

Abstract
BACKGROUND AND PURPOSE
Amyloid-β (Aβ) aggregation into synaptotoxic, prefibrillar oligomers is a major pathogenic event underlying the neuropathology of Alzheimer's disease (AD). The pharmacological and neuroprotective properties of a novel Aβ aggregation inhibitor, SEN1269, were investigated on aggregation and cell viability and in test systems relevant to synaptic function and memory, using both synthetic Aβ1-42 and cell-derived Aβ oligomers.
EXPERIMENTAL APPROACH
Surface plasmon resonance studies measured binding of SEN1269 to Aβ1–42. Thioflavin-T fluorescence and MTT assays were used to measure its ability to block Aβ1–42–induced aggregation and reduction in cell viability. In vitro and in vivo long-term potentiation (LTP) experiments measured the effect of SEN1269 on deficits induced by synthetic Aβ1–42 and cell-derived Aβ oligomers. Following i.c.v. administration of the latter, a complex (alternating-lever cyclic ratio) schedule of operant responding measured effects on memory in freely moving rats.
KEY RESULTS
SEN1269 demonstrated direct binding to monomeric Aβ1–42, produced a concentration-related blockade of Aβ1–42 aggregation and protected neuronal cell lines exposed to Aβ1–42. In vitro, SEN1269 alleviated deficits in hippocampal LTP induced by Aβ1–42 and cell-derived Aβ oligomers. In vivo, SEN1269 reduced the deficits in LTP and memory induced by i.c.v. administration of cell-derived Aβ oligomers.
CONCLUSIONS AND IMPLICATIONS
SEN1269 protected cells exposed to Aβ1–42, displayed central activity with respect to reducing Aβ-induced neurotoxicity and was neuroprotective in electrophysiological and behavioural models of memory relevant to Aβ-induced neurodegeneration. It represents a promising lead for designing inhibitors of Aβ-mediated synaptic toxicity as potential neuroprotective agents for treating AD.
Keywords: Alzheimer's disease, aggregated Aβ, Aβ oligomers, long-term potentiation, memory, pharmacotherapy, synaptic toxicity
Introduction
Progressive accumulation of amyloid-β (Aβ) assemblies is considered fundamental to the development of neurodegenerative pathology and inflammation that contribute to Alzheimer's disease (AD) (Tanzi et al., 2004). The key pathogenic event in the onset of AD appears to be the formation of extracellular soluble Aβ peptide into synaptotoxic, prefibrillar oligomers (Verdile et al., 2004; Walsh and Selkoe, 2004a,b), and accumulating evidence supports a cogent argument that compounds preventing the generation of toxic Aβ assemblies could provide successful new treatments for AD (Estrada and Soto, 2007). Several studies indicate that AD severity correlates closely with accumulation of soluble oligomers of Aβ (Lue et al., 1999; McLean et al., 1999; Wang et al., 1999). Consequently, pharmacotherapies that block initial Aβ assembly prior to the stage of oligomerization (Selkoe and Schenk, 2003; Walsh and Selkoe, 2004b), so as to prevent oligomerization and allow natural neurophysiological clearance mechanisms to effect the removal of Aβ (Golde, 2006; Walsh and Selkoe, 2007) and preserve normal synaptic function, might provide an effective treatment strategy.
AD is considered a product of initial synaptic failure that precedes neuronal degeneration, and Aβ-induced dysfunction of synaptic plasticity appears to contribute to early memory loss (Haass and Selkoe, 2007; Shankar et al., 2008). Long-term potentiation (LTP), a well-established model of synaptic plasticity related to memory, involves a sustained increase in excitatory synaptic transmission; and inhibition of LTP by Aβ may mimic an early manifestation of the memory loss seen in AD (Ondrejcak et al., 2010). Cultured cells that express human Aβ precursor protein (APP) generate and secrete soluble oligomers of human Aβ, which contain the Aβ1–40 and Aβ1–42 species (Podlisny et al., 1995), and LTP induction in the hippocampus in vivo is inhibited by their conditioned medium (7PA2 cell conditioned medium, 7PA2 CM) that contains naturally secreted human Aβ oligomers (Walsh et al., 2002; Townsend et al., 2006a). In normal adult rats and mice, synaptotoxic Aβ oligomers have been found to potently inhibit hippocampal LTP in vitro (Walsh et al., 2000) and in vivo (Walsh et al., 2002). Support for the role of low-n Aβ oligomers as a key toxic species has come from Cleary et al. (2005), who demonstrated that defined molecular species of the Aβ protein cause acute cognitive deficits in the rat. Specifically, Cleary et al. and others have shown that soluble oligomeric forms of Aβ are both necessary and sufficient to disrupt learned behaviour (Cleary et al., 2005; Townsend et al., 2006b; Poling et al., 2008).
Preventing Aβ aggregation is a therapeutically attractive neuroprotective approach to preserve synaptic function because this process is believed to be an exclusively pathological event, and drugs targeting this mechanism are likely to have a better safety profile than other pharmacotherapeutic approaches (Amijee and Scopes, 2009). Small molecule starting points for inhibitors of Aβ aggregation are scarce, and the vast majority of compounds claimed as inhibitors of Aβ aggregation are unsuitable in terms of their biological profile (Rishton, 2008). RS-0406 (Figure 1) is a non-peptide small molecule that has been shown to be an effective inhibitor of Aβ toxicity and is also capable of inhibiting Aβ1–42 aggregation (Nakagami et al., 2002). Our group has used the RS-0406 structure to engineer modifications that improve potency, specificity and bioavailability to provide inhibitors of Aβ toxicity as improved pharmacological tools to better understand the neuroprotection afforded by such compounds. The novel 5-aryloxypyrimidine, SEN1269 (Figure 1), shows greater cell penetration and an improved in vitro ADME profile (Scopes et al., 2008). The greater cell penetration may additionally allow SEN1269 to act on intracellular Aβ oligomer-producing pathways (LaFerla et al., 2007). Here, we demonstrate for the first time that a pharmacologically active, non-peptide, small molecule was protective against Aβ-induced cell death, synaptotoxicity in vitro and in vivo, and, consequently, prevented the deficit in cognition induced by 7PA2 CM in a model of memory in freely moving intact animals.
Figure 1.
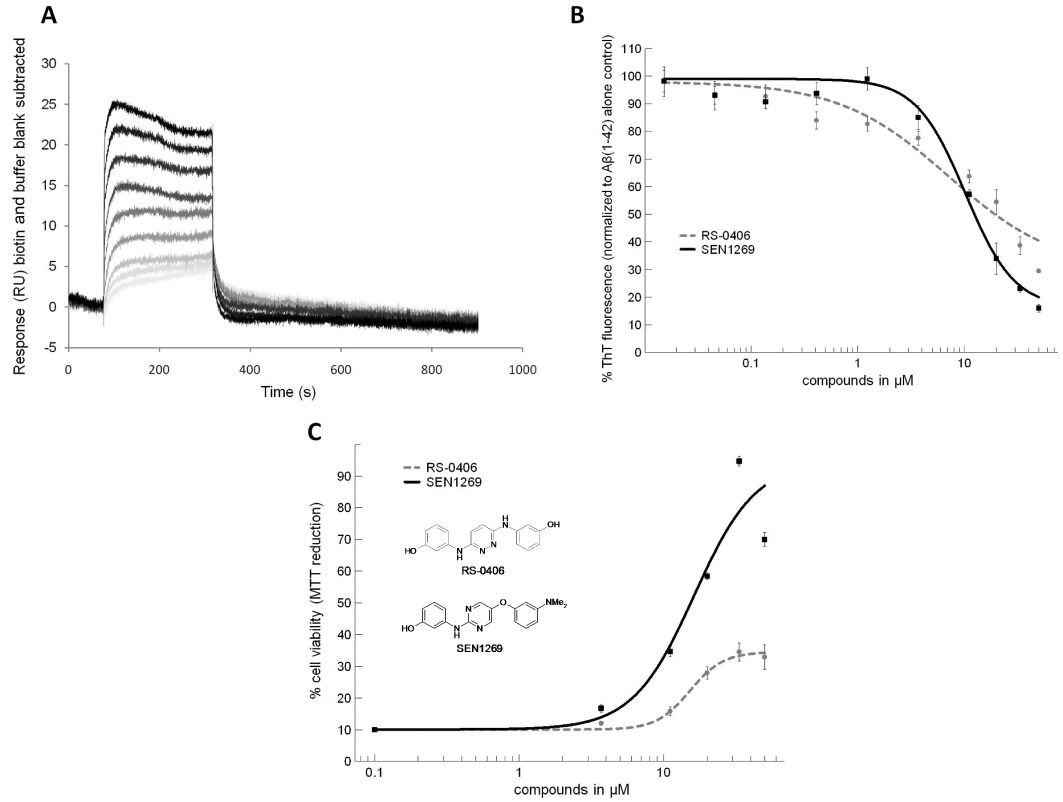
SEN1269 inhibits Aβ aggregation, protects cell viability against Aβ1–42-induced insults and binds to Aβ1–42. (A) Surface plasmon resonance response curves elicited by binding of SEN1269 (0.5, 1, 3, 5, 10, 15, 20 and 30 µM) to monomeric Aβ1–42. (B) Thioflavin-T fluorescence concentration–response curves of compounds incubated with 10 µM Aβ1–42 for 24 h. (C) Concentration–response curves of compounds incubated with 10 µM Aβ1–42 for 24 h on SHSY5Y cells using MTT colorimetric assay (cell viability level for 10 µM Aβ1–42 was 10%).
Methods
Aβ1–42 binding, aggregation and cell viability assays
Surface plasmon resonance was used to measure binding of SEN1269 to Aβ1–42 (see Item S1). Biotinylated Aβ1–42 was prepared in 50% dimethyl sulfoxide (DMSO) and pulse injected into a streptavidin (SA) chip surface until the immobilization level was established (Biocore T-100 biosensor); 1 mM biotin was injected into a second flow cell to allow saturation of the SA chip as control. Binding of SEN1269 in the flow phase onto immobilized monomeric Aβ1–42 was established, and KD data relative to SEN1269 according to a 1:1 model were determined. A thioflavin-T assay (LeVine, 1999) was used to quantify Aβ fibrillogenesis and to measure SEN1269 inhibition of Aβ1–42 aggregation (see Item S1). Aβ1–42 HCl salt was dissolved in hexafluoroisopropanol, with brief sonification and vortexing. This solution was freeze-dried then dissolved in DMSO. The ability of SEN1269 (or RS-0406 for comparison) to inhibit aggregation induced by 10 µM Aβ1–42 was assessed by incubation with Aβ1–42 at 37°C for 24 h in PBS. At 24 h, a 50 µL aliquot was dispensed into a black 96-well plate, and 50 µL thioflavin T (40 µM in 50 mM glycine buffer) was added to each well. The plate was shaken, and fluorescence was recorded using excitation and emission filters of 440 ± 15 and 485 ± 10 nm, respectively. An MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium) assay (Shearman et al., 1995) was used to determine the effect of SEN1269 on the deficit in SH-SY5Y cell viability caused by Aβ1–42 (see Item S1). Cell wells were prepared with 3 µL of SEN1269 (or 3 µL RS-0406 for comparison) in DMSO (mixed with 294 µL Opti-MEM), then 3 µL of Aβ1–42 (2 mM) was added and mixed thoroughly. Fifty microlitres of this solution was aspirated and dispensed into wells containing 50 µL of media opti-MEM + SH-SY5Y cells (Aβ1–42 final concentration 10 µM), and the wells were then incubated for 24 h. Fifteen microlitres of MTT was added to the cell wells, which were then incubated in 5% CO2 at 37° for 4 h. One hundred microlitres of stop/solubilization solution was added, and the wells were placed in a humidified box at room temperature overnight before reading.
In vitro LTP
Extracellular field excitatory post-synaptic potential (fEPSP) recordings were made from 400 µm thick transverse hippocampal slices prepared from male Sprague–Dawley rats humanely killed by cervical dislocation. After a minimum of 1 h recovery period in aCSF at room temperature, slices were transferred to an interface chamber, warmed to 30 ± 1°C and perfused with aCSF. Schaffer collaterals were stimulated (7–28 V, 0.1 ms pulse width, 0.05 Hz) every 20 s with a concentric bipolar electrode, and fEPSPs were recorded from the stratum radiatum of the CA1 region (Axoclamp 2A amplifier and software; pClamp 8.2, Axon, MDS Analytical Technologies, Sunnyvale, CA, USA) using a glass microelectrode. Stimulation intensity was set to evoke fEPSPs of approximately 50% of the maximum amplitude. A minimum 10 min stable baseline period was recorded before the administration of test substances in aCSF (30 min application period), 10 min before three periods of high frequency stimulation (HFS; 1 s of 100 Hz) at 10 min intervals. Aβ1–42 experimental manipulations (see Item S1) consisted of exposure of the slices to aggregated 1 µM Aβ1–42[33 µM in Tris–HCl + 20 µL DMSO (stock solution) agitated at 37°C for 24 h using an orbital shaker], or 20 µL of 5 mM SEN1269 added to the Aβ1–42 stock solution and agitated. The control consisted of 20 µL DMSO in 1 mL Tris–HCl agitated. These agitated solutions were then diluted to 33 mL in aCSF immediately before they were applied to the hippocampal slices. 7PA2 CM experimental manipulations (see Item S1) consisted of slice exposure to 7PA2 CM or CHO CM (control) with DMSO; or 60 µM SEN1269 added to 7PA2 CM or CHO CM, equilibrated for 1 h at room temperature and diluted to 20 mL with aCSF prior to slice application (giving a final SEN1269 concentration of 3 µM). The fEPSPs were recorded for 80 min after the final HFS stimulation, and the final 10 min of recording (30 sweeps) were selected and averaged for group comparisons using the unpaired t-test. 7PA2 CM contains a total human Aβ concentration of 3223 ± 1217 pg·mL−1, which is similar to that found in human CSF (Walsh et al., 2002), and the oligomeric Aβ species comprises approximately 10–20% of the total immunoprecipitate of 7PA2 CM (Podlisny et al., 1995). Using elisa, it was found that the concentration of total Aβ in the 7PA2 CM used in the LTP and behavioural studies was in the range of 2–5 nM.
In vivo LTP
Animals were kept on a 12 h light/dark cycle with food and water available ad libitum. Extracellular fEPSPs were recorded from the hippocampi of 36 anaesthetized (urethane i.p. 1 mL 100 g−1 assessed by cardiovascular responses to paw-pinch and the stability of measured cardiovascular variables with supplements i.v. of 0.2 mL 100 g−1, 12% solution) adult male Sprague–Dawley rats. Core body temperature was maintained at 37°C using a homeothermic blanket, and polyethylene catheters were inserted into the right femoral artery and vein for monitoring arterial blood pressure and for anaesthetic drug administration, respectively. The head was mounted in a stereotaxic frame before lowering a concentric bipolar stimulating electrode and carbon fibre recording electrode vertically into the CA1 area of the hippocampus. Stimulating electrode coordinates (Paxinos and Watson, 1998): bregma −3.5 mm, lateral 2.6 mm and 2.0–2.5 mm below the pial surface, recording electrode coordinates; bregma −4.0 mm, lateral 3.0 mm and 2.0–2.5 mm below the pial surface. Subsequently, a preloaded 30 gauge stainless steel i.c.v. cannula was lowered into the lateral ventricle: bregma +0.5 mm, lateral 1.5 mm and 3.6 mm below the pial surface with a 15–17° rostro-caudal angle. Electrical stimulation (0.1 ms pulse width, 10–100 V, 0.14 Hz) was used to identify and optimise fEPSPs, and an input/output curve was created to determine maximal fEPSP amplitude and the voltage required to generate fEPSPs of 30–40% of maximum amplitude. Stimulation parameters were maintained at 0.03 Hz, and after a stable fEPSP had been recorded for 8 min, 7PA2 or CHO CM with/without SEN1269 (5 µL of 10 or 100 µM SEN1269 added to 495 µL 7PA2/CHO CM, making final concentrations of 100 nM or 1 µM SEN1269) was administered i.c.v., followed by further baseline recording (28 min) before HFS of the Schaffer–collateral pathway. This comprised of three periods (inter-period interval 30 s) of HFS (10 trains of 10 stimuli; 200 Hz; inter-train interval 50 ms). The fEPSPs were recorded for 90 min after HFS and the final 10 min of recording (20 sweeps) were selected and averaged for group comparisons using the unpaired t-test.
Behavioural test
Behavioural data were collected using an alternating-lever cyclic ratio (ALCR) schedule of food reinforced operant behaviour, which is recognized as an appropriate and sensitive experimental analysis of behaviour technique (Cleary et al., 2005). Eighty-four male Sprague–Dawley rats (Harlan, Oxford, UK), weighing 220–250 g at the beginning of the experiment, were maintained at 90% of their free-feeding body weights and housed individually with water available at libitum in the home cage. The temperature in the vivarium was maintained at 23°C under a 12 h light/12 h dark cycle (lights on 0800 h). The rats were trained and tested in two-lever rat test chambers (Med Associates Inc., St. Albans, NJ, USA) enclosed in sound-proof compartments. Reinforcers were 45 mg sucrose pellets (BioServ, Frenchtown, NJ, USA), which were delivered into a tray situated midway between the two operant levers, and ALCR training was according to the procedure previously reported (Cleary et al., 2005). Briefly, rats were initially trained to press both levers, in a two-lever operant chamber, according to an alternating-lever fixed ratio 1 (FR-1) schedule for reinforcement. During the training, the active lever was extended into the chamber and illuminated by a light, while the inactive lever was retracted (forced choice). When the rats were able to obtain 100 reinforcers in 30 min, they were reinforced according to a hierarchical series of alternating-lever FR schedules (FR-2, FR-4, FR-6 and FR-8). They were then trained under a forced choice arithmetic ALCR schedule, comprising of an ascending followed by a descending sequence of the ratio values 3, 6, 9, 12, 15 and 18, over six cycles, until performance showed no change in trends. They were then trained under a forced choice ALCR schedule comprising of an ascending followed by a descending sequence of the ratio values 2, 6, 12, 20, 30, 42 and 56, presented over six cycles. When the rats were capable of completing this forced choice ALCR schedule in 50 min, the forced choice conditions were removed, both levers were extended into the chamber and the light cues were extinguished. When the rats were capable of completing the terminal ALCR schedule in 50 min, they were anaesthetized with fentanyl citrate (0.4 mL·kg−1), placed in a sterotaxic frame and fitted with a permanent indwelling cannula (23 gauge) aimed at the lateral ventricle of the brain. Half of the rats received left lateral ventricle cannula implants, and half received right cannula implants. With the incisor bar set 3.0 mm below the interaural line, stereotaxic coordinates (Paxinos and Watson, 1998) were −1.5 mm lateral, 1.0 mm posterior to bregma and 3.0 mm below the pial surface. The animals were allowed 10 days for recovery before experimental testing, and cannula placement was verified by the observation of vigorous drinking (>5.0 mL in 20 min) following i.c.v. injection of 5.0 µL (0.5 µg·mL−1) angiotensin II (Johnson and Epstein, 1974). Following recovery from surgery, the animals were randomly assigned to one of six groups. Group 1 received an i.c.v. injection of 10 µL 7PA2 CM, followed 2 min later by i.c.v. injection of 10 µL DMSO (the vehicle SEN1269 was dissolved in), to establish the untreated Aβ oligomer behavioural deficit. Group 2 received i.c.v. injection of 10 µL CHO CM, followed 2 min later by i.c.v. injection of 10 µL DMSO, to establish response parameters in the absence induction of a behavioural deficit or treatment. Dose-response parameters were then established in groups 3, 4 and 5, where i.c.v. injection of 10 µL 7PA2 CM was followed 2 min later by i.c.v. injection of (group 3) 10 µL of 100 nM SEN1269, (group 4) 1.0 µM SEN1269 or (group 5) 10 µM SEN1269. In order to determine any behavioural effects specific to SEN1269, group 6 was injected i.c.v. with 10 µL CHO CM, followed 2 min later by 10 µL of 10 µM SEN1269. ALCR data were collected 2 h after i.c.v. injections (Cleary et al., 2005), mean baseline error rates (lever switching errors and incorrect lever perseverations) were established and data were analysed as % of overall baseline performance. To test for motivational or peripheral motor effects induced by the experimental manipulations, the slopes of functions were obtained by plotting response rates against reinforcement rates (motivation) and running response rates against ratio values (motor retardation) were compared. Data were analysed by one-way anova followed by Fisher's post hoc least significant difference tests.
All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). All studies using animals were conducted under Home Office License (UK), the UK Animals (Scientific Procedures) Act, 1986 and with Ethics Committee approval from the contributing institutions.
Results
Aβ1–42 binding, aggregation and cell viability assays
Using surface plasmon resonance (see Item S1), immobilized Aβ1–42 was characterized using A11, OC, 6E10 and 4G8 antibodies: A11 and OC (Aβ oligomer and Aβ fibril specific, respectively) showed no binding; whereas 6E10 and 4G8 showed strong binding responses. SEN1269 bound to monomeric Aβ1–42 in a concentration-related manner (Figure 1A) with a KD of 4.4 µM, which suggests that its mode of action involves direct interaction with Aβ1–42. SEN1269 also bound to Aβ1–42 oligomers (data not shown). Incubation of SEN1269 with Aβ1–42 reduced the fluorescence exhibited by thioflavin-T (LeVine, 1999) (see Item S1) in a concentration-related manner (IC50= 11 µM), indicating blockade of the Aβ aggregation process, and SEN1269 was more effective in this regard than the parent compound RS-0406 (Figure 1B). In the MTT assay (Shearman et al., 1995) (see Item S1), SEN1269 was effective in protecting neuronal cell lines against an Aβ1–42 insult (IC50= 15 µM) and was considerably more effective than the parent compound (Figure 1C). As a positive comparator in the thioflavin-T and MTT assays, the Praecis pentapeptide aggregation inhibitor PPI-1019 (Findeis, 2002) displayed IC50 values of 1.5 and 7 µM, respectively. The cell permeability of SEN1269 was 20.7 and 17.8 × 10−6 cm·s−1 for apical-basolateral and basolateral-apical permeability, respectively. RS-0406 showed corresponding values of 0.9 and 1.8 × 10−6 cm·s−1, indicating that biological membranes are highly permeable to SEN1269.
In vitro LTP
The LTP of fEPSPs in the CA1 region of hippocampal slices was reduced following application of Aβ1–42, from a control increase of 66.8 ± 10.9% to an increase of 17.5 ± 8.9% after Aβ1–42 application (P < 0.05 vs. control). This effect was completely reversed by co-incubation of Aβ1–42 with SEN1269, with LTP under these conditions amounting to 77.2 ± 9.6% (P < 0.05 vs. Aβ1–42 alone; Figure 2A–C). SEN1269 (3 µM) was also effective at reducing the deficit in LTP caused by application of cell-derived Aβ oligomers (7PA2 CM) to hippocampal slices (Figure 2D–F). Following HFS of the Schaffer collaterals, the LTP of fEPSPs in control amounted to an increase of 66.3 ± 6.3% of baseline fEPSP amplitude. In the presence of 7PA2 CM, the amplitude of fEPSPs following HFS was lower than that observed prior to HFS, being reduced to 93.7 ± 9.7% (P < 0.05 vs. control) of baseline fEPSPs amplitude (100%). This inhibition of LTP was almost completely reversed following co-incubation of 7PA2 CM with SEN1269, the magnitude of LTP being restored to 56.9% ± 11.9% (P < 0.05 vs. 7PA2 CM alone).
Figure 2.
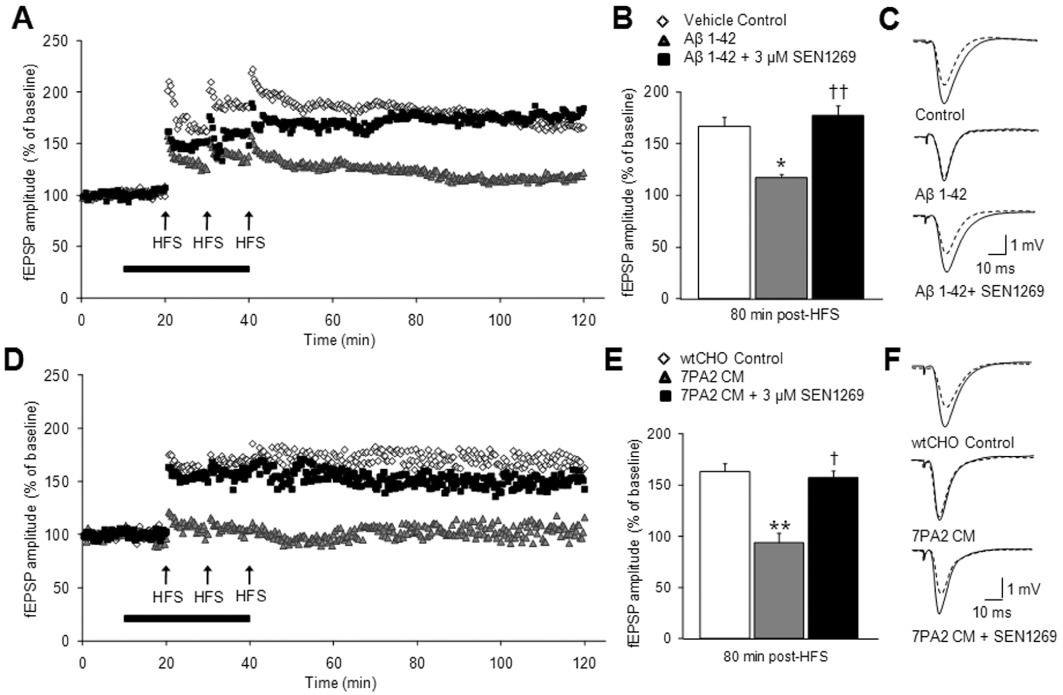
Inhibition of Aβ aggregation and fibrillogenesis by SEN1269 protects against synthetic Aβ1–42 and 7PA2 CM-induced deficits in LTP in vitro. (A) Scatter plot showing the effect of co-administration of SEN1269 (3 µM) + synthetic Aβ1–42 (n= 4), Aβ1–42 alone (n= 7), vehicle control (n= 4) on the enhancement of fEPSP amplitude evoked by HFS on hippocampal slices. (B) Histograms showing the level of LTP recorded 80–90 min post-HFS. **P < 0.01 compared to control, ††P < 0.01 compared to Aβ1–42, †P < 0.05 compared to Aβ1–42. (C) Typical fEPSPs before and 80 min after HFS for the three treatment groups. Traces shown are the average of 10 sweeps. (D) Scatter plot showing the effect of co-administration of SEN1269 (3 µM) + 7PA2 CM (n= 4), 7PA2 CM alone (n= 4) CHO CM alone (n= 4) on the enhancement of fEPSP amplitude evoked by HFS on hippocampal slices. (E) Histograms showing the level of LTP recorded 80 min post HFS. **P < 0.01 compared to CHO CM, ††P < 0.01 compared to 7PA2 CM, †P < 0.05 compared to 7PA2 CM. (F) Typical fEPSPs before and 80 min after HFS for the three treatment groups. Traces shown are the average of 10 sweeps.
In vivo LTP
In anaesthetized rats, SEN1269 was also effective at preventing deficits in LTP induced by 7PA2 CM. After i.c.v. administration of 7PA2 CM, 30 min before HFS, only a modest LTP of 19.9 ± 8.9% was observed, significantly less than that produced after administration of control CHO CM; 97.2 ± 12.3% (P < 0.05 vs. control; Figure 3A–C). This depression of LTP induced by 7PA2 CM was partly prevented when it was co-administered with 100 nM SEN1269; the magnitude of LTP recorded under these conditions amounting to 75.5 ± 13.3% of baseline fEPSPs (P < 0.05 vs. 7PA2 CM alone; Figure 3A–C). In a separate set of experiments, i.c.v. co-administration of 1 µM SEN1269 with 7PA2 CM 30 min before HFS also significantly prevented the depression of LTP induced by administration of 7PA2 CM alone. LTP of fEPSPs observed after 7PA2 CM administration was depressed, amounting to an increase in fEPSP amplitude of 21.3 ± 7.7% prevented by co-administration with 1 µM SEN1269, where LTP amounted to an increase of 98.6 ± 16.0% (P < 0.05 vs. 7PA2 CM alone; Figure 3D–F). In vehicle experiments, after i.c.v. administration of CHO CM, HFS evoked LTP of 111.2 ± 14.2%. These data show that SEN1269 protects against the synaptotoxocity induced by 7PA2 CM.
Figure 3.
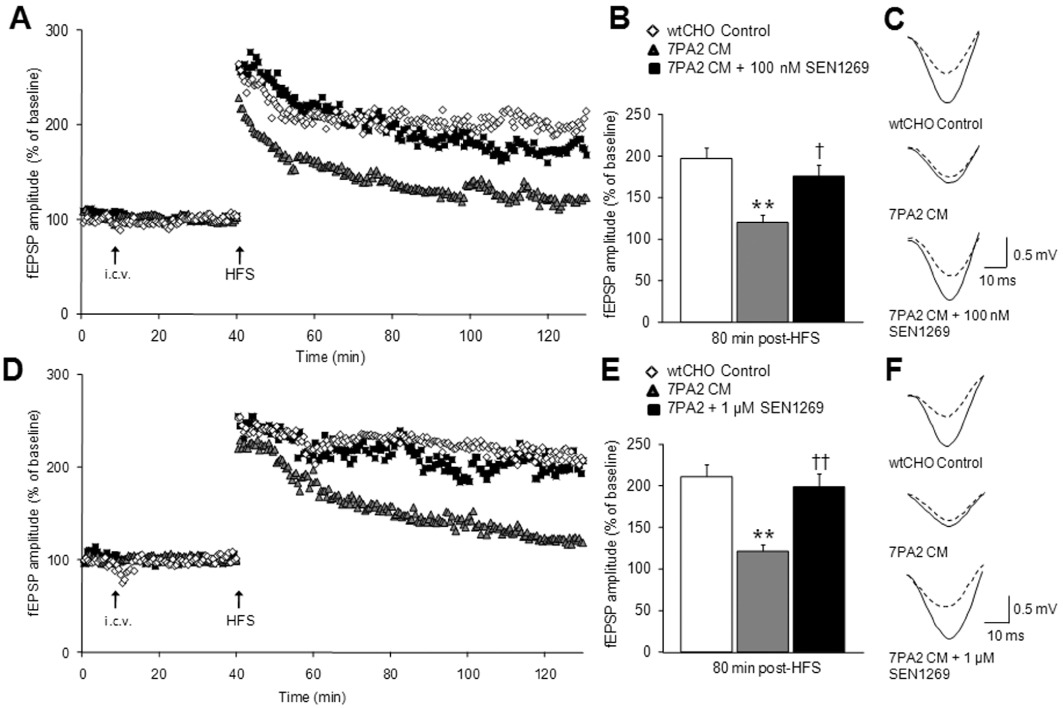
Inhibition of Aβ aggregation and fibrillogenesis by SEN1269 protects against 7PA2 CM-induced deficits in LTP in vivo. (A) Scatter plot showing the effect of co-administration of SEN1269 (100 nM) + 7PA2 CM (n= 6), 7PA2 CM alone (n= 4) and CHO CM (n= 4) on the enhancement of fEPSP amplitude evoked by HFS in the CA1 region of the hippocampus in an anaesthetized rat. (B) Histograms showing the level of LTP recorded 90 min post-HFS. **P < 0.01 compared to CHO CM, †P < 0.05 compared to 7PA2 CM. (C) Typical fEPSPs before and 80 min after HFS for the three treatment groups. Traces shown are the average of 10 sweeps. (D) Scatter plot showing the effect of co-administration of SEN1269 (1 µM) + 7PA2 CM (n= 6), 7PA2 CM alone (n= 10) and CHO CM (n= 5) on the enhancement of fEPSP amplitude evoked by HFS in the CA1 region of the hippocampus in an anaesthetized rat. (E) Histograms showing the level of LTP recorded 90 min post-HFS. **P < 0.01 compared to CHO CM, ††P < 0.01 compared to 7PA2 CM. (F) Typical fEPSPs before and 80 min after HFS for the three treatment groups. Traces shown are the average of 10 sweeps.
Behaviour
Following i.c.v. injection of 7PA2 CM in rats trained under the ALCR schedule, there was a significant overall treatment effect on lever switching errors 120 min post injection (F5,78= 3.460, P= 0.0071). The 7PA2 CM injected rats exhibited significantly more lever switching errors as compared with the vehicle-injected group (P < 0.05). The 7PA2 CM-injected groups treated with SEN1269 (100 nM, P= 0.532; 1 µM, P= 0.046; and 10 µM, P < 0.029) demonstrated significantly fewer lever switching errors than the 7PA2 CM-injected subjects that received vehicle alone (Figure 4A). With respect to incorrect lever perseverations, there was a significant overall treatment effect on incorrect lever perseverations 120 min following 7PA2 CM injections (F5,78= 8.213, P < 0.0001). The 7PA2 CM-injected rats exhibited significantly more incorrect lever perseverations as compared with the vehicle-injected group (P < 0.0001). The 7PA2 CM-injected groups treated with SEN1269 (100 nM, P= 0.019; 1 µM, P < 0.009; and 10 µM, P < 0.0001) demonstrated significantly fewer incorrect lever perseverations than the 7PA2 CM-injected subjects that received vehicle alone, and this effect was dose-dependent (Figure 4B). These findings indicate that SEN1269, which inhibited the toxic effects of 7PA2 CM on LTP in vitro and in vivo, was also protective against 7PA2 CM-induced cognitive deficits in vivo. The ALCR schedule also provides data enabling evaluation of the effects of experimental manipulations on the ability to track the changing parameters of the operant schedule, changes in the motivational state of the animal and peripheral effects on motor ability. Analysis of the data indicated no significant effect of 7PA2 CM or SEN1269 (P-values < 0.05); consequently, the data reported are strongly indicative of authentic central effects of the experimental manipulations. These data show that SEN1269 also pharmacologically protects against synaptotoxocity induced by 7PA2 CM to alleviate Aβ oligomer-induced memory deficits in vivo.
Figure 4.
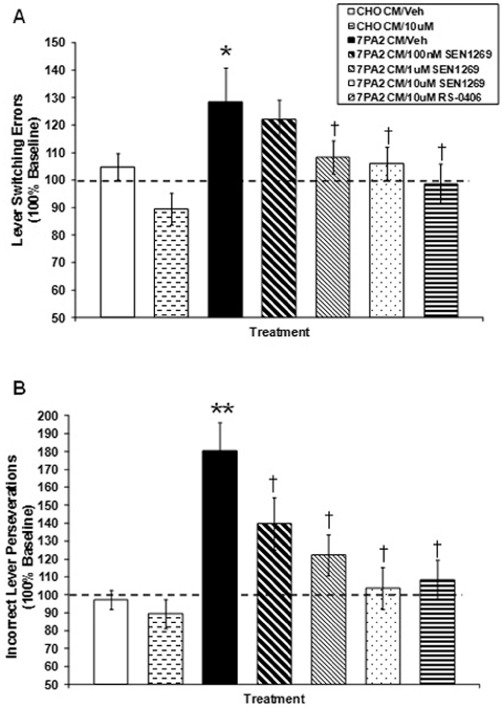
Effect of SEN1269 on 7PA2 CM-induced memory deficits (n= 14 for each study arm). (A) Effect of SEN1269 treatment on 7PA2 CM-induced lever switching errors. *P < 0.05 compared to CHO-CM, †P < 0.05 compared to 7PA2 CM. (B) Effect of SEN1269 treatment on 7PA2 CM-induced incorrect lever perseverations. **P < 0.0001 compared to CHO-CM, †P < 0.05 compared to 7PA2 CM. RS-0406 data obtained using the identical behavioural protocol included for comparison (from O'Hare et al., 2010).
Discussion and conclusions
Available therapies only treat the symptoms of AD (Melnikova, 2007; Shah et al., 2008; Wilcock, 2008); however, disease-modifying pharmacological interventions preventing the generation of toxic Aβ assemblies may have the potential to slow, or even halt AD progression. RS-0406, a non-peptide small molecule, has been shown to be an inhibitor of Aβ (Nakagami et al., 2002), to protect primary hippocampal neurons against Aβ1–42-induced cytotoxicity, to rescue synthetic Aβ1–42-induced impairment of LTP in hippocampal slices, to prevent Aβ-mediated block of LTP by specifically inhibiting oligomer formation (Walsh et al., 2005) and to arrest Aβ oligomer-induced behavioural deterioration in the rat (O'Hare et al., 2010). The current study demonstrates that a modified version of RS-0406, SEN1269, is pharmacologically active and is an improved research tool for in vitro and in vivo use to better understand the relative acute synaptotoxicity caused by Aβ. SEN1269 is a novel, non-peptide small molecular weight compound that binds to Aβ and offers significant neuroprotection biochemically and in electrophysiological and behavioural models of memory relevant to synaptic dysfunction caused by Aβ. Thus, surface plasmon resonance studies illustrated that SEN1269 binds directly to Aβ1–42. The thioflavin-T assay indicated that SEN1269 produced a concentration-related blockade of Aβ1–42 aggregation, and the MTT assay demonstrated that SEN1269 protected neuronal cell lines exposed to Aβ1–42. These results suggest that mechanistically SEN1269 acts by binding to Aβ to prevent its assembly and fibrillogenesis, properties of considerable interest given that the key pathogenic event in AD is believed to be aggregation of the Aβ peptide into synaptotoxic, prefibrillar oligomers (Verdile et al., 2004; Walsh and Selkoe, 2004a,b).
Electrophysiological studies confirmed that in vitro and in vivo LTP of synaptic transmission, considered to underlie synaptic plasticity associated with learning and memory, was compromised by synthetic Aβ1–42 or cell-derived Aβ oligomers (7PA2 CM); and that these effects were reversed by incubation of SEN1269 with Aβ1–42 or 7PA2 CM. Thus, SEN1269 protects against Aβ-induced synaptotoxicity. Low-n oligomers are present in 7PA2 CM at low- to sub-nanomolar concentrations, similar to those of Aβ in human CSF (Walsh et al., 2002), and it has been established that these cell-derived Aβ oligomers are composed of N- and C-terminally heterogeneous human Aβ peptides, including the Aβ1–40 and Aβ1–42 species that occur in the human brain and extracellular fluids (Podlisny et al., 1995; 1998; Walsh et al., 2002; 2005). Furthermore, in freely moving intact animals trained to respond under a complex schedule of food reinforced behaviour, SEN1269 significantly reduced numbers of lever switching errors and incorrect lever perseverations induced by i.c.v. 7PA2 CM injections. There were no significant differences in reinforcement rates or running response rates between the 7PA2 CM-injected groups treated with SEN1269 and the 7PA2 CM-injected group that received vehicle, demonstrating that SEN1269 neither increased nor decreased the motivational state or peripheral motor output of the animals, and indicating that the behavioural effect of SEN1269 was central and rescued cognitive processes.
A number of studies have established that Aβ1–42 is the more neurotoxic form of Aβ (McGowan et al., 2005), Aβ1–40 and Aβ1–42 possess different biochemical properties, and Aβ1–42 is considered to be the major aetiological agent in the pathogenesis of AD due to its more aggressive aggregation or oligomerization properties (Burdick et al., 1992; Jarrett et al., 1993). Compelling evidence indicates that the toxic amyloid species underlying the cognitive deficit in AD are soluble low-n oligomers of the Aβ peptide, which subsequently form insoluble amyloid fibrils via association of the peptide β-strands (Walsh and Selkoe, 2007; Klyubin et al., 2008), and Aβ dimers have been identified as the smallest synaptotoxic species in Alzheimer's brains (Shankar et al., 2008). Oligomeric Aβ is able to permeate biological membranes (Glabe, 2006) and the current study used conditioned medium from 7PA2 cells, which contains low-n Aβ oligomers, to induce electrophysiological and behavioural deficits. Our pharmacological observations lend support to the biological hypothesis that inhibiting the process of Aβ peptide aggregation centrally may be a viable neuroprotective strategy to potentially treat AD. Moreover, SEN1269 is the first non-peptide small molecule (molecular weight 321) that binds to Aβ monomer and oligomers, inhibits Aβ fibrillogenesis, maintains cell viability against a toxic insult of Aβ1–42, prevents the deficit in LTP caused by synthetic Aβ1–42 or cell-derived oligomers of Aβ and is effective in protecting against the deficit in cognition induced by cell-derived oligomers of Aβ in a behavioural model of memory relevant to synaptic dysfunction caused by Aβ. As synaptic loss underlies the neuropathology of AD, these new acute data on SEN1269 provide support for the design of further molecules in this structural class, which are orally bioavailable, CNS penetrating and that have therapeutically useful half-lives suitable for testing in chronic animal models that are thought to be relevant to the neuropathology of AD. In the future, development of an orally bioavailable analogue of SEN1269 would be of interest as a potential disease-modifying treatment of AD.
Acknowledgments
We thank DJ Selkoe (Brigham and Women's Hospital and Harvard Medical School) for 7PA2 cells, DM Walsh (Brigham and Women's Hospital and Harvard Medical School) for helpful discussions and E Nerou (Senexis Limited) for assistance with cell culture experiments. We are grateful to BTG plc and The Wellcome Trust for funding.
Glossary
- ALCR
alternating-lever cyclic-ratio
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- Aβ
amyloid-β
- CM
conditioned medium
- DMSO
dimethyl sulfoxide
- fEPSP
extracellular field excitatory post-synaptic potential
- HFS
high-frequency stimulation
- LTP
long-term potentiation
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium
- SA
streptavidin
Conflicts of interest
D S, H A and M T are employees of Senexis Limited and hold share options in the Company. E O'H holds share options in Senexis Limited.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Item S1 Preparation of stock solutions for biological assays. Full procedures for thioflavin-T and cell viability assays and surface plasmon resonance studies. Compound sample preparation and storage for LTP experiments.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Amijee H, Scopes DIC. The quest for small molecules as amyloid inhibiting therapies for Alzheimer's disease. J Alzheimers Dis. 2009;17:33–47. doi: 10.3233/JAD-2009-1044. [DOI] [PubMed] [Google Scholar]
- Burdick D, Soreghan B, Kwon M, Kosmoski J, Knauer M, Henschen A, et al. Assembly and aggregation properties of synthetic Alzheimer's A4/beta amyloid peptide analogs. J Biol Chem. 1992;267:546–554. [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Estrada LD, Soto C. Disrupting β-amyloid aggregation for Alzheimer's disease treatment. Curr Top Med Chem. 2007;7:115–126. doi: 10.2174/156802607779318262. [DOI] [PubMed] [Google Scholar]
- Findeis MA. Peptide inhibitors of beta amyloid aggregation. Curr Top Med Chem. 2002;2:417–423. doi: 10.2174/1568026024607508. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Golde TE. Disease modifying therapy for AD. J Neurochem. 2006;99:689–707. doi: 10.1111/j.1471-4159.2006.04211.x. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- Johnson AK, Epstein AN. The cerebral ventricles as the avenue for the dipsogenic action of intracranial angiotensin. Brain Res. 1974;86:399–418. doi: 10.1016/0006-8993(75)90891-4. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, et al. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer's disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- LeVine H., III Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuler SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Melnikova I. Therapies for Alzheimer's disease. Nat Rev Drug Discov. 2007;6:341–342. doi: 10.1038/nrd2314. [DOI] [PubMed] [Google Scholar]
- Nakagami Y, Nishimura S, Murasugi T, Kaneko I, Meguro M, Marumoto S, et al. A novel β-sheet breaker, RS-0406, reverses amyloid β-induced cytotoxicity and impairment of long-term potentiation in vitro. Br J Pharmacol. 2002;137:676–682. doi: 10.1038/sj.bjp.0704911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hare E, Scopes DI, Treherne JM, Norwood K, Spanswick D, Kim EM. RS-0406 arrests amyloid-beta oligomer-induced behavioural deterioration in vivo. Behav Brain Res. 2010;210:32–37. doi: 10.1016/j.bbr.2010.01.044. [DOI] [PubMed] [Google Scholar]
- Ondrejcak T, Klyubin I, Hu NW, Barry AE, Cullen WK, Rowan MJ. Alzheimer's disease amyloid beta-protein and synaptic function. Neuromolecular Med. 2010;12:13–26. doi: 10.1007/s12017-009-8091-0. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. New York: Academic Press; 1998. [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, et al. Aggregation of secreted amyloid beta-protein into sodium dodecyl sulphate-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- Podlisny MB, Walsh DM, Amorante P, Ostaszewski BL, Stimson ER, Maggio JE, et al. Oligomerization of endogenous and synthetic amyloid beta-protein at naomolar levels in cell culture and stabilization of monomer by Congo red. Biochemistry. 1998;37:3602–3611. doi: 10.1021/bi972029u. [DOI] [PubMed] [Google Scholar]
- Poling A, Morgan-Paisley K, Panos JJ, Kim EM, O'Hare E, Cleary JP, et al. Oligomers of the amyloid-beta protein disrupt working memory: confirmation with two behavioural procedures. Behav Brain Res. 2008;193:230–234. doi: 10.1016/j.bbr.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishton GM. Aggregator compounds confound amyloid fibrillization assay. Nat Chem Biol. 2008;4:159–160. doi: 10.1038/nchembio0308-159. [DOI] [PubMed] [Google Scholar]
- Scopes DI, Amijee H, Nerou E, Treherne JM, O'Hare E, Jeggo R, et al. SEN1269: a novel inhibitor of amyloid beta toxicity. Alzheimers Demen. 2008;4:P2–358. [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–548. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Shah RS, Lee HG, Xiongwei Z, Perry G, Smith MA, Castellani RJ. Current approaches in the treatment of Alzheimer's disease. Biomed Pharmacother. 2008;62:199–207. doi: 10.1016/j.biopha.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearman MS, Hawtin SR, Tailor VJ. The intracellular component of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction is specifically inhibited by beta-amyloid peptides. J Neurochem. 1995;65:218–227. doi: 10.1046/j.1471-4159.1995.65010218.x. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006a;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Cleary JP, Mehta T, Hofmeister J, Lesné S, O'Hare E, et al. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann Neurol. 2006b;60:668–676. doi: 10.1002/ana.21051. [DOI] [PubMed] [Google Scholar]
- Verdile G, Fuller S, Atwood CS, Laws SM, Gandy SE, Martins RN. The role of beta amyloid in Alzheimer's disease: still a cause of everything or the only one who got caught? Pharmacol Res. 2004;50:397–409. doi: 10.1016/j.phrs.2003.12.028. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004a;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004b;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Aβ oligomers – a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydell RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, et al. Certain inhibitors of synthetic amyloid β-peptide (Aβ) fibrillogenesis block oligomerisation of natural Aβ and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dickson DW, Trojanowski JQ, Lee VM. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer's disease from normal and pathologic aging. Exp Neurol. 1999;158:328–337. doi: 10.1006/exnr.1999.7085. [DOI] [PubMed] [Google Scholar]
- Wilcock GK. The pharmacological treatment of Alzheimer's disease with cholinesterase inhibitors and memantine. In: Cuello AC, editor. Pharmacological Mechanisms in Alzheimer's Therapeutics. New York: Springer; 2008. pp. 36–49. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.