

Abstract
β-amyloid (Aβ) is widely accepted to be one of the major pathomechanisms underlying Alzheimer's disease (AD), although there is presently lively debate regarding the relative roles of particular species/forms of this peptide. Most recent evidence indicates that soluble oligomers rather than plaques are the major cause of synaptic dysfunction and ultimately neurodegeneration. Soluble oligomeric Aβ has been shown to interact with several proteins, for example glutamatergic receptors of the NMDA type and proteins responsible for maintaining glutamate homeostasis such as uptake and release. As NMDA receptors are critically involved in neuronal plasticity including learning and memory, we felt that it would be valuable to provide an up to date review of the evidence connecting Aβ to these receptors and related neuronal plasticity. Strong support for the clinical relevance of such interactions is provided by the NMDA receptor antagonist memantine. This substance is the only NMDA receptor antagonist used clinically in the treatment of AD and therefore offers an excellent tool to facilitate translational extrapolations from in vitro studies through in vivo animal experiments to its ultimate clinical utility.
Keywords: Alzheimer's disease, memantine, glutamate, β -amyloid, NMDA, LTP
Pathophysiology of Alzheimer's disease
The pathophysiology of Alzheimer's disease (AD) is characterized by chronic, progressive neurodegeneration. The precise aetiology of AD is still not fully clarified but is known to be complex and multifactorial, with a notable overlap between familial and non-familial forms but also with different forms of dementia such as vascular dementia. The neurodegeneration seen in AD involves early synaptotoxicity and loss of neurophil, neurotransmitter disturbances, accumulation of extracellular β-amyloid (Aβ) deposits (amyloid/senile plaques) and intracellular neurofibrils (neurofibrillary tangles, NFTs), gliosis and only at later stages overt loss of neurons and associated brain atrophy (Yankner, 1996; Heininger, 1999; Bell and Claudio Cuello, 2006; Citron, 2010). At early stages of the disease, the entorhinal cortex and hippocampus are particularly affected, and this is associated with deficits in cognition/memory (Braak et al., 1993). Over the course of AD, up to 80% of neurons in the hippocampus die, and the progressive symptoms of AD manifest themselves as cognitive disturbances, reduced ability to cope with everyday life and worsening of clinical global impression score (Morris, 1986).
Aβ
As described by Alois Alzheimer himself (Alzheimer, 1907), one of the key histopathological hallmarks of the AD brain is the presence of extracellular ‘amyloid/senile plaques’ around neurons and glia. Such amyloid plaques are insoluble, quasi-crystalline deposits (Lesne et al., 2006), the main component of which is Aβ– a peptide (most commonly 40–42 amino acids in length) that is formed by enzymatic cleavage of the transmembrane amyloid precursor protein (APP) (Hardy and Higgins, 1992; Citron, 2010). Due to its neurotoxic effects and accumulation in AD, Aβ is believed to be a crucial pathogenic factor in disease development, both in familial and non-familial forms. Aβ is produced by the enzymatic cleavage of APP by β-secretase (extracellular cleavage) and γ-secretase (cuts in the middle of the membrane), whereas cleavage by α-secretase precludes formation of Aβ. The 42-amino-acid form, Aβ1–42, has a higher tendency to aggregate than Aβ1–40 and has been ascribed to be the main pathogenic form of this peptide (Citron, 2010) – but see also (Schlenzig et al., 2009). Aβ is continually released from neurons and glial cells into the extracellular environment where, at very low nM concentrations and possibly in monomeric form it may also play a physiological role (Puzzo et al., 2008).
Soluble Aβ oligomers
More recent evidence indicates that soluble oligomeric forms of Aβ, rather than the insoluble deposits, are primarily responsible for both the neurodegeneration and especially the impairment of synaptic function in AD (Barghorn et al., 2005; Ferreira et al., 2007; 2011; Lacor et al., 2007; Parsons et al., 2007; Demuro et al., 2010; Xia, 2010; Ferreira and Klein, 2011; Wilcox et al., 2011). For example, Aβ1–42 and Aβ oligomers were recently reported to be dramatically increased in the soluble fraction of Alzheimer's disease brain extracts, with oligomer levels 20-fold higher in aqueous compared with detergent extracts. Multiple oligomeric forms, including small oligomers, 56 and 200 kDa assemblies were found and proposed by the authors to contribute to synaptic dysfunction (Sokolow et al., 2011). However, contradictory findings have also been reported by others (e.g. van Helmond et al., 2010).
APP transgenic (TG) mice expressing the E693Delta mutation, which is reported to cause AD by enhanced Aβ oligomerization without fibrillization, displayed age-dependent accumulation of intraneuronal Aβ oligomers starting at 8 months but no extracellular amyloid deposits even at 24 months (Tomiyama et al., 2010). These mice indeed already showed deficits in synaptic plasticity, learning, synaptic markers, microglial activation and tau phosphorylation at 8 months, indicating that they might be a useful model of Aβ oligomer-induced pathology in the absence of amyloid plaques (Tomiyama et al., 2010). Soluble Aβ associated with (Q22, Dutch) or (G22, Arctic) mutant APP peptides was approximately 100-fold more potent than wild-type Aβ in inhibiting long-term potentiation (LTP) (Klyubin et al., 2004).
These soluble Aβ oligomers are thought to promote disturbances in glutamatergic neurotransmission and also increase the phosphorylation of tau (De Felice et al., 2007b). For example, chronic treatment with nanomolar concentration of Aβ oligomers was recently reported to induce NMDA receptor-dependent inward calcium ion (Ca2+) currents, mitochondrial Ca2+ overload/membrane depolarization, oxidative stress and apoptotic cell death in primary dissociated entorhinal cortex/hippocampal organotypic cultures (Alberdi et al., 2010; Bieschke et al., 2011).
Aβ oligomers are now believed to impair neuronal function and cognition, even before the appearance of overt toxicity (Lesne et al., 2006). However, the exact pathogenic role of deposits versus soluble forms and, in the latter case especially the major oligomeric species of Aβ involved (e.g. dimer, trimer or dodecamer), is still controversial (Barghorn et al., 2005; Selkoe, 2008; Bao et al., 2011). In contrast to soluble oligomeric forms of Aβ, this peptide in its soluble monomeric form has recently even been ascribed a physiological function and can enhance LTP at low pM concentrations (Puzzo et al., 2008), increase synaptic release probability (Abramov et al., 2009) and even protect against excitotoxic insults (Giuffrida et al., 2009).
It is not the intention of the present review to deepen this discussion; rather, we took as our starting point the present widely supported hypothesis that the soluble oligomeric forms of Aβ are primarily responsible for Aβ pathology and, what is perhaps more pertinent to this review, dysfunction of synaptic plasticity, which is expressed in patients as cognitive deficits.
Toxicity
Most early studies used high concentrations of synthetic Aβ (e.g. Aβ1–40, Aβ1–42 or even the truncated toxic fragment Aβ25–35) for studies on toxic effects. For example, Aβ1–42 (40 µM) enhanced glutamate neurotoxicity in human cerebral cortical cell cultures, whilst scrambled peptide was without effect (Mattson et al., 1992). Aβ1–42 was ineffective when applied alone; the insult required prolonged incubation for 4 days to develop and reflected general compromised ability of the neurons to handle elevated intracellular calcium levels following various glutamate agonists and even a calcium ionophore. All effects were associated with changes in intracellular Ca2+ levels (Mattson et al., 1992). Similarly, the selective NMDA receptor antagonist d-amino-phosponovaleric acid (D-APV, D-AP5) completely blocked Aβ1–42 (15–30 µM) uptake/internalization and subsequent up-regulation of cathepsin D and activation of microglia in organotypic hippocampal cultures (Bi et al., 2002).
However, more recently, it has been accepted by many working in the field that only much lower concentrations of Aβ are really relevant for chronic toxic effects in AD. For example, oligomeric Aβ1–42 (1 µM) induced reactive oxygen species (ROS) production from cultured cortical neurons through activation of NADPH oxidase. ROS derived from NADPH oxidase led to activation of ERK1/2, phosphorylation of cytosolic phospholipase A(2)α[cPLA(2)α] and arachidonic acid (AA) release (Shelat et al., 2008). The involvement of NMDA receptors in mediating these effects was shown by their reversibility by D-APV (10 µM) and memantine (5 µM) (Shelat et al., 2008).
In mixed neuronal-glial cultures from rat cerebellum, 250 nM Aβ1–42 induced about 30% loss of neurons and synapses after 2 to 3 days of treatment, whereas reverse peptide had no effect. There was no signal for overt apoptosis or necrosis, but Aβ1–42 rather increased the phagocytic capacity of microglia (Neniskyte et al., 2011).
Pathophysiologically relevant, even lower concentrations (pM) of naturally secreted Aβ oligomers (dimers and trimers, but not monomers) extracted directly from the cerebral cortex of subjects with AD produced a progressive loss of hippocampal synapses in organotypic hippocampal cultures (Shankar et al., 2007). Since this was prevented by the competitive NMDA receptor antagonist 3-[(R)-2-carboxypiperazin-4-yl]-prop-2-enyl-1-phosphonic acid (CPP, 20 µM) and associated with decreased Ca2+ influx, the involvement of NMDA receptors was postulated (Shankar et al., 2007).
Neurofibrillary tangles
Another characteristic histopathological feature of AD is the deposition of NFTs within neurons (Alzheimer, 1907; Braak et al., 1994). These abnormal protein bundles consist of flame-shaped, helical deposits of hyperphosphorylated tau protein. Under normal physiological conditions, phosphorylation of the tau protein (at five epitopes) helps to maintain cytoskeletal structure. The balance of phosphorylated and unphosphorylated tau regulates the stability of microtubules in the cytoskeleton, which act as an intracellular transport system and maintain the axoplasmic flow (Goedert, 1993; Goedert et al., 2006). In AD, there is probably an imbalance between phosphorylating protein kinases and dephosphorylating protein phosphatases, leading to excessive tau phosphorylation (at up to 21 epitopes), microtubule instability and, consequently, cell death (Noble et al., 2003; Goedert et al., 2006). Glycogen synthase kinase 3β (GSK-3β) and cell division protein kinase 5 (CDK5) seem to play pivotal roles in this hyperphosphorylation (Gong and Iqbal, 2008). The hyperphosphorylated tau protein accumulates inside the cell, dimerizing to paired helical filaments, which aggregate to form the typical NFTs seen in AD. The role of tau will not be addressed further, but readers are referred to recent reviews (Churcher, 2006; Goedert et al., 2006).
The glutamatergic neurotransmitter system
Glutamate is the major fast excitatory neurotransmitter and is involved in almost all CNS functions, especially in cortical and hippocampal regions – 70% of all excitatory synapses in the CNS utilize glutamate as a neurotransmitter (Watkins and Evans, 1981; Danysz et al., 1995; Parsons et al., 1998; 2002). Ionotropic glutamate receptors are ligand-gated ionic channels permeable to the monovalent cations Na+ and K+ and, depending on the subtype, also to the divalent cation Ca2+. AMPA receptors show very fast activation/inactivation kinetics, are largely postsynaptic, impermeable to Ca2+ and participate in most forms of fast excitatory synaptic neurotransmission (Watkins and Evans, 1981; Shinozaki, 1988; Parsons et al., 2002). In contrast, NMDA receptors are normally only synaptically activated under certain physiological conditions, for example during the induction of synaptic plasticity (Cotman et al., 1988; Collingridge and Singer, 1990).
Synaptic plasticity
The hippocampus, with its high density of glutamate receptors and in particular NMDA receptors, is known to be extremely important for some forms of learning and memory. Glutamatergic synapses can show pronounced plasticity in terms of the number and strength of individual synapses and are also characterized by their ability to express LTP – a long-lasting strengthening of synaptic transmission (Cotman et al., 1988; Collingridge and Singer, 1990). This remodelling at the cellular and molecular level is widely accepted to be an underlying synaptic mechanism for learning and memory (Collingridge and Singer, 1990; Butterfield and Pocernich, 2003). Signal cascades triggered by the activation of postsynaptic NMDA receptors are fundamentally important for LTP induction and, thereby, for neuronal plasticity.
The NMDA receptor has three cardinal features that permit its ‘co-incidence’ detector function in Hebbian synaptic plasticity: high permeability to Ca2+ ions, voltage-dependent block by magnesium ions (Mg2+) and relatively slow ligand gated kinetics. The resting membrane potential of a healthy neuron is normally around −70 mV, and the Ca2+ channel of the NMDA receptor is blocked by Mg2+ ions. As a consequence, normal resting conditions are associated with a low background level of postsynaptic intracellular Ca2+. Even during normal fast excitatory glutamatergic neurotransmission, postsynaptic intracellular Ca2+ levels remain low due to the above discussed biophysical properties of NMDA receptors. Only during, for example the induction of LTP does the stronger/more prolonged pulsatile glutamate release and a more pronounced influx of Na+ ions into the postsynaptic neuron via AMPA receptors decrease membrane potential for long enough to remove the block of the NMDA receptor channel by Mg2+ at which stage, Ca2+ ions can freely enter the cell via the NMDA receptor channel and trigger a cascade of second messenger processes that are involved in the fixation of increased synaptic strength.
At this juncture, one should emphasize the crucial physiological role of endogenous Mg2+ ions in this process which function as a switch to keep NMDA receptors blocked under resting or normal fast synaptic transmission conditions but allow Ca2+ ion influx when the pattern of activation has features characteristic for those required for learning processes, that is temporal and spatial convergence (cooperativity). This transient influx of Ca2+ is clearly distinguished against the low levels of background Ca2+ noise and, through downstream second-messenger processes, leads to detection of the neuronal plasticity/‘learning’ signal.
Glutamate excitotoxicity
Consistent with the involvement of the glutamatergic system in learning and memory, disturbances in glutamate neurotransmission have been linked with the pathophysiological processes underlying AD (Hardy and Cowburn, 1987; Greenamyre and Young, 1989; Palmer and Gershon, 1990; Cacabelos et al., 1999; Francis, 2003; Wenk et al., 2006). Chronic, mild activation of NMDA receptors ultimately leads to neurodegeneration – an effect termed chronic ‘excitotoxicity’ (Greenamyre and Young, 1989; Mattson et al., 1989; Braak et al., 1994; Dodd et al., 1994; Holscher, 1998; Butterfield and Pocernich, 2003). Notably, in this regard, the Mg2+ blockade of the NMDA receptor channel can be lifted by even moderate depolarization of the cell plasma membrane as well as by other factors discussed below. This triggers the pathological influx of Ca2+ ions into postsynaptic neurons. The prolonged Ca2+ overload leads first to loss of synaptic function, followed by synaptotoxicity and ultimately cell death, which correlates with the loss of memory function and learning ability in AD patients (Parsons et al., 1998; Danysz and Parsons, 2003; Miguel-Hidalgo et al., 2003; Wenk et al., 2006).
Factors that can influence the sensitivity of the glutamatergic system
Various pathologies such as the deposition of Aβ in plaques, soluble Aβ oligomers, hyperphosphorylated tau protein in NFTs, oxidative stress, mitochondrial dysfunction, energy deficits, chronically elevated concentrations of glutamate and neuronal inflammation have been associated with increased sensitivity and/or activity of the glutamatergic system, resulting in neuronal dysfunction and cell death in AD (Gray and Patel, 1995; Mattson et al., 1999; Wenk, 2006; Wenk et al., 2006; De Felice et al., 2007a; Parihar and Brewer, 2007; Gasparini and Dityatev, 2008; Parameshwaran et al., 2008).
Resting levels of glutamatergic agonists
The numerous factors that can influence the levels of endogenous glutamate receptor agonists and their downstream effects are presented in a schematic way in Figure 1. Some of these factors are outside of the scope of the present review, but the reader is recommended to refer to one of the following overviews for more information (Greenamyre and Young, 1989; Maragakis and Rothstein, 2001; Butterfield and Pocernich, 2003; Francis, 2003; Wenk et al., 2006; Jacob et al., 2007; Parsons et al., 2007; Bojarski et al., 2008).
Figure 1.
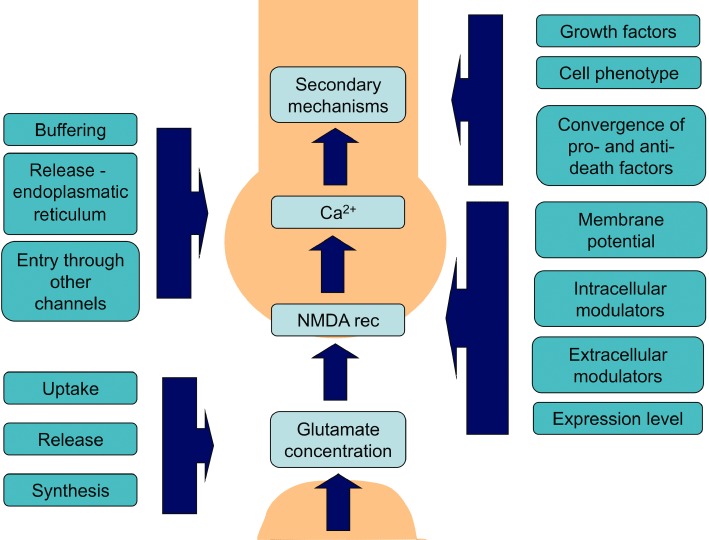
Schematic illustrating the factors directly involved in normal physiological NMDA receptor-mediated synaptic transmission/plasticity and associated processes/factors that can modulate such NMDA receptor activation/transmission both under physiological, but more importantly also under disturbed pathological conditions. For simplification, the roles of other receptors (e.g. AMPA) and feedback inhibition in synaptic plasticity have been omitted from this cartoon (see also Figure 2A). The points where such secondary factors interact with this signalling cascade are indicated by the vertical blue boxes with associated arrows pointing to the light blue boxes.
Resting glutamate concentrations under physiological conditions are normally in the low micromolar range. Only during synaptic transmission do these levels transiently reach mM concentrations for a few milliseconds (Clements et al., 1992). These low background levels of extracellular glutamate are normally regulated following transient physiological synaptic glutamate release by tightly controlled, very efficient re-uptake processes and intracellular metabolism to glutamine by glutamine synthetase in, for example, glial cells that serves to recycle glutamate released at these synapses. Glutamine passively diffuses to the presynaptic button where it is recycled into glutamate by glutaminase (Fagg et al., 1986; Danbolt, 2001).
Glutamate uptake/recycling mechanisms can be severely impaired in AD due to deficits in glutamate transporter expression. For example, glutamate transporter capacity (Bmax, Kd) and protein expression is reduced in the frontal/temporal cortex of AD patients, and there is also a selective loss of the vesicular glutamate transporter (VGluT) (Masliah et al., 1996; 2000; Li et al., 1997; Kirvell et al., 2006). It has also recently been reported that the excitatory amino acid transporter 2 (EAAT2), which is concentrated in perisynaptic astrocytes, also undergoes disease- and pathology-specific changes, with relatively greater expression of splice variants with reduced function in AD (Scott et al., 2010).
Glutamate is not the only endogenous agonist for NMDA receptors, and the levels of other endogenous agonists have been reported to be tonically elevated in AD. One clear example is homocysteic acid. This, probable non-neurotransmitter, endogenous amino acid is an agonist at both NMDA and metabotropic glutamate receptor 5 (mGluR5) receptors, and its levels have been reported to be chronically elevated in AD due to deficits in folic acid metabolism (Bleich et al., 2003).
NMDA receptor sensitivity
One prominent feature of NMDA receptors is their ability to be directly modulated by numerous endogenous factors. Probably, the most physiologically relevant are the co-agonists glycine/d-serine, the continuous (i.e. non-synaptically released) presence of which is an absolute prerequisite for NMDA receptor activation by glutamate (Kleckner and Dingledine, 1988; Danysz and Parsons, 1998). d-serine is the predominant endogenous co-agonist of the NMDA receptor in the forebrain and may be involved in controlling the extent of NMDA receptor-mediated neurotoxic insults observed in CNS disorders, including AD (Danysz and Parsons, 1998). Serine racemase knockout mice showed an approximately 90% decrease in forebrain d-serine content and reduced neurotoxicity induced by both NMDA and Aβ1–42 injections into the forebrain in vivo (Inoue et al., 2008). Conditioned medium from Aβ1–42 (15 µM) treated microglia also contained elevated levels of d-serine. This conditioned media was toxic to cultured hippocampal neurons, an effect that could be blocked by the NMDA receptor glycine site antagonist 5,7-dicholorokynurenic acid and by enzymatic degradation of d-amino acids by d-amino acid oxidase (Wu et al., 2004). Serine racemase mRNA levels were reported to be elevated in both these microglia cultures as well as in AD hippocampus (Wu et al., 2004). In contrast, others have reported that Aβ25–35 (3 nmol i.c.v.) – induced learning deficits in spontaneous alternation and step-down passive avoidance were reversed by the NMDA glycine site partial agonist d-cycloserine (1–30 mg·kg−1 i.p.) and the glycine prodrug milacemide (3–100 mg·kg−1 i.p.) (Maurice et al., 1996).
However, other factors have a somewhat more subtle but still very important influence on NMDA receptor function. Some examples would be endogenous polyamines like spermine (Williams, 1997), which have multiple effects on NMDA receptors, the most important of which is their ability to positively modulate NMDA receptors containing the NR2B subunit. Other important factors are free radicals, redox potential, inflammation, pH, etc., which clearly change during pathological processes such as those occurring in AD (Wenk et al., 2006).
The sensitivity of NMDA receptors to detect physiological/pathological signals does not just depend on the presence of agonists/modulators. One obviously important factor is their expression level. In general, the overall expression level of NMDA receptors in AD is reduced rather than increased, but this probably reflects compensatory reactions of the biological system in an attempt to compensate the pathological changes (i.e. receptor down-regulation as an adaptive change), as well as blatant loss of neuronal cells and synapses expressing these receptors as a long term consequence of chronic excitotoxicity (Geddes et al., 1986; Procter et al., 1989; Ninomiya et al., 1991; Hynd et al., 2001).
The synaptic/non-synaptic distribution of NMDA receptors is also of paramount importance in determining which receptors are available for physiological activation and which might rather be available for excitotoxic processes. In this regard, also the subunit composition is of importance as, for example, NR2B subunit containing receptors have been deemed by some to be extra synaptic ‘death’ receptors (Hardingham et al., 2002; Bordji et al., 2011). The postsynaptic localization and thereby the ability of NMDA receptors to be activated by physiologically, synaptically released glutamate also depends on their association with postsynaptic anchoring proteins such as PSD-95. The phosphorylation status of NMDA receptors can also influence their sensitivity to both activation by agonists (Zheng et al., 1997) and modulation by the endogenous channel blocker Mg2+ (Chen and Huang, 1992) (Figure 1).
Perhaps one of the most important factors, however is the resting membrane potential. The NMDA receptor is almost unique in its combined ligand and voltage-gating properties (Nowak et al., 1984) (i.e. physiological sensitivity to synchronise transient changes in both neurotransmitter concentration and membrane potential). Precisely, these properties render NMDA receptors their ability to act as coincidence detectors, essential for their role in synaptic plasticity (Cotman et al., 1988; Collingridge and Singer, 1990). The caveat is that this voltage dependency can be a burden in chronic disease states such as those occurring in AD. Factors that disturb the normal resting membrane potential of neurons can have severe impact on the normal function of NMDA receptors as these can lead to a tonic relief of their voltage-dependent modulation by Mg2+.
Specific issues to be addressed in this review
The ultimate goal of this review is to try to address the following questions:
How does Aβ affect homeostasis of the glutamatergic system?
How does Aβ impact directly on NMDA receptor function?
What is the resulting effect of Aβ on synaptic function/plasticity, partially independent from overt toxicity?
What is the evidence that NMDA receptor antagonists like memantine may reverse/prevent these negative effects of Aβ?
How does Aβ affect homeostasis of the glutamatergic system (Table 1)?
Table 1.
Effect of Aβ on glutamate homeostasis (studies showing effects leading to decrease in synaptic glutamate are in bold text)
| Experimental system | Aβ type/dose | Effect of Aβ | Reference |
|---|---|---|---|
| Hippocampal slices | Aβ25–35 (Macromolecular Analysis Lab) dissolved in water, 10 µM | Enhanced depolarization-stimulated glutamate release- stronger in aged animals – by Aβ pre-incubated for 1 h. in slices. Added acute, did not have any effect. | Arias et al. (1995) |
| Xenopus oocytes expressing heterologous proteins | Aβ1–42 (40 µM, Sigma) and Aβ25–35 (590 µM, Sigma) in 100 mM CH3COOH stock stored at −20°C | Aβ1–42 (1 µM) inhibited the ATPase Na+/K+ pump and glutamate transporter EAAC1 | Gu et al. (2004) |
| Glial cultures | Aβ25–35 (Bachem) 100 µM dissolved immediately before experiment | Inhibited glial glutamate uptake | Harris et al. (1996) Harris et al. (1995) |
| Rat primary glial cultures | Aβ25–35 (Univ. of Iowa), 7 days incubation | Astrocytes exposed for 7 days to Aß showed reduced glutamate uptake | ParpuraGill et al. (1997) |
| Cultured macrophages | Aβ1–40, Aβ25–35, Aβ40-1 (Bachem) dissolved in water and stored at −20oC, | Aβ1–40 (from 10 µM) but not reverse Aβ40-1 or the Aβ25–35 sub fragment enhanced glutamate and oxygen free radical (after 15 min) production by cultured macrophages | Klegeris and McGeer (1997) |
| Human cortical cultures | Aβ1–38 or 25–35 (Bachem), stored in water or DMSO at −20oC, 1 and more days incubation in culture | Aβ at 40 µM enhanced the toxicity of NMDA and starting at 20 µM of glutamate. | Mattson et al. (1992) |
| Microglia cultures | Aβ25–35 (Peptide Inst, Osaka) (stored at −80°C until use) 5 µM final concentration | Reversed glutamate transporter activity | Noda et al. (1999) |
| Cultured neurons and astrocytes | Aβ25–35 (Neosystem) in water, allowed to aggregate (confirmed by microscope) | Aβ25–35 (0.25–15 µM) applied for 30 min to cultured neurons and astrocytes increased glutamate levels in media but astrocytes were more sensitive. These effects were also associated with overt toxicity, but surviving neurons showed enhanced uptake of glutamate. | Fernandez-Tome et al. (2004) |
| Rat cortical synaptosomes | Aβ1–42 pre incubation for 24 h in 37oC in PBS | Aβ1–42 also increases HNE conjugation to the glutamate transporter resulting in uptake inhibition | Lauderback et al. (2001) |
| Primary microglia cultures | sAPP released from cells | Aβ stimulated microglia release glutamate | Barger and Basile (2001) |
| Primary hippocampal rat astrocytes | Aβ25–35 | At 100 µM inhibition by 50% was observed at 30 min. Prevented by scavengers. | Lauderback et al. (1999) |
| Primary rat telencephalonic astrocytes in vitro | Aβ25–35 custom synthesis | Aβ25–35 (100 µM) for 24 h caused depolarization and inhibition of glutamate uptake. | Harkany et al. (2000) |
| Rat magnocellular nucleus basalis (MBN) in vivo | Aβ1–42 or Aβ25–35 custom synthesis (200 µM)- 1 µL into NBM | Aβ infusion via microdialysis caused increased extracellular concentrations of excitatory amino acid neurotransmitters within 20–30 min | Harkany et al. (2000) |
| Primary cortical rat astrocytes and neurons | Aβ1–40 and Aβ1–42 (Tokyo Metropolitan Inst. Of Gerontology, Tokyo), prepared in basic conditions to prevent aggregation | Aβ1–42 (20 µM) incubated for 12–48 h increased glutamate uptake activity in primary cultures of rat cortical astrocytes and neurons (Ikegaya et al., 2002). This effect was associated with an increase in cell-surface expression of the glial glutamate transporter GLAST. Only modest effects were seen with the less toxic species Aβ1–40 | Ikegaya et al. (2002) |
| Rat cortical astrocytes | Aβ25–35, Aβ1–42, Aβ1–40 (Sigma) | Aβ1–42 or Aβ25–35 (20 µM) for 24–48 h increased expression levels of the glutamate transporter GLAST and uptake of glutamate from the culture media | Abe and Misawa (2003) |
Aβ influences glutamate concentrations in the synaptic cleft
Most studies on the effects of Aβ on glutamate transport mechanisms have been performed with toxic sub fragments of Aβ. Studies in hippocampal slices indicate that glutamate uptake is impaired in the aged hippocampus, and that Aβ25–35 augments the release and/or inhibits the uptake of glutamate and aspartate, especially in aged animals (Arias et al., 1995; ParpuraGill et al., 1997). Aβ25–35 (0.25–15 µM) applied for 30 min to cultured neurons and astrocytes increased glutamate levels in media by decreasing glutamate uptake (Harris et al., 1996; Fernandez-Tome et al., 2004). These effects were associated with overt toxicity, but surviving neurons showed enhanced uptake of glutamate, possibly as a reactive protective mechanism (Fernandez-Tome et al., 2004).
A possible mechanism for this effect is the fact that glutamate transporters are inhibited by oxidative damage from ROS and lipid peroxidation products such as 4-hydroxy-2-nonenal (HNE). Aβ1–42 has been reported to increase HNE conjugation to the glutamate transporter resulting in inhibition of glutamate uptake (Lauderback et al., 2001).
When natural length Aβ has been investigated, until recently, extremely high concentrations of up to 100 µM were normally used to demonstrate inhibition of glutamate uptake (Lauderback et al., 1999). However, synaptic glutamate uptake was recently reported to be strongly decreased by much lower levels of soluble Aβ from several sources (synthetic, cell culture, human brain extracts) (Li et al., 2009). This in turn resulted in enhancement of NR2B-containing NMDA receptor currents and extra synaptic responses, an effect mimicked by the glutamate reuptake inhibitor dl-threo-β-benzyloxyaspartic acid (Li et al., 2011).
The important role of inflammatory process through the cascade Aβ > microglia > TNF-α > NMDA should also be considered (Wenk et al., 2006). For example, the Aβ25–35 subfragment has even been reported to causes reverse glutamate transport by microglia (Noda et al., 1999). Although only Aβ1–40 but not the Aβ25–35 subfragment caused a moderate enhancement of glutamate and oxygen free radical production by cultured macrophages (Klegeris and McGeer, 1997), both potentiated the stimulatory effect of phorbol myristate acetate (PMA) when used as priming agents (Klegeris and McGeer, 1997).
The physiological Aβ precursor protein APP has been reported to have ‘positive’ effects on glutamate transport (Mattson et al., 1993; Masliah et al., 1996; 1998). In contrast, microglia activated by soluble APP (sAPP) have been shown to release excitotoxic levels of glutamate, probably as a consequence of auto protective antioxidant glutathione production within the microglia, ultimately causing synaptic degeneration and neuronal death via NMDA receptor activation (Barger and Basile, 2001).
The opposite effect of Aβ on glutamate levels has also been reported, for example, Aβ1–42 (20 µM) incubated for 12–48 h increased glutamate uptake activity in primary cultures of rat cortical astrocytes and neurons (Ikegaya et al., 2002). This effect was associated with an increase in cell-surface expression of the glial glutamate transporter (GLAST). Only modest effects were seen with the less toxic species Aβ1–40. Similarly, treatment of astrocytic cultures with Aβ1–42 or Aβ25–35 (20 µM) for 24–48 h increased expression levels of the glutamate transporter GLAST and uptake of glutamate from the culture media (Abe and Misawa, 2003). However, it is possible that this increased uptake/expression was a secondary protective mechanism of cells surviving toxicity (Fernandez-Tome et al., 2004).
Excitotoxins released into media from brain mononuclear phagocytes (macrophages and microglia) following activation by co-culture with neuronal cells expressing wild-type APP or familial AD-linked APP mutants were neurotoxic, induced ROS and were able to evoke inward currents in Xenopus oocytes heterologously expressing NMDA receptors (Ikezu et al., 2003). Similarly, conditioned media from αAPPs/p3- and βAPPs/Aβ-stimulated cultured human monocyte-derived macrophages directly induced inward currents through NR1a/NR2B receptors expressed in Xenopus oocytes that were blocked by the NMDA receptor antagonist D-APV (50 µM) but not by the AMPA receptor antagonist CNQX (20 µM) (Xiong et al., 2004).
Taken together, the different effects of toxic Aβ and physiological APP on glutamate transport mechanisms seems likely to contribute to excitotoxicity and the neuronal degeneration observed in AD, whereby Aβ may increase availability/residence of glutamate in the synaptic cleft through inhibition, and even reversal, of uptake mechanisms (Gegelashvili and Schousboe, 1997). This evidence linking Aβ to disturbed homeostasis of glutamate levels has been listed in Table 1. However, one caveat that should be noted is that most studies used very high concentrations of Aβ and/or toxic subfragments of this peptide.
Intracellular signalling cascades
Apart from synaptic consequences of Aβ, certain intracellular mechanisms may also be changed by this peptide leading either to enhancement or inhibition of downstream effects of glutamate receptor activation as illustrated in Figure 1. For example, Aβ1–42 oligomers (Aβ derived diffusible ligands ADDLs) have also been shown to induce overexpression of the Arc gene (associated with memory function), leading to a loss of NMDA receptors and altered cell morphology (Klein et al., 2007)
In primary neuronal cell culture and hippocampal slices, Aβ oligomers impaired LTP and spontaneous network activity and induced retraction of synaptic contacts long before major cytotoxic effects were visible (Ronicke et al., 2010). In this same study, the second messenger Jacob was shown to couple extra synaptic NMDA receptor activity to CREB protein dephosphorylation and accumulated in the nucleus after Aβ oligomer administration. The NR2B-containing NMDA receptor antagonists ifenprodil and Ro 25–6981 both blocked all of these effects (Ronicke et al., 2010).
Other factors influencing NMDA receptor second messenger effects include alterations of intracellular Ca2+ concentration, buffering, release from intracellular stores and sequestration as well as changes in expression/function of Ca2+ target proteins like Ca2+/calmodulin-dependent protein kinases II (CamKII) (Mattson et al., 1993; Koizumi et al., 1998; Brzyska and Elbaum, 2003; Zhao et al., 2004; Cheung et al., 2008). Intracellular mechanisms are not intended as a primary focus of the present review and were previously discussed in an excellent review (Ferreira et al., 2010).
Effects of Aβ on NMDA receptor function
There are many indications that Aβ may directly affect NMDA receptor function (Figure 2 and Table 2). For example, (+)MK-801 or removal of extracellular Ca2+ reduced Aβ1–40-induced Ca2+ transients, NO production and neurotoxicity in cultured neuroblastoma (MES 23.5) cells (Le et al., 1995). (+)MK-801 partially prevented the decrease in cell viability and the energy impairment in HEK293 cells transiently expressing NR1/NR2A or NR1/NR2B subunits exposed to Aβ1–42 (Domingues et al., 2007). Aβ1–40 treatment of cultured cerebellar granule cells induced a time- and concentration-dependent activation of NF-κB, which was inhibited by (+)MK-801 (10 µM) (Kawamoto et al., 2007). These authors suggested that Aβ activates NF-κB by an NMDA-Src-Ras-like protein through MAPK and PI3K pathways (Kawamoto et al., 2007).
Figure 2.
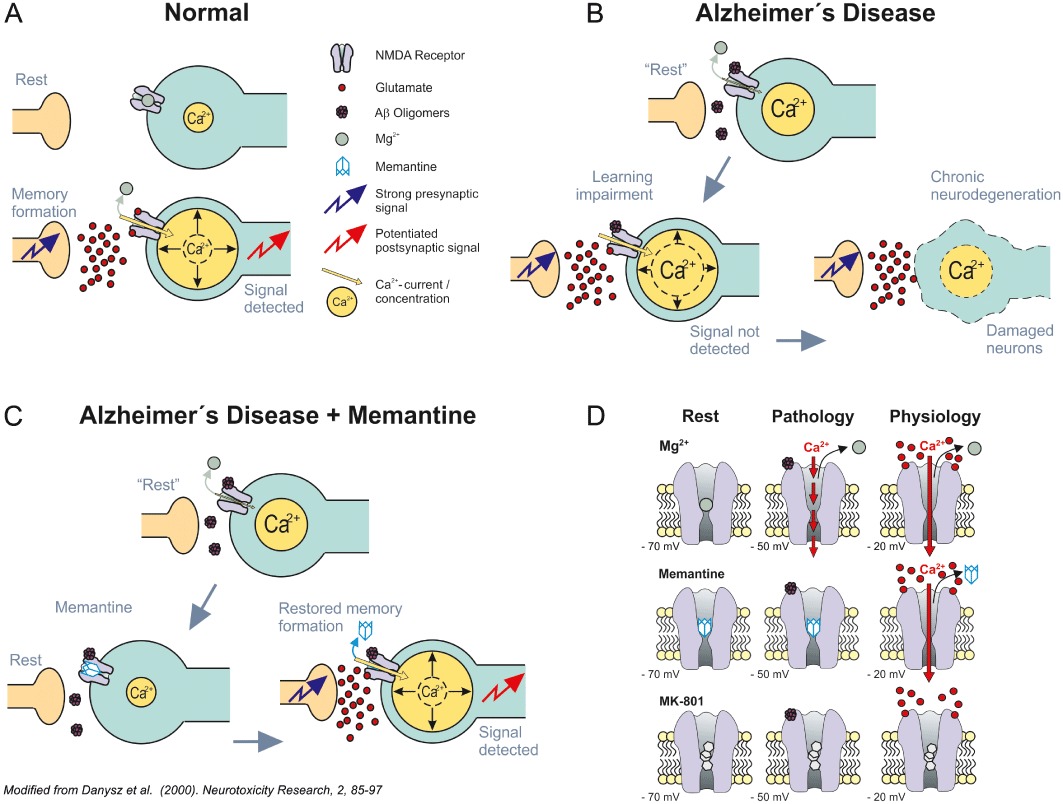
(A) Under normal physiological conditions, synaptic plasticity/learning depends on the detection of a relevant (sufficiently strong) synaptic signal over background noise (here referring to transient high vs. prolonged moderate intracellular Ca2+ levels), resulting in a sufficient signal-to-noise ratio. Intracellular Ca2+ concentrations at any one time point are represented by different sizes of the yellow Ca2+ containing circles. Jagged blue arrow indicates arrival of a presynaptic signal. Jagged red arrow indicates detection of the postsynaptic signal. Mg2+ and glutamate are illustrated by large green and small red circles respectively. For simplification, the roles of other receptors (e.g. AMPA) and feedback inhibition have been omitted from this cartoon (please refer to figure 3 in Parsons et al., 2007 for a more detailed depiction of the processes believed to underlie such synaptic plasticity). (B) The signal-to-noise ratio hypothesis assumes that in Alzheimer's disease (AD), due to a tonic over activation of NMDA receptors by, for example soluble β-amyloid oligomers, Mg2+ is no longer effective enough to play its ‘filtering’ function. In turn, synaptic noise rises, impairing detection of the relevant synaptic signal required for learning/plasticity. The light blue straight arrows indicate the proposed course of events (i.e. first symptomatic disturbance of synaptic plasticity) followed by synaptotoxicity and ultimately neuronal death. Soluble β-amyloid oligomers represented as mauve aggregates of small circles – here binding directly to NMDA receptors for simplification, but probably interacting more directly with anchoring protein complexes and thereby affecting the function of their associated proteins such as NMDA receptors. Other symbols have the same meaning as in panel A. (C) Schematic illustrating memantine's proposed MoA in AD based on the signal-to-noise hypothesis. Memantine is able to serve as a more effective filter than Mg2+, blocking pathological ‘noise’ at glutamatergic synapses and thereby allowing detection of the relevant synaptic signal. Synaptic plasticity is restored and synaptotoxicity/ultimate neuronal death is prevented by the same MoA. Memantine illustrated as a simple light blue adamantane cage. For other aspects, see legends to panels A and B. Modified from Danysz et al. (2000). (D) Schematic illustrating the hypothesis explaining how the fast unblocking kinetics of memantine allow this voltage-dependent compound to differentiate between the physiological and pathological activation of NMDA receptors. Under resting therapeutic conditions [i.e. in their continuing presence at −70 mV (left), Mg2+ (top), memantine (middle) and MK-801 (bottom)], all occupy the NMDA receptor channel. Both Mg2+ and memantine are able to leave the NMDA receptor channel upon strong synaptic depolarization (−20 mV, right) due to their pronounced voltage dependency and rapid unblocking kinetics, whereas the slow, potent blocker MK-801 remains trapped. However, memantine – in contrasts to Mg2+– does not leave the channel so easily upon moderate prolonged depolarization during chronic excitotoxic insults caused by soluble β-amyloid oligomers tonically activating NMDA receptors (−50 mV, centre). Transient strong and prolonged moderate Ca2+ influx illustrated by the full and dashed red arrows respectively. Modified after Kornhuber and Weller (1997) and Parsons et al. (1999).
Table 2.
Effect of Aβ on NMDA receptor function (studies showing a decrease of NMDA function after Aβ are indicated by bold text)
| Experimental system | Aβ type/dose | Effect of Aβ observed | Reference |
|---|---|---|---|
| Patch clamp – rat hippocampal slices, dentate gyrus | Aβ1–40 (Bachem, 100–200 nM) | Applied by perfusion or intracellular via the recording pipette enhanced NMDA receptor-mediated synaptic currents | Wu et al. (1995a) |
| Single unit recordings, iontophoretic application to CA1 in vivo | Aβ1–42 mixture of different species (fibrils and protofibrils) | Responses to NMDA were potentiated to 260% but to AMPA decreased | Szegedi et al. (2005) |
| Akt phosphorylation – mice slices or primary cortical/hippocampal cultures | Aß length not specified (California Peptide Research), claimed to be in forms of dimers and trimers | Aß stimulated phosphorylation of Akt and this effect was attenuated by the NR2B antagonist ifenprodil | Abbott et al. (2007) |
| Inward currents, Ca2+ entry, apoptosis – rat cortical primary cultures and organotypic slices from entorhinal-hippocampal region | Aß1–41 (ABX), stated to be soluble Aß at the time of application | Aß stimulated inward currents (whole cell patch clamp) and Ca2+ influx (fluorescence imaging) and these effects were attenuated by AP5, Memantine, (+)MK-801. AMPA antagonist as also active indicating contribution of AMPA receptors. | Alberdi et al. (2010) |
| Axonal trafficking of dense-core vesicles and mitochondria – primary hippocampal cultures | Soluble Aß oligomers (5 µM, incubated for 4 or 18 h) | Aß effect was prevented by AP5, Memantine and (+)MK-801 | Decker et al. (2010b) |
| Aß toxicity (LDH release) in HEK293 cells | Aß1–40 (Bachem, 1 µM) aged for 7 days at 37°C to produce ß-sheet fibrils, incubation for 24–48 h. | Aß-induced increase in toxicity was present in NR1/NR2A but not NR1/NR2B expressing cells, | Domingues et al. (2007) |
| Neurite growth – primary rat cortical neurons | Aß1–42 (Athens), freshly prepared at 5 µM, incubation for 3 days. | Aß-induced decrease in neurite growth was attenuated by memantine | Hu et al. (2007) |
| Inward currents –Xenopus oocytes expressing NMDA receptor subunits | Aß1–42 (ABX, 100 µM), incubated at 4°C for 24 h to allow aggregation. | Aß-induced inward currents were blocked by memantine, AP5 and (+)MK-801. Ca2+ cytosolic increase was attenuated by AP5 but only partially inhibited by ifenprodil (NR2B ant.) | Texido et al. (2011) |
| Spine density – rat organotypic cultures | Aß1–42 containing several oligomeric species, secreted by neurons | Reduction in spine density produced by Aß was attenuated by AP5 | Wei et al. (2009) |
| fEPSPs and neuronal damage – organotypic rat hippocampal cultures | Aß1–42 globular form pre-incubated for 12 h at room temperature. | Memantine (1 µM) attenuated fEPSP deficits produced by Aß (82 nM, globulomer). Similar effects were observed against neuronal damage. | Nimmrich et al. (2010) |
| Apoptosis, cGMP – MES 23.5 cell line | Aß1–42 (Bachem), 10 µM | (+)MK-801 (100 µM) prevented cGMP increase and toxicity produced by Aß | Le et al. (1995) |
| Expression of PSD-95 and synaptophysin – primary rat hippocampal cultures | Aß25–35 (American Peptides) 10 µM used without pre-incubation, low N oligomers expected | Down-regulation of PSD-95 and synaptophysin induced by Aß was attenuated by (+)MK-801 (but note that low doses of NMDA were also preventative) | Liu et al. (2010) |
| Surface expression of insulin receptors – cultures of hippocampal neurones | Aß1–42 (American Peptides) 100 µM, pre-incubated overnight to allow formation of ADDLs followed by centrifugation and supernatant was used as soluble Aß oligomers | Memantine (20 µM) and AP5 (50 µM) prevented Aß-induced insulin receptor loss. | Zhao et al. (2008) |
| NR1a/NR2B receptors expressed in X. oocytes | Conditioned media from aAPPs/p3- and bAPPs/Aβ-stimulated cultured human monocyte-derived macrophages | This media produced induced inward currents that were blocked by the NMDA receptor antagonist D-APV (50 µM) but not AMPA antagonist | Xiong et al. (2004) |
| Rat primary cortical cultures | Aβ1–40 (American Peptides), 0.1–10 µM (claimed to be monomeric to tetrameric) | Caused an NMDA receptor dependent decrease in PSD-95 after 60 but not 15 min. starting at 0.1 µM. Prevented by MK-801 and ifenprodil. NMDA + Aβ was not stronger than Aβ alone. | Roselli et al. (2005) |
| Receptor binding in rat cortical membranes | Aβ25–35 (UCB Bioproducts) dissolved immediately before the experiment | Aβ25–35 (10 µM) inhibited both [3H]glutamate and [3H]glycine binding (by 20 and 70% respectively) and stimulated functional [3H]MK-801 binding | Cowburn et al. (1997) |
| Natural | Aβ dodecameric oligomers co-immunoprecipitate with NR1 and NR2A | Venkitaramani et al. (2007) | |
| Aβ1–42, freshly prepared in 0.1 N NaOH, pH readjusted to 7.4, and then diluted in serum-free culture medium. | Internalization of Aβ is blocked by NMDA receptor antagonists indicating | Bi et al. (2002) | |
| Activation of NMDA receptors by Aβ was proposed to be secondary to microglial activation and production of TNFα NMDA | Wenk et al. (2006) | ||
| Differentiated cultures of hippocampal neurons | Soluble Aβ-derived diffusible ligands (ADDLs) | Aβ bound to glutamatergic but not GABA-ergic neurons and to postsynaptic density complexes containing NMDA receptors. After chronic exposure produced abnormal spine morphology and a decrease in their density. Subsequent consequences such as loss of the spine cytoskeletal protein drebrin were prevented by memantine at a concentration of 5 µM | Lacor et al. (2007) |
| Hippocampal neuronal cultures | Aβ1–42 (California Peptide) oligomers aged overnight at 4°C, and centrifuged to remove insoluble aggregates | Aβ (starting at 300–500 nM) produced oxidative stress and calcium influx that was prevented by memantine (5 or 10 µM) or anti NMDA receptor antibodies. | De Felice et al. (2007a) |
| Aβ co-immunoprecipitated together with NMDA receptor | |||
| Hippocampal neuronal cultures and neuroblastoma cells | Aβ1–42 (California Peptide) oligomers aged overnight at 4°C, and centrifuged to remove insoluble aggregates (ADDLs) | ADDLs stimulated tau phosphorylation at epitopes characteristically hyperphosphorylated in AD | De Felice et al. (2007b) |
| Hippocampal neuronal cultures | Aβ25–35 (Bachem and Sigma), 18 h of incubation | Aβ increased intracellular Ca2+ (starting at 11 µM) which was enhanced by Mg2+ removal and blocked by NMDA receptor antagonists (claimed to be due to enhanced glutamatergic synaptic network activity but not due to direct effects on NMDA receptors) | Brorson et al. (1995) |
| Wistar rats, field excitatory potentials in CA1 | Aβ1-40 (Bachem) freshly prepared | Aβ1-40 caused a long lasting depression of hippocampal EPSPs in vivo 24 h. after i.c.v. injection (1 µL of 3.5 mM) (Cullen et al., 1996). This effect was prevented by the NMDA receptor antagonist CPP. The same effect was seen in the DG of hippocampal slices | Cullen et al. (1996) |
| Hippocampal slices recording in DG | |||
| Cultured cerebellar granule cells | Aβ1-40 (Bachem, pre-incubated in PBS for 5 days at 37°C to allow aggregation, to cells it was applied at 1 or 2 µM for 6, 12, 24 h) | Aβ produced activation of NFkB (1 µM at 12 h) that was inhibited by MK-801 (10 µM) | Kawamoto et al. (2007) |
| Assessment of oxidative stress (2,3-DHB generation) after application through microdialysis probe in the striatum | Aβ1–40 (Bachem) pre-incubation for 4 days at 37°C | Aβ infusion produced oxidative stress that was attenuated by MK-801 | Parks et al. (2001) |
| Patch clam whole cell recording from primary rat cortical cultures | Aβ1–42 after 35 min of incubation | Aβ produced depression of NMDA mediated current related to NMDA receptor endocytosis that was α7 dependent | Snyder et al. (2005) |
| Cortical neuronal cultures from mice | Aβ1–42 (American Peptide) pre-incubation in water for 7 days at 37°C | Conditioned medium from microglia incubated with Aβ1–42 produced neuronal death by oxidative mechanisms. This was prevented by memantine (1 µM) and D-APV (10 µM) | Floden et al. (2005) |
| Single unit recording in vivo in CA1 iontophoretic application | Aβ1–42, Aβ25–35 (own synthesis) sonicated to lessen aggregation | Both Aβ forms enhanced NMDA responses | Molnar et al. (2004) |
| Primary rat cerebral cultures | Aβ1–42 incubated at 4°C for 24 h in 2%DMSO and Ham F-12 medium | 1 µM (30 min) Aβ1–42-induced AA release that was inhibited by D-APV (10 µM) and memantine (5 µM) | Shelat et al. (2008) |
| Primary neocortical cultures neurons and glia (patch clamp) | Aβ (Biopolymer facility at Brigham and Women's Hospital) assemblies in a prefibrillar form | 1 µM Aβ produced neuronal activation that was attenuated in case of prefibrillar form by the NMDA receptor antagonist D-APV but effect produced by fibrillar form was not significantly attenuated (but was by AMPA antagonist) | Ye et al. (2004) |
| Organotypic hippocampal slices | Aβ1–42, freshly prepared in 0.1 N NaOH, pH readjusted to 7.4, and then diluted in serum-free culture medium. | The selective NMDA receptor antagonist D-APV completely blocked Aβ1–42 (15–30 µM) internalization, up-regulation of cathepsin D, and activation of microglia | Bi et al. (2002) |
| Cultured cortical neurons | Supplementary methods not available | Aβ1–42 (1 µM for 1 h) produced a rapid and persistent depression of NMDA receptor mediated currents and synaptic receptor endocytosis | Snyder et al. (2005) |
| Inward currents –Xenopus oocytes expressing NMDA receptors | Aβ1–42, (Bachem) globulomers (dodecamers), 6 h pre-incubation at 37°C, | Aß had no effect on NMDA-induced glutamate currents | Mezler et al. (2011) |
| Toxicity (LDH release) – septal rat primary neurons in culture | Aβ1–42 (Peptide Institute, Osaka) dissolved directly before use | Memantine (up to 10 µM) and (+)MK-801 (10 µM) did not prevent toxicity produced by Aß measured as LDH release | Kimura et al. (2005) |
| Organotypic hippocampal slices | Aβ, naturally secreted dimmers and trimers | Oligomers decrease Ca2+ influx and produced loss of hippocampal synapses that was prevented by 20 µM NMDA antagonists CPP, which at lower concentrations produced synapse loss on its own | Shankar et al. (2007) |
Neuronal activation in primary neocortical cultures was selectively dependent on the assembly state of Aβ. Protofibril (PF)-induced activity was specifically attenuated by the NMDA receptor antagonist D-APV. In contrast, the non-NMDA ionotropic glutamate receptor antagonist, NBQX, preferentially reduced Aβ fibril (FB)-induced activity. Removal of Mg2+ from the medium, increased both PF- or FB-induced activation, but D-APV was more effective in attenuating PF-induced excitatory activity (Ye et al., 2004).
Further evidence of Aβ/NMDA receptor interactions is the fact that natural Aβ dodecameric oligomers co-immunoprecipitate with NR1 and NR2A (Venkitaramani et al., 2007). Moreover, Aβ1–42 oligomers (ADDLs) bind to glutamatergic neurons expressing NR1 and NR2B but not GABA-ergic neurons (Lacor et al., 2007). Aβ25–35 (10 µM) inhibited both [3H]glutamate and [3H]glycine binding (by 20% and 70%, respectively) and stimulated functional [3H]MK-801 binding (Cowburn et al., 1997). These authors concluded that Aβ25–35 shows moderate affinity for the agonist recognition sites of the NMDA receptor, but not for other excitatory amino acid receptor types or for L-type voltage-dependent calcium channels, and that this fragment enhances NMDA receptor function (Cowburn et al., 1997). However, others have failed to detect binding of Aβ1–42 to any known recognition sites on glutamate receptors (Von Euler et al., 2008).
Most recent evidence indicates that such effects of Aβ1–42 on NMDA receptors may be secondary to Aβ1–42 binding to postsynaptic anchoring proteins like PSD-95 (De Felice et al., 2007a; Lacor et al., 2007). Exposure of cultured cortical neurons to soluble oligomers of Aβ1–40 (0.1–10 µM) reduced levels of the synaptic PSD-95 and AMPA receptors in a concentration- and time-dependent manner (Roselli et al., 2005). This effect was prevented by the NMDA receptor channel blocker (+)MK-801 and the NR2B site antagonist ifenprodil but was not increased by combining Aβ with NMDA indicating possible direct activation of NMDA receptors by Aβ (Roselli et al., 2005). Soluble oligomeric Aβ1–42 also down-regulated the levels of PSD-95 and synaptophysin, and that this effect was also blocked by (+)MK-801 and ifenprodil (Liu et al., 2010). The authors proposed that Aβ leads to a loss of these associated synaptic proteins subsequent to binding to PSD-95 and indirect suppression of NR2A function but activation of NR2B function that, in turn, induces caspase-8 and caspase-3 activity (Liu et al., 2010). Indeed, selective enhancement of NR2A activity and/or reduction of NR2B activity has been suggested to be a useful therapeutic approach in AD (Liu et al., 2010).
Similarly, although NMDA receptor knock-down using an amplicon vector abolished Aβ1–42 oligomer (ADDLs) binding to dendrites, and associated neuronal oxidative stress, both oligomer-attacked and non-attacked control neurons exhibited similar levels of NMDA receptor surface expression (Decker et al., 2010a). Moreover, insulin treatment down-regulated Aβ1–42 oligomer-binding sites in the absence of a parallel reduction in surface levels of NMDA receptors. (Decker et al., 2010a)
Recently, single particle tracking of quantum dot-labelled Aβ1–42 membrane-attached oligomers (ADDLs) revealed that, whilst initially moving freely, their diffusion was hindered upon accumulation at synapses (Renner et al., 2010). Concomitantly, individual metabotropic glutamate receptors (mGluR5) also showed reduced lateral diffusion, aberrant clustering at synapses and caused an elevation of intracellular calcium and synaptic loss which was prevented by an mGluR5 antagonist (Renner et al., 2010). In this regard, it should be noted that NMDA and mGluR5 receptors are closely associated with each other in postsynaptic complexes (Tu et al., 1999).
Others have reported that Aβ25–35 induced increases in intracellular Ca2+ in cultured hippocampal neurones, and that this effect was enhanced by Mg2+ removal and blocked by NMDA receptor antagonists (Brorson et al., 1995). However, this was proposed to be due to enhanced glutamatergic synaptic network activity rather than direct activation of NMDA receptors (Brorson et al., 1995). Aβ1–42 has also been reported to inhibit the sodium-potassium adenosine triphosphatase (ATPase Na+/K+) pump and could thereby cause membrane depolarization and relief of Mg2+ blockade of NMDA receptors (Gu et al., 2004). However, application of Aβ1–40 by extracellular perfusion (200 nM) or intracellularly via the recording pipette (100 nM) resulted in a gradual enhancement of NMDA receptor-mediated synaptic currents in granule cells in the rat dentate gyrus (DG) in vitro with no effect on AMPA receptor-mediated transmission, resting membrane potential or input resistance (Wu et al., 1995a).
There are also multiple effects of Aβ seen in vivo that can be attributed to over activation of NMDA receptors since they are blocked by antagonists of this receptor type (Table 2). In vivo responses of single hippocampal neurons to local microiontophoretic NMDA were potentiated by local application of Aβ1–42, whereas those of AMPA/kainate receptors were decreased (Molnar et al., 2004; Szegedi et al., 2005). Oxidative stress seen in vivo after Aβ application is also blocked by NMDA receptor antagonists (Parks et al., 2001).
Others have reported that Aβ1–42 (1 µM for 1 h) produced a rapid and persistent depression of NMDA receptor-mediated currents and synaptic receptor endocytosis secondary to activation of protein phosphatase 2B (PP2B) and striatal enriched tyrosine phosphatase (STEP) (Snyder et al., 2005). Pre-incubation of primary neuronal cultures with synthetic Aβ1–42 oligomers decreased NR2B-immunoreactive synaptic spines and surface expression of NR2B containing NMDA receptors in vitro (Dewachter et al., 2009). Prolonged exposure of primary cortical neurons to Aβ1–42 oligomers exhibited toxic effects (mitochondrial dysfunction and production of ROS) associated with an attenuation of NMDA receptor-mediated Ca2+ influx and inhibition of NMDA-induced AA release (He et al., 2011). However, such changes could reflect reactive endocytosis of NMDA receptors subsequent to their tonic pathological activation (Bi et al., 2002; Snyder et al., 2005; Hsieh et al., 2006).
Neurons from a genetic mouse model of AD also rather showed reduced expression of surface NMDA receptors and dephosphorylation of the NMDA receptor subunit NR2B at Tyr1472, which correlated with receptor endocytosis (Snyder et al., 2005). Decreased concentrations of the NMDA receptor subunit NR2B and PSD-95, impaired NMDA-dependent LTP and decreased NMDA and AMPA receptor currents in hippocampal Cornu ammonis area 1 (CA1) region have also been reported in APP[V717I] transgenic mice (Dewachter et al., 2009). Similar, probably reactive effects of glutamatergic and cholinergic neurotransmitter systems have also been reported at the level of PKB/Akt phosphorylation (Abbott et al., 2007).
In conclusion, the majority of the evidence listed above (see also Table 2) indicates increased tonic NMDA receptor stimulation in the presence of soluble forms of Aβ. However, one caveat is that most earlier studies used high concentrations of Aβ, which makes extrapolation of these data to the true disease conditions in vivo challenging.
Effects of glutamatergic transmission on Aβ processing/levels
Already two decades ago dysregulation of neuronal calcium homeostasis has been implicated to enhance the production of Aβ in AD (Mattson et al., 1993). Recent results indicate that Ca2+ stimulates the formation of oligomers of Aβ (Itkin et al., 2011). It therefore seems plausible that prolonged Ca2+ influx via pathological activation of NMDA receptors could also promote the intracellular generation of toxic Aβ oligomers – a kind of positive feedback viscious circle. Also, internalization of Aβ1–42 itself has been reported to be blocked by NMDA receptor antagonists (Bi et al., 2002).
Sub-toxic activation of NMDA receptors increases the proportion of Kunitz protease inhibitor domain containing APP that, in turn, favours β-secretase over α-secretase processing resulting in enhanced Aβ production. Prolonged activation of extrasynaptic NMDA receptors, but not synaptic NMDA receptors, was recently reported to increase the neuronal production of Aβ (Bordji et al., 2010). This effect was preceded by a shift from APP695 to Kunitz protease inhibitory domain (KPI) containing APPs (KPI-APPs), isoforms exhibiting greater amyloidogenic potential. Somewhat in line with this notion is the concept that calcium influx through synaptic NMDA receptors actually promotes non-amyloidogenic α-secretase-mediated APP processing (Hoey et al., 2009). In rat organotypic slices, acute overproduction and synaptic release of either axonal or dendritic Aβ reduced spine density and plasticity at nearby dendrites (Wei et al., 2009). However, in this case, only the synaptotoxic effects, but not Aβ production, was sensitive to NMDA receptor blockade by D-APV (Wei et al., 2009).
Others have proposed that general synaptic activity-dependent modulation of endogenous Aβ production/secretion may normally rather participate in a negative feedback loop to depresses excitatory synaptic transmission to keep neuronal hyperactivity in check and that disruption of this feedback system could contribute to disease progression in AD (Kamenetz et al., 2003). More recently, Cirrito et al. (2005; 2008) showed, using brain microdialysis in vivo, that synaptic activity increases endocytosis of APP and also subsequently increases Aβ levels in the brain extracellular fluid through increased Aβ exocytosis. However, the relationship between synaptic activity and increase in pathogenic Aβ is still controversial. The Gouras group showed that synaptic activity promotes APP intracellular transport to the synapses, decreases intracellular Aβ due to neprilysin activity and protects against Aβ related synaptic changes (Tampellini and Gouras, 2010).
Functional consequences of Aβ (LTP, learning) and the role of NMDA receptors
Effect of Aβ on LTP indicative of a role for NMDA receptors
There are numerous reports that incubation of rodent hippocampal slices with small diffusible Aβ1–42 oligomers strongly inhibits the induction LTP in the CA1 and dentate gyrus, but not NMDA receptor-independent LTP – see schematic in Figure 2 (Table 3). These effects occur via interactions with NMDA receptors as they are blocked by many different NMDA receptor antagonists and manifest themselves well before any signs of overt excitotoxicity (Lambert et al., 1998; Wang et al., 2002; Chen et al., 2002b; Rowan et al., 2003; Wang et al., 2004a; Walsh et al., 2005; Puzzo and Arancio, 2006; Townsend et al., 2006; Martinez-Coria et al., 2010) (Table 3).
Table 3.
Effect of Aβ on LTP. Majority of studies show disruption of LTP, few studies show an enhancement of LTP by Aβ (indicated by bold text)
| Experimental system | Aβ type/dose | Effect of Aβ observed | Reference |
|---|---|---|---|
| LTP in hippocampal slices, CA1 | Aβ1–42 (Bachem), incubation for 6 h and then for 18 h after dilution at 37°C to produce globular forms | Completely blocked LTP at 42 nM given 80–120 min before tetanus | Barghorn et al. (2005) |
| LTP in vivo, CA1 | Aβ25–35 (Bachem) 10 and 100 nM i.c.v. | At 5 min, 100 but not 10 nM impaired LTP. At 1 h, both were active. The effects of Aβ on LTP were probably mediated via a postsynaptic mechanism because they did not affect paired pulse facilitation | Freir et al. (2001) |
| LTP in vivo, CA1 | Aβ1–42 (Bachem)- stock solution in 0.1% NH4+OH-, centrifuged, supernatant stored at −80°C. | Aβ1–42 (80 pmol i.c.v.) impaired LTP in vivo and this effect was reversed by NR2B and TNFα antagonists | Hu et al. (2009) |
| LTP in vitro, CA1, from transgenic APP (V717I) mice | Aß1–42 (Bachem, 100 µM), incubated for 24 h at 4°C for oligomers and 24 h at 37°C in DMSO) | APP Tg mice had impaired LTP in CA1 region (NMDA dependent) | Dewachter et al. (2009) |
| LTP in vivo, CA1 | Aβ1–42 (Bachem)- stock solution in 0.1% NH4+OH-, centrifuged, supernatant stored at −80°C. | Aβ1–42 (80 pmol i.c.v.) impaired LTP in vivo and this effect was reversed by low doses of memantine | Klyubin et al. (2011). |
| LTP rat hippocampal slices CA1 | Aβ1–42 (AnaSpec), pre-incubated at room temp. for 1–2-days | 1 µM Aβ (3 h) moderately inhibited LTP. Co-treatment with a sub-threshold concentration of glutamate (30 µM) strongly impaired LTP. | Nakagami et al. (2002); Nakagami and Oda (2002) |
| LTP rat hippocampal slices CA1 | Aβ1–42 and Aβ25–35 (1 µM) (Peptide Institute, Osaka) | LTP was markedly reduced by i.c.v. Aβ25–35 (10 nmol) and completely blocked by Aβ25–35 (100 nmol). Aβ did not affect NMDA EPSP. Not NMDA involvement but downstream mechanisms. | Nomura et al. (2005) |
| LTP rat hippocampal slices, CA1 | Aβ1–42, Aβ1–40 (Keck Peptide Synthesis Lab.) or their active fragment Aβ25–35 (Bachem) applied for 20 min before HFS | Aβ25–35 (10 µM) inhibited both [3H]glutamate and [3H]glycine binding (by 20 and 70% respectively) and stimulated functional [3H]MK-801 binding | Chen et al. (2000) |
| LTP hippocampal slices DG | Aβ1–42 (Bachem) applied 20 min before HFFS, no pre-aggregation | Aβ at 200 but not 20 nM inhibited LTP, involving calcineurin mechanisms. Bath application of Aβ1–42 (1 µM, 10 min) reduced NMDA receptor-mediated EPSCs. | Chen et al. (2002b) |
| LTP rat hippocampal slices | Diffusible Aβ1–42 derived diffusible ligands (ADDLs) | Prevented LTP induction. | Puzzo and Arancio (2006) |
| LTP rat hippocampal slices CA1 | Aβ1–40 (200 nM) (US Peptides) | Caused rapid inhibition of LTP and a modest short term inhibition (approx. 25%) of NMDA EPSP but no long-term effect on normal synaptic transmission or LTD. | Raymond et al. (2003) |
| LTP rat hippocampal slices CA1 | Aβ dimmers isolated from human brain | Suppressed LTP and enhanced LTD | Shankar et al. (2008) |
| LTP in vivo | Aβ naturally secreted from microsomes from hamster ovary | The negative effects of i.c.v. Aβ oligomers on hippocampal LTP in vivo were prevented by monoclonal Aβ antibodies. Monomers were inactive. | Walsh et al. (2002) |
| LTP rat hippocampal slices CA1 | Aβ1–42 (Bachem) globulomers prepared according to Barghorn | Aβ1–42 (42 nM) completely blocked LTP and this was reversed by memantine 1 µM | Martinez-Coria et al. (2010) |
| LTP rat hippocampal slices CA1 | Aβ1–42 (Bachem)- soluble Aβ-derived oligomers (ADDLs) | Aβ1–42 (1–50 nM), concentration-dependently blocked LTP and this was reversed by memantine 1 µM as well as by NR2B and mGluR5 receptor negative allosteric modulators | Rammes et al. (2011) |
| LTP rat hippocampal slices CA1 | Aβ1–42 (MoBiTec) HFIP aliquots dissolved (100 µM) in DMSO, diluted to 20 µM in F12/DMEM and incubated at 4°C for 24 h | Aβ1–42 (500 nM) strongly inhibited LTP and this was reversed by the NR2B receptor negative allosteric modulators ifenprodil and Ro25-6981 | Ronicke et al. (2010) |
| LTP rat hippocampal slices CA1 | Aβ1–42 (American Peptide), incubated at 4°C for 24–30 h. | Aβ inhibited LTP but not LTD | Wang et al. (2002) |
| LTP hippocampal slices DG | Aβ naturally secreted (CHO cells expressing human APP751) and synthetic Aβ1–42 (Bachem) | Inhibited the induction of LTP (natural at 1 µM and synthetic at 100–200 nM). mGluR5 antagonists blocked this effect of Aβ | Wang et al. (2004b) |
| LTP rat hippocampal slices CA1 | Aβ25–35 | Aβ25–35 impaired both PTP and LTP | Costello and Herron (2004) |
| LTP hippocampal slices DG | Aβ25–35 (Bachem) perfused for 40 min (no pre-incubation) | Aβ (500 nM) inhibited induction of NMDA dependent LTP (involving superoxide), but not induction of NMDA-independent LTP or long-term depression (LTD) | Wang et al. (2004a) |
| LTP hippocampal slices DG | Aβ (Bachem, type not specified) perfused for 40 min (no pre-incubation) | Inhibited LTP induction which involved the TNFα and metabotropic glutamate receptors (mGluR5) | Wang et al. (2005) |
| LTP rat hippocampal slices CA1 | 0.1 mM of the short Aβ fragment Aβ31–35 and Aβ25–35 | Suppressed the induction of LTP of PS in a similar manner to the longer fragment Aβ25–35. Had no effect on NMDA receptor mediated multiple PS in Mg2+-free medium, suggesting that these Aβ fragments suppressed the induction of LTP through an NMDA receptor-independent pathway | Ye and Qiao (1999) |
| LTP in mouse hippocampal slices, CA1 | Aβ, soluble oligomers derived from CHO cells expressing human APPv717F, 20 min before HFS | Aβ inhibited LTP | Townsend et al. (2006) |
| LTP hippocampal slices D | Aβ1–42 (Bachem) no pre-aggregation applied 20 min before HFS | Aβ (final concentration not known) inhibited LTP | Zhao et al. (2004) |
| LTP hippocampal slices DG | Aβ1–40 (Bachem) stored as water solution at −20°C | At 200 nM, Aβ enhanced LTP but not basal responses 25 min after application | Wu et al. (1995a,b) |
| LTP in vivo, CA1 | Aβ1–42 (Bachem) | Facilitated the induction of LTD and depotentiation of LTP in the an NMDA receptor-dependent manner | Kim et al. (2001) |
For example, soluble Aβ oligomers from different sources (cultured cells, AD cortex or synthetic peptide) consistently inhibit LTP in murine and rat hippocampal slices, and this inhibition can be prevented by the NR2B negative allosteric modulators ifenprodil and Ro 25–6981 (Hu et al., 2009; Ronicke et al., 2010; Li et al., 2011; Rammes et al., 2011). Additionally, Aβ1–42 (42 nM) incubated under somewhat artificial conditions – that is, in the presence of SDS to produce stable globular ‘dodecameric’ forms – bound specifically to dendritic processes/spines of neurons but not glia in hippocampal cell cultures and completely blocked LTP in the CA1 region in hippocampal slices (Barghorn et al., 2005; Albrecht et al., 2008; Martinez-Coria et al., 2010). Similar results were seen when Aβ1–42 was prepared under conditions presumably more closely resembling the pathophysiological situation (e.g. lacking SDS, ADDLs) (Lacor et al., 2007). Aβ1–42 (1–50 nM) concentration-dependently blocked LTP, with strong effects already seen at the lowest concentration of Aβ tested (1 nM) (Rammes et al., 2011). Under both conditions, the LTP deficits induced by Aβ1–42 were completely reversed by 1 µM memantine, as well as by NR2B negative allosteric modulators and partial knockout of NR2B subunits (Rammes et al., 2011).
Such effects of Aβ1–42 are normally not reflected in effects on baseline AMPA receptor-mediated EPSPs, but one study showed a reduction in the amplitude of isolated NMDA receptor-mediated synaptic currents in dentate granule cells in vitro via a postsynaptic mechanism (Chen et al., 2002b).
There are indications that the inhibitory effects of Aβ1–42 on LTP involves reactive oxygen and nitrogen species (Wang et al., 2004a), and the pro-inflammatory cytokine TNF-α; as such, suppression is not seen in TNF-α-deficient mice (Wang et al., 2005). Further evidence that TNF-α mediates this deleterious action was recently provided by the ability of TNF-α antagonists to prevent Aβ1–42 inhibition of LTP in vivo and the abrogation of a similar disruptive effect of TNFα using the NR2B selective NMDA receptor antagonist Ro 25–6981 (Hu et al., 2009). Similarly, stimulation of the kinases JNK, CDK5 and p38 MAPK; TNF-α; metabotropic glutamate receptors (mGluR5) and CREB protein have been proposed to be involved in Aβ-induced deficits in LTP (Wang et al., 2004b; 2005; Li et al., 2011).
Aβ1–42 also inhibited LTP and associated phosphorylation processes in DG of rat hippocampal slices (Zhao et al., 2004). The authors suggested that activity-dependent CaMKII autophosphorylation and AMPA receptor phosphorylation are essential for LTP in this region and that disruption of such mechanisms could directly contribute to Aβ-induced deficits in hippocampal synaptic plasticity and memory. Similarly, Aβ1–42 (200 nM) inhibited LTP of EPSPs and population spikes (PS) in the same region in vitro (Chen et al., 2000). Interestingly, in this same study, even the often reported less toxic Aβ1–40 blocked LTP of EPSPs at the same relatively low concentration, but was less effective against PS (Chen et al., 2000). In contrast, some authors have reported that Aβ1–40 actually enhanced LTP but not basal responses in the DG of hippocampal slices (Wu et al., 1995a,b).
Negative effects of Aβ1–42 on LTP were also reported for Schaffer-collateral projecting to CA1 in vivo (e.g. Kim et al., 2001; Hu et al., 2009). Such effects of i.c.v. Aβ1–42 oligomers on hippocampal LTP in vivo were completely prevented by co-administration of monoclonal Aβ antibodies (Kim et al., 2001; Walsh et al., 2002).
In some studies, pretreatment of rat hippocampal slices with Aβ1–42 alone only moderately inhibited LTP, but co-treatment with a sub-threshold concentration of glutamate agonists impaired LTP more strongly, implying an interplay between Aβ and the glutamatergic system (Nakagami et al., 2002).
Smaller fragments of the Aβ peptide usually have similar effects. Aβ25–35 was also found to impair both post-tetanic potentiation (PTP) and LTP in the hippocampal CA1 in vitro and, in agreement with (Wang et al., 2004a,b), these effects were proposed to involve activation of the JNK signalling pathway (Costello and Herron, 2004). Similarly, 0.1 µM of the short Aβ fragment Aβ31–35 suppressed the induction of LTP of PSs in CA1 of rat hippocampal slices in a similar manner to the longer fragment Aβ25–35, whereas neither treatment changed the amplitude of the baseline PS. These fragments had no effect on NMDA receptor-mediated multiple PSs when recorded in Mg2+-free medium, which was taken to imply that these Aβ fragments suppress the induction of LTP through a NMDA receptor-independent pathway (Ye and Qiao, 1999). A similar conclusion was drawn by others (Nomura et al., 2005).
In vivo LTP was also markedly reduced by i.c.v. Aβ25–35 at 10 nmol and completely blocked at 100 nmol (Freir et al., 2001). The effects of this Aβ fragment on LTP were probably mediated via a postsynaptic mechanism because they did not affect paired pulse facilitation (Freir et al., 2001).
Aβ1–42 has also been reported to facilitate the induction of LTD and depotentiation of LTP in the CA1 area of the rat hippocampus in vivo in an NMDA receptor-dependent manner (Kim et al., 2001). Similarly, soluble Aβ oligomers from several sources (synthetic, cell culture, human brain extracts) facilitated NMDA- and mGluR-dependent LTD in the CA1 region of hippocampal slices (Li et al., 2009). This Aβ-facilitated LTD was mimicked by the glutamate reuptake inhibitor threo-β-benzyloxyaspartic acid (TBOA) and prevented by an extracellular glutamate scavenger system (Li et al., 2009).
In another study, Aβ1–40 caused a long-lasting (2–5 days) depression of CA1 hippocampal EPSPs in vivo after i.c.v. injection (1 µL of 3.5 nM) that was prevented by the NMDA receptor antagonist CPP 7 g·kg−1 i.p. twice, but there was no change in the ability to induce LTP (Cullen et al., 1996). The same effect was seen in the DG of hippocampal slices (Cullen et al., 1996). In contrast, others have reported that application of Aβ1–40 by extracellular perfusion (200 nM) or intracellularly via the recording pipette (100 nM) to the same region in vitro resulted in a gradual enhancement of the NMDA receptor-mediated synaptic currents which did not reverse upon washout (Wu et al., 1995a,b).
Pathophysiologically relevant concentrations of naturally secreted dimeric Aβ extracted directly from the cerebral cortex of subjects with AD potently (pM) inhibited LTP, enhanced LTD and reduced dendritic spine density in normal rat hippocampal slices (Shankar et al., 2008). NMDA receptors were required for the spine loss. Insoluble amyloid plaque cores from AD cortex did not impair LTP unless they were first solubilized to release Aβ dimers, suggesting that plaque cores are largely inactive but sequester Aβ dimers that are synaptotoxic. This same extracted Aβ also disrupted memory in normal rats (Shankar et al., 2008). Similary, i.c.v. injection of Aβ-containing aqueous extracts of AD brain robustly inhibited LTP without significantly affecting baseline excitatory synaptic transmission in the rat hippocampus in vivo (Barry et al., 2011).
Effects of Aβ on NMDA receptor-dependent learning
In contrast to in vitro experiments, it is much more difficult in behavioural studies to provide evidence supporting a specific effect of Aβ on NMDA receptor-dependent learning. One type of evidence is based on studies showing that impairment of learning/memory produced by Aβ is attenuated by NMDA receptor antagonists. However, the authors are aware of the fact that it is not really strong evidence since correction of the deficit may be achieved by a different mechanism than that causing the deficit. One example could be the fact that some effects of Aβ1–40 are also attenuated by AChE inhibitors (AChEIs) (Yamada et al., 2005). Another type of supporting evidence, even less direct, comes from data obtained in transgenic animals. Here, we have to assume that the learning/memory deficit comes solely from overproduction of Aβ. Being aware of these limitations we provide below selected evidence. We should mention that a review on the effects of exogenous Aβ on behavioural parameters, in general, has been recently published by our group (Chambon et al., 2011).
The only evidence not based on the use of NMDA receptor antagonists, but an agonist, was generated by Sipos et al. (2007). They showed that Aβ1–42 injected bilaterally into the entorhinal cortex of rats did not affect spatial working memory (alternation task, 10–17 days later) but produced deficits in recognition memory in an object recognition task and the Morris water maze (MWM), where a hidden platform has to be found. This pattern of behavioural deficits mirrored the effects of NMDA administration supporting that similar mechanisms could play a role.
The NMDA receptor channel blocker (+)MK-801 (2.5 mg·kg−1) applied 2 h before Aβ1–42 injected into the nucleus basalis of Meynert (NBM) prevented passive avoidance learning deficits assessed 2 weeks later (Harkany et al., 1999). It should, however, be stressed that the dose of (+)MK-801 used was very high, and saturation of NMDA receptors would be expected with (+)MK-801 below 0.5 mg·kg−1. The evidence supporting the role of NMDA receptors using memantine against deficits caused by i.c.v. injection of Aβ in transgenic animals has been discussed in the sections ‘Effects of memantine on the toxic actions of Aβin vivo’ and ‘Effects of memantine in transgenic models of AD’ respectively. Nevertheless, in summary, in the majority of studies memantine has been shown to correct many of the deficits, presumably resulting from Aβ administration or overproduction, giving support for the role of NMDA receptors in learning deficits in transgenic AD animals.
Is the net effect hypo- or hyperactivity of NMDA receptor system?
Although it is still not clear whether NMDA receptors are directly or indirectly activated/modulated by Aβ, enhanced NMDA receptor sensitivity might, intuitively, rather be expected to enhance LTP. However, we propose similar mechanisms as discussed above for the impairment of synaptic plasticity/learning during conditions of chronic, non-phasic activation of NMDA receptors. This has been demonstrated, for example, for LTP following reduction in Mg2+ concentration (Coan et al., 1989), application of non-toxic concentrations of NMDA (see above) (Parsons et al., 2007) as well as in glutamate transporter 1 (GLT-1) knockout mice (Katagiri et al., 2001).
The direct and indirect effects of Aβ on NMDA receptor function are likely to keep the ion channel tonically open in AD. This chronic pathological activation would be expected to cause a constant mild influx of Ca2+, even under resting conditions, greatly increasing the level of background noise at the postsynaptic terminal. As a result, incoming physiological signals may not be distinguished against this raised background noise and, consequently, synaptic plasticity, LTP and learning/memory could be impaired. Ultimately, the excessive influx of Ca2+ ions could cause death of the postsynaptic neuron via associated effects, such as the formation of free radicals, changes in nuclear chromatin and DNA breakage (Danysz et al., 2000; De Felice et al., 2007a; Parsons et al., 2007) – see Figure 1.
Prolonged exposure to Aβ1–42 probably also induces a reactive endocytosis of both NMDA and AMPA receptors subsequent to their tonic pathological activation (Bi et al., 2002; Snyder et al., 2005; Hsieh et al., 2006). Even following such reactive receptor down-regulation, the remaining receptors are likely to be sensitized and continue mediating negative effects of Aβ as concentrations of glutamate may also increase due to inhibition of glial uptake (Harkany et al., 2000).
These aspects provide a rational for therapeutic intervention focusing on inhibition of NMDA receptors.
Caveats to most data presented in Tables 1–3
It should be stressed that the vast majority of studies on Aβ suffer from technical obstacles. The most problematic is related to the fact that therapeutically relevant extracellular Aβ concentrations are actually only within the high pM to low nM range. Unfortunately, such concentrations do not usually produce negative effects on neuronal function in the short time frame used for experiments, in particular when the system is not additionally challenged with other insults (e.g. NMDA agonists, oxidative stress, etc.). In turn, much higher concentrations of Aβ are typically used (see Tables 1–3), usually in the 10–100 µM range, to achieve faster effects on neurons. The caveat is that such high concentrations are probably not so relevant for the in vivo situation in AD as different aggregation pathways/target protein interactions are likely to be operative. The same caveats apply to the frequently used ‘toxic’ sub-fragments of Aβ such as the sequence from amino acids 25 to 35.
Memantine
Strong support for the clinical relevance of such interactions between Aβ, glutamate and NMDA receptors in AD is provided by the NMDA receptor antagonist memantine. This substance is the only NMDA receptor antagonist used clinically in the treatment of AD and therefore offers an excellent tool to facilitate translational extrapolations from in vitro studies through in vivo animal experiments to its ultimate clinical utility.
Mechanism of action of the NMDA receptor antagonist memantine
Memantine is an uncompetitive NMDA receptor antagonist with strong voltage dependency and rapid unblocking kinetics (Kornhuber et al., 1989; Chen et al., 1992; Parsons et al., 1993). These properties have been proposed to allow memantine to prevent the tonic pathological influx of Ca2+ and oxidative stress in postsynaptic neurons, whilst preserving the transmission of strong transient physiological signals, which can then be detected against reduced levels of background noise (Parsons et al., 1993; 2007) – see Figure 2. Therefore, as for the Mg2+ block in the healthy brain, memantine block of the NMDA receptor channel is transiently relieved during temporally and/or spatially convergent/co-operative activation of glutamatergic synapses – for example, during learning and memory processes.
Memantine has recently been shown to selectively target extrasynaptic ‘death’ receptors, which are mainly composed of NR2B subunits and coupled to different signalling pathways than the physiologically more relevant subsynaptic receptors (Okamoto et al., 2009; Xia et al., 2010) and may be of particular relevance for pathologically processes in AD (Bordji et al., 2010; Rammes et al., 2011). This moderate selectivity probably has little to do with true pronounced subtype selectivity of memantine at NR2B receptors (see e.g. Bresink et al., 1996), as discussed in these studies, but rather relates to the more moderate but prolonged membrane depolarization at these receptor loci and the voltage dependency of memantine.
Effects of memantine on the toxic actions of Aβin vivo
In vivo, infusion of memantine prevented the development of errors in the delayed non-matching to sample lever pressing task produced by i.c.v. infusion of Aβ1–40 in rats (Yamada et al., 2005). I.c.v. infusion of Aβ25–35 in rodents decreased the level of immunoreactive somatostatin and substance P in the hippocampus prior to neuronal loss or caspase activation, which was correlated with the loss of spine density and activation of inducible NOS (iNOS) (Arif and Kato, 2009; Arif et al., 2009). Memantine, attenuated these Aβ25–35-induced changes of neuropeptides, their metabolizing enzymes, glial marker proteins and activation of iNOS.
In the hippocampus, s.c. semi-chronic infusions of memantine (15 mg·kg·day−1) prevented neuronal cell loss and apoptosis induced by the direct injection of Aβ1–40 into this structure (Miguel-Hidalgo et al., 2002). Memantine treatment decreased lesions, glial fibrillary acidic protein (GFAP) staining, ED1-labelled Aβ deposits and the number of pyknotic/fragmented cell nuclei in the hippocampus, indicating that reduction of apoptotic cell death involved in this effect. This study was later extended to demonstrate clear attenuation of apoptosis and active avoidance learning deficits (Miguel-Hidalgo et al., 2006).
Memantine (10, 20 mg·kg·day−1 s.c. infusion for 6 weeks) starting 24 h before Aβ1–40 injection into the rat hippocampus (followed 2 days later by ibotenic acid) significantly prevented learning deficits in the MWM, which emerged 5 weeks after Aβ1–40 injection (Nakamura et al., 2006). A lower dose of memantine (5 mg·kg·day−1) and relatively high doses of (+)MK-801 (0.312, 0.624 mg·kg·day−1) were without beneficial effect. Neuronal damage in the hippocampus, assessed via elevation in levels of the peripheral-type benzodiazepine-binding site (a gliosis marker for neuronal damage) and Cresyl violet staining, was significantly attenuated by memantine (10, 20 mg·kg·day−1) and (+)MK-801 (0.624 mg·kg·day−1) (Nakamura et al., 2006). In naïve rats, (+)MK-801 produced a significant learning impairment in the MWM task at a dose of 0.624 mg·kg·day−1, whilst memantine (20 mg·kg·day−1 s.c. infusion) did not (Nakamura et al., 2006). These results suggest that whilst both memantine and (+)MK-801 exert protective effects on progressive neuronal damage caused by Aβ, only memantine prevents memory impairment in hippocampal-lesioned rats.
Local injection of Aβ into the NBM caused a conformation-dependent enhancement of cortical NOS activity, which was blocked by the NR2B selective antagonist ifenprodil (OMahony et al., 1998). Memantine also rescued the neocortical cholinergic fibres originating from NBM cholinergic neurons, attenuated microglial activation around the intracerebral lesion sides, and improved attention and memory in object recognition and passive avoidance tasks after i.c.v. Aβ1–42 injection (Nyakas et al., 2011). In this study, Aβ1–42 was claimed to consist mainly of monomers and tetramers. Memantine is also protective against various other toxic insults to this cholinergic structure including NMDA, mitochondrial toxins and LPS (Wenk et al., 1995; 1996; 1997).
Systemic treatment with memantine (1 mg·kg−1) prevented the Aβ1–42 induced perseveration errors in a food reinforced lever pressing task (Klyubin et al., 2011). Interestingly, in this study Aβ1–42 was injected i.c.v. 2 h before testing whilst memantine was injected 45 min before testing, meaning that the effect observed was reversal, rather than prevention, of Aβ1–42-induced deficits.
Effects of memantine on the toxic actions of Aβin vitro
Memantine has been shown to attenuate the deleterious action of Aβin vitro in numerous studies. Early data indicated that the toxic effects of Aβ1–40 in cultured cortical neurons were blocked for 48 h by brief exposure to memantine (Tremblay et al., 2000). Similarly, Aβ1–42-induced reduction of neurite outgrowth in neuronal cultures was attenuated by memantine (Hu et al., 2007). The vicious circle of Aβ activating NMDA receptors, which then cause a further increase in Aβ production, was blocked by low, 1 µM concentrations memantine (Floden et al., 2005). Memantine also concentration-dependently inhibited extrasynaptic NMDAR-induced KPI-APPs expression as well as neuronal production and release of Aβ (Bordji et al., 2010)
It has also been claimed that the ability of memantine to protect rat cortical cultured neurons against Aβ-induced toxicity is a secondary consequence of attenuating tau phosphorylation (Song et al., 2008). Thus, in primary mouse cortical neurons, calmodulin-dependent protein kinase β (CaMKK β) activation of AMPK in response to Aβ1–42 leads to increased phosphorylation of tau at S262/S356 and S396 (Thornton et al., 2011). This effect was blocked by memantine providing a possible mechanism of action (MoA) for its positive effects on the pathophysiological phosphorylation of tau observed in patients (Thornton et al., 2011).
In contrast, neither memantine nor (+)MK-801 showed any neuroprotective effect against overt toxicity in cultured septal cholinergic neurones following 1 week treatment with a high concentration of Aβ1–42 (5 µM), whereas donepezil concentration-dependently reduced LDH efflux (Kimura et al., 2005).
More recent attention has been placed on the toxic role of low nM concentrations of oligomeric Aβ species in vitro. Chronic exposure of hippocampal cultures to soluble Aβ1–42 oligomers (ADDLs) produced abnormal spine morphology and a decrease in their density and the formation of ROS (Lacor et al., 2007). Associated consequences of this synaptic deterioration including loss of the spine cytoskeletal protein drebrin were completely prevented by memantine 5 µM (Lacor et al., 2007; Lambert et al., 2007). The same group showed that memantine also completely protected against Aβ-induced ROS (De Felice et al., 2007a).
NMDA and oligomeric Aβ1–42 induce ROS production from cortical neurons through activation of NADPH oxidase. ROS derived from NADPH oxidase leads to activation of ERK1/2, phosphorylation of cPLA(2)α and AA release. Aβ1–42-induced AA release was inhibited by both memantine and D-APV, providing strong support that these toxic effects of Aβ are mediated via NMDA receptors (Shelat et al., 2008). Memantine, as well as two further NMDA receptor antagonists (D-APV, (+)MK-801), prevented the disruption of axonal trafficking of dense-core vesicles and mitochondria by 500 nM Aβ1–42 oligomers (ADDLs) (Decker et al., 2010b). Signal transduction by neuronal insulin receptors (IR) is also strikingly sensitive to disruption by soluble Aβ1–42 oligomers (ADDLs) in mature cultures of hippocampal neurons, an effect that was completely blocked by relatively high concentrations of memantine (20 µM, lower concentrations unfortunately were not tested) (Zhao et al., 2008).
Recently, Aβ oligomers were reported to induce inward non-desensitizing currents in Xenopus oocytes expressing the NMDA receptor subunits NR1/NR2A and NR1/NR2B that were blocked in the presence of memantine, D-APV and (+)MK-801 (Texido et al., 2011). Interestingly, the responses to Aβ oligomers was greater for NR1/NR2A heteromers than for NR1/NR2B heteromers. Similar increases in the cytosolic concentration of Ca2+ induced by Aβ oligomers in cortical neurons were only slightly attenuated by the NR2B preferring NMDA receptor antagonist ifenprodil, indicating that Aβ oligomers directly activate NMDA receptors, particularly those with the NR2A subunit (Texido et al., 2011). In contrast, others found no effect of oligomeric Aβ on glutamate-induced currents in Xenopus oocytes expressing NMDA receptors under conditions where clear effects of voltage-gated P/Q-type calcium channels were seen (Mezler et al., 2011).
Vesicular zinc released during neurotransmission has been reported to be critical for targetting synaptically released Aβ1–42 oligomers to extrasynaptic NR2B containing NMDA receptors, an effect blocked by memantine (10 µM) as well as by other NMDA receptor antagonists (Deshpande et al., 2009).
To examine the specific effects of memantine on Aβ-induced deficits in LTP, extracellular field EPSPs (fEPSPs) were obtained from the dendritic region of the CA1 region of hippocampal slices from adult mice (Albrecht et al., 2008; Martinez-Coria et al., 2010). Under control conditions, fEPSPs were found to be potentiated 60 min after the stimulus was applied (100 Hz, 1 s). Administration of memantine alone (1 µM) did not affect this synaptic plasticity. After washing in Aβ1–42 globulomers (42 nM)(Barghorn et al., 2005), the same stimulus produced only short-term potentiation (returning to baseline after 60 min). However, when memantine was applied together with the Aβ globulomers, LTP was completely developed. Therefore, at a clinically relevant concentration, memantine was able to reverse the complete block of LTP by Aβ oligomeric globulomers.
Bell-shaped dose response relationship of memantine
Similar results were recently published for hippocampal LTP both in the DG in vitro (memantine 1 µM) and in the CA1 in vivo, although the therapeutic window in vivo in this study was quite narrow as effects were lost at higher doses of memantine (acute at 10 but not at 20 mg·kg−1 i.p. was effective) (Klyubin et al., 2011). Systemic administration of a sub threshold dose of memantine (1 mg·kg−1 i.p.) in an operant learning task also prevented the Aβ1–42-mediated increase in perseveration errors in this same study (Klyubin et al., 2011). Low concentrations of memantine (1 µM) prevented Aβ1–42 oligomer (82 nM)-induced deficits in neurotransmission in organotypic hippocampal slice cultures, and this effect was also lost at higher concentrations (3 and 10 µM) (Nimmrich et al., 2010).
Such bell-shaped dose–response relationships are often seen with memantine and seem to be inherent to its MoA; that is, positive effects are seen following moderate negative modulation of NMDA receptor function but lost if the NMDA receptor is blocked more strongly by higher concentrations of memantine, which then also block synaptic activation by higher mM concentrations of glutamate (Zajaczkowski et al., 1997; Frankiewicz and Parsons, 1999; Zoladz et al., 2006). Our previous study showed that at the doses producing positive effect there is c.a. 30% NMDA receptor occupancy in vivo (More et al., 2008).
Effects of memantine on levels of Aβ
Treatment of human neuroblastoma (SK-N-SH) cells with memantine (50 nM–50 mM) increased the levels of sAPPα in the conditioned media without affecting the levels of total intracellular APP (Chen et al., 2002a). This increase in sAPPα secretion by memantine, without a concomitant increase in the cellular APP levels, was taken to suggest that memantine may enhance the α-secretase (non-amyloidogenic) pathway. Memantine treatment increased cell viability and metabolic activity. In contrast, others have reported that memantine (10 µM) decreased the levels of the secreted form of sAPP, sAPPα and Aβ1–40 in similar cells (Ray et al., 2010). Similarly, memantine (1–4 µM) decreased levels of secreted APP, Aβ1–40 and Aβ1–42 and also lowered Aβ1–42 secretion in neuroblastoma cells and primary cultures of cortical neurons (Alley et al., 2009). It is unclear how memantine could affect levels of Aβ and related peptides (see also next section), but this effect could be unrelated to NMDA receptor antagonism and rather associated with the known lysotrophic properties of this amphiphlic molecule (Honegger et al., 1993).
Effects of memantine in transgenic models of AD
In APP/presenilin 1(PS1) double transgenic (TG) mice, memantine (30 mg·kg−1 p.o. daily for 2–3 weeks) significantly improved memory acquisition in the MWM but did not affect swimming speed, locomotor activity or aggressive behaviour (Minkeviciene et al., 2004). These data indicate that memantine improves hippocampus-based spatial learning without inducing non-specific effects on locomotion or exploratory activity. In the same TG mice, oral dosing of memantine (20 mg kg−1 day−1 for 8 days) significantly reduced the elevated cortical levels of soluble Aβ1–42 (Alley et al., 2009).
Memantine has also shown beneficial effects in a study in 4–month-old TG APP23 mice, which develop amyloid plaques, accompanied by astrogliosis and microgliosis, at the age of just 6 months, thus presumably reflecting the prodromal stage of AD (Sturchler-Pierrat et al., 1997; Sturchler-Pierrat and Staufenbiel, 2000). At the age of 6 weeks (i.e. before plaque formation), the mice received an s.c. infusion of memantine at various doses for 2 months or saline. After a wash-out phase of 3 weeks, their learning ability and cognitive function was investigated in the MWM. Treatment with memantine significantly improved both learning (distance covered during training) and memory (accuracy with which the platform was found)(Van Dam and De deyn, 2006). This effect of memantine treatment was observed 3 weeks after treatment termination, thus indicating disease modification (Van Dam and De deyn, 2006).
Long-term administration of memantine (10 and 20 mg kg−1 day−1 for 6 months) to Tg2576 mice was associated with a significant decrease in Aβ plaque deposition, increases in synaptic density and lowered appearance of degenerating axons (Dong et al., 2008). Administration of a lower dose of memantine (5 mg·kg−1) was also associated with a significant decrease in Aβ plaque deposition and a significant increase in synaptic density, but no significant effect on degenerating axons was observed (Dong et al., 2008). However, memantine did not significantly improve behavioural deficits in these mice in a fear-conditioning paradigm at any dose. Others have reported that memantine treatment reduced the total cortical levels of membrane-bound APP(45–55%) in both Tg2576 mice and non-transgenic mice in vivo (Unger et al., 2006).
Tg2576 mice (8-month-old) treated with memantine (30 mg kg−1 day−1) with or without folic acid (8 mg kg−1 day−1) for 4 months showed learning improvements in the MWM and less neuronal damage accompanied by an up-regulation of CNS genes involved in neurogenesis, neural differentiation, memory and neurotransmission (Chen et al., 2010).
The effects of sub-chronic memantine administration on spatial and non-spatial learning as well as exploratory activity and nest-building in APP/PS1 mutant mice have also been assessed (Filali et al., 2011). Memantine (10 mg·kg−1, i.p.) improved reversal of left–right discrimination but had no effect in the MWM, passive avoidance learning or non-learned behaviours such as elevated plus-maze exploration and nest building. APP/PS1 TG mice treated with memantine for a period of 4 months starting at 3 months of age performed as well as wild-type control mice in a novel object recognition task (Scholtzova et al., 2008). Memantine-treated TG mice had a reduced plaque burden detected with µMRI, which correlated with the improvement in cognitive performance (Scholtzova et al., 2008).
Most recently, LaFerla's group (Martinez-Coria et al., 2010) examined the therapeutic and neuroprotective effects of memantine in triple TG AD mice (APP, PS1 and tau mutations). In the therapeutic arm of the study, adult mice (aged 9 months), with established plaque and NFT pathology, were treated with memantine (30 mg kg−1 day−1 via drinking water) or placebo for 3 months (model of established AD). In these animals, memantine significantly improved spatial memory (MWM) and fear conditioning, but not novel object recognition. In the preventive arm of the study, young mice (aged 2 months), with no detectable pathology or behavioural deficits, were treated with memantine (30 mg kg−1 day−1 in drinking water) or ‘placebo’ for 10 months (model of developing AD). In these animals, memantine significantly improved fear conditioning and retention of spatial memory, but not acquisition of spatial memory or novel object recognition. Interestingly, the same treatment with memantine again resulted in a decrease in plaque load (Luhrs et al., 2006; Martinez-Coria et al., 2010). These results indicate that memantine is able to slow cognitive decline in younger transgenic mice, as well as to reverse established cognitive deficits in older transgenic mice.
Secondary pathological processes blocked by memantine
As introduced above, various processes overlap and influence one another in AD pathogenesis (e.g. Aβ accumulation, excitotoxicity at NMDA receptors, formation of tau neurofibrils, disturbance of mitochondrial function and neuroinflammation) (Peng and Greenamyre, 1998; Duchen, 2000; De Felice et al., 2007a). Neuroprotective effects have been described for memantine in numerous preclinical models of various chronic neurodegenerative diseases asides from AD (Danysz et al., 2000; Rosi et al., 2006). Secondary mitochondrial function disturbances may contribute to the neuronal damage observed in AD. As with the NMDA-induced lesions, long-term treatment with memantine has been shown to protect against the damaging effects of mitochondrial toxins and hypoxia (Schulz et al., 1996; Wenk et al., 1996; Karanian et al., 2006; Volbracht et al., 2006). Inflammatory processes are also thought to contribute to the neurodegenerative changes during AD pathogenesis (Katsuura et al., 1989; Rothwell and Strijbos, 1995; Hanisch et al., 1997; Emerit et al., 2004) and may be linked with the enhanced activation of NMDA receptors [reviewed by (Wenk et al., 2006; Parsons et al., 2007)]. In an animal model of neuroinflammation, memantine (at the therapeutically relevant dose in rats of 20 mg kg−1 day−1 s.c.) also protected cholinergic neurons from inflammatory processes (Willard et al., 2000). I.c.v. streptozotocin treated rats showed memory deficits and significantly decreased p-GSK3β levels in both the hippocampus and PFC. The memory impairment was reversed by memantine (5 mg·kg−1), but no changes in p-GSK3β levels were seen (Ponce-Lopez et al., 2011).
Neuroprotection in the clinical situation
Whilst numerous preclinical studies have shown the strong neuroprotective potential of memantine, such has been very difficult to translate to the clinical situation, largely due to the difficulty in designing/performing such trials that, in the case of AD, would have to be very large, start early in the prodromal stage of the disease, last several years and include a wash-out phase. Nonetheless, there are hints from retrospective analyses that memantine does indeed have disease modifying potential in AD patients (Wilkinson and Andersen, 2007; Atri et al., 2008)
Conclusions
The reviewed evidence suggests that the glutamatergic system in general, and NMDA receptors in particular, may play a significant role in the execution of synaptic dysfunction and neuronal death triggered by Aβ in AD. This implies that NMDA receptor antagonists with special features may prevent/attenuate these pathological processes. In fact, memantine, which is an uncompetitive NMDA receptor antagonist with fast, voltage-dependent blocking properties, is able to selectively block pathological tonic NMDA receptor activation in the presence of soluble Aβ oligomers, e.g. (Parsons et al., 2007; Albrecht et al., 2008) whilst preserving their physiological transient synaptic activation. Memantine is hypothesized to provide both symptomatic improvements in, for example, cognition and long-term neuroprotective effects by this same MoA. In fact, several clinical trials have proven such beneficial effects of memantine on symptoms of AD (Reisberg et al., 2003; Tariot et al., 2004; Peskind et al., 2006), and meta-analysis of several trials suggests potential to reduce clinical worsening (Wilkinson and Andersen, 2007; Weiner et al., 2011).
Glossary
- AA
arachidonic acid
- Aβ
β-amyloid
- AChEI
AChEI inhibitor
- AD
Alzheimer's disease
- ADDLs
Aβ-derived diffusible ligands
- AMPK
AMP-activated protein kinase
- APP
amyloid precursor protein
- CA1
Cornu ammonis area 1
- CamKII
Ca2+/calmodulin-dependent protein kinases II
- CaMKK
β, calmodulin-dependent protein kinase-β
- CDK5
cell division protein kinase 5
- CPP
3-[(R)-2-carboxypiperazin-4-yl]-prop-2-enyl-1-phosphonic acid
- CREB
cAMP response element-binding
- D-APV
D-AP5, D-amino-phospovaleric acid
- DG
dentate gyrus
- EAAT2
excitatory amino acid transporter 2
- FB
Aβ
- fibril
GFAP, glial fibrillary acidic protein
- GLAST
glial glutamate transporter
- GLT-1
glutamate transporter 1
- GSK-3β
glycogen synthase kinase 3β
- HNE
4-hydroxy-2-nonenal
- iNOS
inducible NOS
- IR
insulin receptor
- KPI
Kunitz protease inhibitory domain
- KPI-APPs
KPI containing APPs
- LTD
long-term depression
- LTP
long-term potentiation
- mGluR5
metabotropic glutamate receptor 5
- MoA
mechanism of action
- MWM
Morris water maze
- NBM
nucleus basalis of Meynert
- NFTs
neurofibrillary tangles
- NR2B
NMDA receptor subunit
- p-
phosphorylated
- PF
protofibril
- PFC
prefrontal cortex
- PLA
phospholipase A
- PMA
phorbol myristate acetate
- PP2B
protein phosphatase 2B
- PS
population spike
- PS1
presenilin 1
- PSD-95
post-synaptic anchoring protein 95
- PTP
post-tetanic potentiation
- ROS
reactive oxygen species
- sAPP
soluble amyloid precursor protein
- ATPase Na+/K+
sodium-potassium adenosine triphosphatase pump
- STEP
striatal enriched tyrosine phosphatase
- TBOA
threo-β-benzyloxyaspartic acid
- TG
transgenic
- VgluT
vesicular glutamate transporter
Conflict of interest
Both authors work for a pharmaceutical company.
The drug/molecular target nomenclature used in this review conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011).
References
- Abbott JJ, Howlett DR, Francis PT, Williams RJ. Abeta(1-42) modulation of Akt phosphorylation via alpha7 nAChR and NMDA receptors. Neurobiol Aging. 2007;29:992–1001. doi: 10.1016/j.neurobiolaging.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Abe K, Misawa M. Amyloid beta protein enhances the clearance of extracellular L-glutamate by cultured rat cortical astrocytes. Neurosci Res. 2003;45:25–31. doi: 10.1016/s0168-0102(02)00190-6. [DOI] [PubMed] [Google Scholar]
- Abramov E, Dolev I, Fogel H, Ciccotosto GD, Ruff E, Slutsky I. Amyloid-beta as a positive endogenous regulator of release probability at hippocampal synapses. Nat Neurosci. 2009;12:1567–1576. doi: 10.1038/nn.2433. [DOI] [PubMed] [Google Scholar]
- Alberdi E, Sanchez-Gomez MV, Cavaliere F, Perez-Samartin A, Zugaza JL, Trullas R, et al. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium. 2010;47:264–272. doi: 10.1016/j.ceca.2009.12.010. [DOI] [PubMed] [Google Scholar]
- Albrecht M, Rammes G, Parsons CG. 2008. Memantine reverses β-amyloid oligomers-induced deficits in long term potentiation (LTP) in murine hippocampal slices. 38th Society for Neuroscience Annual Meeting. Washington, USA. 15-11-2008. Society for Neuroscience Abstracts. 34, #829.21.
- Alexander S, Harmar A, McGrath I. New updated GRAC Fifth Edition with searchable online version Launch of new portal Guide to Pharmacology in association with NC-IUPHAR Transporter-Themed Issue. Br J Pharmacol. 2011;164:1749–1750. doi: 10.1111/j.1476-5381.2011.01751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alley GM, Bailey JA, Chen D, Ray B, Puli LK, Tanila H, et al. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J Neurosci Res. 2009;88:143–154. doi: 10.1002/jnr.22172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Centralblatt Fur Nervenheilkunde Psychiatrie. 1907;30:177–179. [Google Scholar]
- Arias C, Arrieta I, Tapia R. beta-amyloid peptide fragment 25-35 potentiates the calcium-dependent release of excitatory amino acids from depolarized hippocampal slices. J Neurosci Res. 1995;41:561–566. doi: 10.1002/jnr.490410416. [DOI] [PubMed] [Google Scholar]
- Arif M, Kato T. Increased expression of PAD2 after repeated intracerebroventricular infusions of soluble Abeta(25-35) in the Alzheimer's disease model rat brain: effect of memantine. Cell Mol Biol Lett. 2009;14:703–714. doi: 10.2478/s11658-009-0029-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif M, Chikuma T, Ahmed MM, Nakazato M, Smith MA, Kato T. Effects of memantine on soluble Abeta25-35-induced changes in peptidergic and glial cells in AD model rat brain regions. Neuroscience. 2009;164:1199–1209. doi: 10.1016/j.neuroscience.2009.08.063. [DOI] [PubMed] [Google Scholar]
- Atri A, Shaughnessy LW, Locascio JJ, Growdon JH. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2008;22:209–221. doi: 10.1097/WAD.0b013e31816653bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Wicklund L, Lacor PN, Klein WL, Nordberg A, Marutle A. Different beta-amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiol Aging. 2011;33:1–13. doi: 10.1016/j.neurobiolaging.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Nimmrich V, Striebinger A, Krantz C, Keller P, Janson B, et al. Globular amyloid beta-peptide oligomer – a homogenous and stable neuropathological protein in Alzheimer's disease. J Neurochem. 2005;95:834–847. doi: 10.1111/j.1471-4159.2005.03407.x. [DOI] [PubMed] [Google Scholar]
- Barry AE, Klyubin I, Mc Donald JM, Mably AJ, Farrell MA, Scott M, et al. Alzheimer's disease brain-derived amyloid-{beta}-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J Neurosci. 2011;31:7259–7263. doi: 10.1523/JNEUROSCI.6500-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell KF, Claudio Cuello A. Altered synaptic function in Alzheimer's disease. Eur J Pharmacol. 2006;545:11–21. doi: 10.1016/j.ejphar.2006.06.045. [DOI] [PubMed] [Google Scholar]
- Bi X, Gall CM, Zhou J, Lynch G. Uptake and pathogenic effects of amyloid beta peptide 1-42 are enhanced by integrin antagonists and blocked by NMDA receptor antagonists. Neuroscience. 2002;112:827–840. doi: 10.1016/s0306-4522(02)00132-x. [DOI] [PubMed] [Google Scholar]
- Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, et al. Small-molecule conversion of toxic oligomers to nontoxic beta-sheet-rich amyloid fibrils. Nat Chem Biol. 2011;8:93–101. doi: 10.1038/nchembio.719. [DOI] [PubMed] [Google Scholar]
- Bleich S, Bandelow B, Javaheripour K, Muller A, Degner D, Wilhelm J, et al. Hyperhomocysteinemia as a new risk factor for brain shrinkage in patients with alcoholism. Neurosci Lett. 2003;335:179–182. doi: 10.1016/s0304-3940(02)01194-1. [DOI] [PubMed] [Google Scholar]
- Bojarski L, Herms J, Kuznicki J. Calcium dysregulation in Alzheimer's disease. Neurochem Int. 2008;52:621–633. doi: 10.1016/j.neuint.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Bordji K, Becerril-Ortega J, Nicole O, Buisson A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-{beta} production-test. J Neurosci. 2010;30:15927–15942. doi: 10.1523/JNEUROSCI.3021-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordji K, Becerril-Ortega J, Buisson A. Synapses, NMDA receptor activity and neuronal Abeta production in Alzheimer's disease. Rev Neurosci. 2011;22:285–294. doi: 10.1515/RNS.2011.029. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–408. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, Yilmazer D, Devos RAI, Jansen ENH, Bohl J, et al. Amygdala pathology in parkinson's disease. Acta Neuropathol. 1994;88:493–500. doi: 10.1007/BF00296485. [DOI] [PubMed] [Google Scholar]
- Bresink I, Benke TA, Collett VJ, Seal AJ, Parsons CG, Henley JM, et al. Effects of memantine on recombinant rat NMDA receptors expressed in HEK 293 cells. Br J Pharmacol. 1996;119:195–204. doi: 10.1111/j.1476-5381.1996.tb15971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brorson JR, Bindokas VP, Iwama T, Marcuccilli CJ, Chisholm JC, Miller RJ. The ca2+ influx induced by beta-amyloid peptide 25-35 in cultured hippocampal neurons results from network excitation. J Neurobiol. 1995;26:325–338. doi: 10.1002/neu.480260305. [DOI] [PubMed] [Google Scholar]
- Brzyska M, Elbaum D. Dysregulation of calcium in Alzheimer's disease. Acta Neurobiol Exp (Wars) 2003;63:171–183. doi: 10.55782/ane-2003-1465. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Pocernich CB. The glutamatergic system and Alzheimer's disease: therapeutic implications. CNS Drugs. 2003;17:641–652. doi: 10.2165/00023210-200317090-00004. [DOI] [PubMed] [Google Scholar]
- Cacabelos R, Takeda M, Winblad B. The glutamatergic system and neurodegeneration in dementia: preventive strategies in Alzheimer's disease. Int J Geriatr Psychiatry. 1999;14:3–47. doi: 10.1002/(sici)1099-1166(199901)14:1<3::aid-gps897>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Chambon C, Wegener N, Gravius A, Danysz W. Behavioural and cellular effects of exogenous amyloid-beta peptides in rodents. Behav Brain Res. 2011;225:623–641. doi: 10.1016/j.bbr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- Chen D, Alley GM, Ge1 YW, Farlow MR, Banerjee PK, Lahiri DK. 2002a. Memantine and the processing of the beta-amyloid precursor protein. 32nd Society for Neuroscience Meeting. New Orleans, USA. 2-11-2002a. Society for Neuroscience Abstracts. 28.
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002b;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Chen HSV, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, et al. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine – therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang LYM. Protein kinase C reduces Mg(2+) block of NMDA receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid beta-peptides. J Neurosci Res. 2000;60:65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Chen TF, Huang RF, Lin SE, Lu JF, Tang MC, Chiu MJ. Folic acid potentiates the effect of memantine on spatial learning and neuronal protection in an Alzheimer's disease transgenic model. J Alzheimers Dis. 2010;20:607–615. doi: 10.3233/JAD-2010-1396. [DOI] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, et al. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churcher I. Tau therapeutic strategies for the treatment of Alzheimer's disease. Curr Top Med Chem. 2006;6:579–595. doi: 10.2174/156802606776743057. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, et al. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M. Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9:387–398. doi: 10.1038/nrd2896. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RAJ, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Coan EJ, Irving AJ, Collingridge GL. Low-frequence activation of the NMDA receptor system can prevent the induction of LTP. Neurosci Lett. 1989;105:205–210. doi: 10.1016/0304-3940(89)90038-4. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Singer W. Excitatory amino acid receptors and synaptic plasticity. Trends Pharmacol Sci. 1990;11:290–296. doi: 10.1016/0165-6147(90)90011-v. [DOI] [PubMed] [Google Scholar]
- Costello DA, Herron CE. The role of c-Jun N-terminal kinase in the A beta-mediated impairment of LTP and regulation of synaptic transmission in the hippocampus. Neuropharmacology. 2004;46:655–662. doi: 10.1016/j.neuropharm.2003.11.016. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Monaghan DT, Ganong AH. Excitatory amino acid neurotransmission: NMDA receptors and Hebb-type synaptic plasticity. Annu Rev Neurosci. 1988;11:61–80. doi: 10.1146/annurev.ne.11.030188.000425. [DOI] [PubMed] [Google Scholar]
- Cowburn RF, Wiehager B, Trief E, LiLi M, Sundstrom E. Effects of beta-amyloid-(25-35) peptides on radioligand binding to excitatory amino acid receptors and voltage-dependent calcium channels: evidence for a selective affinity for the glutamate and glycine recognition sites of the NMDA receptor. Neurochem Res. 1997;22:1437–1442. doi: 10.1023/a:1021942109490. [DOI] [PubMed] [Google Scholar]
- Cullen WK, Wu JQ, Anwyl R, Rowan MJ. beta-amyloid produces a delayed NMDA receptor-dependent reduction in synaptic transmission in rat hippocampus. Neuroreport. 1996;8:87–92. doi: 10.1097/00001756-199612200-00018. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danysz W, Parsons CG. Glycine and N-methyl-D-aspartate receptors: physiological significance and possible therapeutic applications. Pharmacol Rev. 1998;50:597–664. [PubMed] [Google Scholar]
- Danysz W, Parsons CG. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer's disease preclinical evidence. Int J Geriatr Psychiatry. 2003;18:S23–S32. doi: 10.1002/gps.938. [DOI] [PubMed] [Google Scholar]
- Danysz W, Parsons CG, Bresink I, Quack G. Glutamate in CNS disorders – a revived target for drug development. Drug News Perspect. 1995;8:261–277. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- Danysz W, Parsons CG, Möbius HJ, Stöffler A, Quack G. Neuroprotective and symptomatological action of memantine relevant for Alzheimer's disease – an unified glutamatergic hypothesis on the mechanism of action. Neurotox Res. 2000;2:85–97. doi: 10.1007/BF03033787. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Velasco PT, Lambert MP, Viola KL, Fernandez SJ, Ferreira ST, et al. Abeta oligomers induce neuronal oxidative stress through an NMDA receptor-dependent mechanism that is blocked by the Alzheimer's drug memantine. J Biol Chem. 2007a;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, et al. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol Aging. 2007b;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker H, Jurgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, et al. N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer's toxic Abeta oligomers. J Neurochem. 2010a;115:1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x. [DOI] [PubMed] [Google Scholar]
- Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010b;30:9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A, Kawai H, Metherate R, Glabe CG, Busciglio J. A role for synaptic zinc in activity-dependent A{beta} oligomer formation and accumulation at excitatory synapses. J Neurosci. 2009;29:4004–4015. doi: 10.1523/JNEUROSCI.5980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, Filipkowski RK, Priller C, Ris L, Neyton J, Croes S, et al. Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol Aging. 2009;30:241–256. doi: 10.1016/j.neurobiolaging.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Dodd PR, Scott HL, Westphalen RI. Excitotoxic mechanisms in the pathogenesis of dementia. Neurochem Int. 1994;25:203–219. doi: 10.1016/0197-0186(94)90064-7. [DOI] [PubMed] [Google Scholar]
- Domingues A, Almeida S, da Cruz E, Oliveira CR, Rego AC. Toxicity of beta-amyloid in HEK293 cells expressing NR1/NR2A or NR1/NR2B N-methyl-D-aspartate receptor subunits. Neurochem Int. 2007;50:872–880. doi: 10.1016/j.neuint.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Dong H, Yuede CM, Coughlan C, Lewis B, Csernansky JG. Effects of Memantine on Neuronal Structure and Conditioned Fear in the Tg2576 Mouse Model of Alzheimer's Disease. Neuropsychopharmacology. 2008;34:1322–1329. doi: 10.1038/npp.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomed Pharmacother. 2004;58:39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Fagg GE, Foster AC, Ganong AH. Excitatory amino acid synaptic mechanisms and neurological function. Trends Pharmacol Sci. 1986;7:357–363. [Google Scholar]
- Fernandez-Tome P, Brera B, Arevalo MA, de Ceballos ML. Beta-amyloid25-35 inhibits glutamate uptake in cultured neurons and astrocytes: modulation of uptake as a survival mechanism. Neurobiol Dis. 2004;15:580–589. doi: 10.1016/j.nbd.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Sinjoanu RC, Nicholson A, Kleinschmidt S. Abeta toxicity in primary cultured neurons. Methods Mol Biol. 2011;670:141–153. doi: 10.1007/978-1-60761-744-0_11. [DOI] [PubMed] [Google Scholar]
- Ferreira IL, Resende R, Ferreiro E, Rego AC, Pereira CF. Multiple defects in energy metabolism in Alzheimer's disease. Curr Drug Targets. 2010;11:1193–1206. doi: 10.2174/1389450111007011193. [DOI] [PubMed] [Google Scholar]
- Ferreira ST, Klein WL. The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer's disease. Neurobiol Learn Mem. 2011;96:529–543. doi: 10.1016/j.nlm.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira ST, Vieira MN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases. IUBMB Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- Filali M, Lalonde R, Rivest S. Subchronic memantine administration on spatial learning, exploratory activity, and nest-building in an APP/PS1 mouse model of Alzheimer's disease. Neuropharmacology. 2011;60:930–936. doi: 10.1016/j.neuropharm.2011.01.035. [DOI] [PubMed] [Google Scholar]
- Floden AM, Li S, Combs CK. Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci. 2005;25:2566–2575. doi: 10.1523/JNEUROSCI.4998-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis PT. Glutamatergic systems in Alzheimer's disease. Int J Geriatr Psychiatry. 2003;18:15–21. doi: 10.1002/gps.934. [DOI] [PubMed] [Google Scholar]
- Frankiewicz T, Parsons CG. Memantine restores long term potentiation impaired by tonic N-methyl-D-aspartate (NMDA) receptor activation following reduction of Mg2+ in hippocampal slices. Neuropharmacology. 1999;38:1253–1259. doi: 10.1016/s0028-3908(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Freir DB, Holscher C, Herron CE. Blockade of long-term potentiation by beta-amyloid peptides in the CA1 region of the rat hippocampus in vivo. J Neurophysiol. 2001;85:708–713. doi: 10.1152/jn.2001.85.2.708. [DOI] [PubMed] [Google Scholar]
- Gasparini L, Dityatev A. Beta-amyloid and glutamate receptors. Exp Neurol. 2008;212:1–4. doi: 10.1016/j.expneurol.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Chang-Chui H, Cooper SM, Lott IT, Cotman CW. Density and distribution of NMDA receptors in the human hippocampus in Alzheimer's disease. Brain Res. 1986;399:156–161. doi: 10.1016/0006-8993(86)90611-6. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Schousboe A. High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol. 1997;52:6–15. doi: 10.1124/mol.52.1.6. [DOI] [PubMed] [Google Scholar]
- Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, et al. ß-Amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer's disease. Trends Neurosci. 1993;16:460–465. doi: 10.1016/0166-2236(93)90078-z. [DOI] [PubMed] [Google Scholar]
- Goedert M, Klug A, Crowther RA. Tau protein, the paired helical filament and Alzheimer's disease. J Alzheimers Dis. 2006;9:195–207. doi: 10.3233/jad-2006-9s323. [DOI] [PubMed] [Google Scholar]
- Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008;15:2321–2328. doi: 10.2174/092986708785909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CW, Patel AJ. Neurodegeneration mediated by glutamate and beta-amyloid peptide: a comparison and possible interaction. Brain Res. 1995;691:169–179. doi: 10.1016/0006-8993(95)00669-h. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Young AB. Excitatory amino acids and Alzheimer's disease. Neurobiol Aging. 1989;10:593–602. doi: 10.1016/0197-4580(89)90143-7. [DOI] [PubMed] [Google Scholar]
- Gu QB, Zhao JX, Fei J, Schwarz W. Modulation of Na(+),K(+) pumping and neurotransmitter uptake by beta-amyloid. Neuroscience. 2004;126:61–67. doi: 10.1016/j.neuroscience.2004.03.022. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Neuhaus J, Rowe W, van RD, Moller T, Kettenmann H, et al. Neurotoxic consequences of central long-term administration of interleukin-2 in rats. Neuroscience. 1997;79:799–818. doi: 10.1016/s0306-4522(97)00040-7. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cowburn R. Glutamate neurotoxicity and Alzheimer's disease. Trends Neurosci. 1987;10:406. [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Harkany T, Mulder J, Sasvari M, Abraham I, Konya C, Zarandi M, et al. N-methyl-D-aspartate receptor antagonist MK-801 and radical scavengers protect cholinergic nucleus basalis neurons against beta-amyloid neurotoxicity. Neurobiol Dis. 1999;6:109–121. doi: 10.1006/nbdi.1998.0230. [DOI] [PubMed] [Google Scholar]
- Harkany T, Abraham I, Timmerman W, Laskay G, Toth B, Sasvari M, et al. beta-Amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci. 2000;12:2735–2745. doi: 10.1046/j.1460-9568.2000.00164.x. [DOI] [PubMed] [Google Scholar]
- Harris ME, Carney JM, Cole PS, Hensley K, Howard BJ, Martin L, et al. beta-amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: implications for alzheimer's disease. Neuroreport. 1995;6:1875–1879. doi: 10.1097/00001756-199510020-00013. [DOI] [PubMed] [Google Scholar]
- Harris ME, Wang YN, Pedigo NW, Hensley K, Butterfield DA, Carney JM. Amyloid beta peptide (25-35) inhibits na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem. 1996;67:277–286. doi: 10.1046/j.1471-4159.1996.67010277.x. [DOI] [PubMed] [Google Scholar]
- He Y, Cui J, Lee JC, Ding S, Chalimoniuk M, Simonyi A, et al. Prolonged exposure of cortical neurons to oligomeric amyloid-beta impairs NMDA receptor function via NADPH oxidase-mediated ROS production: protective effect of green tea (-)-epigallocatechin-3-gallate. ASN Neuro. 2011;3:e00050. doi: 10.1042/AN20100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heininger K. A unifying hypothesis of Alzheimer's disease. II. Pathophysiological processes. Hum Psychopharmacol Clin Exp. 1999;14:525–581. [Google Scholar]
- van Helmond Z, Miners JS, Kehoe PG, Love S. Higher soluble amyloid beta concentration in frontal cortex of young adults than in normal elderly or Alzheimer's disease. Brain Pathol. 2010;20:787–793. doi: 10.1111/j.1750-3639.2010.00374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoey SE, Williams RJ, Perkinton MS. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J Neurosci. 2009;29:4442–4460. doi: 10.1523/JNEUROSCI.6017-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holscher C. Possible causes of Alzheimer's disease: amyloid fragments, free radicals, and calcium homeostasis. Neurobiol Dis. 1998;5:129–141. doi: 10.1006/nbdi.1998.0193. [DOI] [PubMed] [Google Scholar]
- Honegger UE, Quack G, Wiesmann UN. Evidence for lysosomotropism of memantine in cultured human cells – cellular kinetics and effects of memantine on phospholipid content and composition, membrane fluidity and beta-adrenergic transmission. Pharmacol Toxicol. 1993;73:202–208. doi: 10.1111/j.1600-0773.1993.tb01564.x. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, et al. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Schurdak ME, Puttfarcken PS, El Kouhen R, Gopalakrishnan M, Li J. High content screen microscopy analysis of Abeta(1-42)-induced neurite outgrowth reduction in rat primary cortical neurons: neuroprotective effects of alpha7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007;1151:227–235. doi: 10.1016/j.brainres.2007.03.051. [DOI] [PubMed] [Google Scholar]
- Hu NW, Klyubin I, Anwyl R, Rowan MJ. GluN2B subunit-containing NMDA receptor antagonists prevent Abeta-mediated synaptic plasticity disruption in vivo. Proc Natl Acad Sci USA. 2009;106:20504–20509. doi: 10.1073/pnas.0908083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynd MR, Scott HL, Dodd PR. Glutamate(NMDA) receptor NR1 subunit mRNA expression in Alzheimer's disease. J Neurochem. 2001;78:175–182. doi: 10.1046/j.1471-4159.2001.00409.x. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Matsuura S, Ueno S, Baba A, Yamada MK, Nishiyama N, et al. Beta-amyloid enhances glial glutamate uptake activity and attenuates synaptic efficacy. J Biol Chem. 2002;277:32180–32186. doi: 10.1074/jbc.M203764200. [DOI] [PubMed] [Google Scholar]
- Ikezu T, Luo X, Weber GA, Zhao J, McCabe L, Buescher JL, et al. Amyloid precursor protein-processing products affect mononuclear phagocyte activation: pathways for sAPP- and Abeta-mediated neurotoxicity. J Neurochem. 2003;85:925–934. doi: 10.1046/j.1471-4159.2003.01739.x. [DOI] [PubMed] [Google Scholar]
- Inoue R, Hashimoto K, Harai T, Mori H. NMDA- and beta-amyloid1-42-induced neurotoxicity is attenuated in serine racemase knock-out mice. J Neurosci. 2008;28:14486–14491. doi: 10.1523/JNEUROSCI.5034-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itkin A, Dupres V, Dufrene YF, Bechinger B, Ruysschaert JM, Raussens V. Calcium ions promote formation of amyloid beta-peptide (1-40) oligomers causally implicated in neuronal toxicity of Alzheimer's disease. PLoS ONE. 2011;6:e18250. doi: 10.1371/journal.pone.0018250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, et al. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer's disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, et al. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Karanian DA, Baude AS, Brown QB, Parsons CG, Bahr BA. 3-Nitropropionic acid toxicity in hippocampus: protection through N-methyl-D-aspartate receptor antagonism. Hippocampus. 2006;16:834–842. doi: 10.1002/hipo.20214. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci. 2001;14:547–553. doi: 10.1046/j.0953-816x.2001.01664.x. [DOI] [PubMed] [Google Scholar]
- Katsuura G, Gottschall PE, Dahl RR, Arimura A. Interleukin-1 beta increases prostaglandin E2 in rat astrocyte cultures: modulatory effect of neuropeptides. Endocrinology. 1989;124:3125–3127. doi: 10.1210/endo-124-6-3125. [DOI] [PubMed] [Google Scholar]
- Kawamoto EM, Lepsch LB, Boaventura MF, Munhoz CD, Lima LS, Yshii LM, et al. Amyloid beta-peptide activates nuclear factor-kappaB through an N-methyl-D-aspartate signaling pathway in cultured cerebellar cells. J Neurosci Res. 2007;86:845–860. doi: 10.1002/jnr.21548. [DOI] [PubMed] [Google Scholar]
- Kim JH, Anwyl R, Suh YH, Djamgoz MB, Rowan MJ. Use-dependent effects of amyloidogenic fragments of (beta)-amyloid precursor protein on synaptic plasticity in rat hippocampus in vivo. J Neurosci. 2001;21:1327–1333. doi: 10.1523/JNEUROSCI.21-04-01327.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Komatsu H, Ogura H, Sawada K. Comparison of donepezil and memantine for protective effect against amyloid-beta(1-42) toxicity in rat septal neurons. Neurosci Lett. 2005;391:17–21. doi: 10.1016/j.neulet.2005.08.036. [DOI] [PubMed] [Google Scholar]
- Kirvell SL, Esiri M, Francis PT. Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem. 2006;98:939–950. doi: 10.1111/j.1471-4159.2006.03935.x. [DOI] [PubMed] [Google Scholar]
- Kleckner NW, Dingledine R. Requirement for glycine in activation of NMDA receptors expressed in Xenopus oocytes. Science. 1988;214:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- Klegeris A, McGeer PL. beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res. 1997;49:229–235. doi: 10.1002/(sici)1097-4547(19970715)49:2<229::aid-jnr11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Klein WL, Lacor PN, De Felice FG, Ferreira ST. 2007. pp. 154–179. Molecules that disrupt memory circuits in Alzheimer's disease: the attack on synapses by Aâ oligomers (ADDLs). In Memories: Molecules and Circuits.
- Klyubin I, Walsh DM, Cullen WK, Fadeeva JV, Anwyl R, Selkoe DJ, et al. Soluble Arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivo. Eur J Neurosci. 2004;19:2839–2846. doi: 10.1111/j.1460-9568.2004.03389.x. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Wang Q, Reed MN, Irving EA, Upton N, Hofmeister J, et al. Protection against Abeta-mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol Aging. 2011;32:614–623. doi: 10.1016/j.neurobiolaging.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Ishiguro M, Ohsawa I, Morimoto T, Takamura T, Inoue K, et al. The effect of a secreted form of beta-amyloid-precursor protein on intracellular Ca2(+) increase in rat cultured hippocampal neurones. Br J Pharmacol. 1998;123:1483–1489. doi: 10.1038/sj.bjp.0701712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornhuber J, Weller M. Psychotogenicity and n-methyl-d-aspartate receptor antagonism: implications for neuroprotective pharmacotherapy. Biol Psychiatry. 1997;41:135–144. doi: 10.1016/S0006-3223(96)00047-9. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Bormann J, Retz W, Hubers M, Riederer P. Memantine displaces [<3>H]MK-801 at therapeutic concentrations in postmortem human frontal cortex. Eur J Pharmacol. 1989;166:589–590. doi: 10.1016/0014-2999(89)90384-1. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, et al. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, et al. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Lauderback CM, Harris-White ME, Wang Y, Pedigo NW, Carney JM, Butterfield DA. Amyloid beta-peptide inhibits Na+-dependent glutamate uptake. Life Sci. 1999;65:1977–1981. doi: 10.1016/s0024-3205(99)00459-2. [DOI] [PubMed] [Google Scholar]
- Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, et al. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Le WD, Colom LV, Xie WJ, Smith RG, Alexianu M, Appel SH. Cell death induced by beta-amyloid 1-40 in MES 23.5 hybrid clone: the role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res. 1995;686:49–60. doi: 10.1016/0006-8993(95)00450-5. [DOI] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, et al. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Li S, Mallory M, Alford M, Tanaka S, Masliah E. Glutamate transporter alterations in Alzheimer disease are possibly associated with abnormal APP expression. J Neuropathol Exp Neurol. 1997;56:901–911. doi: 10.1097/00005072-199708000-00008. [DOI] [PubMed] [Google Scholar]
- Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble A{beta} oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Chang L, Roselli F, Almeida OF, Gao X, Wang X, et al. Amyloid-beta induces caspase-dependent loss of PSD-95 and synaptophysin through NMDA receptors. J Alzheimers Dis. 2010;22:541–556. doi: 10.3233/JAD-2010-100948. [DOI] [PubMed] [Google Scholar]
- Luhrs LB, Banerjee PK, LaFerla FM. 2006. Memantine reverses cognitive deficits in transgenic mice with both amyloid plaques and neurofibrillary tangles.
- Maragakis NJ, Rothstein JD. Glutamate transporters in neurologic disease. Arch Neurol. 2001;58:365–370. doi: 10.1001/archneur.58.3.365. [DOI] [PubMed] [Google Scholar]
- Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, et al. Memantine improves cognition and reduces Alzheimer's-like neuropathology in transgenic mice. Am J Pathol. 2010;176:870–880. doi: 10.2353/ajpath.2010.090452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Alford M, Deteresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol. 1996;40:759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- Masliah E, Raber J, Alford M, Mallory M, Mattson MP, Yang D, et al. Amyloid protein precursor stimulates excitatory amino acid transport. Implications for roles in neuroprotection and pathogenesis. J Biol Chem. 1998;273:12548–12554. doi: 10.1074/jbc.273.20.12548. [DOI] [PubMed] [Google Scholar]
- Masliah E, Alford M, Mallory M, Rockenstein E, Moechars D, Van Leuven F. Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp Neurol. 2000;163:381–387. doi: 10.1006/exnr.2000.7386. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Guthrie PB, Kater SB. A role for Na<+>-dependent Ca<2+> extrusion in protection against neuronal excitotoxicity. FASEB J. 1989;3:2519–2526. doi: 10.1096/fasebj.3.13.2572500. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. Beta-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Cheng B, Lieberburg I, Smithswintosky VL, Rydel RE. ß-amyloid precursor protein metabolites and loss of neuronal ca2+ homeostasis in Alzheimer's disease. Trends Neurosci. 1993;16:409–414. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann N Y Acad Sci. 1999;893:154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x. [DOI] [PubMed] [Google Scholar]
- Maurice T, Lockhart BP, Su TP, Privat A. Reversion of beta(25-35)-amyloid peptide-induced amnesia by NMDA receptor-associated glycine site agonists. Brain Res. 1996;731:249–253. doi: 10.1016/0006-8993(96)00710-x. [DOI] [PubMed] [Google Scholar]
- Mezler M, Barghorn S, Schoemaker H, Gross G, Nimmrich V. Abeta oligomer directly modulates P/Q-type calcium currents in Xenopus oocytes. Br J Pharmacol. 2011;165:1572–1583. doi: 10.1111/j.1476-5381.2011.01646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Neuroprotection by memantine against neurodegeneration induced by beta- amyloid(1-40) Brain Res. 2002;958:210–221. doi: 10.1016/s0006-8993(02)03731-9. [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Alvarez XA, Quack G, Cacabelos R. 2003. Memantine prevents beta-amyloid-induced neurotoxicity and learning impairment in rats. 34th Annual Meeting of the American Society for Neurochemistry. Newport Beach, California, USA. 3-5-2003. 85, 42. [DOI] [PubMed]
- Miguel-Hidalgo JJ, Paul IA, Wanzo V, Banerjee PK. Memantine inhibits the expression of caspase-8 and improves learning in rats injected with [beta]-amyloid (A[beta]sub>140</sub>. FASEB J. 2006;20:A1135. [Google Scholar]
- Minkeviciene R, Banerjee P, Tanila H. Memantine improves spatial learning in a transgenic mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2004;311:677–682. doi: 10.1124/jpet.104.071027. [DOI] [PubMed] [Google Scholar]
- Molnar Z, Soos K, Lengyel I, Penke B, Szegedi V, Budai D. Enhancement of NMDA responses by beta-amyloid peptides in the hippocampus in vivo. Neuroreport. 2004;15:1649–1652. doi: 10.1097/01.wnr.0000134471.06244.d2. [DOI] [PubMed] [Google Scholar]
- More L, Gravius A, Nagel J, Valastro B, Greco S, Danysz W. Therapeutically relevant plasma concentrations of memantine produce significant NMDA receptor occupancy and do not impair learning in rats. Behav Pharmacol. 2008;19:724–734. doi: 10.1097/FBP.0b013e3283123cad. [DOI] [PubMed] [Google Scholar]
- Morris RG. The memory deficits in Alzheimer-type dementia: a review. Q J Exp Psychol. 1986;38A:575–602. doi: 10.1080/14640748608401615. [DOI] [PubMed] [Google Scholar]
- Nakagami Y, Oda T. Glutamate exacerbates amyloid beta1-42-induced impairment of long-term potentiation in rat hippocampal slices. Jpn J Pharmacol. 2002;88:223–226. doi: 10.1254/jjp.88.223. [DOI] [PubMed] [Google Scholar]
- Nakagami Y, Nishimura S, Murasugi T, Kaneko I, Meguro M, Marumoto S, et al. A novel beta-sheet breaker, RS-0406, reverses amyloid beta-induced cytotoxicity and impairment of long-term potentiation in vitro. Br J Pharmacol. 2002;137:676–682. doi: 10.1038/sj.bjp.0704911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Murayama N, Noshita T, Katsuragi R, Ohno T. Cognitive dysfunction induced by sequential injection of amyloid-beta and ibotenate into the bilateral hippocampus; protection by memantine and MK-801. Eur J Pharmacol. 2006;548:115–122. doi: 10.1016/j.ejphar.2006.07.049. [DOI] [PubMed] [Google Scholar]
- Neniskyte U, Neher JJ, Brown GC. Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem. 2011;286:39904–39913. doi: 10.1074/jbc.M111.267583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmrich V, Reymann KG, Strassburger M, Schoder UH, Gross G, Hahn A, et al. Inhibition of calpain prevents NMDA-induced cell death and beta-amyloid-induced synaptic dysfunction in hippocampal slice cultures. Br J Pharmacol. 2010;159:1523–1531. doi: 10.1111/j.1476-5381.2010.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya H, Fukunaga R, Taniguchi T, Fujiwara M, Shimohama S, Kameyama M. Decreased number of NMDA receptors in the brains of Alzheimer type dementia patients. In: Kameyama T, Nabeshima T, Domino EF, editors. NMDA Receptor Related Agents : Biochemistry, Pharmacology and Behavior. Ann Arbor: 1991. pp. 401–408. Npp Books. [Google Scholar]
- Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–565. doi: 10.1016/s0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- Noda M, Nakanishi H, Akaike N. Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience. 1999;92:1465–1474. doi: 10.1016/s0306-4522(99)00036-6. [DOI] [PubMed] [Google Scholar]
- Nomura I, Kato N, Kita T, Takechi H. Mechanism of impairment of long-term potentiation by amyloid beta is independent of NMDA receptors or voltage-dependent calcium channels in hippocampal CA1 pyramidal neurons. Neurosci Lett. 2005;391:1–6. doi: 10.1016/j.neulet.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Nyakas C, Granic I, Halmy LG, Banerjee P, Luiten PG. The basal forebrain cholinergic system in aging and dementia. Rescuing cholinergic neurons from neurotoxic amyloid-beta42 with memantine. Behav Brain Res. 2011;221:594–603. doi: 10.1016/j.bbr.2010.05.033. [DOI] [PubMed] [Google Scholar]
- Okamoto SI, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med. 2009;15:1407–1413. doi: 10.1038/nm.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OMahony S, Harkany T, Rensink AAM, Abraham I, DeJong GI, Varga JL, et al. beta-amyloid-induced cholinergic denervation correlates with enhanced nitric oxide synthase activity in rat cerebral cortex: reversal by NMDA receptor blockade. Brain Res Bull. 1998;45:405–411. doi: 10.1016/s0361-9230(97)00405-x. [DOI] [PubMed] [Google Scholar]
- Palmer AM, Gershon S. Is the neuronal basis of Alzheimer's disease cholinergic or glutamatergic? FASEB J. 1990;4:2745–2752. doi: 10.1096/fasebj.4.10.2165009. [DOI] [PubMed] [Google Scholar]
- Parameshwaran K, Dhanasekaran M, Suppiramaniam V. Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol. 2008;210:7–13. doi: 10.1016/j.expneurol.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Parihar MS, Brewer GJ. Mitoenergetic failure in Alzheimer disease. Am J Physiol Cell Physiol. 2007;292:8–23. doi: 10.1152/ajpcell.00232.2006. [DOI] [PubMed] [Google Scholar]
- Parks JK, Smith TS, Trimmer PA, Bennett JP, Parker WD. Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J Neurochem. 2001;76:1050–1056. doi: 10.1046/j.1471-4159.2001.00112.x. [DOI] [PubMed] [Google Scholar]
- ParpuraGill A, Beitz D, Uemura E. The inhibitory effects of beta-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res. 1997;754:65–71. doi: 10.1016/s0006-8993(97)00043-7. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G. Glutamate in CNS Disorders as a target for drug development: an update. Drug News Perspect. 1998;11:523–569. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G. Memantine is a clinically well tolerated N-methyl-D-aspartate (NMDA) receptor antagonist – a review of preclinical data. Neuropharmacology. 1999;38:735–767. doi: 10.1016/s0028-3908(99)00019-2. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Lodge D. Preface: introduction to glutamate receptors, their function and pharmacology. In: Lodge D, Danysz W, Parsons CG, editors. Ionotropic Glutamate Receptors as Therapeutic Targets. New York: F.P. Graham Publishing Co; 2002. pp. 1–27. [Google Scholar]
- Parsons CG, Stoffler A, Danysz W. Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system – too little activation is bad, too much is even worse. Neuropharmacology. 2007;53:699–723. doi: 10.1016/j.neuropharm.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Peng TI, Greenamyre JT. Privileged access to mitochondria of calcium influx through N-methyl-D-aspartate receptors. Mol Pharmacol. 1998;53:974–980. [PubMed] [Google Scholar]
- Peskind ER, Potkin SG, Pomara N, Ott BR, Graham SM, Olin JT, et al. Memantine treatment in mild to moderate Alzheimer disease: a 24-week randomized, controlled trial. Am J Geriatr Psychiatry. 2006;14:704–715. doi: 10.1097/01.JGP.0000224350.82719.83. [DOI] [PubMed] [Google Scholar]
- Ponce-Lopez T, Liy-Salmeron G, Hong E, Meneses A. Lithium, phenserine, memantine and pioglitazone reverse memory deficit and restore phospho-GSK3beta decreased in hippocampus in intracerebroventricular streptozotocin induced memory deficit model. Brain Res. 2011;1426:73–85. doi: 10.1016/j.brainres.2011.09.056. [DOI] [PubMed] [Google Scholar]
- Procter AW, Stirling JM, Stratmann GC, Cross AJ, Bowen DM. Loss of glycine-dependent radioligand binding to the N-methyl-D-aspartate-phencyclidine receptor complex in patients with Alzheimer's disease. Neurosci Lett. 1989;101:62–66. doi: 10.1016/0304-3940(89)90441-2. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Arancio O. Fibrillar beta-amyloid impairs the late phase of long term potentiation. Curr Alzheimer Res. 2006;3:179–183. doi: 10.2174/156720506777632871. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, et al. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammes G, Hasenjager A, Sroka-Saidi K, Deussing JM, Parsons CG. Therapeutic significance of NR2B-containing NMDA receptors and mGluR5 metabotropic glutamate receptors in mediating the synaptotoxic effects of beta-amyloid oligomers on long-term potentiation (LTP) in murine hippocampal slices. Neuropharmacology. 2011;60:982–990. doi: 10.1016/j.neuropharm.2011.01.051. [DOI] [PubMed] [Google Scholar]
- Ray B, Banerjee PK, Greig NH, Lahiri DK. Memantine treatment decreases levels of secreted Alzheimer's amyloid precursor protein (APP) and amyloid beta (Abeta) peptide in the human neuroblastoma cells. Neurosci Lett. 2010;470:1–5. doi: 10.1016/j.neulet.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Ireland DR, Abraham WC. NMDA receptor regulation by amyloid-beta does not account for its inhibition of LTP in rat hippocampus. Brain Res. 2003;968:263–272. doi: 10.1016/s0006-8993(03)02269-8. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, et al. Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronicke R, Mikhaylova M, Ronicke S, Meinhardt J, Schroder UH, Fandrich M, et al. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol Aging. 2010;32:1558–1497. doi: 10.1016/j.neurobiolaging.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, et al. Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci. 2005;25:11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosi S, Vazdarjanova A, Ramirez-Amaya V, Worley PF, Barnes CA, Wenk GL. Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience. 2006;142:1303–1315. doi: 10.1016/j.neuroscience.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ, Strijbos PJLM. Cytokines in neurodegeneration and repair. Int J Dev Neurosci. 1995;13:179–185. doi: 10.1016/0736-5748(95)00018-c. [DOI] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Cullen WK, Anwyl R. Synaptic plasticity in animal models of early Alzheimer's disease. Philos Trans R Soc Lond B Biol Sci. 2003;358:821–828. doi: 10.1098/rstb.2002.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenzig D, Manhart S, Cinar Y, Kleinschmidt M, Hause G, Willbold D, et al. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry. 2009;48:7072–7078. doi: 10.1021/bi900818a. [DOI] [PubMed] [Google Scholar]
- Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, et al. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer's-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res. 2008;86:2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Matthews RT, Henshaw DR, Beal MF. Neuroprotective strategies for the treatment of lesions produced by mitochondrial toxins: implications for neurodegenerative diseases. Neuroscience. 1996;71:1043–1048. doi: 10.1016/0306-4522(95)00527-7. [DOI] [PubMed] [Google Scholar]
- Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR. Glutamate transporter variants reduce glutamate uptake in Alzheimer's disease. Neurobiol Aging. 2010;32:e1–11. doi: 10.1016/j.neurobiolaging.2010.03.008. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelat PB, Chalimoniuk M, Wang JH, Strosznajder JB, Lee JC, Sun AY, et al. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A(2) in cortical neurons. J Neurochem. 2008;106:45–55. doi: 10.1111/j.1471-4159.2008.05347.x. [DOI] [PubMed] [Google Scholar]
- Shinozaki H. Pharmacology of glutamate receptors. Prog Neurobiol. 1988;30:399–435. doi: 10.1016/0301-0082(88)90009-3. [DOI] [PubMed] [Google Scholar]
- Sipos E, Kurunczi A, Kasza A, Horvath J, Felszeghy K, Laroche S, et al. Beta-amyloid pathology in the entorhinal cortex of rats induces memory deficits: implications for Alzheimer's disease. Neuroscience. 2007;147:28–36. doi: 10.1016/j.neuroscience.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W, et al. AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol Aging. 2011;33:1545–1555. doi: 10.1016/j.neurobiolaging.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Rauw G, Baker GB, Kar S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci. 2008;28:1989–2002. doi: 10.1111/j.1460-9568.2008.06498.x. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Staufenbiel M. Pathogenic mechanisms of Alzheimer's disease analyzed in the APP23 transgenic mouse model. Ann N Y Acad Sci. 2000;920:134–139. doi: 10.1111/j.1749-6632.2000.tb06915.x. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegedi V, Juhasz G, Budai D, Penke B. Divergent effects of Abeta1-42 on ionotropic glutamate receptor-mediated responses in CA1 neurons in vivo. Brain Res. 2005;1062:120–126. doi: 10.1016/j.brainres.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Tampellini D, Gouras GK. Synapses, synaptic activity and intraneuronal abeta in Alzheimer's disease. Front Aging Neurosci. 2010;2:1–5. doi: 10.3389/fnagi.2010.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291:317–324. doi: 10.1001/jama.291.3.317. [DOI] [PubMed] [Google Scholar]
- Texido L, Martin-Satue M, Alberdi E, Solsona C, Matute C. Amyloid beta peptide oligomers directly activate NMDA receptors. Cell Calcium. 2011;49:184–190. doi: 10.1016/j.ceca.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Thornton C, Bright NJ, Sastre M, Muckett PJ, Carling D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to beta-amyloid exposure. Biochem J. 2011;15:503–512. doi: 10.1042/BJ20101485. [DOI] [PubMed] [Google Scholar]
- Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, et al. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Cleary JP, Mehta T, Hofmeister J, Lesne S, O'Hare E, et al. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006;60:668–676. doi: 10.1002/ana.21051. [DOI] [PubMed] [Google Scholar]
- Tremblay R, Chakravarthy B, Hewitt K, Tauskela J, Morley P, Atkinson T, et al. Transient NMDA receptor inactivation provides long-term protection to cultured cortical neurons from a variety of death signals. J Neurosci. 2000;20:7183–7192. doi: 10.1523/JNEUROSCI.20-19-07183.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- Unger C, Svedberg MM, Yu WF, Hedberg MM, Nordberg A. Effect of subchronic treatment of memantine, galantamine, and nicotine in the brain of Tg2576 (APPswe) transgenic mice. J Pharmacol Exp Ther. 2006;317:30–36. doi: 10.1124/jpet.105.098566. [DOI] [PubMed] [Google Scholar]
- Van Dam D, De deyn PP. Cognitive evaluation of disease-modifying efficacy of Galantamine and Memantine in the APP23 model. Eur Neuropsychopharmacol. 2006;16:59–69. doi: 10.1016/j.euroneuro.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, et al. Beta-amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volbracht C, van Beek J, Zhu C, Blomgren K, Leist M. Neuroprotective properties of memantine in different in vitro and in vivo models of excitotoxicity. Eur J Neurosci. 2006;23:2611–2622. doi: 10.1111/j.1460-9568.2006.04787.x. [DOI] [PubMed] [Google Scholar]
- Von Euler G, Norman J, Radesäter A-C, Dahlqvist C, Sandegren A, Svensson S, et al. Soluble Aβ42 oligomers do not cause synaptic degeneration through direct binding to NMDA receptor sites for glutamate, glycine, and non-competitive blockers. Society for Neuroscience Abstracts. 2008;34:#829.07. [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, et al. The role of cell-derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, et al. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- Wang Q, Rowan MJ, Anwyl R. Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci. 2004a;24:6049–6056. doi: 10.1523/JNEUROSCI.0233-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004b;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Wu J, Rowan MJ, Anwyl R. Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci. 2005;22:2827–2832. doi: 10.1111/j.1460-9568.2005.04457.x. [DOI] [PubMed] [Google Scholar]
- Watkins JC, Evans RH. Excitatory amino acid transmitters. Annu Rev Pharmacol Toxicol. 1981;21:165–204. doi: 10.1146/annurev.pa.21.040181.001121. [DOI] [PubMed] [Google Scholar]
- Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2009;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner MW, Sadowsky C, Saxton J, Hofbauer RK, Graham SM, Yu SY, et al. Magnetic resonance imaging and neuropsychological results from a trial of memantine in Alzheimer's disease. Alzheimers Dement. 2011;7:425–435. doi: 10.1016/j.jalz.2010.09.003. [DOI] [PubMed] [Google Scholar]
- Wenk GL. Neuropathologic changes in Alzheimer's disease: potential targets for treatment. J Clin Psychiatry. 2006;67(Suppl. 3):3–7. [PubMed] [Google Scholar]
- Wenk GL, Danysz W, Mobley SL. MK-801, memantine and amantadine show neuroprotective activity in the nucleus basalis magnocellularis. Eur J Pharmacol. 1995;293:267–270. doi: 10.1016/0926-6917(95)00028-3. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Danysz W, Roice DD. The effects of mitochondrial failure upon cholinergic toxicity in the nucleus basalis. Neuroreport. 1996;7:1453–1456. doi: 10.1097/00001756-199606170-00001. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Zajaczkowski W, Danysz W. Neuroprotection of acetylcholinergic basal forebrain neurons by memantine and neurokinin B. Behav Brain Res. 1997;83:129–133. doi: 10.1016/s0166-4328(97)86056-1. [DOI] [PubMed] [Google Scholar]
- Wenk GL, Parsons CG, Danysz W. Potential role of N-methyl-D-aspartate receptors as executors of neurodegeneration resulting from diverse insults: focus on memantine. Behav Pharmacol. 2006;17:411–424. doi: 10.1097/00008877-200609000-00007. [DOI] [PubMed] [Google Scholar]
- Wilcox KC, Lacor PN, Pitt J, Klein WL. Abeta Oligomer-Induced Synapse Degeneration in Alzheimer's Disease. Cell Mol Neurobiol. 2011;31:939–948. doi: 10.1007/s10571-011-9691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D, Andersen HF. Analysis of the effect of memantine in reducing the worsening of clinical symptoms in patients with moderate to severe Alzheimer's disease. Dement Geriatr Cogn Disord. 2007;24:138–145. doi: 10.1159/000105162. [DOI] [PubMed] [Google Scholar]
- Willard LB, Hauss-Wegrzyniak B, Danysz W, Wenk GL. The cytotoxicity of chronic neuroinflammation upon basal forebrain cholinergic neurons of rats can be attenuated by glutamatergic antagonism or cyclooxygenase-2 inhibition. Exp Brain Res. 2000;134:58–65. doi: 10.1007/s002210000446. [DOI] [PubMed] [Google Scholar]
- Williams K. Modulation and block of ion channels: a new biology of polyamines. Cell Signal. 1997;9:1–13. doi: 10.1016/s0898-6568(96)00089-7. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Anwyl R, Rowan MJ. beta-amyloid selectively augments NMDA receptor-mediated synaptic transmission in rat hippocampus. Neuroreport. 1995a;6:2409–2413. doi: 10.1097/00001756-199511270-00031. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Anwyl R, Rowan MJ. Beta-amyloid-(1-40) increases long-term potentiation in rat hippocampus in vitro. Eur J Pharmacol. 1995b;284:R1–R3. doi: 10.1016/0014-2999(95)00539-w. [DOI] [PubMed] [Google Scholar]
- Wu SZ, Bodles AM, Porter MM, Griffin WS, Basile AS, Barger SW. Induction of serine racemase expression and d-serine release from microglia by amyloid beta-peptide. J Neuroinflammation. 2004;1:1–11. doi: 10.1186/1742-2094-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W. Brain amyloid beta protein and memory disruption in Alzheimer's disease. Neuropsychiatr Dis Treat. 2010;6:605–611. doi: 10.2147/NDT.S7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong H, McCabe L, Costello J, Anderson E, Weber G, Ikezu T. Activation of NR1a/NR2B receptors by soluble factors from APP-stimulated monocyte-derived macrophages: implications for the pathogenesis of Alzheimer's disease. Neurobiol Aging. 2004;25:905–911. doi: 10.1016/j.neurobiolaging.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Yamada K, Takayanagi M, Kamei H, Nagai T, Dohniwa M, Kobayashi K, et al. Effects of memantine and donepezil on amyloid beta-induced memory impairment in a delayed-matching to position task in rats. Behav Brain Res. 2005;162:191–199. doi: 10.1016/j.bbr.2005.02.036. [DOI] [PubMed] [Google Scholar]
- Yankner BA. Mechanisms of neuronal degeneration in alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- Ye C, Walsh DM, Selkoe DJ, Hartley DM. Amyloid beta-protein induced electrophysiological changes are dependent on aggregation state: N-methyl-D-aspartate (NMDA) versus non-NMDA receptor/channel activation. Neurosci Lett. 2004;366:320–325. doi: 10.1016/j.neulet.2004.05.060. [DOI] [PubMed] [Google Scholar]
- Ye L, Qiao JT. Suppressive action produced by beta-amyloid peptide fragment 31-35 on long-term potentiation in rat hippocampus is N-methyl-D-aspartate receptor-independent: it's offset by (-)huperzine A. Neurosci Lett. 1999;275:187–190. doi: 10.1016/s0304-3940(99)00795-8. [DOI] [PubMed] [Google Scholar]
- Zajaczkowski W, Frankiewicz T, Parsons CG, Danysz W. Uncompetitive NMDA receptor antagonists attenuate NMDA-induced impairment of passive avoidance learning and LTP. Neuropharmacology. 1997;36:961–971. doi: 10.1016/s0028-3908(97)00070-1. [DOI] [PubMed] [Google Scholar]
- Zhao D, Watson JB, Xie CW. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol. 2004;92:2853–2858. doi: 10.1152/jn.00485.2004. [DOI] [PubMed] [Google Scholar]
- Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008;22:246–260. doi: 10.1096/fj.06-7703com. [DOI] [PubMed] [Google Scholar]
- Zheng X, Zhang L, Wang AP, Bennett MVL, Zukin RS. Ca2+ influx amplifies protein kinase C potentiation of recombinant NMDA receptors. J Neurosci. 1997;17:8676–8686. doi: 10.1523/JNEUROSCI.17-22-08676.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoladz PR, Campbell AM, Park CR, Schaefer D, Danysz W, Diamond DM. Enhancement of long-term spatial memory in adult rats by the noncompetitive NMDA receptor antagonists, memantine and neramexane. Pharmacol Biochem Behav. 2006;85:298–306. doi: 10.1016/j.pbb.2006.08.011. [DOI] [PubMed] [Google Scholar]