, 1991; Sainz et al., 2004; Scotland et al., 2005), a technique analogous to clinic BP measurement. A similar absence of difference is also evident in some studies using the more accurate and sensitive radiotelemetric method (analogous to 24 h ABPM) in either rats (e.g. Calhoun et al., 1995; Haywood & Hinojosa-Laborde, 1997; Sampson et al., 2008) or mice (e.g. Xue et al., 2005). However, in a recent study, telemetric measurement of BP in rats was sufficiently sensitive to expose ∼4 mmHg higher resting BP in males compared with females (Sartori-Valinotti et al., 2008). Interestingly, in male mice lacking a functional oestrogen receptor β (ERβ), one of the two ER subtypes that govern oestrogen activity, telemetric assessment of BP demonstrated raised pressures compared with wild-type controls (Zhu et al., 2002). These latter two studies suggest that the relative absence of data demonstrating sex differences in healthy animals may simply be a reflection of the limitations of the technology used. Such studies highlight the importance of using radiotelemetry when investigating subtle differences in BP. To make reasonable comparisons between animal models and the clinical situation, in this review, we will focus our discussion on studies conducted in conscious animals, as there is now general acceptance that the measurement of baseline BP in anaesthetized animals is not a good reflection of awake BP. In addition, where possible, we will attempt to limit our discussion to studies using the most sensitive technique of BP measurement: radiotelemetry. However, in some instances, our discussion will inevitably focus on tail-cuff BP measurements where radiotelemetric evidence is not yet available.
Despite a relative absence of reliable data demonstrating sex differences in baseline BP in healthy animals, it is clear that the picture is entirely different in rodent models of hypertension (Table 1). For the sake of brevity, we have limited our discussion to the most commonly used mouse and rat models. For a broader description of the different models of hypertension available across the species, we refer readers to an excellent review on the subject (Lerman et al., 2005). There are several important caveats to consider in our discussion. Human hypertension is a complex disease influenced by both environmental factors and genetic variability, and it is a combination of these that underlies the condition. Very few animal models combine these two factors, and therefore most animal models provide information on discrete aspects of hypertension that may be relevant to humans. In addition, in terms of the sex differences in hypertension and the influence of sex hormones, the normal menstrual cycle of rodents lasts ∼4 days, and rodents do not naturally enter menopause. These differences should be considered when extrapolating observations from animal models to the clinical setting.
Table 1.
Sexual dimorphism of blood pressure in animal models of hypertension measured in conscious animals
| Type of hypertension | Magnitude in blood pressure shift of hypertensive males over females | References |
|---|---|---|
| Spontaneously hypertensive rat | MAP ↑∼10 mmHg | Sullivan et al. (2010) |
| SBP ↑∼35 mmHg | Chen and Meng (1991) | |
| SBP ↑∼25 mmHg | Reckelhoff et al. (1998) | |
| DOCA-salt | SBP ↑∼25 mmHg | Ouchi et al. (1987) |
| Dahl salt-sensitive | SBP ↑∼30 mmHg | Dahl et al. (1975) |
| Renal-wrap + salt | MAP ↑ 30 mmHg | Haywood and Hinojosa-Laborde (1997) |
| Transgenic mRen2(27) | MAP ↑∼30 mmHg | Lee et al. (1996) |
| mRen2.Lewis | MAP ↑∼50 mmHg | Pendergrass et al. (2008) |
| l-NAME-treated rats | SBP ↑∼30 mmHg, | Sainz et al. (2004) |
| MAP ↑27 mmHg | ||
| eNOS KO | SBP ↑∼22 mmHg | Scotland et al. (2005) |
| eNOS/COX-1 KO | SBP ↑∼21 mmHg | Scotland et al. (2005) |
| AngII-induced | MAP ↑30 mmHg | Xue et al. (2005) |
| MAP ↑∼20 mmHg | Sampson et al. (2008) | |
| Developmentally induced | MAP ↑∼20 mmHg | Alexander (2003) |
In all models described, there is a greater elevation of blood pressure in males compared with females that matches closely to the clinical scenario. Several animal models that have not been discussed within the text are included here to highlight the consistency observed amongst different hypertensive animal models.
AngII, angiotensin II; DOCA, deoxycorticosterone acetate; COX-1 KO, COX 1 knockout; eNOS KO, eNOS knockout; l-NAME, nitro-l-arginine methyl ester; MAP, mean arterial pressure; SBP, systolic blood pressure.
SHR
Sex differences
The SHR is the most commonly used model of primary hypertension and is thought to be a consequence of a number of genetic variants (see Doggrell and Brown, 1998; Pinto et al., 1998), although it is important to appreciate that the genetic variants in these animal models may not be identical to those identified in human essential hypertension to date. A recent large meta-analysis of data collected from two models of genetic hypertension including the SHR has identified a number of genetic associations. It is noteworthy that there were no genes identified that were common to those genes highlighted in genome-wide associations for human essential hypertension and BP. However, there were a number of genes revealed in rats that exhibit transcriptional differences in pathways that are also altered and could be relevant in human essential hypertension and variants that similarly have been implicated in smaller studies investigating genetic links with human essential hypertension, for example, UCP3 and the natriuretic peptide genes NPPA and NPPB (see Marques et al., 2010).
The SHR model is characterized by progressively increasing arterial pressure, vascular dysfunction and eventual left ventricular hypertrophy. Importantly, SHRs show clear sex differences in many aspects of cardiovascular function including the degree of hypertension (Sullivan et al., 2010). Both sexes are severely hypertensive with animals being pre to mildly hypertensive for the first 5–6 weeks of life with conscious systolic BPs generally in the range of 120–150 mmHg (Beierwaltes et al., 1982; Harrap et al., 1990; Touyz et al., 1999). However, the rise in BP from 8 to 20 weeks is, in general, more rapid in onset and substantially greater in males than in females (Chen and Meng, 1991; Reckelhoff et al., 1998). Recent evidence demonstrates that while this sexual dimorphism is evident for up to 9 months that by 12 months of age females have similar BP to males, with peak systolic BPs in the region of 200 mmHg; pressures evident in males often by 6 months of age (Chan et al., 2011).
Role of sex hormones
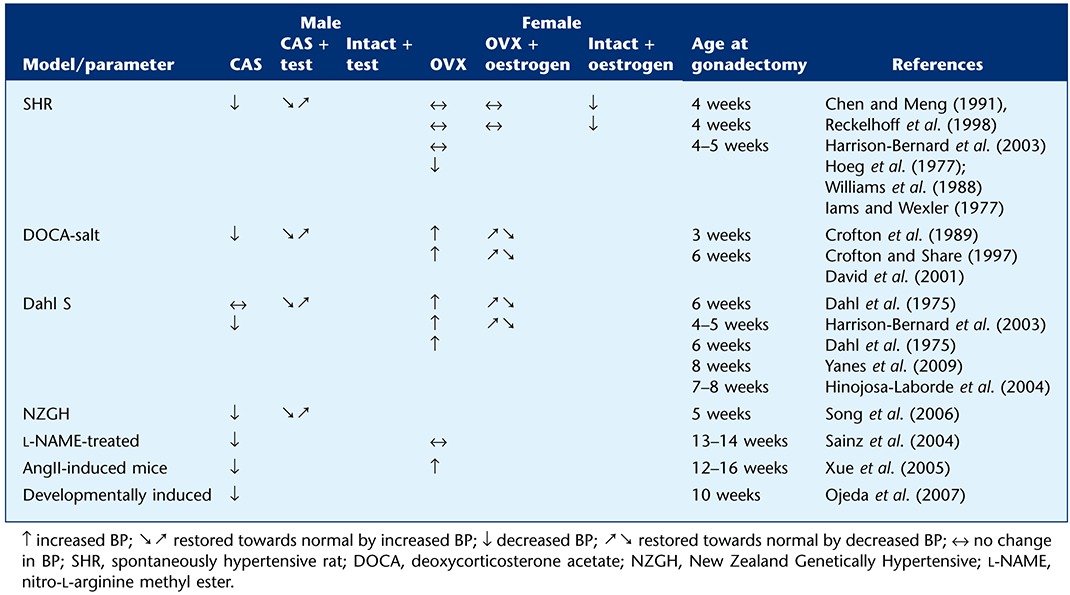
In the SHR, it seems that it is predominantly the male sex hormones that underlie the BP differences between the sexes (Kienitz and Quinkler, 2008). Castration of SHR males prior to adulthood decreased the extent of BP rise with age (Table 2), an effect overcome by testosterone replacement (Chen and Meng, 1991; Reckelhoff et al., 1998). In contrast, several studies show no differences in BP between intact or ovariectomized (OVX) females, or OVX females with oestrogen replacement (Chen and Meng, 1991; Harrison-Bernard et al., 2003; Reckelhoff et al., 1998; Wassmann et al., 2001). Yet chronic oestrogen treatment in intact females results in lower BP in some studies (Hoeg et al., 1977; Williams et al., 1988). However, caution needs to be exercised when interpreting data using hormone treatment and/or gonadectomy in animal models. In many studies, gonadectomy is conducted soon after weaning while in some studies, surgery is conducted in adulthood, such differences resulting of course in differences in the length of exposure of animals to sex hormones, which in turn may result in differences in sensitivity to hypertensive stimuli. Indeed, there is some evidence implicating ovarian hormones in rising BP, where removal of ovaries from young SHR prevented the development of hypertension (Iams and Wexler, 1977). In addition, the ‘replacement’ of hormones in gonadectomized animals is commonly used as a method to restore the circulating levels of sex hormones following ovariectomy or castration but also often on top of normal physiological levels. In the study of Hoeg et al. mentioned earlier, oestrogen treatment was administered to intact females at doses of ∼300–350 µg·kg−1·day−1. The more commonly used doses found within the literature of 200–1000 µg·kg−1·day−1, administered using osmotic mini-pumps or subcutaneous pellets, produce circulating levels of oestrogen 4–10 times greater than normal physiological levels. In a recent investigation examining the effects of chronic oestrogen administration in mice, it was demonstrated that doses of 17β-oestradiol (20 µg·kg−1·day−1 equating to ∼0.4 µg−1·day−1) resulting in physiological levels (∼100 pg·mL−1) induced cardioprotective effects, yet higher doses exerted detrimental effects (Zhan et al., 2008). Therefore, both gonadectomy and hormone replacement investigations need to be interpreted carefully with the above caveats in mind.
Table 2.
Effect of gonadal hormones on blood pressure in animal models
 |
Mechanisms of effects
In terms of the specific pathways involved in the differences between the sexes in many studies, the renin-angiotensin system (RAS) has been identified as playing an important role. Evidence demonstrates that blockade of the RAS pathway attenuates the hypertension evident in both sexes (Reckelhoff et al., 2000). However, chronic angiotensin (Ang)II infusion in SHR elevates BP to a greater extent in males compared with females (Figure 1; Yanes et al., 2006). This difference in sensitivity to AngII has been attributed to increased AngII receptor type 1 (AT1R) expression and activation since in SHR males treatment with the AT1R blocker normalized BP (Yanes et al., 2006). Moreover, recent studies demonstrate greater renal AT1R mRNA expression in males compared with females (Sullivan et al., 2010). In contrast, it has been suggested that enhanced AngII receptor type 2 (AT2R) in females might, in part, underlie the reduced pressor responses to AngII in females because AngII-mediated vasodilatation and the ratio of AT2R : AT1R is higher in female compared with male SHR (Silva-Antonialli et al., 2004). The authors of this paper suggested that up-regulation of this vasodilator-arm of the RAS may underlie the reduced susceptibility of females to hypertension. In SHR, ovariectomy increased AT1R mRNA and, accordingly, AT1R antagonism prevented endothelial dysfunction, although this was not related to BP (Wassmann et al., 2001). Studies exploring the effect of gonadectomy or hormone treatment in animals on the RAS pathway suggest substantial similarities with humans. Indeed, while oestrogen promotes elevation of angiotensinogen mRNA levels in both animals and humans, it also attenuates plasma renin levels, angiotensin converting enzyme (ACE) activity and AT1R expression. In contrast, testosterone also promotes angiotensinogen mRNA, but it also increases renin levels and activity and does not appear to have any inhibitory effects (for a review, see Fischer et al., 2002). Interestingly, there is some evidence to suggest that in aging SHR, the hypertension evident in females is related to endothelin (ET) 1 bioactivity because BP was decreased by ETA receptor blockers in ‘postmenopausal’ but not young female rats (Yanes et al., 2010). Together, this evidence supports a role for testosterone in driving the RAS, an effect that likely underlies the differences between the sexes in SHR with an increasing role for ET-1 in the aging female.
Figure 1.
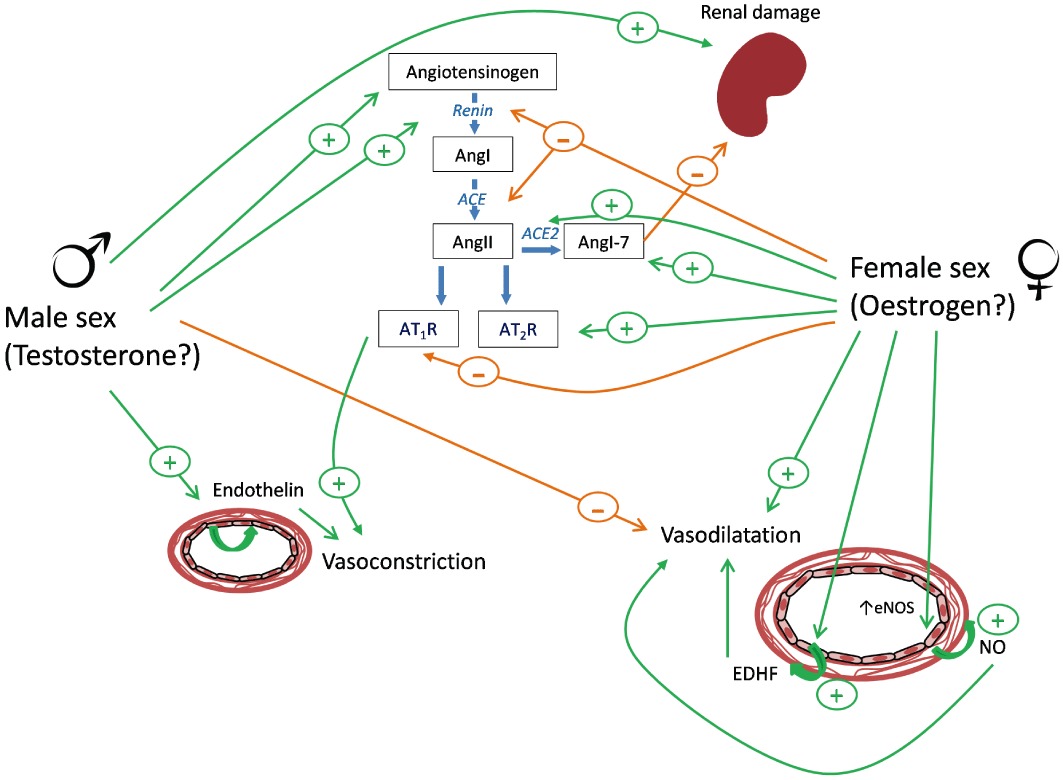
Potential mechanisms involved in the sex differences of hypertension. Several pathways have been implicated in underlying the differences in blood pressure evident between males and females in the various models of hypertension. RAS, the endothelin pathway and the vasodilating endothelium-dependent pathways are all thought to play prominent roles.
Deoxycorticosterone acetate (DOCA)-salt
Sex differences
The DOCA-salt rat is considered a model of secondary hypertension and is characterized by endothelial dysfunction, cardiac hypertrophy, proteinuria and glomerulosclerosis. It is the only model where targeted treatment of the RAS is ineffective in reducing BP (Pinto et al., 1998). This may be related to the fact that a feature of this model is the presence of abnormally low plasma renin activity assessed by measurement of AngI generation, that is, 0.32 ± 0.11 in DOCA-salt rats compared with 2.05 ± 0.35 ng AngI·mL−1·h−1 in control rats (Lariviere et al., 1993a).
The hypertension that develops in this model has been attributed to increased synthesis of the potent vasoconstrictor ET (Lariviere et al., 1993a,b). Accordingly, blockade of the ET receptors ETA and ETB reduce BP in this model (Bird et al., 1995; Schiffrin et al., 1995). This model involves uninephrectomy in early adulthood, followed by treatment with DOCA and excess salt intake (i.e. 1% NaCl in drinking water). This results in increased antidiuretic hormone (vasopressin) causing both water retention leading to increased blood volume and vasoconstriction. While these effects result in an increase in systolic BP in both sexes, the magnitude of this increase is much greater in males (a rise of ∼60 mmHg) than in females (rise of ∼35 mmHg; Ouchi et al., 1987). DOCA-salt treatment in mice similarly causes greater increases in mean arterial BP in males than in females when measured by radiotelemetry, sex differences that were not evident when BP was measured via an implanted catheter in anaesthetized mice (Karatas et al., 2008). This observation highlights the essential need for determination of BP in conscious animals.
Role of sex hormones and mechanisms
Similarly to the SHR, in DOCA-salt hypertension, the rise in systolic BP is not only attenuated by castration but also, in contrast to SHR, exacerbated by the removal of the ovaries (Crofton et al., 1989). These changes were reversed by testosterone and oestrogen treatment in gonadectomized males and females respectively (Crofton and Share, 1997). Interestingly, oestrogen treatment of intact males also attenuated systolic BP, yet testosterone treatment in females did not cause a rise in systolic BP in intact females (Crofton and Share, 1997). The authors speculated that this difference may reflect either a relative absence of testosterone receptors in the vasculature or CNS in female animals, or that there may be relatively higher aromatase activity in females. The irreversible synthesis of oestrogens from androgens is catalysed by the cytochrome P450 aromatase (Simpson et al., 2002). Disruption of the single gene, Cyp19, which encodes cytochrome P450 aromatase, results in prevention of oestrogen production from androgens (Fisher et al., 1998). Within the cardiovascular system, aromatase expression in endothelial cells is thought to underlie a substantial component of androgen-derived oestrogen (Mukherjee et al., 2002). Thus, any administered testosterone could feasibly be converted to oestrogen. Further support for a protective effect of oestrogen in DOCA-salt rats is provided (by David et al., 2001) where OVX elevated systolic BP, and it returned to normal with oestrogen treatment. A role for oestrogen is supported by studies using a slightly different model of hypertension, aldosterone + salt. In this model, ER agonists including the non-selective 17β-oestradiol and both the ERα and the ERβ selective agonists, 16αLE2 and 8βVE2, respectively, caused lowering of BP in OVX female animals (Arias-Loza et al., 2007).
The DOCA-salt rat is a model of hypertension dependent upon enhanced activity of the ET-1 pathway. In particular, alterations in ETA/ETB receptor signalling have been implicated. In vitro investigations demonstrate increased contractile sensitivity to ET-1 in endothelium-denuded aortas from male but not from female DOCA-salt rats. These effects are associated with up-regulated aortic mRNA expression of ET-1 and ETB receptors (David et al., 2002). Thus, the DOCA model of hypertension appears to be a model predominantly associated with enhanced ET-1 activity, and females, relative to their male counterparts, are protected due to a relative decrease in ET receptor expression and activity (Figure 1).
Dahl salt-sensitive (Dahl S) rats
Sex differences
The Dahl S rat is a genetic model of salt-sensitive hypertension demonstrating an acute sensitivity to salt loading (8%NaCl vs. <0.5% for normal salt diet) that accelerates the rate of BP rise. Like the SHR, and indeed humans, both sexes of the Dahl S rats develop high BP eventually (∼200 mmHg systolic BP); however, the onset and rate of rise in pressures is considerably delayed and slower in females (Dahl et al., 1975).
Role of sex hormones and mechanisms
It has been suggested that the difference between the sexes relates to oestrogen-induced suppression of AngII binding to AT1R. Indeed, Hinojosa-Laborde et al. (2004) demonstrated that as oestrogen levels decreased with ageing, AT1R binding was increased, and this corresponded with higher mean arterial pressures, a process accelerated by removal of the ovaries. This view is further supported by findings demonstrating that 0.5% salt-fed OVX Dahl S female animals exhibit higher systolic BP (measured using tail-cuff) than sham-operated Dahl S females. Moreover, treatment with the AT1R antagonist candesartan or with oestrogen decreased BP, in the former, to below the levels of sham-operated intact females (30–40 mmHg decrease in systolic BP; Harrison-Bernard et al., 2003). The authors suggested that these findings support the contention that oestrogen acts as an AT1R inhibitor. It has also been suggested that the reduced rise in BP in females may relate to an enhanced vasodilator effect of the RAS pathway in this sex. AngII can also be converted by ACE2 to form Ang1-7, which causes sustained reductions in BP (measured using tail-cuff) of hypertensive female but not male rats (Eatman et al., 2001). In a second publication, these beneficial effects of Ang(1–7) in this model were blocked by antagonism of the Mas receptor (Bayorh et al., 2002).
Similarly to DOCA-salt rats, OVX Dahl S rats have higher systolic BP than high-salt intact (Dahl et al., 1975) or normal-salt sham-operated animals (Harrison-Bernard et al., 2003), suggesting that it is the female sex hormones that provide protection against hypertension. Similarly, prolonged BP measurement over 10 months in female Dahl S rats on a low-salt diet demonstrated substantially greater BP in OVX animals that was prevented with oestrogen supplementation (Hinojosa-Laborde et al., 2004). Moreover, in this study, the authors demonstrated that over the 10 months of measurement, a natural drop in oestrogen levels occurred with age in intact females and that this progressive decrease was associated with a progressive increase in mean arterial BP (Hinojosa-Laborde et al., 2004). However, animals were treated with a dose of 17-β-oestradiol (5 mg implanted subcutaneously in silastic tubing and replaced every 12 weeks) that resulted in circulating levels of oestrogen exceeding normal maximum levels of oestrogen found in the reproductive cycle by ∼4 times. It is uncertain whether such dosing regimens accurately reflect the actions of oestrogen under physiological conditions.
Whether testosterone has any role to play in this sexual dimorphism is not completely clear. In contrast to the SHR, castration of males does not alter the development of hypertension, measured using tail-cuff (Dahl et al., 1975). In contrast, a very recent study using radiotelemetry has challenged these findings demonstrating that castrated Dahl S rats, fed a high-salt diet, have lower BP than intact males, and moreover, that replacement of testosterone (to physiological levels) reversed these effects (Yanes et al., 2009).
It has been suggested that augmentation of intrarenal angiotensinogen contributes to the rise in BP in male Dahl S rats on a high-salt diet (Kobori et al., 2003); however, this is not the case in females, nor in castrated males. Thus, testosterone appears to be important in mediating increased renal angiotensinogen. With the absence of any alterations to intrarenal renin gene expression, it is likely that AngII production would be increased in the high-salt Dahl S males over females, but this was not measured (Yanes et al., 2009).
AngII-induced hypertension
Sex differences
To take advantage of knockout (KO) technology, there has been considerable effort invested in establishing models of hypertension in mice that might be relevant for the human condition. In this respect, models based upon the continuous release of the potent vasoconstrictor AngII have emerged, and it is clear that the same sexual diversity also extends to mice. In particular, studies using constant infusion of AngII (800 ng·kg−1·min−1 by osmotic mini pump) demonstrate a rapidly developing and persistent rise in radiotelemetric BP that is greater in males compared with females (mean arterial BP increased by ∼50 mmHg in males and ∼15 mmHg in females) (Xue et al., 2005).
Role of sex hormones and mechanisms
Interestingly, the degree of hypertension in mice treated with AngII was substantially attenuated in gonadectomized males and enhanced in OVX females (Xue et al., 2005), the latter being reversed by oestrogen ‘replacement’. The authors implicated oestrogen receptors in these responses because there was a ∼4-fold greater elevation in mean arterial BP in OVX mice treated with oestrogen plus ICI-182 780, a non-selective oestrogen receptor antagonist (Xue et al., 2007). Furthermore, it is likely that the ERα receptor subtype plays an important role because arterial BP was substantially greater in ERα KO females compared with intact wild-type females but similar to OVX wild-type animals (Xue et al., 2007). This effect of AngII is also evident in rats where chronic AngII infusion (400–500 ng·kg−1·min−1) causes greater increases in arterial pressure in males than in females (Tatchum-Talom et al., 2005; Sampson et al., 2008). This was associated with endothelial dysfunction and up-regulation of smooth muscle vasoconstrictor responses (Tatchum-Talom et al., 2005). Interestingly, infusion of a low concentration of AngII (50 ng·kg−1·min−1) did not alter BP in males, yet in females, mean arterial BP was reduced, and this was dependent on AT2R activation.
Together, these studies suggest that a clear sexual dimorphism exists in responses to AngII, in both rats and mice that likely involve distinct differences in AT receptor subtype expression.
Hypertension produced by disrupted endothelium-derived vasodilator function
Sex differences
The three major endothelium-derived vasodilators, NO, prostacyclin and endothelium-derived hyperpolarizing factor (EDHF), play a substantial role in the regulation of peripheral vascular resistance and therefore BP. Sex differences in the contribution of these dilators to vascular function have been observed. In particular, many of the vasoprotective effects of female sex have been attributed to oestrogen-induced enhancement of NO generation and/or activity within the blood vessel wall (reviewed in Orshal and Khalil, 2004; Villar et al., 2008). As an example of the impact of enhanced vascular NO function on BP are the studies of Sainz and co-workers in Wistar rats. In this study, the effect of chronic administration of nitro-l-arginine methyl ester (l-NAME), an inhibitor of NOS, administered in the drinking water (0.5–0.75 mg·mL−1) for 2 weeks on BP was measured. The authors found that blockade of NOS activity elevated BP (measured using tail-cuff) in both sexes; however, a greater increase was evident in male animals (∼Δ35 mmHg) compared with females (∼Δ20 mmHg, a difference further exacerbated after 5 weeks treatment (to 60 and 30 mmHg respectively; Sainz et al., 2004). Orchidectomy of males decreased mean arterial BP by 30 mmHg, implicating testosterone, while ovariectomy had no significant effect. The authors suggested that this effect of l-NAME was due to enhanced activity of the RAS because the increases in BP were likewise associated with increases in plasma renin activity. Curiously, the treatment of intact males with oestrogen reduced l-NAME-induced hypertension, but testosterone treatment in females had no effect. This latter finding is reminiscent of the studies in DOCA-salt rats (Crofton and Share, 1997) and provides further support for the suggestion of a deficit of testosterone receptors in intact females.
Greater dependence of male BP on eNOS-derived NO is also supported by our own studies using mice with genetic ablation of eNOS, where we demonstrated elevated systolic BP (tail-cuff) in males but not in females (Scotland et al., 2005). Although in another study, BP (tail-cuff) was shown to be elevated in eNOS KO mice (an effect associated with increases in plasma renin activity), but there was no evident difference between the sexes (Shesely et al., 1996). The reason for the difference between these studies is uncertain but may be explained by differences in the source of the eNOS deficient mice, an explanation proposed for other divergent findings in the literature (Sharp et al., 2002). Interestingly, we also demonstrated that while BP was similar in both sexes in wild-type and COX-1 KO mice (i.e. mice deficient in vascular prostacyclin) that male mice deficient in both eNOS and COX-1 were hypertensive but females were not (Scotland et al., 2005). In addition, we demonstrated a loss of both endothelium-dependent vasorelaxation of isolated resistance arteries and bradykinin-induced hypotension in male but not in female eNOS−/−/COX-1−/− mice. These studies support a role for EDHF in regulating vascular tone in females, while in males, endothelial NO is the main source for this vasodilator regulation. It is noteworthy that loss of endothelium-derived NO bioactivity is thought to characterize and contribute to the phenomenon of ‘endothelial dysfunction’. Endothelial dysfunction describes the change of endothelium phenotype from vasodilator and anti-inflammatory to vasoconstrictor and pro-inflammatory, which is thought to play an important role in the pathogenesis of hypertension (Schulz et al., 2011). Of course, if females rely less on endothelial NO to regulate vascular tone, as proposed in the above studies, this might provide at least some explanation for the relative protection from hypertension that females enjoy, at least during the premenopausal period (Figure 1).
Developmental models
Emerging evidence supports the concept that disease susceptibility can be determined during the developmental stages in utero. There is a growing interest in investigating this concept to explore how diseases can be ‘programmed’ to occur later in life whether that be in utero or in early post-natal life (Gluckman et al., 2008). Interestingly, elevated BP and/or predisposition to develop cardiovascular complications have been common occurrences in the offspring of many of the animal models used to investigate an adverse intrauterine environment. Like other models of hypertension, many of these developmental adversities result in a sex bias in the extent of CVD (for a review, see Gilbert and Nijland, 2008). Reduced uterine perfusion, a model of placental insufficiency, caused greater hypertension in male compared with female offspring (Alexander, 2003). Interestingly, this form of hypertension was associated with testosterone, as placenta-restricted males had elevated serum testosterone compared with control males, an effect reversed by castration (Ojeda et al., 2007). Protein restriction during pregnancy resulted in hypertensive male (Woods et al., 2001) but normotensive female offspring at 6 months of age (Woods et al., 2005). A low-calorie diet for the duration of gestation resulted in an earlier onset of hypertension in males than in females, although both sexes were eventually affected to the same degree (Ozaki et al., 2001).
The common theme among these animal models is that males are affected to a greater degree than females. Although, mild hypertension during pregnancy in rabbits led to elevated mean arterial BP in female offspring, but no difference in BP was evident between males born to hypertensive or normotensive mothers (Denton et al., 2003). This is supportive of data from humans, where females born to mothers with preeclampsia were at an increased risk of systolic blood pressure greater than 140 mmHg compared with controls, yet male offspring had no greater hypertensive risk than controls (Seidman et al., 1991). In addition, rats fed a high-fat diet during pregnancy produced female but not male offspring with elevated systolic BP at 1 year (Khan et al., 2003). It is noteworthy that in this study, systolic BP was ∼140 mmHg in females of fat-fed dams compared with ∼125 mmHg in controls; however, systolic BP was ∼140 mmHg in males of both groups. It is possible, therefore, that this higher baseline in males may account for, or contribute to, the sex differences observed. These preclinical studies are of particular current interest because the emerging picture intimates important epigenetic consequences of maternal nutrition on ensuing adult health, including increased susceptibility to hypertension. Such models should prove useful in ascertaining the importance/risk of consumption of certain dietary constituents.
A role for sex chromosomes?
While gonadal hormones clearly underlie a major component of the sex differences in BP, there is also good evidence that the sex chromosomes per se are involved (for a review, see Charchar et al., 2003; Sampson et al., 2012). Cross-breeding of SHR and Wistar Kyoto rats, to produce the Y consomic rat model, first demonstrated a clear association of higher BP with the SHR Y chromosome (Ely and Turner, 1990). Further studies in SHR demonstrated that the SRY locus, the sex-determining (testis formation) region of the Y chromosome, plays an important role in the raised BP evident in males in this model (Turner et al., 2009). However, the development of the four core genotype (FCG) mouse model suggests that perhaps other chromosomal loci play a role. In the FCG mice, the SRY gene that is deleted from the Y chromosome has been transferred to an autosome. This creates an XY-Sry male mouse, which, when mated with normal females, gives rise to the FCG progeny, which include XX and XY mice with ovaries (female) and XX and XY mice with testes (male). Interestingly, gonadectomy of either the male or the female XX mice resulted in elevation of mean arterial BP in response to AngII infusion compared with XY mice. This suggests that the X chromosome carries no protective effect against BP elevation (Ji et al., 2010). Yet the location on the X chromosome of the genes for both AT2R (Koike et al., 1994) and ACE2 (Tipnis et al., 2000), both of which are involved in the antihypertensive actions of the RAS and are altered in sex-dependent hypertension (Pendergrass et al., 2008), is surely not a coincidence. Such studies suggest that loci other than the SRY may also be important for BP regulation.
II. Atherosclerosis
As with hypertension, atherosclerotic disease is less prevalent in premenopausal women compared with age-matched men. In 2008 in the UK, of the deaths due to CVD in individuals aged up to 55, only ∼18% of the deaths due to coronary heart disease and 45% due to stroke were in women: differences lost after menopause. The reasons for these sex differences are clearly multiple and include issues pertaining to differences in awareness and understanding of disease as well as differences in susceptibility to risk factors between the sexes. As an example of the latter recent evidence suggests that the detrimental effects of smoking with respect to coronary events is greater in women than in men (Huxley and Woodward, 2011). However, in addition, it is clear that differences in the molecular pathways involved in both suppression and stimulation of plaque development between the sexes play a role, and it is this difference, in rodent models of atherosclerosis, that we focus upon in this review.
In contrast to hypertension, where most evidence implicates testosterone in males as underpinning sex differences, in atherosclerotic disease, the weight of evidence implicates oestrogen as the effector of protection in females. This view is supported by a plethora of experimental studies in animals and observational data in women (for a review, see Mikkola and Clarkson, 2002; Dubey et al., 2004; Perez-Lopez et al., 2010). However, prospective studies investigating the effect of oestrogen administration to postmenopausal women are equivocal and have shown a decrease (Stampfer et al., 1991), no change (Grady et al., 2002) or an increase in cardiovascular risk (Wilson et al., 1985; Rossouw et al., 2002). These divergent findings have caused a great deal of concern regarding the predictive value of preclinical studies, since in vivo animal models have consistently demonstrated a reduction in atherosclerotic lesion size in response to oestrogen of between 30 and 75%, (Bourassa et al., 1996; Elhage et al., 1997; Marsh et al., 1999). Closer examination of the studies in women has, however, identified beneficial anti-atherosclerotic effects of oestrogen treatment in specific groups of women, particularly those with few postmenopausal years (Manson et al., 2007). These observations have led to the proposal of the ‘timing hypothesis’ where it is suggested that hormone treatment of women in the early stages of menopause and therefore with minimal CVD burden is protective. In contrast, following menopause CVD burden rapidly increases and exposure of women with significant atherosclerotic burden and therefore several postmenopausal years exposes the pro-thrombotic activity of oestrogens.
Several different animal models have been used over the years to study atherosclerosis. Most of the models utilize dietary feeding of high cholesterol (1–2%) to stimulate or perpetuate plaque formation. In mouse models of atherosclerosis, variations of the ‘Paigen’ high-fat diets (1.25% cholesterol, 0.5% cholic acid, 15% cocoa butter and 1% corn oil) are more commonly used with the ‘Western’ diet being the diet of choice (21% milk fat, 0.2% cholesterol ±1% corn oil; Smith and Breslow, 1997). The atherosclerosis susceptible New Zealand White rabbit (Buja et al., 1983) and the Watanabe heritable hyperlipidemic (WHHL) rabbit (Tanzawa et al., 1980) were the first main laboratory animals to be used for studying the effects of sex on atherosclerosis. As in human disease, the onset and extent of plaque formation in these rabbit models is both less and delayed in females compared with males (Hayashi et al., 1995), and attributed to the protective actions of oestrogen (Hayashi et al., 2006). Similar effects have also been demonstrated in primates (Adams et al., 1985; Roger et al., 2011).
The advent of mouse models of atherosclerosis has, however, massively accelerated research in this field, and these are now the most commonly used models of atherosclerosis. This is largely due to a number of benefits pertaining to cost and size over some of the classical models of atherosclerosis in larger species. The two most commonly used murine models are the apolipoprotein E (ApoE) KO and low-density lipoprotein receptor (LDLr) KO mouse models. The remainder of this review will focus on observations noted in these two mouse models only.
ApoE KO mouse
Sex differences
Hypercholesterolaemia is a major risk factor for atherosclerosis (Bhatnagar et al., 2008) and is defined as a cholesterol greater than 240 mg·dL−1 (6.2 mmol·L−1, American Heart Association). Male and female ApoE KO mice fed a normal chow diet have total cholesterol levels between the ranges of 3.1–15.7 mmol·L−1 (118–606 mg·dL−1) (Nakashima et al., 1994; Grimsditch et al., 2000; Teupser et al., 2004; Maeda et al., 2007; Surra et al., 2010) and 2.4–9.7 mmol·L−1 (92–374 mg·dL−1) (Grimsditch et al., 2000; Teupser et al., 2004; Maeda et al., 2007; Surra et al., 2010) respectively. However, when ApoE KO mice are fed a Western-type diet, cholesterol levels rise to ∼38.9 mmol·L−1 (1500 mg·dL−1) in both sexes (Nakashima et al., 1994; Grimsditch et al., 2000). Feeding a Western diet, generally for 3 months, accelerates formation and increases lesion size, for example, at 40 weeks plaque coverage of the aorta is just under 15% in chow fed mice but ∼40% in mice fed a Western diet (Tabibiazar et al., 2005). The use of such diets, therefore, saves both time and inevitably costs associated with long-term maintenance of animals. The larger plaque coverage is also particularly useful when interrogating the potential of novel anti-atherosclerotic therapeutics. It is important to note that the discussion earlier relates to animals bred on a C57BL/6 background, as different strains of mice have varying degrees of susceptibility to atherosclerosis (Grimsditch et al., 2000; Teupser et al., 2003; Surra et al., 2010).
Despite comparable cholesterol levels, lesion formation is generally greater in female compared with male ApoE KO mice fed a chow or Western diet (Grimsditch et al., 2000; Teupser et al., 2004; Maeda et al., 2007; Smith et al., 2010; Surra et al., 2010). Lesion formation in mice tends to occur along the entire length of the aorta with a high incidence at the aortic root and arch (Nakashima et al., 1994; Bourassa et al., 1996; Maeda et al., 2007). Commonly, it is within these particularly susceptible regions that sex differences are evident in the size of lesion. Although in one study, a trend toward males having more extensive lesions has also been demonstrated (Tangirala et al., 1995). Importantly, the influence of sex appears to change with age and while in studies lasting 3 months, females have increased atherosclerosis, in mice fed a chow diet for 18 months, males had larger lesions than aged-matched females (Caligiuri et al., 1999; Pereira et al., 2010), corresponding well with human observational studies. In addition, this difference was accompanied by a twofold increase in plasma cholesterol levels in the males (Pereira et al., 2010).
Role of sex hormones
Despite this rather mixed picture regarding the differences between males and females, the evidence for a cardioprotective effect of oestrogens in this mouse model is unequivocal. OVX female mice exhibit enhanced atherosclerotic lesion area in comparison with sham-operated females, despite both groups expressing comparable cholesterol levels (Bourassa et al., 1996; Marsh et al., 1999). Furthermore, low-dose oestrogen administration (0.5 µg·day−1) reduces lesion area with no change in cholesterol levels (Elhage et al., 1997). Importantly, the effects of oestrogen are consistently independent of any effects on cholesterol and, as such, intimate that cholesterol level is not the sole driver of atherosclerotic lesion formation. Indeed, atherosclerosis is a chronic inflammatory disorder (Ross, 1999), and several key inflammatory processes/mediators have been implicated in pathogenesis (Libby et al., 2010). As such, the possibility that sex may influence the inflammatory responses has generated some interest and is discussed later in this review.
LDLr-KO mice
Sex differences
As with the ApoE KO mouse, lesion formation in the LDLr KO mouse occurs along the length of the aorta. The densest plaque formation is found at the arch and in branches off the arch including the brachiocephalic artery. Plaque size then decreases down the length of the aorta and is predominantly evident at sites of bifurcation (Nakashima et al., 1994). The total cholesterol levels in these mice are lower than in the ApoE KO mouse at ∼2.6 mmol·L−1 (100 mg·dL−1) in both sexes when fed a chow diet (Arai et al., 1999; Teupser et al., 2004). Similarly to the ApoE KO mice, studies using LDLr KO mice in general utilize a Western-type diet to accelerate the atherosclerotic process due to minimal atherosclerosis in mice fed a chow diet (Teupser et al., 2003). Under such conditions, cholesterol levels are raised to approximately 38.9 mmol·L−1 (1500 mg·dL−1) in both sexes (Nakashima et al., 1994; Grimsditch et al., 2000; VanderLaan et al., 2009).
Role of sex hormones
In LDLr KO mice, the evidence is less consistent with data demonstrating that males have a tendency toward more lesions than females (Tangirala et al., 1995) and vice versa in spite of similar plasma cholesterol levels (Teupser et al., 2003; VanderLaan et al., 2009). However, ovariectomy of female mice enhances atherosclerotic lesion area by ∼100% in comparison with sham-operated females (Marsh et al., 1999).
Influence of sex on inflammation in atherosclerosis
As noted earlier, the lack of effect of oestrogen on cholesterol levels has led some researchers to investigate the possibility that the inflammatory processes key to the development of atherosclerosis may be the target for this sex hormone. The approach that has been taken to address this possibility has been to compare lesion size in mice that are deficient not only in ApoE or LDLr but also in some of the key inflammatory adhesion molecules and chemokines that have been implicated in these models.
In this respect, a number of different key adhesion molecules have been targeted. LDLr KO mice have been compared with mice also deficient for the adhesion molecule P-selectin. P-selectin is an essential molecule involved in the early ‘cell-rolling’ stages of inflammatory cell recruitment (Mayadas et al., 1993), and we have shown more recently that lower levels of P-selectin expression underlie reduced cell recruitment induced by the inflammatory cytokines IL-1β or TNFα in females compared with males (Villar et al., 2011). Interestingly, in male LDLr/P-selectin KO, mice lesion size is reduced by ∼60% in comparison with LDLr KO mice after 8 weeks of high-fat diet (Johnson et al., 1997). In contrast, no difference in lesion size between the genotypes is evident in female mice (Johnson et al., 1997), suggesting an essential role for P-selectin in the atherosclerotic process in males but not in females. It is noteworthy that leukocyte recruitment in response to inflammatory stimuli is suppressed in female compared with male mice and that, in part, this difference relates to increased expression of P-selectin in males compared with females (Villar et al., 2011). However, in ApoE KO mice, the beneficial effects of 17β-oestradiol on plaque formation have shown to be independent of P-selectin or the adhesion molecule, intercellular adhesion molecule 1, the latter of which plays an important role in inflammatory cell adhesion (Gourdy et al., 2003). In this study, however, 17β-oestradiol treatment caused a 31% reduced expression level of the adhesion molecule vascular cell adhesion molecule 1 (VCAM-1), an adhesion molecule involved in firm adhesion of leukocytes. VCAM-1 is also raised in human coronary intimal neovasculature with a strong correlation to intimal leukocyte accumulation (O'Brien et al., 1996).
Thus, the deletion of a single adhesion molecule appears to have little effect upon lesion formation in female mice. Interestingly, however, mice doubly deficient in both P- and E-selectin on a LDLr KO background after 8 weeks of high-fat diet show reduced lesion size in both sexes, albeit with a greater reduction observed in male compared with female mice (Dong et al., 1998). This study suggests that perhaps some redundancy exists with respect to the inflammatory responses in atherosclerosis. It is interesting to note, however, that the females deficient in P- and E-selectin developed cutaneous infections starting at ∼16 weeks of age, limiting the study of sex differences in the longer term (Dong et al., 1998). Such a finding highlights a potential issue with using animals in which multiple pro-inflammatory targets have been deleted, potentially rendering animals in an immunocompromized state.
In addition to the selectin adhesion molecules, a number of chemokines have been implicated in the beneficial effects of female sex hormones. Fractalkine (CX3CL1), a chemokine implicated in cell adhesion and transmigration, is expressed in human atherosclerotic lesions (Lucas et al., 2003). Accordingly, fractalkine-deficient male and female ApoE and LDLr KO mice expressed fewer and smaller lesions associated with fewer macrophages (Teupser et al., 2004). In this study, peripheral blood leukocyte, neutrophil, lymphocyte and monocyte numbers were similar in both sexes in wild-type and KO animals (Teupser et al., 2004). Nonetheless, it has been shown that fractalkine-deficient animals have decreased numbers of blood leukocytes expressing the cell surface marker F4/80, a marker used for the identification of monocytes (Cook et al., 2001). Such an observation raises the possibility that the reduction in lesion size observed is due to a reduction in circulating monocyte numbers, hence less available for plaque initiation and development, rather than a specific block of inflammatory cell recruitment at the lesion site. Because a plethora of chemokines have been identified as having dual functions with respect to both local inflammatory responses as well as haematopoiesis (Youn et al., 2000), we suggest that it is paramount to assess circulating cell numbers in doubly deficient animals in atherosclerosis to ensure that the effects seen do not simply relate to alteration in haematopoiesis.
Another key chemokine implicated in atherosclerosis is monocyte chemoattractant protein (MCP)-1, which plays a pivotal role in monocyte recruitment and is expressed in human atherosclerotic coronary arteries (Nelken et al., 1991). Male and female double MCP-1/ApoE KO mice exhibit 64% and 71% reduced atherosclerotic lesion size, respectively, compared with ApoE KO mice, again with no change in cholesterol levels (Dawson et al., 1999). This study suggests that MCP-1 is not influenced by sex; however, postmenopausal women taking 1 mg 17β-oestradiol supplement demonstrated a 17% reduction in serum MCP-1 levels after 12 months (Stork et al., 2002), indicating that oestrogen can interfere with MCP-1 generation.
In line with an important role for inflammation in the pathogenesis of atherosclerosis, several studies suggest that infection increases susceptibility to CVD (reviewed in Rosenfeld and Campbell, 2011). Multiple pathogens have been implicated in the pathogenesis of atherosclerosis, although whether sexual dimorphism exists in this phenomenon has been largely untested. In one study, ApoE heterozygote mice infected with Porphyromonas gingivalis, a bacterium associated with periodontal disease, exhibited no sex difference in relation to aortic atheroma lesion score (Champagne et al., 2009). Whether this might be the case across the broad range of infectives implicated in atherosclerosis is, as of yet, unknown.
It is clear that sex differences in the extent of atherosclerosis exist, both in humans and in mice. Moreover, the evidence supports the view that oestrogen is a driving factor for these differences. However, the exact molecular pathways underlying these sex differences are unclear, although emerging data have highlighted differences in inflammatory responses. Hopefully, further research encouraged by these early studies investigating the link between sex, inflammation and atherosclerosis will enhance our understanding of the processes involved in atherosclerosis development in both sexes and thereby potentially identify novel therapeutic targets.
Conclusion
In summary, animal models are essential for elucidating the mechanisms involved in the sex differences clearly evident in both hypertension and atherosclerosis. For both of these conditions, there are a number of models available for use, but not all models are suitable for every experimental protocol. In particular, when attempting to dissect the mechanisms underlying any sex differences, it is essential to carefully design the studies to account for physiological changes and compensatory regulation and redundancy in response to altering hormone levels or removal of genes. With continued investigation in this field, it is likely that novel targets for the development of therapeutics will be identified to aid in the battle of our worldwide epidemic of CVD.
Acknowledgments
KB and RK are funded by The British Heart Foundation. This work forms part of the research themes contributing to the translational research portfolio of the National Institute for Health Research Cardiovascular Biomedical Research Unit at Barts and the London School of Medicine and Dentistry.
Glossary
- ABPM
ambulatory blood pressure monitoring
- ACE
angiotensin converting enzyme
- Ang
angiotensin
- ApoE
apolipoprotein E
- AT1R
angiotensin II receptor type 1
- AT2R
angiotensin II receptor type 2
- BP
blood pressure
- CVD
cardiovascular disease
- DOCA
deoxycorticosterone acetate
- EDHF
endothelium-derived hyperpolarizing factor
- ER
oestrogen receptor
- ET
endothelin
- KO
knockout
- LDLr
low density lipoprotein receptor
- l-NAME
nitro-l-arginine methyl ester
- MCP
monocyte chemoattractant protein
- OVX
ovariectomized
- RAS
renin-angiotensin system
- SHR
spontaneously hypertensive rats
- VCAM-1
vascular cell adhesion molecule 1
Conflicts of interest
None.
References
- Adams MR, Kaplan JR, Clarkson TB, Koritnik DR. Ovariectomy, social status, and atherosclerosis in cynomolgus monkeys. Arteriosclerosis. 1985;5:192–200. doi: 10.1161/01.atv.5.2.192. [DOI] [PubMed] [Google Scholar]
- Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension. 2003;41:457–462. doi: 10.1161/01.HYP.0000053448.95913.3D. [DOI] [PubMed] [Google Scholar]
- Arai T, Wang N, Bezouevski M, Welch C, Tall AR. Decreased atherosclerosis in heterozygous low density lipoprotein receptor-deficient mice expressing the scavenger receptor BI transgene. J Biol Chem. 1999;274:2366–2371. doi: 10.1074/jbc.274.4.2366. [DOI] [PubMed] [Google Scholar]
- Arias-Loza PA, Hu K, Dienesch C, Mehlich AM, Konig S, Jazbutyte V, et al. Both estrogen receptor subtypes, alpha and beta, attenuate cardiovascular remodeling in aldosterone salt-treated rats. Hypertension. 2007;50:432–438. doi: 10.1161/HYPERTENSIONAHA.106.084798. [DOI] [PubMed] [Google Scholar]
- Ashraf MS, Vongpatanasin W. Estrogen and hypertension. Curr Hypertens Rep. 2006;8:368–376. doi: 10.1007/s11906-006-0080-1. [DOI] [PubMed] [Google Scholar]
- Ashton N, Balment RJ. Sexual dimorphism in renal function and hormonal status of New Zealand genetically hypertensive rats. Acta Endocrinol (Copenh) 1991;124:91–97. doi: 10.1530/acta.0.1240091. [DOI] [PubMed] [Google Scholar]
- Barrett-Connor E. Sex differences in coronary heart disease. Why are women so superior? The 1995 Ancel Keys Lecture. Circulation. 1997;95:252–264. doi: 10.1161/01.cir.95.1.252. [DOI] [PubMed] [Google Scholar]
- Bayorh MA, Eatman D, Walton M, Socci RR, Thierry-Palmer M, Emmett N. 1A-779 attenuates angiotensin-(1-7) depressor response in salt-induced hypertensive rats. Peptides. 2002;23:57–64. doi: 10.1016/s0196-9781(01)00579-4. [DOI] [PubMed] [Google Scholar]
- Beierwaltes WH, Arendshorst WJ, Klemmer PJ. Electrolyte and water balance in young spontaneously hypertensive rats. Hypertension. 1982;4:908–915. doi: 10.1161/01.hyp.4.6.908. [DOI] [PubMed] [Google Scholar]
- Bhatnagar D, Soran H, Durrington PN. Hypercholesterolaemia and its management. BMJ. 2008;337:a993. doi: 10.1136/bmj.a993. [DOI] [PubMed] [Google Scholar]
- Bird JE, Moreland S, Waldron TL, Powell JR. Antihypertensive effects of a novel endothelin-A receptor antagonist in rats. Hypertension. 1995;25:1191–1195. doi: 10.1161/01.hyp.25.6.1191. [DOI] [PubMed] [Google Scholar]
- Bourassa PA, Milos PM, Gaynor BJ, Breslow JL, Aiello RJ. Estrogen reduces atherosclerotic lesion development in apolipoprotein E-deficient mice. Proc Natl Acad Sci U S A. 1996;93:10022–10027. doi: 10.1073/pnas.93.19.10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buja LM, Kita T, Goldstein JL, Watanabe Y, Brown MS. Cellular pathology of progressive atherosclerosis in the WHHL rabbit. An animal model of familial hypercholesterolemia. Arteriosclerosis. 1983;3:87–101. doi: 10.1161/01.atv.3.1.87. [DOI] [PubMed] [Google Scholar]
- Burt VL, Whelton P, Roccella EJ, Brown C, Cutler JA, Higgins M, et al. Prevalence of hypertension in the US adult population. Results from the Third National Health and Nutrition Examination Survey, 1988–1991. Hypertension. 1995;25:305–313. doi: 10.1161/01.hyp.25.3.305. [DOI] [PubMed] [Google Scholar]
- Calhoun DA, Zhu ST, Chen YF, Oparil S. Gender and dietary NaCl in spontaneously hypertensive and Wistar-Kyoto rats. Hypertension. 1995;26:285–289. doi: 10.1161/01.hyp.26.2.285. [DOI] [PubMed] [Google Scholar]
- Caligiuri G, Nicoletti A, Zhou X, Tornberg I, Hansson GK. Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis. 1999;145:301–308. doi: 10.1016/s0021-9150(99)00081-7. [DOI] [PubMed] [Google Scholar]
- Champagne C, Yoshinari N, Oetjen JA, Riche EL, Beck JD, Offenbacher S. Gender differences in systemic inflammation and atheroma formation following Porphyromonas gingivalis infection in heterozygous apolipoprotein E-deficient mice. J Periodontal Res. 2009;44:569–577. doi: 10.1111/j.1600-0765.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- Chan V, Fenning A, Levick SP, Loch D, Chunduri P, Iyer A, et al. Cardiovascular changes during maturation and ageing in male and female spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2011;57:469–478. doi: 10.1097/FJC.0b013e3182102c3b. [DOI] [PubMed] [Google Scholar]
- Charchar FJ, Tomaszewski M, Strahorn P, Champagne B, Dominiczak AF. Y is there a risk to being male? Trends Endocrinol Metab. 2003;14:163–168. doi: 10.1016/s1043-2760(03)00032-8. [DOI] [PubMed] [Google Scholar]
- Chen YF, Meng QC. Sexual dimorphism of blood pressure in spontaneously hypertensive rats is androgen dependent. Life Sci. 1991;48:85–96. doi: 10.1016/0024-3205(91)90428-e. [DOI] [PubMed] [Google Scholar]
- Cook DN, Chen SC, Sullivan LM, Manfra DJ, Wiekowski MT, Prosser DM, et al. Generation and analysis of mice lacking the chemokine fractalkine. Mol Cell Biol. 2001;21:3159–3165. doi: 10.1128/MCB.21.9.3159-3165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofton JT, Share L. Gonadal hormones modulate deoxycorticosterone-salt hypertension in male and female rats. Hypertension. 1997;29((Pt 2)):494–499. doi: 10.1161/01.hyp.29.1.494. [DOI] [PubMed] [Google Scholar]
- Crofton JT, Share L, Brooks DP. Gonadectomy abolishes the sexual dimorphism in DOC-salt hypertension in the rat. Clin Exp Hypertens A. 1989;11:1249–1261. doi: 10.3109/10641968909038168. [DOI] [PubMed] [Google Scholar]
- Dahl LK, Knudsen KD, Ohanian EV, Muirhead M, Tuthill R. Role of the gonads in hypertension-prone rats. J Exp Med. 1975;142:748–759. doi: 10.1084/jem.142.3.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaei G, Finucane MM, Lu Y, Singh GM, Cowan MJ, Paciorek CJ, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country-years and 2.7 million participants. Lancet. 2011;378:31–40. doi: 10.1016/S0140-6736(11)60679-X. [DOI] [PubMed] [Google Scholar]
- David FL, Carvalho MH, Cobra AL, Nigro D, Fortes ZB, Reboucas NA, et al. Ovarian hormones modulate endothelin-1 vascular reactivity and mRNA expression in DOCA-salt hypertensive rats. Hypertension. 2001;38((Pt 2)):692–696. doi: 10.1161/01.hyp.38.3.692. [DOI] [PubMed] [Google Scholar]
- David FL, Montezano AC, Reboucas NA, Nigro D, Fortes ZB, Carvalho MH, et al. Gender differences in vascular expression of endothelin and ET(A)/ET(B) receptors, but not in calcium handling mechanisms, in deoxycorticosterone acetate-salt hypertension. Braz J Med Biol Res. 2002;35:1061–1068. doi: 10.1590/s0100-879x2002000900006. [DOI] [PubMed] [Google Scholar]
- Dawson TC, Kuziel WA, Osahar TA, Maeda N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. 1999;143:205–211. doi: 10.1016/s0021-9150(98)00318-9. [DOI] [PubMed] [Google Scholar]
- Denton KM, Flower RL, Stevenson KM, Anderson WP. Adult rabbit offspring of mothers with secondary hypertension have increased blood pressure. Hypertension. 2003;41((Pt 2)):634–639. doi: 10.1161/01.HYP.0000052949.85257.8E. [DOI] [PubMed] [Google Scholar]
- Doggrell SA, Brown L. Rat models of hypertension, cardiac hypertrophy and failure. Cardiovasc Res. 1998;39:89–105. doi: 10.1016/s0008-6363(98)00076-5. [DOI] [PubMed] [Google Scholar]
- Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD. The combined role of P- and E-selectins in atherosclerosis. J Clin Invest. 1998;102:145–152. doi: 10.1172/JCI3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey RK, Imthurn B, Zacharia LC, Jackson EK. Hormone replacement therapy and cardiovascular disease: what went wrong and where do we go from here? Hypertension. 2004;44:789–795. doi: 10.1161/01.HYP.0000145988.95551.28. [DOI] [PubMed] [Google Scholar]
- Eatman D, Wang M, Socci RR, Thierry-Palmer M, Emmett N, Bayorh MA. Gender differences in the attenuation of salt-induced hypertension by angiotensin (1-7) Peptides. 2001;22:927–933. doi: 10.1016/s0196-9781(01)00404-1. [DOI] [PubMed] [Google Scholar]
- Elhage R, Arnal JF, Pieraggi MT, Duverger N, Fievet C, Faye JC, et al. 17 beta-estradiol prevents fatty streak formation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 1997;17:2679–2684. doi: 10.1161/01.atv.17.11.2679. [DOI] [PubMed] [Google Scholar]
- Ely DL, Turner ME. Hypertension in the spontaneously hypertensive rat is linked to the Y chromosome. Hypertension. 1990;16:277–281. doi: 10.1161/01.hyp.16.3.277. [DOI] [PubMed] [Google Scholar]
- Fischer M, Baessler A, Schunkert H. Renin angiotensin system and gender differences in the cardiovascular system. Cardiovasc Res. 2002;53:672–677. doi: 10.1016/s0008-6363(01)00479-5. [DOI] [PubMed] [Google Scholar]
- Fisher CR, Graves KH, Parlow AF, Simpson ER. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U S A. 1998;95:6965–6970. doi: 10.1073/pnas.95.12.6965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JS, Nijland MJ. Sex differences in the developmental origins of hypertension and cardiorenal disease. Am J Physiol Heart Circ Physiol. 2008;295:R1941–R1952. doi: 10.1152/ajpregu.90724.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourdy P, Mallat Z, Castano C, Garmy-Susini B, Mac Gregor JL, Tedgui A, et al. The atheroprotective effect of 17 beta-estradiol is not altered in P-selectin- or ICAM-1-deficient hypercholesterolemic mice. Atherosclerosis. 2003;166:41–48. doi: 10.1016/s0021-9150(02)00322-2. [DOI] [PubMed] [Google Scholar]
- Grady D, Herrington D, Bittner V, Blumenthal R, Davidson M, Hlatky M, et al. Cardiovascular disease outcomes during 6.8 years of hormone therapy: heart and Estrogen/progestin Replacement Study follow-up (HERS II) JAMA. 2002;288:49–57. doi: 10.1001/jama.288.1.49. [DOI] [PubMed] [Google Scholar]
- Grimsditch DC, Penfold S, Latcham J, Vidgeon-Hart M, Groot PH, Benson GM. C3H apoE(-/-) mice have less atherosclerosis than C57BL apoE(-/-) mice despite having a more atherogenic serum lipid profile. Atherosclerosis. 2000;151:389–397. doi: 10.1016/s0021-9150(99)00400-1. [DOI] [PubMed] [Google Scholar]
- Harrap SB, Van der Merwe WM, Griffin SA, Macpherson F, Lever AF. Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension. 1990;16:603–614. doi: 10.1161/01.hyp.16.6.603. [DOI] [PubMed] [Google Scholar]
- Harrison-Bernard LM, Schulman IH, Raij L. Postovariectomy hypertension is linked to increased renal AT1 receptor and salt sensitivity. Hypertension. 2003;42:1157–1163. doi: 10.1161/01.HYP.0000102180.13341.50. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Fukuto JM, Ignarro LJ, Chaudhuri G. Gender differences in atherosclerosis: possible role of nitric oxide. J Cardiovasc Pharmacol. 1995;26:792–802. doi: 10.1097/00005344-199511000-00017. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Esaki T, Sumi D, Mukherjee T, Iguchi A, Chaudhuri G. Modulating role of estradiol on arginase II expression in hyperlipidemic rabbits as an atheroprotective mechanism. Proc Natl Acad Sci U S A. 2006;103:10485–10490. doi: 10.1073/pnas.0603918103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood JR, Hinojosa-Laborde C. Sexual dimorphism of sodium-sensitive renal-wrap hypertension. Hypertension. 1997;30((Pt 2)):667–671. doi: 10.1161/01.hyp.30.3.667. [DOI] [PubMed] [Google Scholar]
- Hinojosa-Laborde C, Craig T, Zheng W, Ji H, Haywood JR, Sandberg K. Ovariectomy augments hypertension in aging female Dahl salt-sensitive rats. Hypertension. 2004;44:405–409. doi: 10.1161/01.HYP.0000142893.08655.96. [DOI] [PubMed] [Google Scholar]
- Hoeg JM, Willis LR, Weinberger MH. Estrogen attenuation of the development of hypertension in spontaneously hypertensive rats. Am J Physiol. 1977;233:H369–H373. doi: 10.1152/ajpheart.1977.233.3.H369. [DOI] [PubMed] [Google Scholar]
- Huxley R, Woodward M. Cigarette smoking as a risk factor for coronary heart disease in women compared with men: a systematic review and meta-analysis of prospective cohort studies. Lancet. 2011;378:1297–1305. doi: 10.1016/S0140-6736(11)60781-2. [DOI] [PubMed] [Google Scholar]
- Iams SG, Wexler BC. Retardation in the development of spontaneous hypertension in SH rats by gonadectomy. J Lab Clin Med. 1977;90:997–1003. [PubMed] [Google Scholar]
- Ji H, Zheng W, Wu X, Liu J, Ecelbarger CM, Watkins R, et al. Sex chromosome effects unmasked in angiotensin II-induced hypertension. Hypertension. 2010;55:1275–1282. doi: 10.1161/HYPERTENSIONAHA.109.144949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RC, Chapman SM, Dong ZM, Ordovas JM, Mayadas TN, Herz J, et al. Absence of P-selectin delays fatty streak formation in mice. J Clin Invest. 1997;99:1037–1043. doi: 10.1172/JCI119231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel WB, Hjortland MC, McNamara PM, Gordon T. Menopause and risk of cardiovascular disease: the Framingham study. Ann Intern Med. 1976;85:447–452. doi: 10.7326/0003-4819-85-4-447. [DOI] [PubMed] [Google Scholar]
- Kapil V, Milsom AB, Okorie M, Maleki-Toyserkani S, Akram F, Rehman F, et al. Inorganic nitrate supplementation lowers blood pressure in humans: role for nitrite-derived NO. Hypertension. 2010;56:274–281. doi: 10.1161/HYPERTENSIONAHA.110.153536. [DOI] [PubMed] [Google Scholar]
- Karatas A, Hegner B, de Windt LJ, Luft FC, Schubert C, Gross V, et al. Deoxycorticosterone acetate-salt mice exhibit blood pressure-independent sexual dimorphism. Hypertension. 2008;51:1177–1183. doi: 10.1161/HYPERTENSIONAHA.107.107938. [DOI] [PubMed] [Google Scholar]
- Kaushik M, Sontineni SP, Hunter C. Cardiovascular disease and androgens: a review. Int J Cardiol. 2010;142:8–14. doi: 10.1016/j.ijcard.2009.10.033. [DOI] [PubMed] [Google Scholar]
- Khan IY, Taylor PD, Dekou V, Seed PT, Lakasing L, Graham D, et al. Gender-linked hypertension in offspring of lard-fed pregnant rats. Hypertension. 2003;41:168–175. doi: 10.1161/01.hyp.0000047511.97879.fc. [DOI] [PubMed] [Google Scholar]
- Kienitz T, Quinkler M. Testosterone and blood pressure regulation. Kidney Blood Press Res. 2008;31:71–79. doi: 10.1159/000119417. [DOI] [PubMed] [Google Scholar]
- Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension. 2003;41:592–597. doi: 10.1161/01.HYP.0000056768.03657.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike G, Horiuchi M, Yamada T, Szpirer C, Jacob HJ, Dzau VJ. Human type 2 angiotensin II receptor gene: cloned, mapped to the X chromosome, and its mRNA is expressed in the human lung. Biochem Biophys Res Commun. 1994;203:1842–1850. doi: 10.1006/bbrc.1994.2402. [DOI] [PubMed] [Google Scholar]
- Lariviere R, Day R, Schiffrin EL. Increased expression of endothelin-1 gene in blood vessels of deoxycorticosterone acetate-salt hypertensive rats. Hypertension. 1993a;21((Pt 2)):916–920. doi: 10.1161/01.hyp.21.6.916. [DOI] [PubMed] [Google Scholar]
- Lariviere R, Thibault G, Schiffrin EL. Increased endothelin-1 content in blood vessels of deoxycorticosterone acetate-salt hypertensive but not in spontaneously hypertensive rats. Hypertension. 1993b;21:294–300. doi: 10.1161/01.hyp.21.3.294. [DOI] [PubMed] [Google Scholar]
- Lee MA, Bohm M, Paul M, Bader M, Ganten U, Ganten D. Physiological characterization of the hypertensive transgenic rat TGR(mREN2)27. Am J Physiol. 1996;270((Pt 1)):E919–E929. doi: 10.1152/ajpendo.1996.270.6.E919. [DOI] [PubMed] [Google Scholar]
- Lerman LO, Chade AR, Sica V, Napoli C. Animal models of hypertension: an overview. J Lab Clin Med. 2005;146:160–173. doi: 10.1016/j.lab.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Lerner DJ, Kannel WB. Patterns of coronary heart disease morbidity and mortality in the sexes: a 26-year follow-up of the Framingham population. Am Heart J. 1986;111:383–390. doi: 10.1016/0002-8703(86)90155-9. [DOI] [PubMed] [Google Scholar]
- Libby P, Okamoto Y, Rocha VZ, Folco E. Inflammation in atherosclerosis: transition from theory to practice. Circ J. 2010;74:213–220. doi: 10.1253/circj.cj-09-0706. [DOI] [PubMed] [Google Scholar]
- Lucas AD, Bursill C, Guzik TJ, Sadowski J, Channon KM, Greaves DR. Smooth muscle cells in human atherosclerotic plaques express the fractalkine receptor CX3CR1 and undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1) Circulation. 2003;108:2498–2504. doi: 10.1161/01.CIR.0000097119.57756.EF. [DOI] [PubMed] [Google Scholar]
- Maeda N, Johnson L, Kim S, Hagaman J, Friedman M, Reddick R. Anatomical differences and atherosclerosis in apolipoprotein E-deficient mice with 129/SvEv and C57BL/6 genetic backgrounds. Atherosclerosis. 2007;195:75–82. doi: 10.1016/j.atherosclerosis.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia G, De Backer G, Dominiczak A, Cifkova R, Fagard R, Germano G, et al. 2007 Guidelines for the Management of Arterial Hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) J Hypertens. 2007;25:1105–1187. doi: 10.1097/HJH.0b013e3281fc975a. [DOI] [PubMed] [Google Scholar]
- Manson JE, Allison MA, Rossouw JE, Carr JJ, Langer RD, Hsia J, et al. Estrogen therapy and coronary-artery calcification. N Engl J Med. 2007;356:2591–2602. doi: 10.1056/NEJMoa071513. [DOI] [PubMed] [Google Scholar]
- Marques FZ, Campain AE, Yang YH, Morris BJ. Meta-analysis of genome-wide gene expression differences in onset and maintenance phases of genetic hypertension. Hypertension. 2010;56:319–324. doi: 10.1161/HYPERTENSIONAHA.110.155366. [DOI] [PubMed] [Google Scholar]
- Marsh MM, Walker VR, Curtiss LK, Banka CL. Protection against atherosclerosis by estrogen is independent of plasma cholesterol levels in LDL receptor-deficient mice. J Lipid Res. 1999;40:893–900. [PubMed] [Google Scholar]
- Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. 1993;74:541–554. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- Mikkola TS, Clarkson TB. Estrogen replacement therapy, atherosclerosis, and vascular function. Cardiovasc Res. 2002;53:605–619. doi: 10.1016/s0008-6363(01)00466-7. [DOI] [PubMed] [Google Scholar]
- Mukherjee TK, Dinh H, Chaudhuri G, Nathan L. Testosterone attenuates expression of vascular cell adhesion molecule-1 by conversion to estradiol by aromatase in endothelial cells: implications in atherosclerosis. Proc Natl Acad Sci U S A. 2002;99:4055–4060. doi: 10.1073/pnas.052703199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovasc Res. 2007;75:478–486. doi: 10.1016/j.cardiores.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88:1121–1127. doi: 10.1172/JCI115411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien KD, McDonald TO, Chait A, Allen MD, Alpers CE. Neovascular expression of E-selectin, intercellular adhesion molecule-1, and vascular cell adhesion molecule-1 in human atherosclerosis and their relation to intimal leukocyte content. Circulation. 1996;93:672–682. doi: 10.1161/01.cir.93.4.672. [DOI] [PubMed] [Google Scholar]
- Ojeda NB, Grigore D, Yanes LL, Iliescu R, Robertson EB, Zhang H, et al. Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am J Physiol Heart Circ Physiol. 2007;292:R758–R763. doi: 10.1152/ajpregu.00311.2006. [DOI] [PubMed] [Google Scholar]
- Orshal JM, Khalil RA. Gender, sex hormones, and vascular tone. Am J Physiol Heart Circ Physiol. 2004;286:R233–R249. doi: 10.1152/ajpregu.00338.2003. [DOI] [PubMed] [Google Scholar]
- Ouchi Y, Share L, Crofton JT, Iitake K, Brooks DP. Sex difference in the development of deoxycorticosterone-salt hypertension in the rat. Hypertension. 1987;9:172–177. doi: 10.1161/01.hyp.9.2.172. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Nishina H, Hanson MA, Poston L. Dietary restriction in pregnant rats causes gender-related hypertension and vascular dysfunction in offspring. J Physiol. 2001;530((Pt 1)):141–152. doi: 10.1111/j.1469-7793.2001.0141m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendergrass KD, Pirro NT, Westwood BM, Ferrario CM, Brosnihan KB, Chappell MC. Sex differences in circulating and renal angiotensins of hypertensive mRen(2). Lewis but not normotensive Lewis rats. Am J Physiol Heart Circ Physiol. 2008;295:H10–H20. doi: 10.1152/ajpheart.01277.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira TM, Nogueira BV, Lima LC, Porto ML, Arruda JA, Vasquez EC, et al. Cardiac and vascular changes in elderly atherosclerotic mice: the influence of gender. Lipids Health Dis. 2010;9:87–96. doi: 10.1186/1476-511X-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Lopez FR, Larrad-Mur L, Kallen A, Chedraui P, Taylor HS. Gender differences in cardiovascular disease: hormonal and biochemical influences. Reprod Sci. 2010;17:511–531. doi: 10.1177/1933719110367829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto YM, Paul M, Ganten D. Lessons from rat models of hypertension: from Goldblatt to genetic engineering. Cardiovasc Res. 1998;39:77–88. doi: 10.1016/s0008-6363(98)00077-7. [DOI] [PubMed] [Google Scholar]
- Reckelhoff JF, Zhang H, Granger JP. Testosterone exacerbates hypertension and reduces pressure-natriuresis in male spontaneously hypertensive rats. Hypertension. 1998;31((Pt 2)):435–439. doi: 10.1161/01.hyp.31.1.435. [DOI] [PubMed] [Google Scholar]
- Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension. 2000;35:480–483. doi: 10.1161/01.hyp.35.1.480. [DOI] [PubMed] [Google Scholar]
- Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics–2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld ME, Campbell LA. Pathogens and atherosclerosis: update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb Haemost. 2011;106:858–867. doi: 10.1160/TH11-06-0392. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- Sainz J, Osuna A, Wangensteen R, de Dios Luna J, Rodriguez-Gomez I, Duarte J, et al. Role of sex, gonadectomy and sex hormones in the development of nitric oxide inhibition-induced hypertension. Exp Physiol. 2004;89:155–162. doi: 10.1113/expphysiol.2003.002652. [DOI] [PubMed] [Google Scholar]
- Sampson AK, Moritz KM, Jones ES, Flower RL, Widdop RE, Denton KM. Enhanced angiotensin II type 2 receptor mechanisms mediate decreases in arterial pressure attributable to chronic low-dose angiotensin II in female rats. Hypertension. 2008;52:666–671. doi: 10.1161/HYPERTENSIONAHA.108.114058. [DOI] [PubMed] [Google Scholar]
- Sampson AK, Jennings GL, Chin-Dusting JP. Y are males so difficult to understand?: a case where ‘X’ does not mark the spot. Hypertension. 2012;59:525–531. doi: 10.1161/HYPERTENSIONAHA.111.187880. [DOI] [PubMed] [Google Scholar]
- Sartori-Valinotti JC, Iliescu R, Yanes LL, Dorsett-Martin W, Reckelhoff JF. Sex differences in the pressor response to angiotensin II when the endogenous renin-angiotensin system is blocked. Hypertension. 2008;51:1170–1176. doi: 10.1161/HYPERTENSIONAHA.107.106922. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL, Sventek P, Li JS, Turgeon A, Reudelhuber T. Antihypertensive effect of an endothelin receptor antagonist in DOCA-salt spontaneously hypertensive rats. Br J Pharmacol. 1995;115:1377–1381. doi: 10.1111/j.1476-5381.1995.tb16626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz E, Gori T, Munzel T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens Res. 2011;34:665–673. doi: 10.1038/hr.2011.39. [DOI] [PubMed] [Google Scholar]
- Scotland RS, Madhani M, Chauhan S, Moncada S, Andresen J, Nilsson H, et al. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase-1 double-knockout mice: key role for endothelium-derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation. 2005;111:796–803. doi: 10.1161/01.CIR.0000155238.70797.4E. [DOI] [PubMed] [Google Scholar]
- Seidman DS, Laor A, Gale R, Stevenson DK, Mashiach S, Danon YL. Pre-eclampsia and offspring's blood pressure, cognitive ability and physical development at 17-years-of-age. Br J Obstet Gynaecol. 1991;98:1009–1014. doi: 10.1111/j.1471-0528.1991.tb15339.x. [DOI] [PubMed] [Google Scholar]
- Sharp BR, Jones SP, Rimmer DM, Lefer DJ. Differential response to myocardial reperfusion injury in eNOS-deficient mice. Am J Physiol Heart Circ Physiol. 2002;282:H2422–H2426. doi: 10.1152/ajpheart.00855.2001. [DOI] [PubMed] [Google Scholar]
- Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, et al. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 1996;93:13176–13181. doi: 10.1073/pnas.93.23.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Antonialli MM, Tostes RC, Fernandes L, Fior-Chadi DR, Akamine EH, Carvalho MH, et al. A lower ratio of AT1/AT2 receptors of angiotensin II is found in female than in male spontaneously hypertensive rats. Cardiovasc Res. 2004;62:587–593. doi: 10.1016/j.cardiores.2004.01.020. [DOI] [PubMed] [Google Scholar]
- Simpson ER, Clyne C, Rubin G, Boon WC, Robertson K, Britt K, et al. Aromatase – a brief overview. Annu Rev Physiol. 2002;64:93–127. doi: 10.1146/annurev.physiol.64.081601.142703. [DOI] [PubMed] [Google Scholar]
- Smith DD, Tan X, Tawfik O, Milne G, Stechschulte DJ, Dileepan KN. Increased aortic atherosclerotic plaque development in female apolipoprotein E-null mice is associated with elevated thromboxane A2 and decreased prostacyclin production. J Physiol Pharmacol. 2010;61:309–316. [PMC free article] [PubMed] [Google Scholar]
- Smith JD, Breslow JL. The emergence of mouse models of atherosclerosis and their relevance to clinical research. J Intern Med. 1997;242:99–109. doi: 10.1046/j.1365-2796.1997.00197.x. [DOI] [PubMed] [Google Scholar]
- Song J, Kost CK, Jr, Martin DS. Androgens augment renal vascular responses to ANG II in New Zealand genetically hypertensive rats. Am J Physiol Heart Circ Physiol. 2006;290:R1608–R1615. doi: 10.1152/ajpregu.00364.2005. [DOI] [PubMed] [Google Scholar]
- Stampfer MJ, Colditz GA, Willett WC, Manson JE, Rosner B, Speizer FE, et al. Postmenopausal estrogen therapy and cardiovascular disease. Ten-year follow-up from the nurses' health study. N Engl J Med. 1991;325:756–762. doi: 10.1056/NEJM199109123251102. [DOI] [PubMed] [Google Scholar]
- Stork S, von Schacky C, Angerer P. The effect of 17beta-estradiol on endothelial and inflammatory markers in postmenopausal women: a randomized, controlled trial. Atherosclerosis. 2002;165:301–307. doi: 10.1016/s0021-9150(02)00242-3. [DOI] [PubMed] [Google Scholar]
- Sullivan JC, Bhatia K, Yamamoto T, Elmarakby AA. Angiotensin (1-7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension. 2010;56:658–666. doi: 10.1161/HYPERTENSIONAHA.110.153668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surra JC, Guillen N, Arbones-Mainar JM, Barranquero C, Navarro MA, Arnal C, et al. Sex as a profound modifier of atherosclerotic lesion development in apolipoprotein E-deficient mice with different genetic backgrounds. J Atheroscler Thromb. 2010;17:712–721. doi: 10.5551/jat.3541. [DOI] [PubMed] [Google Scholar]
- Tabibiazar R, Wagner RA, Ashley EA, King JY, Ferrara R, Spin JM, et al. Signature patterns of gene expression in mouse atherosclerosis and their correlation to human coronary disease. Physiol Genomics. 2005;22:213–226. doi: 10.1152/physiolgenomics.00001.2005. [DOI] [PubMed] [Google Scholar]
- Tangirala RK, Rubin EM, Palinski W. Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res. 1995;36:2320–2328. [PubMed] [Google Scholar]
- Tanzawa K, Shimada Y, Kuroda M, Tsujita Y, Arai M, Watanabe H. WHHL-rabbit: a low density lipoprotein receptor-deficient animal model for familial hypercholesterolemia. FEBS Lett. 1980;118:81–84. doi: 10.1016/0014-5793(80)81223-3. [DOI] [PubMed] [Google Scholar]
- Tatchum-Talom R, Eyster KM, Martin DS. Sexual dimorphism in angiotensin II-induced hypertension and vascular alterations. Can J Physiol Pharmacol. 2005;83:413–422. doi: 10.1139/y05-012. [DOI] [PubMed] [Google Scholar]
- Teupser D, Persky AD, Breslow JL. Induction of atherosclerosis by low-fat, semisynthetic diets in LDL receptor-deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement) Arterioscler Thromb Vasc Biol. 2003;23:1907–1913. doi: 10.1161/01.ATV.0000090126.34881.B1. [DOI] [PubMed] [Google Scholar]
- Teupser D, Pavlides S, Tan M, Gutierrez-Ramos JC, Kolbeck R, Breslow JL. Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc Natl Acad Sci U S A. 2004;101:17795–17800. doi: 10.1073/pnas.0408096101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Endemann D, He G, Li JS, Schiffrin EL. Role of AT2 receptors in angiotensin II-stimulated contraction of small mesenteric arteries in young SHR. Hypertension. 1999;33((Pt 2)):366–372. doi: 10.1161/01.hyp.33.1.366. [DOI] [PubMed] [Google Scholar]
- Turner ME, Farkas J, Dunmire J, Ely D, Milsted A. Which Sry locus is the hypertensive Y chromosome locus? Hypertension. 2009;53:430–435. doi: 10.1161/HYPERTENSIONAHA.108.124131. [DOI] [PubMed] [Google Scholar]
- Vaccarino V, Badimon L, Corti R, de Wit C, Dorobantu M, Hall A, et al. Ischaemic heart disease in women: are there sex differences in pathophysiology and risk factors? Position paper from the working group on coronary pathophysiology and microcirculation of the European Society of Cardiology. Cardiovasc Res. 2011;90:9–17. doi: 10.1093/cvr/cvq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanderLaan PA, Reardon CA, Thisted RA, Getz GS. VLDL best predicts aortic root atherosclerosis in LDL receptor deficient mice. J Lipid Res. 2009;50:376–385. doi: 10.1194/jlr.M800284-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar IC, Hobbs AJ, Ahluwalia A. Sex differences in vascular function: implication of endothelium-derived hyperpolarizing factor. J Endocrinol. 2008;197:447–462. doi: 10.1677/JOE-08-0070. [DOI] [PubMed] [Google Scholar]
- Villar IC, Scotland RS, Khambata RS, Chan M, Duchene J, Sampaio AL, et al. Suppression of endothelial P-selectin expression contributes to reduced cell trafficking in females: an effect independent of NO and prostacyclin. Arterioscler Thromb Vasc Biol. 2011;31:1075–1083. doi: 10.1161/ATVBAHA.111.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmann S, Baumer AT, Strehlow K, van Eickels M, Grohe C, Ahlbory K, et al. Endothelial dysfunction and oxidative stress during estrogen deficiency in spontaneously hypertensive rats. Circulation. 2001;103:435–441. doi: 10.1161/01.cir.103.3.435. [DOI] [PubMed] [Google Scholar]
- Wiinberg N, Hoegholm A, Christensen HR, Bang LE, Mikkelsen KL, Nielsen PE, et al. 24-h ambulatory blood pressure in 352 normal Danish subjects, related to age and gender. Am J Hypertens. 1995;8((Pt 1)):978–986. doi: 10.1016/0895-7061(95)00216-2. [DOI] [PubMed] [Google Scholar]
- Williams SP, Shackelford DP, Iams SG, Mustafa SJ. Endothelium-dependent relaxation in estrogen-treated spontaneously hypertensive rats. Eur J Pharmacol. 1988;145:205–207. doi: 10.1016/0014-2999(88)90231-2. [DOI] [PubMed] [Google Scholar]
- Wilson PW, Garrison RJ, Castelli WP. Postmenopausal estrogen use, cigarette smoking, and cardiovascular morbidity in women over 50. The Framingham Study. N Engl J Med. 1985;313:1038–1043. doi: 10.1056/NEJM198510243131702. [DOI] [PubMed] [Google Scholar]
- Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res. 2001;49:460–467. doi: 10.1203/00006450-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Heart Circ Physiol. 2005;289:R1131–R1136. doi: 10.1152/ajpregu.00037.2003. [DOI] [PubMed] [Google Scholar]
- Xue B, Pamidimukkala J, Hay M. Sex differences in the development of angiotensin II-induced hypertension in conscious mice. Am J Physiol Heart Circ Physiol. 2005;288:H2177–H2184. doi: 10.1152/ajpheart.00969.2004. [DOI] [PubMed] [Google Scholar]
- Xue B, Pamidimukkala J, Lubahn DB, Hay M. Estrogen receptor-alpha mediates estrogen protection from angiotensin II-induced hypertension in conscious female mice. Am J Physiol Heart Circ Physiol. 2007;292:H1770–H1776. doi: 10.1152/ajpheart.01011.2005. [DOI] [PubMed] [Google Scholar]
- Yanes LL, Romero DG, Iles JW, Iliescu R, Gomez-Sanchez C, Reckelhoff JF. Sexual dimorphism in the renin-angiotensin system in aging spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2006;291:R383–R390. doi: 10.1152/ajpregu.00510.2005. [DOI] [PubMed] [Google Scholar]
- Yanes LL, Sartori-Valinotti JC, Iliescu R, Romero DG, Racusen LC, Zhang H, et al. Testosterone-dependent hypertension and upregulation of intrarenal angiotensinogen in Dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2009;296:F771–F779. doi: 10.1152/ajprenal.90389.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanes LL, Romero DG, Iliescu R, Zhang H, Davis D, Reckelhoff JF. Postmenopausal hypertension: role of the renin-angiotensin system. Hypertension. 2010;56:359–363. doi: 10.1161/HYPERTENSIONAHA.110.152975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn BS, Mantel C, Broxmeyer HE. Chemokines, chemokine receptors and hematopoiesis. Immunol Rev. 2000;177:150–174. doi: 10.1034/j.1600-065x.2000.17701.x. [DOI] [PubMed] [Google Scholar]
- Zhan E, Keimig T, Xu J, Peterson E, Ding J, Wang F, et al. Dose-dependent cardiac effect of oestrogen replacement in mice post-myocardial infarction. Exp Physiol. 2008;93:982–993. doi: 10.1113/expphysiol.2008.042788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Bian Z, Lu P, Karas RH, Bao L, Cox D, et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science. 2002;295:505–508. doi: 10.1126/science.1065250. [DOI] [PubMed] [Google Scholar]