

Abstract
Potassium 1-(alkoxy/acyloxy)alkyltrifluoroborates have been synthesized through a copper-catalyzed diboration of aldehydes and subsequent conversion of the resulting potassium 1-(hydroxy)alkyltrifluoroborates. The palladium-catalyzed Suzuki-Miyaura reaction employing the potassium 1-(benzyloxy)alkyltrifluoroborates with aryl and heteroaryl chlorides provides access to protected secondary alcohols in high yields. The β-hydride elimination pathway is avoided through use of the benzyl protecting group, which is proposed to stabilize the diorganopalladium intermediate by coordination of the arene to the metal center. This cross-coupling is stereospecific with complete retention of stereochemistry.
INTRODUCTION
Secondary benzylic alcohols represent an important class of organic compounds whose inherent reactivity makes them both important synthetic intermediates and components in biologically active molecules. The vast majority of routes to such systems rely upon conversion of the sp2 prochiral center of a carbonyl to the sp3 stereocenter containing the hydroxyl functional group, the main disconnections for which are summarized in Scheme 1. Thus, the addition of an aryl or alkyl organometallic reagent to the requisite aldehyde comprises one general protocol to access alcohols via carbon-carbon bond formation (route a).1 Another method for the generation of the product involves reduction of a ketone (route b),2 which can be carried out by a variety of methods3 (e.g., transition metal catalyzed hydrogenation,3a transfer hydrogenation,3b hydrosilylation, 3b catalytic hydroboration,3b reduction with metal hydrides or boranes,2 or enzymatic methods3c).
Scheme 1.

Disconnections to Access Secondary Alcohols.
The vast majority of these methods provide excellent product yields, and of course each of these protocols possesses individualized sets of advantages and limitations. Perhaps most importantly, however, these approaches can be limited in terms of their access to enantioenriched secondary alcohols with reliably high asymmetric induction (>97% ee) across a broad class of substructures. Thus, the levels of enantiomeric excess can be highly variable within a subset of molecules, as transformations relying on conversion of planar, prochiral systems to tetrahedral configurations are often highly substrate dependent and substrate variable, even within groups of reactants that are structurally quite similar.
In terms of C-C bond-forming routes to benzylic alcohols, the high nucleophilicity of Grignards and organolithiums prevents the preparation of highly functionalized reagents, and enanti-oselective additions to aldehyde substrates with consistently high ees are virtually unknown.4
The asymmetric addition of diorganozinc species to aldehydes, in contrast, is an efficient method to generate enantioenriched secondary alcohols, as several systems allow access to the desired products in high yield and with a range of ees up to 99%. However, there remains a challenge of obtaining high enantioselectivity in the addition of organozincs to alkyl aldehydes, which often requires the use of a Lewis acid in addition to the catalyst. The addition of external Lewis acids is also common when employing less reactive diorganozinc species, such as dimethylzinc. Additionally, these methods suffer from the need to generate the active organozinc species in situ, and excess organozinc reagents are often required. Furthermore, the synthesis of many organozinc species often requires the use of pyrophoric diethylzinc and/or trialkylboranes.4
Triarylboranes5 and triarylalanes6 have been also employed in enantioselective additions to both alkyl and aryl aldehydes with reasonable success. However, these reagents are not readily available, they deliver only a single aryl group, and they are pyrophoric, greatly diminishing their appeal. The more readily available and easily handled arylboronic acids have been employed in Ru(II)-catalyzed reactions with aryl aldehydes,7 but to the best of our knowledge addition to alkanals have not been examined.
The asymmetric reduction of prochiral ketones provides another approach to enantioenriched secondary alcohols in high yield and ee, but again this approach has its own set of limitations. For example, the use of excess reducing agent commonly employed provides a drawback for large-scale reactions in terms of separation and purification.3 Additionally, although aryl alkyl ketones are able to undergo addition with reasonable stereochemical induction, their diaryl ketone counterparts are more challenging. Thus, the reduction of diaryl ketones requires electronically differentiated aryl groups or ortho-substituted aryl rings,4 because the enantiomeric excess of the reaction is mainly determined by the difference in steric bulk of the two substituents.
One means to avoid these drawbacks would be to employ a stereospecific cross-coupling between an enantiopure, configurationally stable 1-(alkoxy)alkyl organometallic species and an aryl halide (route c). This strategically complementary mode of reactivity would move the challenge of asymmetric synthesis to a reliable early stage synthesis of enantiopure reagents, and would alleviate the vagaries associated with the late-stage conversion of prochiral centers to stereocenters, permitting the direct insertion of chirality into molecules with configurational integrity and reliability.
To the best of our knowledge, only one such cross-coupling based on this bond connection has been reported in the literature.8 Thus, a Stille reaction requiring the use of organostannanes, prepared using 4 equiv of tributyltin hydride and 4 equiv of diethylzinc,9 has been previously reported. The published research outlines the cross-coupling of 3-phenyl-1-(tri-n-butylstannyl)propyl 4-(trifluoromethyl)benzoate (TFMB) with an array of (hetero)aryl and alkenyl iodides and bromides, with yields ranging from 52–86%. The coupling of enantioenriched α-(acyloxy)-tri-n-butylstannanes, such as 3-phenyl-1-(tri-n-butylstannyl)propyl benzoate, was shown to proceed with complete retention of stereochemistry.
To reduce exposure to potentially toxic reagents and to use a more atom economical protecting group that can be cleaved under mild reaction conditions, the synthesis and cross-coupling of potassium 1-(alkoxy)alkyltrifluoroborates was envisioned as a general approach to secondary benzylic alcohols via a complementary bond connection. The Suzuki-Miyaura cross-coupling reaction has become one of the most powerful transformations in organic synthesis for the generation of carbon-carbon bonds.10 Organotrifluoroborates11 are often preferred coupling reagents because of their favorable properties, which include: low toxicity, air- and water stability, and functional group compatibility.
In optimizing the cross-coupling reaction, we sought to find a system where high stereochemical fidelity could be translated from the starting organometallic to the cross-coupled product. In addition to the value of a new synthetic approach to optically enriched benzylic alcohols, the development of the cross-coupling of an enantioenriched potassium 1-(alkoxy)alkyltrifluoroborate would be of interest to elucidate further the contributing factors that lead to retention or inversion of configuration at the boron center in cross-coupling of such systems. Prior research by Crudden12 and Suginome13a revealed that cross-coupling of α-methylbenzylboronic esters and α-(acylamino)alkylboronic esters (the latter in the presence of a Lewis acid), respectively, proceeds with retention of configuration through a conventional four-membered transition structure, whereas prior research by our group14 and Suginome13b demonstrated that cross-coupling of potassium β-trifluoroboratoamides and α-(acylamino)alkylboronic esters (the latter in the absence of Lewis acids), respectively, proceeds with inversion of configuration. The inversion phenomenon has been ascribed to the presence of a strongly coordinating group to bind the boronate during the reaction.
Reported herein is the synthesis of potassium 1-(hydroxy)alkyltrifluoroborates and their conversion to potassium 1-(alkoxy)alkyl- and 1-(acyloxy)alkyltrifluoroborates, the former of which have been successfully cross-coupled stereo-specifically with diverse aryl and heteroaryl chlorides. By extending the paradigm of employing a hemilabile ligand to avoid β-hydride elimination, this method demonstrates the development of an effective and straightforward route to obtain protected secondary alcohols in enantiopure form.
RESULTS AND DISCUSSION
Seeking to develop a general method for the synthesis of enantioenriched potassium 1-(alkoxy)alkyltrifluoroborates, our initial efforts focused on a directed lithiation sequence, utilizing a carbamate-directed borylation to form potassium 1-(N,N-diisopropylcarbamoyl)-3-phenylpropyltrifluoroborate 2 (eq 1).15
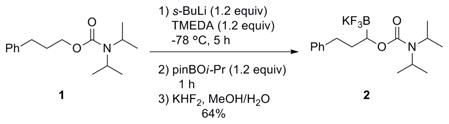 |
(1) |
This synthetic route would allow an enantioenriched organotrifluoroborate to be synthesized by replacing TMEDA with (−)-sparteine.15 Although successful, the directed lithiation route does not permit diverse organotrifluoroborates to be synthesized because it requires a directing group and employs very basic conditions that limit functional group tolerance. Therefore, attention was turned to a copper-catalyzed diboration of aldehydes.16 The original Sadighi protocol for this transformation called for the reaction to be run for 22 h in benzene and required an oxygen and moisture-free atmosphere for the work-up and isolation of the unstable products. Isolated alkyl-, aryl-, and heteroaryldibora compounds with yields up to 95% were reported. It was anticipated that this method could be directly applied to access the desired organotrifluoroborates, as quenching this reaction with aqueous KHF2 would not only transform the pinacol boronate to the corresponding trifluoroborate, but would also cleave the borate O-B bond, converting the dibora compounds to the corresponding potassium 1-(hydroxy)-alkyltrifluoroborate, thus allowing the work-up and isolation to be performed outside a glovebox.
The protocol for the previously reported diboration of aldehydes17 and ketones18 did not involve the addition of methanol. In the absence of methanol, intermediate 3c (Scheme 2) undergoes transmetalation with a molecule of bis(pinacolato)diboron in the rate determining step to generate the desired dibora compound 3d and regenerate the active catalyst 3a. Because methanol has been shown to facilitate protolytic turnover of the catalyst in other diboration reactions,19 the original Sadighi diboration was optimized by performing the reaction in toluene with the addition of methanol, thereby greatly accelerating the reaction, resulting in complete conversion in only 1.5 h. The proposed catalytic cycle (Scheme 2) starts with the active copper complex 3a, generated by subsequent transmetalation with NaOt-Bu and B2pin2. Coordination of the carbonyl of the aldehyde to copper results in the formation of 3b, where the boron then undergoes addition to the aldehyde to provide intermediate 3c. Protonation of this complex with methanol yields the 1-(hydroxy)alkylboronic acid pinacol ester from the aldehyde as well as 3e. Transmetalation with another molecule of B2pin2 regenerates the active catalyst 3a, completing the catalytic cycle.
Scheme 2.

Proposed Catalytic Cycle for Diboration of Aldehydes with Methanol
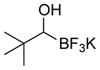
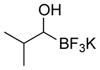
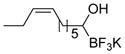
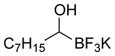
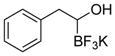
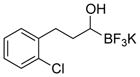
This one-pot procedure, starting from aldehydes, provided access to the desired products (4a–4t) after subsequent treatment of the 1-(hydroxy)alkyl pinacolboronates with KHF2 (Table 1), and both aryl and alkyl aldehydes were successfully employed to provide the corresponding trifluoroborates in modest to excellent yield. Unlike the corresponding dibora compounds 3d, which oxidize back to the aldehyde starting material upon prolonged exposure to air,16 the organotrifluoroborates 4 are all solids that can be stored on the bench-top without particular precautions. Alkyl aldehydes of various carbon chain lengths (entries 1–13, 18–20) as well as substituted aryl (entries 14–16) or naphthyl (entry 17) aldehydes can be used in this reaction with yields of the desired products as high as 98%. Both sterically hindered alkyl and aryl aldehydes (entries 4, 16) provide the desired trifluoroborates in good yields. Terminal and internal alkenes, as well as protected alcohols and amines, do not interfere with the diboration, as evident by the generation of the desired product in high yields from substrates bearing these functional groups (entries 6, 7, 9, 19, 20). The diboration of chiral aldehydes was examined, resulting in the diastereoselective formation of the corresponding trifluoroborates. Garner’s aldehyde afforded a single diastereomer (entry 20), whereas (S)-(−)-citronellal provided a 1:1 mixture of diastereomers (entry 19), and 2-phenylpropionaldehyde reacted with a 4:1 diastereoselectivity as determined by 1H NMR spectroscopy (entry 18). Substitution beta to the aldehyde, as in the diboration of 3-phenylbutanal (entry 12) results in a 1:1 mixture of diastereomers as determined by 1H NMR spectroscopy.
Table 1.
Preparation of Potassium 1-(Hydroxy)alkyltrifluoroborates.

| |||
|---|---|---|---|
| Entry | RBF3K | Isolated Yield (%) | |
| 1 |

|
4a | 98 |
| 2 |

|
4b | 89 |
| 3 |
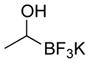
|
4c | 42 |
| 4 |
|
4d | 96 |
| 5 |
|
4e | 74 |
| 6 |

|
4f | 61e |
| 7 |
|
4g | 79e |
| 8 |
|
4h | 79e |
| 9 |
|
4i | 94 |
| 10 |
|
4j | 81e |
| 11 |
|
4k | 87 |
| 12 |
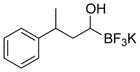
|
4l | 87 |
| 13 |

|
4m | 77 |
| 14 |
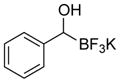
|
4n | 34a |
| 15 |

|
4o | 50a |
| 16 |

|
4p | 77 |
| 17 |

|
4q | 95 |
| 18 |
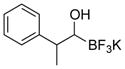
|
4r | 80b |
| 19 |
|
4s | 64c |
| 20 |
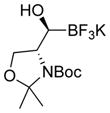
|
4t | 85d |
Reaction conditions: 1.0 equiv of aldehyde, 1.0 equiv of B2pin2, equiv of base/Cu ratio = 2, toluene, rt, 1.5 h.
Reaction stirred overnight at rt.
Isolated as a diasteromeric mixture (4:1 by 1H NMR).
Isolated as a diastereomeric mixture (1:1 by 1H NMR).
Isolated as a single diastereomer by 1H NMR.
Reaction performed on 1 g scale.
The next step in performing the desired cross-coupling would be the installation of a suitable protecting group on the alcohol of the potassium 1-(hydroxy)alkyltrifluoroborates. This group should be readily cleavable, but should also be capable of complexation with the intermediate diorganopalladium species to prevent β-hydride elimination. Although many advances have been made in C(sp2)–C(sp3) bond-forming reactions, the possibility of β-hydride elimination remains an inherent obstacle in cross-coupling of alkyl compounds.20 After transmetalation of the nucleophilic partner, β-hydride elimination competes with the product-forming reductive elimination (Scheme 3).
Scheme 3.

Possible pathways after transmetalation of the organoboron
To insure success in such an alkyl cross-coupling reaction, a means must be found to favor reductive elimination over β-hydride elimination. Prior research has demonstrated that one successful paradigm for accomplishing this is to perform cross- couplings and related reactions in the presence of electron-donating groups21 that can serve as hemilabile ligands for the intermediate organometallics. These hemilabile ligands can serve several roles. First, they donate electron density to the coordinatively unsaturated metal center, thereby minimizing the electron deficiency on the metal and reducing agostic interactions with neighboring hydrogens that would subsequently lead to β-hydride elimination. Complexing groups incorporated within the substrate can also enforce conformations that inhibit syn β-hydride elimination. For example, the success of the cross-coupling of alkyl β-trifluoroboratoamides14 has been credited in part to stabilization provided by the carbonyl,8,13 which complexes to the diorganopalladium intermediate after transmetalation (Scheme 4). Calculations have revealed that the hydrogens on the most substituted carbon undergo β-hydride elimination most readily.22 Upon coordination of the stabilizing carbonyl group, the alkyl chain is rotated in such a way that the β-hydrogens at the most substituted carbons are no longer able to become co-planar, thus preventing β-hydride elimination.14 Finally, electron-withdrawing groups in the substrate have also been found to inhibit β-hydride elimination. The success of the aforementioned cross-coupling of β-trifluoroboratoamides can also be attributed to some degree to the inductive effect of the amide substituent in the organo--palladium intermediate. The carbonyl of the amide withdraws electron density from the hydrogens β to the palladium, disfavoring β-hydrogen agostic interactions with the metal, thereby further inhibiting β-hydride elimination.22
Scheme 4.

Proposed Mechanism for the Coupling of Alkyl β-Trifluoroboratoamides
Potassium 1-(acyloxy)alkyltrifluoroborates possess a structural motif analogous to that of the β–trifluoroboratoamides. Consequently, an inherent resistance to β–hydride elimination was expected in these substrates owing to the same factors found in the β–trifluoroboratoamides (i.e., complexation of the carbonyl to the organopalladium as well as an electron withdrawing inductive effect). Therefore, initial efforts focused on the synthesis and palladium-catalyzed coupling reactions of such organotrifluoroborates with aryl halides. From the readily available 1-(hydroxy)alkyltrifluoroborates, representative examples of 1-(acyloxy)alkyltrifluoroborates were synthesized (Table 2) by treatment with acyl chlorides (entries 1–3) or anhydrides (entry 4). All of these trifluoroborates were isolated as colorless, free-flowing solids, capable of being stored open to air indefinitely without undergoing protodeboronation or other side reactions. Thus, because of their tetracoordinate nature, they demonstrate the same inherent stability as all other organotrifluoroborates.
Table 2.
Preparation of Potassium 1-(Acyloxy)alkyltrifluoroborates.

| |||
|---|---|---|---|
| Entry | RBF3K | Isolated Yield (%) | |
| 1 |
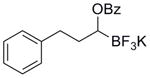
|
5a | 87 |
| 2 |
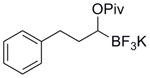
|
5b | 86 |
| 3 |
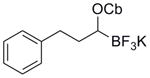
|
5c | 95 |
| 4 |

|
5d | 92a |
Reaction conditions: 1.0 equiv of trifluoroborate, 1.5 equiv of KH, 1.5 equiv of electrophile, THF, rt, o/n.
K2CO3 as base, 0 °C to rt, Ac2O in place of AcCl. (Cb = CONiPr2)
The first reaction screened was the cross-coupling of 5c with 4-chloroanisole (eq 2). Electron donation from both the nitrogen and oxygen of the carbamate was anticipated to increase the electron density on the carbonyl oxygen. This increased electron density was expected to aid in the complexation of the carbonyl moiety to palladium after transmetalation of the organoboron reagent, thereby preventing β-hydride elimination. The carbamate can also be viewed as an electron-withdrawing group on the reactant, which would also help disfavor β-hydride elimination.
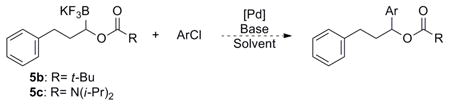 |
(2) |
Initial screening of the coupling reaction led to a set of conditions that appeared promising, but which led to an interesting product. Instead of observing the desired cross-coupled product, low yields of the oxidized ketone 6 of the expected product were obtained. Because of the high temperature and amount of base required for the Suzuki-Miyaura reaction, the protecting group was hydrolyzed, and the corresponding alcohol was oxidized under the reaction conditions. The mechanistic pathway to this product was envisioned to transpire via deprotonation of the deprotected alcohol and formation of an alkoxypalladium species, with subsequent β-hydride elimination forming the ketone. Oxidation of secondary alcohols in the presence of palladium and aryl chlorides has been reported in the literature.23 The feasibility of this oxidative process under the conditions utilized was confirmed upon subjection of an authentic sample of the alcohol to the initially developed reaction conditions (eq 3).
 |
(3) |
As the carbamate was deprotected under the reaction conditions, attention was turned to the cross-coupling of the pivalate-protected trifluoroborate 5b with 4-chloroanisole (eq 2). Prior research had shown that an alkyltrifluoroborate with a terminal alcohol protected with a pivalate could be cross-coupled in 10:1 toluene/H2O with Pd(OAc)2, RuPhos, and K2CO3 as base.24 However, attempts to couple 5b with 4-chloroanisole did not result in product formation, presumably owing to deprotection and oxidation as observed for the carbamate. It became clear from these results that carbonyl-containing protecting groups could not be employed as a stabilizing moiety because of their facile hydrolysis under the reaction conditions.
Based on prior reports of the ability of aromatic rings to coordinate palladium through an ipso-carbon or η2 binding mode,25 it was believed that a benzyl group could act as a stabilizing entity (Figure 1). The benzyloxy group is also inductively electron withdrawing, and therefore would further aid in disfavoring β-hydride elimination.
Figure 1.
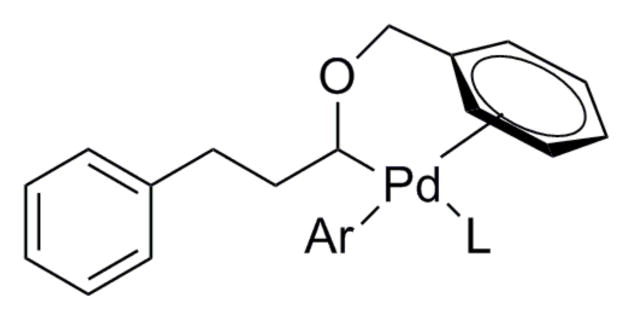
Proposed coordination from benzyl stabilizing group.
In fact, Fu reported a nickel-catalyzed coupling of homobenzylic bromides, citing the high levels of enantiomeric induction as being attributed to a secondary interaction between the benzyl substituent and the organonickel intermediate (eq 4).26 Although unmentioned, it seems likely that the aromatic substituent also stabilizes the diorganometallic intermediate, preventing β-hydride elimination.
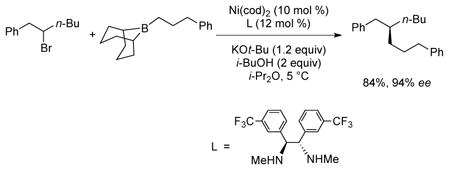 |
(4) |
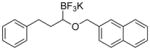
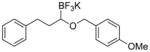
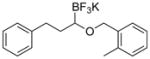
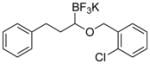
Therefore, potassium 1-(benzyloxy)-3-phenylpropyltrifluoroborate 7a was synthesized. Initial screening results of the cross-coupling of 7a with 4-chloroanisole were promising, providing evidence that this stabilization was aiding in the prevention of β-hydride elimination during the cross-coupling reaction. After these encouraging initial results, several representative 1-(arylmethyleneoxy)- and 1-(heteroarylmethyleneoxy)alkyltrifluoroborates were assembled (Table 3). 1-(Arylmethyleneoxy)alkyltrifluoroborates bearing electron withdrawing (entries 2 and 3) and electron donating (entries 5–8) groups were synthesized in high yields. Aryl rings with both ortho- and meta-substituents were also employed in the reaction, providing the desired products with yields up to 81% (entries 6, 8, 9). It is important to note that the para-methoxybenzyl (PMB) protected alcohol can be prepared in 90% yield (entry 7). Heteroaryl-containing trifluoroborates were also prepared in moderate yields (entries 10 and 11). Other ether-containing alkyl trifluoroborates were synthesized by reacting 4a with allyl bromide or iodomethane to provide the corresponding ethers in excellent yield (entries 12 and 13).
Table 3.
Preparation of Potassium 1-(Alkoxy)alkyltrifluoroborates.

| |||
|---|---|---|---|
| Entry | RBF3K | Isolated Yield (%) | |
| 1 |
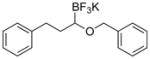
|
7a | 97 |
| 2 |

|
7b | 81 |
| 3 |
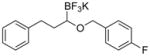
|
7c | 77 |
| 4 |
|
7d | 84 |
| 5 |

|
7e | 93 |
| 6 |
|
7f | 81 |
| 7 |
|
7g | 90 |
| 8 |
|
7h | 71 |
| 9 |
|
7i | 80 |
| 10 |
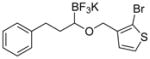
|
7j | 53 |
| 11 |
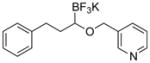
|
7k | 45 |
| 12 |

|
7l | 92a |
| 13 |
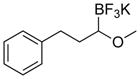
|
7m | 94a,b |
Reaction conditions: 1.0 equiv of trifluoroborate, 1.5 equiv of KH, 1.5 equiv of electrophile, THF, 50 °C, overnight.
rt, overnight.
MeI in place of R-Br.
The Suzuki-Miyaura cross-coupling reaction of 7a with 4-chloroanisole was subsequently optimized, aided by high throughput experimentation.27 Many screens were performed, a few of which are detailed below. For each screen, the reactions were run for 24 h at either 105 °C or 110 °C. The purpose of the first screen was to investigate general conditions to determine which type of solvent, co-solvent, ligand, and base would be best for the reaction. Therefore, a 96-well plate was run with 4 different bases, 4 different ligands, and 6 different solvents. The results of the screen are displayed in Figure 2.
Figure 2.

Graphical summary of Screen #1. Reaction conditions: 1.0 equiv of 7a, 1.0 equiv of 4-chloroanisole, 0.1 equiv of palladium, 110 °C, 3.0 equiv of Cs2CO3, 24 h. (Pd-L = L-Pd-G2, Cat A = cataCXium A, tamylOH = tert-amyl alcohol)
The first screen indicated that alcoholic solvents were not ideal for this reaction, even as a co-solvent, and water as a co-solvent provided the product in the highest yield. This screen conveyed that cataCXium A [di-(1-adamantyl)-n-butylphosphine] is the ligand that worked the best, and also showed that Cs2CO3 and K3PO4 were effective bases for this reaction. Based on these conclusions, another screen was conducted that focused solely on cataCXium A as the ligand and used the two best bases from the previous screen. An array of palladium sources was analyzed, and the ratio of the co-solvent was changed to determine the importance of water in the reaction.28 The results of this screen are depicted in Figure 3.
Figure 3.

Graphical summary of Screen #2. Reaction conditions: 1.0 equiv of 7a, 1.0 equiv of 4-chloroanisole, 0.1 equiv of palladium, 0.2 equiv of cataCXium A, 3.0 equiv of Cs2CO3, 110 °C, 24 h.
An analysis of the screen results revealed that an increased amount of water in the reaction led to an increase of the desired product, and therefore, subsequent screens focused on a 1:1 solvent/H2O ratio. An increase in the amount of water often increases the rate of cross-coupling relative to protodeboronation of the organotrifluoroborate.28
Through this second screen, it was determined that the nature of the palladium precursor is very important for the coupling. The palladium source resulting in the best conversion and yield was the cataCXium A derivative of Buchwald’s 2nd generation catalyst 9 (cataCXium A-Pd-G2).29 This catalyst benefits from a rapid reductive elimination to form carbazole and concomitant formation of the active L1Pd(0) complex, which is believed to be the most reactive species for cross-coupling.30 This precatalyst was synthesized from the aminobiphenyl palladium μ-Cl dimer 8 in 92% isolated yield (eq 5).31
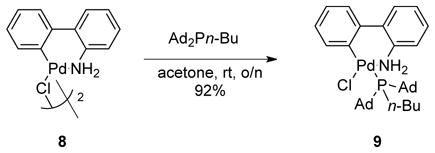 |
(5) |
After determining the optimal solvent, source of palladium, and base, the choice of ligand was the only variable left to be investigated. Examination of 48 different ligands with [Pd(allyl)Cl]2, 3 equiv of Cs2CO3, and a solvent system of 1:1 THF/H2O was carried out. For those ligands which were also precatalysts, [Pd(allyl)Cl]2 was not added to the reaction.
The results of this screen, as shown in Figure 4, confirmed that the ideal ligand for the desired transformation was cataCXium A. Although some other ligands provided product, there were only two reactions where there was both high conversion and enhanced product formation. These two systems both employed cataCXium A as the ligand. The difference between the top two hits from the screen was the palladium source, for which the top result was from cataCXium A-Pd-G2 9, and the second hit utilized [Pd(allyl)Cl]2 as the source of palladium. At this point, the initial optimization of the reaction was deemed complete. This extensive screening process led us to the following conditions: a 1:1 cyclopentyl methyl ether (CPME)/H2O solvent mixture, a temperature of 105 °C, cataCXium A-Pd-G2 precatalyst 9 (7.5 mol %), and Cs2CO3 (3 equiv).
Figure 4.

Graphical summary of Screen #3. Only the top ten ligands are shown. Reaction conditions: 1.0 equiv of 7a, 1.0 equiv of 4-chloroanisole, 0.1 equiv of palladium, 0.2 equiv of L, 3.0 equiv of Cs2CO3, 110 °C, 24 h. (Pd-L = L-Pd-G2)
Attempts to apply these conditions to a collection of aryl chlorides resulted in yields ranging from 53–75%.32 An evaluation of the crude reactions revealed that the low and variable yields were a result of incomplete conversion of the starting material. To avoid increasing the temperature and/or reaction time, optimization continued, and it was soon realized that simply replacing the base with CsOH•H2O greatly increased the yield of the reaction. Jutand and Hartwig have revealed that for couplings wherein transmetalation is the rate-determining step, hydroxide bases are most effective, as the higher concentration of hydroxide leads to the more active ArPd(OH)L complex, which undergoes transmetalation faster than the corresponding ArPd(X)L complex.33,34 To demonstrate the importance of CsOH•H2O, one last screen was completed that probed the amount and type of base. As CsOH•H2O is very hygroscopic, 3 and 5 equiv of base were compared in the reaction. The palladium source, both with and without the addition of extra ligand, was also examined.
Analysis of this screen (Figure 5) confirmed the importance of cataCXium A-Pd-G2 precatalyst (9), which led to the highest amount of product independent of the nature or amount of base in the reaction. Also evident is that excess cataCXium A impedes the reaction. CsOH•H2O was a much better base than Cs2CO3, and increasing the equiv of base from 3 to 5 led to a better conversion and product formation. Although several different ethereal solvents can be used, it is clear that when 5 equiv of CsOH•H2O is employed, the best solvent is CPME.
Figure 5.

Graphical summary of Screen #4. Reaction conditions: 1.0 equiv of 7a, 1.0 equiv of 4-chloroanisole, 0.075 equiv of palladium, 105 °C, 24 h. (A = [Pd(allyl)Cl]2 with cataCXium A, B = cataCXium A-Pd-G2, 1:0/1:1/1:2 = palla-dium:ligand ratio)
Based on these results, a 1:1 CPME/H2O solvent mixture, reaction temperature of 105 °C, cataCXium A-Pd-G2 9 catalyst system (7.5 mol %), and CsOH•H2O (5 equiv) base were adopted as the standard conditions for the subsequent studies and applied to representative aryl and heteroaryl chlorides.
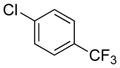
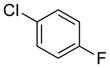
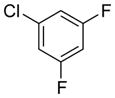
To investigate the method fully, both electron-poor (Table 4, entries 1–6) and electron-rich (entries 7–20) aryl chlorides were examined. All of these aryl chlorides were successfully cross-coupled, providing the desired benzyl-protected secondary alcohols with yields of 39–95%. Both 1- and 2-chloronaphthalene were successfully engaged in this reaction with yields of 95% and 85%, respectively (entries 19 and 20).
Table 4.
Scope of the Cross-Coupling with Aryl Chlorides.
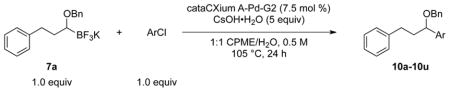
| |||
|---|---|---|---|
| Entry | ArCl | Isolated Yield (%) | |
| 1 |
|
10a | 84 |
| 2 |
|
10b | 77 |
| 3 |
|
10c | 74 |
| 4 |
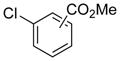
|
m:10d | 52a |
| 5 | p: 10e | 57a | |
| 6 |
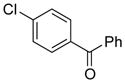
|
10f | 64 |
| 7 |
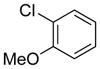
|
10g | 73 |
| 8 |
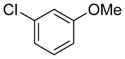
|
10h | 77 |
| 9 |
|
X = Cl:10i | 87 (79b) |
| 10 | X = Br:10i | 60 | |
| 11 | I:10i | 35 | |
| 12 |
|
10j | 67 |
| 13 |

|
10k | 67 |
| 14 |

|
10l | 84 |
| 15 |

|
10m | 79 |
| 16 |
|
o: 10n | 76 |
| 17 | m: 10o | 81 | |
| 18 | p: 10p | 85 | |
| 19 |

|
10q | 95 |
| 20 |

|
10r | 85 |
| 21 |

|
10s | 76 |
| 22 |
|
10t | 57 |
| 23 |
|
10u | 39 |
Reaction conditions: 1.0 equiv of trifluoroborate, 1.0 equiv of electrophile, 7.5 mol % Pd, 5.0 equiv of base, 1:1 CPME/H2O, 105 °C, 24 h.
3 equiv of Cs2CO3 as base.
3 mmol scale, 3.75 mol % Pd.
Electron donating groups can be present in any position of the aryl chloride, as evident by the couplings of ortho-, meta-, and para-chloroanisole as well as ortho-, meta- and para-chlorotoluene (entries 7–9, 16–18). Several common functional groups were tolerated, including carbamates (entry 13), amines (entries 14, 15), and ketones (entry 6). Two methyl ester-substituted aryl chlorides were successfully cross-coupled by changing the base to Cs2CO3 to avoid hydrolysis of the ester by CsOH•H2O (entries 4, 5).
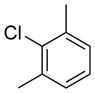
The effects of steric hindrance were tested with mono- or di-ortho substituted electrophiles (entries 7, 16, 22, 23). The standard conditions proved to be very efficient for mono-ortho substituted electrophiles, as both 2-chloroanisole and 2-chlorotoluene provided the product in high yield. 2-Chloro-5-methoxy-1,3-dimethylbenzene as well as 2-chloro-1,3-dimethylbenzene are known to be difficult substrates, owing to the presence of two methyl groups in the ortho positions;35 however, both were coupled in modest yields of 57% and 39%, respectively.
The ability to conduct the reaction with various electrophilic partners was then analyzed (entries 9–11). 4-Bromoanisole and 4-iodoanisole afforded the desired product in modest yields. 4-Methoxyphenyl methanesulfonate did not provide any of the desired product under the reaction conditions. Only unreacted starting material was detected after 24 h.
The cross-coupling with 4-chloroanisole was performed on a 1 g scale by reducing the catalyst loading in half to provide the desired product in 79% isolated yield (entry 9).
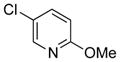
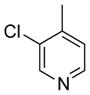
To expand the range of electrophiles, the potassium 1-(benzyloxy)-3-phenylpropyltrifluoroborate (7a) was coupled with a variety of heteroaryl chlorides (Table 5). The optimized conditions proved to be efficient for these couplings, resulting in yields of up to 88% for nine heteroaryl chlorides. A variety of substrates was examined, including pyridine, with and without substituents, as well as isomeric chloropyridines (entries 1–3, 9). Chloroquinolines were also efficient electrophiles for this reaction, as the chloride could be located at the 4-, 6-, and 8-positions. Substitution of the quinoline did not seem to impact the reaction (entry 4–6). A sulfur-containing heterocycle can also be coupled, albeit in a lower yield of 41% (entry 8).
Table 5.
Scope of the Cross-Coupling with Heteroaryl Chlorides.
| |||
|---|---|---|---|
| Entry | HetArCl | Isolated Yield (%) | |
| 1 |

|
11a | 87 |
| 2 |
|
11b | 74 |
| 3 |
|
11c | 42 |
| 4 |
|
11d | 74 |
| 5 |
|
11e | 53 |
| 6 |
|
11f | 53 |
| 7 |

|
11g | 89 |
| 8 |
|
11h | 41 |
| 9 |
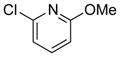
|
11i | 63 |
Reaction conditions: 1.0 equiv of trifluoroborate, 1.0 equiv of electrophile, 7.5 mol % Pd, 5.0 equiv of base, 1:1 CPME/H2O, 105 °C, 24 h.
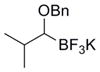
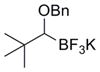
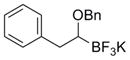
To demonstrate the versatility of this method, other potassium 1-(benzyloxy)alkyltrifluoroborates were synthesized using the same conditions for the synthesis of 7a (Table 6). Conversion of 1-(hydroxy)alkyltrifluoroborates yielded the desired products after heating overnight at 50 °C. The products were isolated as colorless solids, with yields ranging from 52–96%.
Table 6.
Preparation of Various Potassium 1-(Benzyloxy)alkyltrifluoroborates.

| |||
|---|---|---|---|
| Entry | RBF3K | Isolated Yield (%) | |
| 1 |
|
12a | 93 |
| 2 |
|
12b | 52 |
| 3 |

|
12c | 65 |
| 4 |
|
12d | 95 |
| 5 |
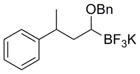
|
12e | 96 |
| 6 |

|
12f | 75 |
| 7 |

|
12g | 85 |
| 8 |
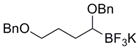
|
12h | 67 |
| 9 |

|
12i | 61 |
| 10 |
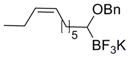
|
12j | 73 |
| 11 |
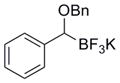
|
12k | 79 |
| 12 |

|
12l | 89 |
Reaction conditions: 1.0 equiv of trifluoroborate, 1.5 equiv of KH, 1.5 equiv of electrophile, THF, 50 °C, o/n
These potassium (1-benzyloxy)alkyltrifluoroborates were subsequently subjected to cross-coupling reactions under the standard reaction conditions (Table 7). Straight chain alkyl groups can be cross-coupled in good yield (entry 1), as well as other substituted alkyltrifluoroborates (entries 2–4). 1-(Benzyloxy)(phenyl)methyltrifluoroborate 12k yielded the desired product in 52% yield with the use of Cs2CO3 as base, while the use of CsOH•H2O resulted primarily in protode-boronation (entry 5). Other potassium 1-(arylmethyleneoxy)-alkyltrifluoroborates were also subjected to the cross-coupling reaction. Substitution at the ortho-, meta-, and para- positions of the aryl ring does not interfere with the cross-coupling, resulting in the desired products with yields ranging from 61–89% (entries 6–8). It is important to note that this method can be extended to other protected alcohols, as it has been shown that the PMB-protected organotrifluoroborate 7g is also an efficient nucleophile for this coupling reaction (entry 8). A limitation of the current method is that branching α to the alkoxy group (e.g., as in organotrifluoroborates 12a–12c) cannot be tolerated in the cross-coupling.
Table 7.
Scope of the Cross-Coupling of Various Potassium 1-(Benzyloxy)alkyltrifluoroborates.
| ||||
|---|---|---|---|---|
| Entry | RBF3K | Product | Isolated Yield (%) | |
| 1 | 12i |
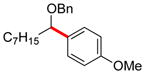
|
13a | 51 |
| 2 | 12g |
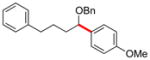
|
13b | 72 |
| 3 | 12e |
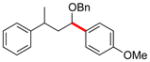
|
13c | 95 |
| 4 | 12d |
|
13d | 66 |
| 5 | 12k |

|
13e | 52a |
| 6 | 7h |
|
13h | 61 |
| 7 | 7f |
|
13i | 89 |
| 8 | 7g |
|
13j | 84 |
Reaction conditions: 1.0 equiv of trifluoroborate, 1.0 equiv of electrophile, 7.5 mol % Pd, 5.0 equiv of base, 1:1 CPME/H2O, 105 °C, 24 h.
Reaction with 3 equiv of Cs2CO3.
For each cross-coupling reaction, regardless of the electrophile or nucleophile, only 1.0 equiv of the starting potassium organotrifluoroborate was engaged. The robust yields achieved indicate that little to no protodeboronation is occurring under the reaction conditions. The high yields of the cross-coupled products reveal that the β-hydride elimination pathway is virtually non-existent as well.
An intramolecular Suzuki-Miyaura cross-coupling was also successful with the potassium 1-(arylmethyleneoxy)-3-phenylpropyltrifluoroborates. Thus, 7i underwent an intramolecular coupling, wherein the trifluoroborate acted as the nucleophile and the aryl chloride on the same molecule served as the electrophile, providing access to a 1,3-dihydroisobenzofuran derivative 14 through an unusual bond connection (eq 6). Substrate 12f underwent a similar intramolecular cross-coupling to yield a five membered carbocycle 15 in 73% yield (eq 7). This umpoled route to cycloalkanol derivatives is complementary to that of intramolecular Barbier-type routes to the same class of molecules.
 |
(6) |
 |
(7) |
To test the importance of the benzyl group on the success of the cross-coupling, the corresponding methyl ether, 16, was subjected to the optimized cross-coupling conditions with 4-chloroanisole (eq 8). Only unreacted aryl chloride and none of the desired product 17 was observed after 24 h, demonstrating the significant role played by the benzyl group as a hemilabile ligand stabilizing the organopalladium intermediate.
 |
(8) |
To validate the method as a means to synthesize secondary alcohols, the benzyl protecting group would need to be removed selectively. Deprotection of a primary benzyl group in the presence of a secondary benzyl group has substantial precedence in the literature.36 Utilizing standard conditions of 10 mol % Pd/C in ethanol under an atmospheric pressure of H2, clean removal of the primary benzyl group was observed, yielding the secondary alcohol 18 in an unoptimized yield of 79% (eq 9).
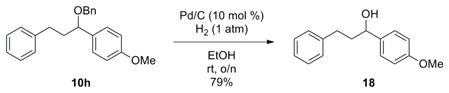 |
(9) |
The importance of the cross-coupling approach to secondary benzylic alcohol synthesis stems from the complementary mode of bond connection compared to those normally employed as well as the availability of organoboron reagents that are configurationally stable and storable. Because a stereogenic center is being incorporated, it was important to determine the stereospecificity of the reaction and whether the transformation occurred with retention or inversion of stereochemistry. To conduct these studies, an enantiomerically enriched potassium (S)-1-(benzyloxy)-3-phenylpropyltrifluoroborate 2237 was synthesized through Matteson homologation chemistry (Scheme 5).38
Scheme 5.

Synthesis of Enantioenriched Potassium (S)-1-(Benzyloxy)-3-phenylpropyltrifluoroborate
The starting (S,S)-DICHED phenethylboronate (19) yielded the (R)-chloride intermediate 20 after the homologation, which underwent inversion of configuration upon displacement by LiOBn. The (4S,5S)-2-((S)-1-(benzyloxy)-3-phenylpropyl)-4,5-dicyclohexyl-1,3,2-dioxaborolane (21) with an ee of greater than 99% can subsequently be converted to its corresponding trifluoroborate 22 by addition of KHF2 dissolved in MeOH/H2O. The ee of 20 and 21 was determined via SFC analysis.
After synthesizing the enantioenriched trifluoroborate 22, it was reacted under the standard reaction conditions with 4-chloroanisole. Examination of the reaction product revealed that the transformation proceeds with complete stereospecificity and retention of stereochemistry (eq 10).39
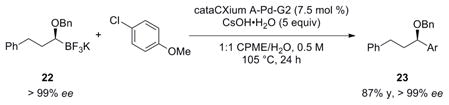 |
(10) |
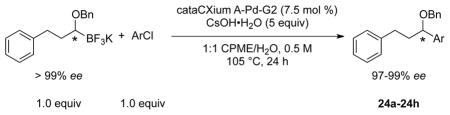
The other enantiomer of 22 was prepared through the same synthetic pathway, except the opposite enantiomer of the 1,2-dicyclohexyl-1,2-ethanediol was utilized as the starting material (Scheme 5). The two enantiomerically enriched organotrifluoroborates were subjected to the cross-coupling with several aryl and heteroaryl chlorides to determine the effect of the aryl halide on the enantiomeric excess (Table 8). The results show that there is little to no erosion of ee based on the aryl halide, as both electron poor (entries 1–2, 5–6) and electron rich (entries 3, 7) aryl chlorides can be subjected to the cross-coupling with high levels of enantiomeric excess. Two different heteroaryl chlorides (entries 4, 8) were also coupled with little, if any, loss of enantioselectivity. These coupling reactions are presumed to proceed with retention of stereochemistry.
Table 8.
Scope of the Cross-Coupling of Enantioenriched 22 with (Hetero)Aryl Chlorides
| |||||
|---|---|---|---|---|---|
| Entry | (x)-22 | ArCl | % ee (x) | Isolated Yield (%) | |
| 1 | (S) |
|
24a | 99 (R) | 81 |
| 2 | (S) |
|
24b | > 99 (R) | 62a |
| 3 | (S) |

|
24e | > 99 (R) | 70 |
| 4 | (S) |
|
24h | > 99 (R) | 86 |
| 5 | (S) |

|
24c | 99 (R) | 75 |
| 6 | (R) |
|
24d | 97 (S) | 77 |
| 7 | (R) |
|
24f | 97 (S) | 78 |
| 8 | (R) |
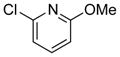
|
24g | 97 (S) | 65 |
Reaction conditions: 1.0 equiv of trifluoroborate, 1.0 equiv of electrophile, 7.5 mol % Pd, 5.0 equiv of base, 1:1 CPME/H2O, 105 °C, 24 h.
Reaction with 5 equiv of Cs2CO3.
The retention of stereochemistry observed is consonant with prior cross-coupling studies of isotopically labeled primary organoborons by Soderquist and Woerpel,40 as well as the cross-coupling of secondary benzylic systems by Crudden (Scheme 6, I)12 but contrasts with prior research in the Molander14 and Suginome groups13 in which inversion of configuration was observed (Scheme 6, II and III). The stereochemically differentiating step of the cross-coupling in these latter studies is the transmetalation, which proceeds with inversion of configuration via an open transition structure. Both examples resulting in inversion of configuration take advantage of intramolecular coordination of a carbonyl to the boron in the transmetalation transition structure. The strong complexation of the carbonyl to the boron species appears to facilitate a process whereby the transmetalation proceeds through an SE2 mechanism, wherein the boron center undergoes electrophilic attack of the L(Ar)PdY species.13a,14 Examples of SE2-type reactions of tetracoordinate boron “ate” complexes with soft electrophiles such as I2 and Br2, proceeding with inversion of configuration, have been reported previously.41 With the approach of the arylpalladium electrophile occurring form the opposite side of boron and reductive elimination transpiring with retention, the result of this transformation is overall inversion of configuration.13,14
Scheme 6.

Stereospecificity of Various Suzuki-Miyaura Cross-Coupling Reactions
Suginome subsequently studied the effect of Lewis acid additives in stereospecific cross-coupling reactions (Scheme 6, IV). The addition of a Lewis acid was postulated to disrupt the intramolecular coordination between the carbonyl and boronate species. The argument was made that with a vacant coordination site on boron, the transmetalation transpired through a four-membered transition structure in which the lone pair of Y (Y = halogen or OH) of the L(Ar)PdY species coordinated to boron. This transformation resulted in retention of configuration for the cross-coupling, as the electrophilic attack of the organopalladium occurred from the same side as boron.13b In a similar manner, the cross-coupling of enantioenriched 22 proceeds with retention of configuration (Scheme 6, V). Because the potassium 1-(benzyloxy)alkyltrifluoroborates lacks a strongly coordinating group to induce SE2 reactivity, the transmetalation proceeds through a four-membered transition structure with retention.
As the reaction developed herein proceeds with near complete stereospecificity, an efficient and facile synthesis of enantioenriched potassium 1-(benzyloxy)alkyltrifluoroborates should prove exceedingly useful for the ultimate synthesis of enantiomerically enriched secondary benzylic alcohols. Investigations on the catalytic asymmetric synthesis of such materials are currently underway. These organotrifluoroborates would serve as metal-centered ‘stereocenters in a bottle’, i.e., enantioenriched nucleophiles that could be stored indefinitely on the benchtop, with a known configuration, allowing the reliable installation of a defined stereocenter independent of the structure of the electrophilic partner.
CONCLUSIONS
The synthesis of potassium 1-(hydroxy)alkyltrifluoroborates and their conversion to potassium 1-(alkoxy)alkyltrifluoroborates and 1-(acyloxy)alkyltrifluoroborates has been reported. Potassium 1-(benzyloxy)alkyltrifluoroborates undergo a palladium catalyzed cross-coupling reaction with a variety of aryl and heteroaryl chlorides in high yields, using stoichiometric quantities of the two partners. The success of this cross-coupling is attributed to the presence of the benzyl protecting group, which is believed to prevent β-hydride elimination in two ways: by coordinating to the palladium center of the diorganopalladium intermediate after transmetalation of the alkyl group and by an electron-withdrawing inductive effect. The cross-coupling proceeds with complete retention of configuration, and therefore, enantioenriched organotrifluoroborates can be used as the nucleophilic species with virtually no loss of enantiomeric excess regardless of the cross-coupling partner. A method for a direct installation of a secondary alcohol through a Suzuki-Miyaura reaction (after deprotection) has thus been developed.
Supplementary Material
Acknowledgments
This research was supported by the NIGMS (R0I GM-081376) and an NSF GOALI grant. Dr. Simon Berritt, Dr. Jason Schmink, and Dr. Cornell Stanciu (University of Pennsylvania) are acknowledged for their assistance in performing the high throughput experiments. BASF is acknowledged for their generous donation of bis(pinacolato)diboron. Dr. Matthew Tudge (Merck) is acknowledged for synthesis of the aminobiphenyl palladium μ-Cl dimer. Dr. Rakesh Kohli (University of Pennsylva-nia) is acknowledged for obtaining HRMS data. Dr. Patrick Car-roll (University of Pennsylvania) is acknowledged for performing X-ray analysis.
Footnotes
ASSOCIATED CONTENT
Experimental procedures, spectral characterization and copies of 1H, 13C, 19F, and 11B NMR spectra for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Carey FA, Sundberg RJ. Advanced Organic Chemistry: Part B: Reaction and Synthesis. 5. Springer; New York: 2007. For a review on the enantioselective alkylation of carbonyls, see: Ramon DJ, Yus M. Alkylation of Carbonyl and Imino Groups. In: Science of Synthesis, Stereoselective Synthesis. De Vries JG, Molander GA, Evans PA, editors. Vol. 2. Georg Thieme Verlag; Stuttgart, Germany: 2011. pp. 349–400.For a review on the enantioselective arylation of carbonyls, see: Kauffman MC, Walsh PJ. Arylation and Alkenylation of Carbonyl and Imino Groups. In: Science of Synthesis, Stereoselective Synthesis. De Vries JG, Molander GA, Evans PA, editors. Vol. 2. Georg Thieme Verlag; Stuttgart, Germany: 2011. pp. 449–496.(d) For reviews on the enantioselective addition of diorganozinc compounds to aldehydes, see: Soai K, Niwa S. Chem Rev. 1992;92:833.Pu L, Yu HB. Chem Rev. 2001;101:757. doi: 10.1021/cr000411y.
- 2.Hartwig J. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito: 2010. [Google Scholar]
- 3.(a) For a review on the enantioselective hydrogenation of prochiral ketones, see: Arai N, Ohmura T. Reduction of Carbonyl Groups: Hydrogenation. In: De Vries JG, Molander GA, Evans PA, editors. Science of Synthesis, Stereoselective Synthesis. Vol. 2. Georg Thieme Verlag; Stuttgart, Germany: 2011. pp. 59–132.Gladiali S, Alberico E. Chem Soc Rev. 2006;35:226. doi: 10.1039/b513396c.(b) For a review on the enantioselective reduction of prochiral ketones, see: Zaidlewicz M, Pakulski MM. Reduction of Carbonyl Groups: Transfer Hydrogenation, Hydrosilylation, Catalytic Hydroboration, and Reduction with Borohydrides, Aluminum Hydrides, or Boranes. In: De Vries JG, Molander GA, Evans PA, editors. Science of Synthesis, Stereoselective Synthesis. Vol. 2. Georg Thieme Verlag; Stuttgart, Germany: 2011. pp. 59–132.Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z.Cho BT. Chem Soc Rev. 2009;38:443. doi: 10.1039/b811341f.De Wilderman S, Sereinig N. Enzymatic Reduction of Carbonyl Groups. In: Science of Synthesis, Stereoselective Synthesis. De Vries JG, Molander GA, Evans PA, editors. Vol. 2. Georg Thieme Verlag; Stuttgart, Germany: 2011. pp. 59–132.For a review on the enzymatic reduction of ketones, see: Moore JC, Pollard DJ, Kosjek B, Devine PN. Acc Chem Res. 2007;40:1412. doi: 10.1021/ar700167a.Kroutil W, Mang H, Edegger K, Faber K. Curr Opin Chem Biol. 2004;8:120. doi: 10.1016/j.cbpa.2004.02.005.
- 4.Binder CM, Singaram B. Org Prep Proced Int. 2011;43:139. [Google Scholar]
- 5.Rudolph J, Schmidt F, Bolm C. Adv Synth Catal. 2004;346:867. [Google Scholar]
- 6.Wu K-H, Gau H-M. J Am Chem Soc. 2006;128:14808. doi: 10.1021/ja062080y. [DOI] [PubMed] [Google Scholar]
- 7.Yamamoto Y, Kurihara K, Miyaura N. Angew Chem, Int Ed. 2009;48:4414. doi: 10.1002/anie.200901395. [DOI] [PubMed] [Google Scholar]
- 8.Goli M, He A, Falck JR. Org Lett. 2011;13:344. doi: 10.1021/ol102863u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He A, Falck JR. Angew Chem Int Ed. 2008;47:6586. doi: 10.1002/anie.200802313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For reviews of the Suzuki-Miyaura Cross-Coupling reaction, see: Miyaura N, Suzuki A. Chem Rev. 1995;95:2457.Bellina F, Carpita A, Rossi R. Synthesis. 2004:2419.Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633.
- 11.For reviews on potassium organotrifluoroborates, see: Molander GA, Figueroa R. Aldrichimica Acta. 2005;38:49.Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q.Stefani HA, Cella R, Vieira AS. Tetrahedron. 2007;63:3623.Darses S, Genet JP. Chem Rev. 2008;108:288. doi: 10.1021/cr0509758.
- 12.Imao D, Glasspoole BW, Laberge VS, Crudden CM. J Am Chem Soc. 2009;131:5024. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]
- 13.For references with the cross-coupling of carbonyl-containing organometallics, see: Ohmura T, Awano T, Suginome M. J Am Chem Soc. 2010;132:13191. doi: 10.1021/ja106632j.Awano T, Ohmura T, Suginome M. J Am Chem Soc. 2011;133:20738. doi: 10.1021/ja210025q.
- 14.Sandrock DL, Jean-Gerard L, Chen CY, Dreher SD, Molander GA. J Am Chem Soc. 2010;132:17108. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beckmann E, Desai V, Hoppe D. Synlett. 2004;13:2275. [Google Scholar]
- 16.Laitar DS, Tsui EY, Sadighi JP. J Am Chem Soc. 2006;128:11036. doi: 10.1021/ja064019z. [DOI] [PubMed] [Google Scholar]
- 17.Zhao H, Dang L, Marder TB, Lin Z. J Am Chem Soc. 2008;130:5586. doi: 10.1021/ja710659y. [DOI] [PubMed] [Google Scholar]
- 18.McIntosh ML, Moore CM, Clark TB. Org Lett. 2010;12:1996. doi: 10.1021/ol100468f. [DOI] [PubMed] [Google Scholar]
- 19.For a review of borylation reactions with B2pin2, see: Burks HE, Morken JP. Chem Commun. 2007:4717. doi: 10.1039/b707779c.
- 20.Lu X. Top Catal. 2005;35:73. [Google Scholar]
- 21.For examples of stabilizing groups in alkylmetallics that inhibit β-hydride elimination see: Wilsily A, Tramutola F, Owston NA, Fu GC. J Am Chem Soc. 2012;134:5794. doi: 10.1021/ja301612y.Lu Z, Wilsily A, Fu GC. J Am Chem Soc. 2011;133:8154. doi: 10.1021/ja203560q.Owston NA, Fu GC. J Am Chem Soc. 2010;132:11098. doi: 10.1021/ja105924f.Weberski JMP, Chen C, Delferro M, Zuccaccia C, Macchioni A, Marks TJ. Organometallics. 2012;31:3773.Giovannini R, Studemann T, Dussin G, Knochel P. Angew Chem Int Ed. 1998;37:2387. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2387::AID-ANIE2387>3.0.CO;2-M.Johnson JB, Rovis T. Angew Chem Int Ed. 2008;47:840. doi: 10.1002/anie.200700278.Luo X, Zhang H, Duan H, Qiang L, Lizheng Z, Zhang T, Lei A. Org Lett. 2009;9:4571. doi: 10.1021/ol701995t.Jensen KH, Sigman MS. Org Biomol Chem. 2008;6:4083. doi: 10.1039/b813246a.Wang A, Jiang H, Chen H. J Am Chem Soc. 2009;131:3846. doi: 10.1021/ja900213d.Urkalan KB, Sigman MS. Angew Chem Int Ed. 2009;48:3146. doi: 10.1002/anie.200900218.Trejos A, Fardost A, Yahiaoui S, Larhed M. Chem Commun. 2009:7587. doi: 10.1039/b918358b.Yahiaoui S, Fardost A, Trejos A, Larhed M. J Org Chem. 2011;76:2433. doi: 10.1021/jo1018188.Sköld C, Kleimark J, Trejos A, Odell LR, Lill SON, Norrby PO, Larhed M. Chem-Eur J. 2012;18:4174. doi: 10.1002/chem.201102678.
- 22.Pudasaini B, Janesko BG. Organometallics. 2011;30:4564. [Google Scholar]
- 23.Zeng M, Du Y, Shao L, Qi C, Zhang X-M. J Org Chem. 2010 doi: 10.1021/jo100089d. [DOI] [PubMed] [Google Scholar]
- 24.Dreher SD, Lim SE, Sandrock DL, Molander GA. J Org Chem. 2009;74:3626. doi: 10.1021/jo900152n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.For X-Ray structures showing coordination of an arene to Pd, see: Yin J, Rainka MP, Zhang XX, Buchwald SL. J Am Chem Soc. 2002;124:1162. doi: 10.1021/ja017082r.Faller JW, Sarantopoulos N. Organometallics. 2004;23:2008.Reddy KR, Surekha K, Lee GH. Organometallics. 2001;20:5557.Yamashita M, Takamiya K, Jin K, Nozaki K. J Organomet Chem. 2006;691:3189.Mikhel IS, Ruegger H, Butti P, Camponovo F, Huber D, Mezzetti A. Organometallics. 2008;27:2937.Marshall WJ, Grushin VV. Organometallics. 2003;22:555.
- 26.Saito B, Fu GC. J Am Chem Soc. 2008;130:6694. doi: 10.1021/ja8013677. [DOI] [PubMed] [Google Scholar]
- 27.See Supporting Information for HTE information.
- 28.Lennox AJJ, Lloyd-Jones GC. J Am Chem Soc. 2012;134:7431. doi: 10.1021/ja300236k. [DOI] [PubMed] [Google Scholar]
- 29.Kinzel T, Zhang Y, Buchwald SL. J Am Chem Soc. 2010;132:14073. doi: 10.1021/ja1073799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.For other references about the synthesis of aminobiphenylpalladium precatalysts, see: Albert J, Granell J, Zafrilla J, Font-Bardia M, Solans X. J Organomet Chem. 2005;690:422.Albert J, Granell J, Luque A, Minguez J, Moragas R, Font-Bardia M, Solans X. J Organomet Chem. 1996;522:87.
- 32.See Supporting Information for coupling with Cs2CO3.
- 33.(a) Amatore C, Jutand A, Le Duc G. Chem Eur J. 2012;18:6616. doi: 10.1002/chem.201200516. [DOI] [PubMed] [Google Scholar]; (b) Amatore C, Jutand A, Le Duc G. Chem Eur J. 2011;17:2492. doi: 10.1002/chem.201001911. [DOI] [PubMed] [Google Scholar]
- 34.Carrow BP, Hartwig JF. J Am Chem Soc. 2011;133:2116. doi: 10.1021/ja1108326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartwig JF. Inorg Chem. 2007;46:1936. doi: 10.1021/ic061926w. [DOI] [PubMed] [Google Scholar]
- 36.For selective debenzylation examples, see: Otera J, Niibo Y, Nozaki H. Tetrahedron. 1991;47:7625.Gracza T, Jaeger V. Synthesis. 1994;12:1359.Krouzelka J, Linhart I, Tobrman T. J Heterocycl Chem. 2008;45:789.Seayad J, Seayad AM, Chai CL. Org Lett. 2010;12:1412. doi: 10.1021/ol902813m.Kitazume T, Yamazaki T. Synth Commun. 1990;20:1469.Pan X, Wilcox CS. J Org Chem. 2010;75:6445. doi: 10.1021/jo101137r.Rahaim RJ, Jr, Maleckza RE., Jr Org Lett. 2011;13:584. doi: 10.1021/ol102757v.Lei S, Xia WJ, Zhan FM, Tu YQ. Synlett. 2002;9:1505.Sharma GVM, Mallesham S. Tetrahedron: Asymmetry. 2010;21:2646.
- 37.For the use of LiOBn as a nucleophile in Matteson homologation chemistry, see: Matteson DS, Man H-W. J Org Chem. 1996:6047.
- 38.For a review of Matteson homologation chemistry, see: Matteson DS. Tetrahedron. 1998;54:10555.
- 39.See Supporting Information for determination of stereoselectivity.
- 40.(a) Ridgway BH, Woerpel KA. J Org Chem. 1998;63:458. doi: 10.1021/jo970803d. [DOI] [PubMed] [Google Scholar]; (b) Matos K, Soderquist JA. J Org Chem. 1998;63:461. doi: 10.1021/jo971681s. [DOI] [PubMed] [Google Scholar]
- 41.(a) Bergbreiter DE, Rainville DP. J Organomet Chem. 1976;121:19. [Google Scholar]; (b) Kabalka GW, Gooch EE., III J Org Chem. 1980;45:3578. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.