

Background: The molecular mechanisms involved in IL-23 up-regulation by PGE2 are not elucidated.
Results: PGE2 induces IL-23p19 through the EP4 cAMP-PKA/EPAC-CREB/C/EBPβ signaling pathway.
Conclusion: PGE2 synergizes with TLR ligands and with proinflammatory cytokines such as TNFα to up-regulate Il23a gene expression.
Significance: Understanding is gained of one of the major functions of PGE2 leading to activation of pathogenic Th17 cells.
Keywords: C/EBP Transcription Factor, Camp, CREB, Dendritic Cells, Inflammation, Prostaglandins, IL-23
Abstract
We reported previously that prostaglandin E2 (PGE2) up-regulates IL-23 in vitro in bone marrow-derived dendritic cells and in vivo in models of collagen-induced arthritis and inflammatory bowel disease, leading to preferential Th17 development and activity. There is very little information on the molecular mechanisms involved in the PGE2-induced up-regulation of Il23a gene expression. In this study we investigated the signaling pathways and transcription factors involved in the stimulatory effect of PGE2. Although PGE2 does not induce IL-23p19 expression by itself, it synergizes with both extra- and intracellular Toll-like receptor ligands and with inflammatory cytokines such as TNFα. We established that the effect of PGE2 in conjunction with either LPS or TNFα is mediated through the EP4 receptor and the cAMP-dependent activation of both protein kinase A (PKA) and exchange protein activated by cAMP (EPAC). Using the EP4 agonist PGE1OH in conjunction with TNFα, we found that PKA-induced phosphorylation of cAMP-response element-binding protein (PCREB) and EPAC-induced phosphorylation of C/AATT enhancer-binding protein β (PC/EBPβ) mediate the stimulatory effect of PGE2 on IL-23p19 expression. This is the first report of CREB and C/EBPβ involvement in Il23a promoter activation. Mutation within the putative CREB and C/EBP sites combined with in vivo DNA binding (ChIP) assays identified the distal CREB site (−1125) and the two proximal C/EBP sites (−274 and −232) as essential for PKA-activated CREB and EPAC-activated C/EBPβ-induced IL-23p19 expression.
Introduction
Prostaglandin E2 (PGE2),2 the most abundant prostanoid generated from the arachidonic acid released from the plasma membrane, is a pleiotropic lipid mediator involved in a variety of physiological functions. In immune cells, in response to inflammatory stimuli such as TLR ligands or proinflammatory cytokines, there is a rapid induction of cyclooxygenase 2 (COX2) and of microsomal PGE2 synthase 1 that results in production and release of PGE2 (for review, see Refs. 1 and 2). The role of PGE2 in inflammation remains paradoxical, with almost equal numbers of reports indicating pro- and anti-inflammatory functions. This presumably results from the expression of various PGE2 receptors (EP1–4) by different cells and from the existence of several independent signaling pathways for some of these receptors (for review, see Refs. 3–5).
In vitro experiments support an anti-inflammatory role, with PGE2 inhibiting the expression of most proinflammatory cytokines and chemokines in innate immune cells, reducing T cell proliferation and inhibiting differentiation into Th1 cells (for reviewed, see Refs. 5 and 6). However, in vivo experiments using mice deficient in the various components of the arachidonic acid → PGE2 axis, such as cytosolic cPLA2α, COX2, microsomal PGE2 synthase 1, and EP4, or wild type mice treated with specific EP receptor agonists or antagonists showed resistance or reduced disease symptoms in models of arthritis, experimental autoimmune encephalomyelitis (EAE), and cerebral ischemia (for review, see Refs. 1 and 7–9).
Th17 effector cells have been shown recently to play an important role in autoimmune diseases such as rheumatoid arthritis and multiple sclerosis (10–12). Although originally Th17 differentiation from naïve CD4+ T cells was thought to depend on IL-6 and TGFβ1, with IL-23 being required only for the expansion and maintenance of the Th17 phenotype, recent developments changed this paradigm. In models of colitis and EAE, a new type of Th17 cells that co-express Rorγt and Tbet and therefore are IL-17+IFNγ+, were generated in the presence of IL-23 and absence of TGFβ1 and shown to accumulate in the intestine and CNS, respectively (13, 14). Recently, most of the IFNγ-producing T cells that migrated to the spinal cord of EAE mice were shown to have originated from T cells that produced IL-17 before their conversion by IL-23 (15). In EAE, in addition to Th17 cells, IL-23 also targets a subset of pathogenic γδT cells that express IL-23R constitutively and suppress the generation and function of Foxp3+ Treg (16). All this recent information points to IL-23 as a central proinflammatory cytokine involved in inflammatory and autoimmune diseases (for review, see Ref. 17).
IL-23 is a heterodimer consisting of the unique p19 subunit and p40, a subunit shared with IL-12 (18). IL-23 is produced primarily by stimulated antigen-presenting cells including macrophages, dendritic cells (DC), monocytes, and microglia through signaling involving PI3K, MAPK, and NFκB (19–21). A number of positive Il23a (p19) transcription factors including c-Rel, AP-1, ATF-2, and SMAD-3 as well as negative regulators such as IRF-1 control IL-23p19 expression in macrophages and/or DC (22–25).
We reported for the first time that PGE2 up-regulated IL-23 while inhibiting IL-12 production in vitro in bone marrow-derived DC and in vivo in models of collagen-induced arthritis and inflammatory bowel disease, resulting in preferential Th17 development and activity (26–29). Up-regulation of IL-23 production by exogenous and endogenous PGE2 and the switch in the IFNγ/IL-17 balance in favor of IL-17 was confirmed in mouse and human DC as well as human T cells (30–33). Little is known about the mechanisms involved in the PGE2 effect on Il23a gene expression. Here, we investigated the PGE2-induced signaling pathways in bone marrow-derived murine DC from EP receptors to transcription factor activation and binding to the p19 promoter.
EXPERIMENTAL PROCEDURES
Mice
6–8-Week-old male B10.A mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and were maintained in the Temple University School of Medicine (Philadelphia, PA) animal facility under pathogen-free conditions. Mice were handled and housed in accordance with the guidelines of the Temple University Animal Care and Use Committee.
Reagents
Prostaglandin E2, LPS (Escherichia coli O26:56), and indomethacin were purchased from Sigma. granulocyte-macrophage colony-stimulating factor and TNFα were purchased from Peprotech Inc. (Rocky Hill, NJ). Butaprost, sulprostone, and PGE1OH were purchased from Cayman (Ann Arbor, MI). Dibutyryl cAMP, the exchange protein activated by cAMP (EPAC)-specific activator 8-CPT-2′OMe-cAMP (8′-CPT), forskolin, H89, LY294002, wortmannin, and KT5720 were purchased from Calbiochem. The PKA peptide inhibitor, PKI-(6–22), amide and antibodies to total CEBPβ and phospho-CEBPβ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to rabbit-CREB (clone D76D11), mouse-phospho-CREB (Ser-133) (1B6), mouse-phospho-AKT, and total AKT were purchased from Cell Signaling Technology (Beverly, MA). Antibodies to rabbit GST were purchased from Abcam Inc. (Cambridge, MA).
Bone Marrow-derived DC (BMDC) and DC2.4 Cells
BMDC were generated in vitro from bone marrow cells as previously described (34). The DC2.4 cell was derived from C57BL/6 bone marrow cells and was generously provided by Dr. Kenneth L. Rock (University of Massachusetts Medical Center, Worcester, MA). The cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 2 mm l-glutamine, 50 μm β-mercaptoethanol, and 1% minimum Eagle's medium nonessential amino acids.
IL-23 and IL-12p70 ELISA
Purified CD11c+ BMDC (2 × 106 cells/ml) were treated as described under “Results.” Supernatants collected 8 h later (for IL-23) and 24 h later (for IL-12p70) were assayed by sandwich ELISA. Antibodies for anti-mouse IL-23p19 and biotin-conjugated anti-mouse IL-12/IL-23p40 were purchased from Ebioscience (San Diego, CA). Antibodies for anti-mouse IL-12p70 and biotin-conjugated anti-mouse IL-12p40 were purchased from BD Biosciences.
Detection of IL-23p19 Initial Transcripts
DC were treated as described under “Results” for 1 h, and 1 μg of RNA was reverse-transcribed. The Moloney murine leukemia virus reverse transcriptase was not added to the “No RT” (see Fig. 2) duplicate sample to control for genomic DNA amplification. Sequences for the primers used for measuring p19 primary transcript were as follows: p19 intron #1 sense 5′-TCCTTGAGTCCTTGTGGGT-3′ and p19 exon #2 antisense 5′-AAACCTTCCCAGTCCTCCAAGTGT-3′.
FIGURE 2.
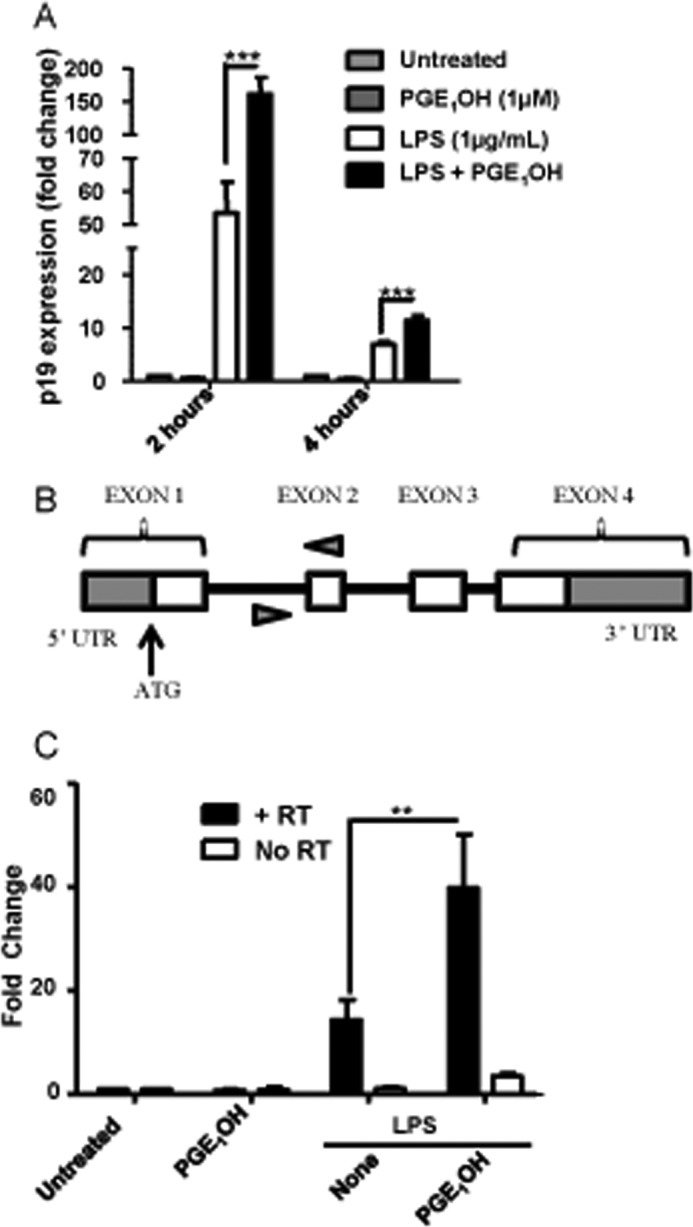
PGE2 affects p19 expression by stimulating de novo mRNA synthesis. A, DC were stimulated with LPS in the presence or absence of PGE1OH. Total RNA was harvested at 2 and 4 h, and expression of IL-23p19 was determined by real-time RT-PCR. B, shown is a schematic of the IL-23p19 primary transcript. Arrows indicate the primers (within intron one and exon two). C, total RNA was extracted from DC stimulated with LPS in the presence or absence of PGE1OH for 1 h and reverse-transcribed. Primers within intron 1 and exon 2 were used for real-time RT-PCR. Control samples with no RT are labeled No RT. **, p < 0.01 compared with LPS alone (C); ***, p < 0.001 compared with LPS alone (A). Data shown are representative of at least two independent experiments.
Real-time PCR
The SYBR Green-based real time PCR technique was used to detect the expression of p19, p40, p35, and COX-2 as previously described (34). Primers used for the real-time PCR reaction were designed using Primer-BLAST. The sequences for each primer are as follows: IL-23p19 sense (5′-TGCTGGATTGCAGAGCAGTAA-3′) and antisense (5′-ATGCAGAGATTCCGAGAGA-3′), IL-12p35 sense (5′-CTGTGCCTTGGTAGCATCTATG-3′) and antisense (5′-GCAGAGTCTCGCCATTATGATTC-3′), IL-12p40 sense (5′-TGGTTTGCCATCGTTTTGCTG-3′) and antisense (5′-ACAGGTGAGGTTCACTGTTTCT-3′), COX-2 sense (5′-CTTAGTTCCGTTTCTCGTGGTCA-3′) and antisense (5′-AACCCAATCAGCGTTTCTCG-3′), and β-actin sense (5′-TCCACCACCACAGCTGAGAGG-3′) and antisense (5′-CAGCTTCTCTTTGATGTCACG-3′). The cycling conditions were 95 °C for 15 s and 60 °C for 1 min for 40 cycles followed by a melting point determination or dissociation curves. The expression level of each gene is indicated by the number of cycles needed for the cDNA amplification to reach a threshold. The amount of DNA is calculated from the number of cycles calibrated to a standard curve to produce copy numbers that are normalized to the housekeeping gene β-actin. Some calculations include an additional normalization to media to show -fold change in expression.
FACS Analysis for Phospho-AKT
Cells were treated as indicated and then fixed, permeabilized, and incubated with anti-rabbit phospho-AKT and anti-rabbit AKT for 40 min at room temperature followed by Alexa-conjugated goat anti-rabbit IgG (Invitrogen) for 30min in the dark at room temperature. Data were collected for 10,000 cells and analyzed by FACS.
Transcription Binding Assay
The protein/DNA array I (Panomics, Santa Clara, CA) was used to assay the levels of 56 different transcription factors (TF) by binding to oligonucleotides that contain the corresponding consensus sequences. Nuclear extracts were isolated by using the nuclear extract kit (Active Motif, Carlsbad, CA) from 8 × 106 DC stimulated with TNFα (100 ng/ml), PGE1OH (1 μm), or a combination of TNFα and PGE1OH for 1 h. 10 μg of nuclear extract proteins were used for the array, as recommended by the manufacturer. Freed probes hybridizing to the array membrane were detected by using IRDye 680 streptavidin antibody (Licor Biosciences, Lincoln, NE), and scanning was done by using the Odyssey Infrared Imaging System (Licor Biosciences).
Chromatin Immunoprecipitation Assay (ChIP)
10−20 million DC were treated and fixed with 1% formaldehyde for 15 min. Fixation was stopped by treatment with 125 mm glycine for 10 min. Cells were washed twice with ice-cold phosphate-buffered saline containing protease inhibitors, collected, and processed as previously described (35). 10% input DNA and immunoprecipitated DNA was PCR-amplified with the following specific primers: distal cAMP-responsive element-binding protein (CREB) site sense (5′-AGCCTGCCGTGTGGTCAT-3′), antisense (5′-TGAAGCTGGGACTCCCCCAACC-3′; proximal CREB site sense (5′-ATAGAAGGCATGACACGGGAACC-3′), antisense (5′-CCGAGGGAAAACCAAAACAGCACTCAT-3′); C/AATT enhancer-binding protein (C/EBP) site −1195 sense (5′- CCAGGGATACACAGAGAAACC-3′), antisense (5′-GTCAGAGTGAGGCCATAGATG-3′); C/EBP site −998 sense (5′-TGGGGTTGGGGGAGTCCCAG-3′), antisense (5′-ACCCGTGCTGGCCTTTCAGC-3′); C/EBP site −496 sense (5′-AGGCATGACACGGGAACCAGACT-3′), antisense (5′-AGAGGCCTAGGTCGGGACACA-3′), C/EBP site −274/−232 sense (5′- TCCCTGCCCCTTCTGCAGTCT), antisense (5′-TAAGGCCCGCCCTTCACACTAG-3′). Results were calculated using ΔΔCt method.
Luciferase Assay
The p19-GL3 vector was a gift from Dr. Youhai H. Chen (University of Pennsylvania). Site-directed mutagenesis of the distal and proximal CREB binding sites and C/EBP sites was performed by using the QuikChange II site-directed mutagenesis kit (Stratagene/Agilent Technologies, Inc. Santa Clara, CA) according to manufacturer's instructions. The CREB dominant negative construct (CREB133) was purchased from Clontech (Mountain View, CA). DC2.4 were transfected with reporter constructs using the GenJet reagent (SignaGen, Rockville, MD). Whole cell lysates were analyzed for luciferase activity using the Dual Luciferase Reporter Assay System (Promega, Madison, WI) according to manufacturer's instructions. Cotransfection with the Renilla luciferase expression vector, pRL-TK (Promega), was used as an internal control. Luciferase units were divided by their renilla control. The value for the empty vector sample was subtracted from the experimental sample values. -Fold change was calculated by dividing experimental values to the untreated (medium) values.
Insertion of Mutations within CREB and C/EBP Binding Sites
The QuikChange® II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to introduce mutations within each CREB and C/EBP binding site according to manufacturer's instructions. Primers designed to introduce the mutations are as follows: distal CREB sense (5′-GGTCATCTATGGCCTCACTCTCAGGACCAGATGGTGC-3′), antisense (5′- GCACCATCTGGTCCTGAGAGTGAGGCCATAGATGACC-3′); proximal CREB sense (5′-AACCAGACTTCGAATCTCAGGACACCATGGGATTTGGAG-3′), antisense (5′-CTCCAAATCCCATGGTGTCCTGAGATTCGAAGTCTGGTT-3′); CEBP −1195 sense (5′-TGAAAGGGGACCCCCATCTCACACTTGTCTCTCAGAGGAAAAG-3′), antisense (5′-TTCCTCTGAGAGACAAGTGTGAGATGGGGGTCCCCTTTCAC-3′); CEBP 998 sense (5′–TGTTACATCATGGAAGGTGCCACTATCAATCAGCCATCAGCGG-3′, antisense (5′-ATGGCTGATTGATAGTGGCACCTTCCATGATGTAACAC-3′); CEBP −496 sense (5′-AAGAGGAAGTGAGGTCGACGGACTGGGGTTGGGTA-3′), antisense (5′-TACCCAACCCCAGTCCGTCGACCTCACTTCCTCTTCCC-3′); CEBP −274 sense (5′-TCTAGCCACAACAACCGAACCGTTCACTTCCCCTGGAACTGAAG-3′, antisense (5′-TTCAGTTCCAGGGGAAGTGAACGGTTCGGTTGTTGTGGCTAGAG-3′); CEBP −232 sense (5′-ATACCTGGGCTCTAGCCTGAGGGAGATGATGTAGGGAG-3′), antisense (5′-TACATCATCTCCCTCAGGCTAGAGCCCAGGTATGCCG-3′).
SDS-PAGE/Western Blot
3–6 million DC were stimulated as indicated under “Results” and lysed with radioimmunoprecipitation assay buffer (50 mm Tris-HCl, pH 8, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS plus Sigma protease inhibitor mixture (Sigma), 5 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, 1 mm sodium fluoride, and 0.2 μm okadaic acid. 20–30 μg of whole protein lysate were mixed with 6× sample buffer and boiled for 7 min. The boiled samples were loaded into 10% SDS-PAGE gels. Separated protein samples were transferred onto polyvinylidene fluoride membranes (Bio-Rad) and probed with primary antibodies at 1:1000 dilutions in Odyssey blocking buffer (LiCor Biosciences). Goat-α-mouse IRDye 800CW and goat-α-rabbit IRDye 680CW antibodies (LICOR Biosciences) were used as secondary antibodies. Transferred proteins were visualized by using the Odyssey infrared image system (LICOR Biosciences).
Statistics
Results are expressed as the mean ± S.D. Comparisons between multiple groups were performed by analysis of variance and followed by the Bonferroni t test. Statistical significance was determined as: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Data were analyzed using Graphpad Prism 5 software.
RESULTS
PGE2 Increases TLR-induced IL-23 in DC
We showed previously that PGE2 can significantly increase LPS-induced IL-23p19 expression and protein production while suppressing IL-12p70 release from bone marrow-derived DC. Consistent with our previous results, we showed here that PGE2 increases p19 and decreases p35 and p40 expression in LPS-stimulated DC (Fig. 1A). Next, we examined the effect of PGE2 on p19 expression in response to other TLR ligands. DC were stimulated with peptidoglycan (TLR2 and -6), poly I:C (TLR3), CpG (TLR9), and LPS (TLR4) in the presence or absence of PGE2. We used PGE2 at 1 μm, the concentration previously determined as optimal (results not shown). Although PGE2 alone did not induce significant p19 expression, it synergized with both extra- and intracellular TLR signaling (Fig. 1B).
FIGURE 1.

PGE2 increases TLR-induced IL-23 in DC. A, purified CD11c+ DC were cultured at a concentration of 3 × 106 cells/ml and stimulated with LPS (1 μg/ml) and PGE2 (1 μm) for 3 and 6 h. Total RNA was isolated, and the expression of IL-23p19 (3 h), IL-12p35 (6 h), and IL-12p40 (6 h) was determined by real-time RT-PCR. B, CD11c+ DC were stimulated with the TLR ligands peptidoglycan (PGN) (40 μg/ml), poly(I:C) (50 μg/ml), LPS (1 μg/ml), and CpG (2 μm) in the presence or absence of PGE2 for 3h. RNA was isolated, and the expression of IL-23p19 was determined by real-time RT-PCR. C, DC were stimulated with LPS in combination with the EP receptor agonists butaprost (10 μm), sulprostone (10 μm), PGE1OH (1 μm), and PGE2 (1 μm) for 3 h. Expression of IL-23p19 was determined by real-time RT-PCR. EP receptor agonist affinities are listed from highest to lowest for each EP receptor. D, DC were stimulated with LPS in the presence or absence of PGE2, PGE1OH, or butaprost. Supernatants were collected 8 h later and subjected to ELISA for IL-23 protein determination. E, DC were stimulated with LPS in the presence or absence of PGE2 or PGE1OH. Supernatants were collected 24 h later and subjected to ELISA for IL-12p70 protein determination. * indicates p < 0.05 compared with LPS alone (D); *** indicates p < 0.001 compared with LPS alone (A, C, D, and E) and corresponding TLR ligand alone (B). Data are representative of at least three independent experiments.
To determine which of the four EP receptors was involved in the effect of PGE2 on p19 expression, we used selective agonists in combination with LPS. We reported previously that murine bone marrow-derived DC expressed primarily EP2 and EP4 (36). Similar to PGE2, treatment with agonists alone did not induce p19 expression. In the presence of LPS, both butaprost and especially PGE1OH led to an increase in p19 expression (Fig. 1C). Butaprost is an EP2-selective agonist, and PGE1OH binds to EP4 and with lower affinity to EP3. Because sulprostone, a selective EP3 agonist, did not increase p19 expression, we concluded that the effect of PGE1OH was mediated through EP4 and that the EP4 receptor was significantly more effective than EP2. Therefore, we used PGE1OH in most of the subsequent experiments. Similar results were obtained in terms of IL-23-secreted protein, with PGE1OH as the major inducer (Fig. 1D). In addition, PGE1OH inhibited IL-12p70 release as the same level as PGE2 (Fig. 1E), supporting the involvement of EP4 in switching the IL-12/IL-23 balance in favor of IL-23. Based on these results, we concluded that the synergistic effect of PGE2 on IL-23 expression and production in LPS-activated DC was mediated primarily through EP4 and to a much lesser degree through EP2.
PGE2 Affects p19 Expression by Stimulating de Novo mRNA Synthesis
Steady-state mRNA levels depend both on de novo synthesis and mRNA stability. Time course experiments using mRNA isolated at 2 versus 4 h showed a rapid and similar decrease in p19 mRNA for LPS and LPS + PGE1OH treatments, arguing against a PGE1OH effect on p19 mRNA stability (Fig. 2A). To investigate if the PGE1OH-induced increase in p19 expression is due to de novo synthesis, DC stimulated with LPS + PGE1OH for 1 h were subjected to total RNA isolation, and the amounts of p19 mRNA initial transcripts were determined by RT-PCR using primers within intron one and exon two of the Il23a gene (Fig. 2B). We observed an increase in p19 initial transcripts after LPS stimulation that was significantly augmented in the presence of PGE1OH (Fig. 2C). To rule out the contribution of genomic DNA, duplicate samples with no reverse transcriptase were used as a control. These data indicate that signaling through the EP4 receptor in addition to TLR stimulation increases the de novo transcription of Il23a in bone marrow-derived DC.
PGE1OH Increases IL-23p19 through Induction of cAMP in TNFα-stimulated DC
LPS signaling through TLR4 involves a complex network of regulatory molecules that makes the identification of signaling pathways initiated by EP4 during the combined LPS + PGE1OH treatment quite difficult. Therefore, we decided on a simplified approach using TNFα stimulation instead of LPS. TNFα has been described as an essential component of the cytokine mixture inducing DC maturation (37). Similar to LPS, the combination of TNFα + PGE2, strongly synergized in terms of p19 induction, induced a slight increase in p40 and did not significantly affect p35 expression (Fig. 3, A and B). It is worth mentioning, however, that the levels of p40 and p35 induced by TNFα alone or in combination with PGE1OH are much lower compared with LPS.
FIGURE 3.

PGE1OH increases IL-23p19 through induction cAMP in TNFα-stimulated DC. A, DC were stimulated with TNFα (100 ng/ml) and PGE1OH (1 μm) for 3 and 6 h. Total RNA was isolated, and the expression of IL-23p19 (3 h), IL-12p35 (6 h), IL-12p40 (6 h) was determined by real-time RT-PCR. B, DC were stimulated with TNFα in combination with EP receptor agonists butaprost (10 μm), sulprostone (10 μm), PGE1OH (1 μm), and PGE2 (1 μm) for 3 h. Expression of IL-23p19 was determined by real-time RT-PCR. C and D, DC were stimulated for 3h with TNFα in the presence or absence of PGE1OH and forskolin (10 μm) (C) or dibutyryl cAMP (dbcAMP) (10 μm, 100 μm) (D). Expression of IL-23p19 was determined by real-time RT-PCR. E, DC were pretreated for 30 min with wortmannin (0.1 μm, 1 μm) or LY294002 (1 μm) followed by stimulation with TNFα in the presence or absence of PGE1OH for 3 h. Expression levels of IL-23p19 were determined by real-time RT-PCR. Data are representative of at least three independent experiments. * indicates p < 0.05 compared with TNFα alone (D), ** indicates p < 0.01 compared with TNFα alone (A and D), and *** indicates p < 0.001 compared with TNFα alone (B and C) and to TNFα + PGE1OH (E). N.S. indicates no statistical difference compared with TNFα alone (A). Results are representative of three independent experiments.
Once again, PGE1OH and butaprost, but not sulprostone, induced p19 expression (Fig. 3B). Similar to LPS, PGE1OH + TNFα induced more initial p19 transcripts than TNFα alone, supporting IL-23p19 up-regulation at the transcriptional level (results not shown).
Next we explored the signaling pathway downstream of the EP4 receptor. The EP4 receptor has been reported to activate both adenylate cyclase and PI3K (4). To analyze the role of adenylate cyclase and cAMP, we used two chemical activators in combination with TNFα. Both forskolin, a chemical activator of adenylate cyclase, and dibutyryl cAMP, a stable analog of cAMP, increased p19 expression (Fig. 3, C and D). Similar results were obtained for LPS-stimulated DC (supplemental Fig. 1). These data show that EP4 receptor activation utilizes a cAMP-dependent signaling pathway to induce IL-23p19 mRNA expression in DC. The second pathway activated by the EP4 receptor involves the recruitment of β-arrestin and activation of the PI3K pathway. To investigate whether the PI3K signaling pathway was involved in the induction of p19 expression, we used two chemical inhibitors of PI3K, wortmannin and LY2940032. Instead of reducing p19 expression, the PI3K inhibitors actually increased TNFα + PGE1OH induced p19 expression (Fig. 3E). We observed the same phenomenon in LPS + PGE1OH-treated DC, although the inhibitors were able to inhibit AKT phosphorylation (supplemental Fig. 2). These data indicate that the EP4-induced PI3K activity does not contribute positively to the induction IL-23p19 mRNA expression in DC. In addition, p38, ERK1/2, and JNK inhibitors did not reduce p19 induction by the TNFα + PGE1OH treatment (supplemental Fig. 3), suggesting that MAPKs do not play an essential role.
EP4 Signals through EPAC and PKA to Mediate PGE1OH Induction of IL-23p19
cAMP activates two independent downstream signaling pathways that involve PKA and a more recently described GTPase named EPAC. To evaluate the contribution of EPAC activation, we used 8-CPT-2′-O-Me-cAMP, a selective activator of EPAC. In combination with TNFα, the EPAC dose-dependently increased p19 expression (Fig. 4A). The role of PKA was determined by using three PKA inhibitors, i.e. the chemical inhibitors H89 and KT5720 and the peptide inhibitor PKI-(6–22). All three inhibitors reduced p19 induction by TNFα + PGE1OH (Fig. 4, B and D). These data indicate that PGE1OH induces IL-23p19 expression through the activation of both EPAC and PKA.
FIGURE 4.

EP4 signaling through EPAC and PKA mediates PGE1OH induction of IL-23p19. A, DC were stimulated with TNFα in the presence or absence of PGE1OH or the EPAC activator 8′-CPT (100 μm, 250 μm). B–D, DC were pretreated with the PKA inhibitors H89 (B), KT5720 (KT) (C), and PKI-(5–24) (PKI) (D) for 3 h. Expression levels of IL-23p19 were determined by real-time RT-PCR. ** indicates p < 0.01 compared with TNFα + PGE1OH (B and C), and *** indicates p < 0.001 compared with TNFα alone (A) and to TNFα + PGE1OH (B–D). Data are representative of three independent experiments.
PGE1OH Induces CREB and C/EBP in DC
Several TFs including c-Rel, AP-1, SMAD-3, and ATF-2, have been reported to bind to the murine p19 promoter and play essential roles in Il23a transcription. We used a protein/DNA array to evaluate changes in the levels of a number of TF after treatment of DC with TNFα + PGE1OH as compared with TNFα alone. Treatment with PGE1OH increased the levels of AP-1, NF-κB, SMAD3/4, STAT3, and IRF-1, previously described as participants in Il23a expression. In addition, several other potential positive TF were identified (Fig. 5A). Among those, the CREB and C/EBP, previously not identified as regulators of Il23a transcription, have predicted binding sites within the p19 promoter (Fig. 5B). CREB is a classical PKA target, and C/EBPβ phosphorylation has been recently linked to EPAC activation (38, 39). Because PGE1OH stimulation of p19 expression appears to be mediated through both PKA and EPAC, we investigated CREB and C/EBP as potential PGE1OH targets in the stimulation of p19 transcription.
FIGURE 5.

PGE1OH induces CREB and C/EBP. A, purified CD11c + DC were stimulated with TNFα in the presence or absence of PGE1OH for 1 h. Nuclear extracts were prepared and subjected to a protein/DNA array as described under “Experimental Procedures.” B, the nucleotide sequence for the IL-23p19 promoter region (NCBI reference sequence) is shown. Putative transcription factor binding sites (boxed-in sequences) were identified by using predictive algorithm-based programs listed under “Experimental Procedures.” Underlined sequences refer to binding sites for transcription factors previously described for IL-23p19 regulation.
PGE1OH-induced CREB Phosphorylation Is Required for Increased p19 Expression
CREB phosphorylation is required for its nuclear translocation and function as a TF. To assess whether PGE1OH induces CREB phosphorylation, we treated DC with PGE1OH, TNFα, or TNFα + PGE1OH and determined the amounts of PCREB by Western blot using an antibody that recognizes CREB phosphorylated at Ser-133. Although both PGE1OH and TNFα induced PCREB, PGE1OH acted at an earlier time point as compared with TNFα (Fig. 6A). Next, we treated DC with PGE1OH in the presence or absence of the PKA inhibitor H89, and as expected observed a significant decrease in CREB phosphorylation in the presence of H89. In contrast to PKA, EPAC activation did not lead to CREB phosphorylation (Fig. 6B). Based on these results we concluded that PGE1OH induces CREB Ser-133 phosphorylation through the activation of PKA.
FIGURE 6.

PGE1OH-induced CREB phosphorylation is required for increased p19 expression. A, DC were treated with TNFα±PGE1OH. Total cell lysates were prepared at different time points and subjected to SDS-PAGE/Western blotting analysis with antibodies specific for Ser-133 phosphorylated CREB and total CREB. Bands were quantified by scanning densitometry. B, DC were treated with PGE1OH with or without the PKA inhibitor H89 (20 μm) or with the EPAC activator, 8′CPT (500 μm), for 15 min. Total cell lysates were subjected to Western blotting with antibodies specific for phosphorylated CREB and total CREB. Bands were quantified by scanning densitometry. C, DC2.4 cells were transfected with CREB dominant negative constructs along with p19-luciferase and Renilla (pRL-TK) constructs. Cells were stimulated with TNFα±PGE1OH for 14 h. Luciferase units were divided by their renilla control, and the empty vector control was then subtracted. -Fold change was calculated by dividing each experimental value by the value for untreated (medium) cells. Reporter activity is presented as -fold change. * indicates p < 0.05 compared with TNFα + PGE1OH, and ** indicates p < 0.01 compared with TNFα alone. Results are representative of at least three independent experiments.
To assess the role of PCREB in the effect of PGE1OH on p19 expression, we transiently transfected the DC2.4 cell line with the p19 reporter construct p19-GL3 that consists of the firefly luciferase gene under the control of the p19 promoter (23). Cotransfections with CREB133, a dominant negative CREB construct that cannot be phosphorylated due to a point mutation changing serine to alanine at position 133, were also performed. TNFα or PGE1OH alone did not induce reporter activity, whereas the TNFα + PGE1OH treatment resulted in significant induction of luciferase (Fig. 6C). Cotransfection with the dominant negative CREB resulted in reduction of reporter activity to almost control levels (Fig. 6C). Altogether, these data indicate that phosphorylated CREB is an essential factor for PGE1OH induction of IL-23p19 expression in DC.
CREB Is Important for IL-23p19 Transcriptional Activity
Through sequence analysis two putative CREB binding sites were identified in the p19 promoter cloned into the GL3 plasmid, i.e. the proximal site at positions −563 to −572 and the distal site at −1125 to −1132. To examine their function, we mutated either site or both, and DC2.4 cells were transfected with the mutated constructs. Mutations of the proximal CREB binding site did not affect reporter activity after treatment with TNFα + PGE1OH. However, mutations within the distal CREB site or in both distal and proximal sites significantly reduced reporter activity (Fig. 7A).
FIGURE 7.

CREB is important for IL-23p19 transcriptional activity. A, DC2.4 cells were transfected with the p19-luciferase construct (p19-GL3), pRL-TK construct, and constructs containing mutations within the distal CREB binding site, the proximal CREB binding site, or both CREB binding sites of p19-GL3. Six hours later the cells were stimulated with TNFα±PGE1OH for 14 h. Luciferase units were divided by their renilla control, and the empty vector control was then subtracted. -Fold change was calculated by dividing each experimental value by the value for untreated (medium) cells. Reporter activity is presented as -fold change. B and C, DC were treated with TNFα in the presence or absence of PGE1OH for 2 h. Cells were fixed, sonicated, and subjected to ChIP analysis using antibodies for CREB (black bar) or control GST (white bar). Precipitated DNA was isolated and evaluated by PCR using specific primers for the proximal (−563) and distal (−1125) CREB binding site. **, p < 0.01 and ***, p < 0.001, compared with TNFα (B) and to p19-GL3 (A). N.S., not significant. Data are representative of four independent experiments.
To examine CREB binding to the IL-23p19 promoter, we performed ChIP assays in primary DC. There was no significant CREB binding to either site upon treatment with TNFα alone, but we observed increased CREB binding to the distal site after treatment with PGE1OH alone. Treatment with TNFα + PGE1OH resulted in significant binding to both sites (Fig. 7B). Taken together, these results indicate that the CREB distal site is an important regulator of PGE1OH-induced IL-23p19 transcription.
EPAC Activates C/EBPβ and Induces in Vivo C/EBPβ Binding to the IL-23p19 Promoter
Results from the protein/DNA array demonstrated that higher levels of C/EBP were bound to DNA after stimulation with TNFα + PGE1OH as compared with TNFα alone. To investigate if activation of EPAC, a downstream effector of PGE1OH, alters C/EBPβ phosphorylation, we performed Western blots. Both TNFα and 8′-CPT (a selective EPAC activator) induced C/EBPβ phosphorylation, although with different kinetics, i.e. maximum effect for TNFα at 5 min as opposed to 8′-CPT, which had a stronger effect at 60 min. The combined treatment resulted in sustained C/EBPβ phosphorylation over the entire 60 min (Fig. 8A). Stimulation with PGE1OH alone or in combination with LPS or TNFα also induced C/EBPβ phosphorylation (supplemental Fig. 4). These data suggest that EPAC stimulation can modify C/EBPβ phosphorylation status in DC.
FIGURE 8.

EPAC stimulates p19 transcriptional activity through activation of C/EBPβ. A, DC were treated with TNFα ± 8′-CPT for 5 and 60 min. Total cell lysates were subjected to SDS-PAGE/Western blotting analysis with antibodies specific for phosphorylated C/EBPβ and β-actin. Bands were quantified by scanning densitometry. B, DC2.4 cells were transfected with empty vector, pRL-TK construct, p19-GL3, and p19-GL3 with mutations within C/EBP binding sites. Six hours later the cells were stimulated with TNFα with or without 8′-CPT for14 h. Luciferase units were calculated as in Figs. 6 and 7. Reporter activity is presented as -fold change. C and D, DC were treated with TNFα and 8′-CPT for 2 h. Cells were fixed, sonicated, and subjected to ChIP analysis using antibodies to C/EBPβ (black bar) or control GST (white bar). Precipitated DNA was isolated and evaluated by PCR using specific primers for C/EBPβ binding sites at −496 (C) and −274 and −232 (D) within the IL-23p19 promoter. **, p < 0.01 and ***, p < 0.001, compared with untreated (C and D) and to p19-GL3 (B) Data are representative of at least three independent experiments.
Five potential C/EBP binding sites were identified in the p19 promoter (Fig. 5B). To investigate their contribution to IL-23p19 transcriptional activity, we mutated each site, and the mutated constructs were transfected into DC2.4. Mutations in the four more proximal C/EBP sites (site 1, −232 to −243; site 2, −274 to −286; site 3, −496 to −509; site 4, −998 to −1011) reduced the reporter activity after TNFα + 8′-CPT treatment to control levels. In contrast, mutations in the most distal site (site 5, −1195 to −1204) had much less of an effect (Fig. 8B). Altogether, these results show that C/EBPβ is an important regulator of IL-23p19 promoter transcriptional activity.
To analyze binding of C/EBP to the IL-23p19 promoter, we performed ChIP assays in primary DC stimulated with TNFα and 8′-CPT. The binding to the two most distal sites (4 and 5) was not consistent. Interestingly, TNFα and 8′-CPT induced C/EBP binding at different sites, i.e. site 3 (−496 to −509) in response to TNFα stimulation and sites 1 and 2 (−232 to −243 and −274 to −286) for 8′-CPT (Fig. 8, C and D).
Endogenous PGE2 Plays a Role in IL-23-p19 Up-regulation
Because LPS stimulation results in production of PGE2, we investigated whether endogenous PGE2 plays a role in IL23p19 up-regulation in DC. DC were stimulated with LPS or TNFα in the presence or absence of the Cox1/2 inhibitor indomethacin, and p19 expression was determined by RT-PCR. In parallel experiments the effect of indomethacin was also tested in DC treated with either LPS + PGE1OH or TNFα + PGE1OH. As expected, in the absence of exogenous PGE1OH, LPS, but not TNFα, induced Cox2 and p19 expression (Fig. 9, A and B). Indomethacin partially inhibited LPS-induced p19 expression, suggesting that LPS-induced endogenous PGE2 plays a role in p19 up-regulation. The addition of exogenous PGE1OH increased the levels of both Cox2 and p19 expression in both LPS- and TNFα-stimulated DC, and again, indomethacin had an inhibitory effect on p19 expression (Fig. 9, A and B). These results support a role for endogenous PGE2 in the up-regulation of p19 expression.
FIGURE 9.
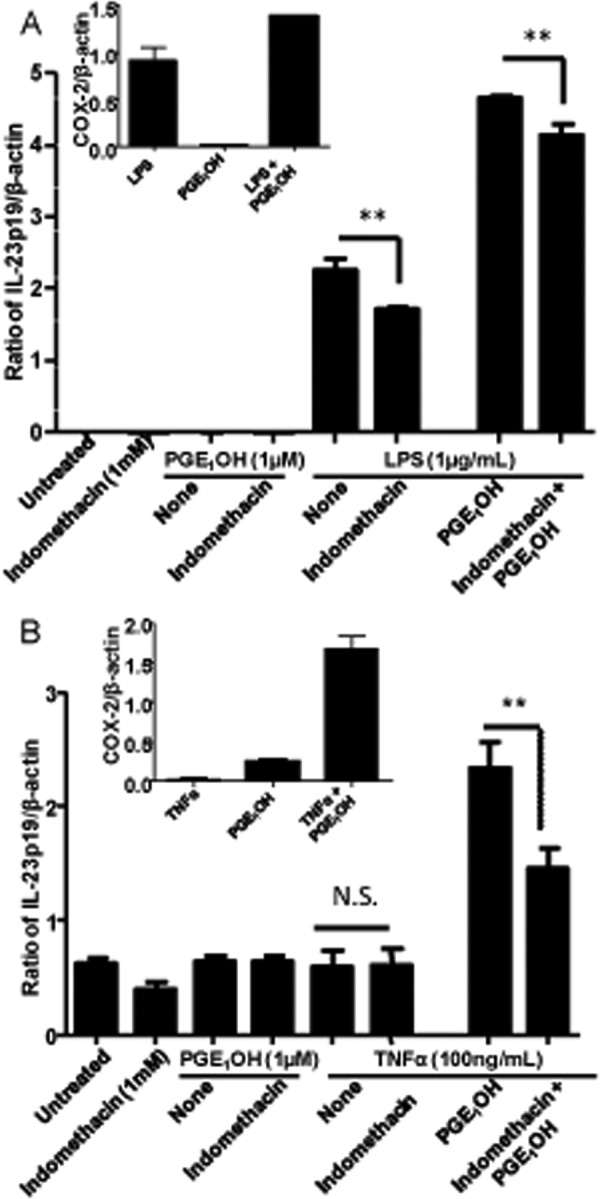
Effect of endogenous PGE2 on IL-23p19 expression. DC were pretreated for 30 min with indomethacin (1 mm). Cells were stimulated with LPS (1 μg/ml) or (A) TNFα (100 ng/ml) (B) in the presence or absence of PGE1OH (1 μm) for 3 h. Expression levels of IL-23p19 and Cox2 were determined by RT-PCR. ** indicates p < 0.01 compared with LPS alone, LPS + PGE1OH, and TNFα + PGE1OH. N.S., not significant. Data are representative of two independent experiments.
DISCUSSION
Previously we reported that PGE2 induced IL-23p19 expression in LPS-stimulated bone marrow-derived murine DC and promoted Th17 amplification in vivo and in vitro (26–29). Similar results were obtained in PGE2-treated splenic DC stimulated through CD40 (33) and in human monocyte-derived DC exposed to Neisseria gonorrhoeae (32). Interestingly, IL-23 production by TLR-activated DC was up-regulated >40-fold in aged mice through increased release of endogenous PGE2 (31). We and others reported that the stimulatory effect of PGE2 on IL-23 induction in DC was mediated primarily through EP4 and cAMP (28, 33).
In this study we sought to identify the signaling pathways and the transcription factors involved in the stimulatory effect of PGE2 on Il23a gene expression. We established that signaling primarily through EP4 results in a cAMP-dependent activation of PKA and EPAC, with both pathways contributing to IL-23p19 induction in DC through the activation of CREB and C/EBP, respectively. This is the first report of CREB and C/EBP involvement in Il23a promoter activation. Mutations within the CREB and C/EBP binding sites combined with ChIP assays identified the distal CREB site (−1125) and the two proximal C/EBP sites (−274 and −232) as essential for PKA-activated CREB- and EPAC-activated C/EBP-induced p19 expression.
Although other signaling pathways involving PI3K, ERK, JNK, and p38 MAPK, have been shown to contribute to IL-23p19 induction in various cell types after stimulation with TLR ligands, IL-17, angiotensin II, or serum amyloid A (19, 20, 40, 41), our results showed no involvement of MAPK or PI3K in IL-23p19 induction after activation of the EP4 receptor.
The effects of PGE2 are mediated through four G-protein coupled receptors, i.e. EP1–4, which use different signaling pathways (4). Immune cells, including DC, express primarily EP4 and EP2 (36). Whereas EP2 receptors are Gs-protein-coupled and signal through activation of adenylate cyclase and increase in cAMP, EP4 uses both Gα protein-mediated adenylate cyclase activation and PI3K activation. In addition, a third pathway is mediated through binding of EP4 receptor-associated protein (EPRAP) to the intracellular EP4 tail resulting in suppression of EP4 phosphorylation and reduction in NFκB activation (42, 43). Activation of adenylate cyclase and PI3K are primarily involved in the proinflammatory effects of PGE2, whereas binding of EP4 receptor-associated protein and inhibition of NFκB appear to be responsible for the anti-inflammatory effect resulting from the inhibition of cytokine and chemokine expression in macrophages, DC, and T cells (for review, see Refs. 5).
Our results indicate that EP4 and to a lesser degree EP2 receptors mediate the PGE2 effect on IL-23p19 expression in DC. The proinflammatory role of EP4 in vivo is supported by numerous reports. EP4-deficient mice showed decreased incidence and disease severity in a model of arthritis (44), and EP4 antagonists were shown to suppress disease in contact hypersensitivity, experimental autoimmune encephalomyelitis, and several models of arthritis by reducing accumulation of both Th1 and Th17 cells (33, 45). In contrast, administration of the PGE2 stable agonist misoprostol or of EP4 selective agonists in models of arthritis and 2,4,6-trinitrobenzenesulfonic acid-induced colitis exacerbated disease increased IL-23p19 expression and promoted Th17 expansion and accumulation in draining lymph nodes and in the affected tissues (27, 29, 45).
EP4 signals through both cAMP and PI3K, and our results indicate that cAMP signaling, but not PI3K activation, is involved in the induction of IL-23p19 by PGE1OH in bone marrow-derived DC. cAMP, but not PI3K, involvement has been also reported for splenic DC (33). Interestingly, both cAMP and PI3K were shown to be involved in the direct effects of PGE2 on T cells, with PI3K facilitating Th1 differentiation in Th1-polarizing conditions and cAMP/PKA facilitating Th17 expansion in the presence of IL-23, presumably through the up-regulation of IL-23R on T cells (33). cAMP activates two major downstream targets, i.e. PKA and EPAC. Our results show that both targets are involved in IL-23p19 expression through the activation of the TF CREB and C/EBPβ, respectively. In contrast, Yao et al. (33) reported that only EPAC contributed to IL-23p19 expression in splenic DC. This could be a characteristic of splenic DC but could also reflect the relative insensitivity of PKA to H89 in DC. Yao et al. (33) did not observe an inhibitory effect for H89 at a concentration of 3 μm. We observed an inhibitory effect only at high H89 concentrations (20 μm), whereas the other two PKA inhibitors were effective over a broader range of concentrations.
Expression of IL-23p19 in activated macrophages or DC has been shown to be controlled by the positive TFs c-Rel, RelA, AP-1, SMAD-3, and ATF-2 and by the negative regulator IRF-1 (22–25, 46). In this study we identified CREB and C/EBP as additional PGE2-induced TFs that enhance IL-23p19 expression in DC activated through TLRs or by the proinflammatory cytokine TNFα. Although PGE2 alone does not induce IL-23p19 expression, it synergizes with LPS or TNFα, suggesting that CREB and C/EBP are part of an enhanceosome consisting of several TFs. Such an enhanceosome consisting of CREB, c-Rel, RelA, NFAT, and SMAD3 has been described for the Foxp3 promoter in T cells (47).
Upon phosphorylation at Ser-133 by kinases such as PKA and MAPK, CREB translocates to the nucleus where it functions as a positive TF for genes whose promoters possess CRE elements (for review, see Refs. 48). CREB can also function as a negative regulator by sequestering CBP or p300, which becomes unavailable for interactions with NFκB, leading to a reduction in NFκB transcriptional activity. The IL-23p19 promoter has two CREB sites, and our results suggest that PGE2 enhances p19 promoter activity through EP4 → cAMP→ PKA → PCREB → binding to the distal CREB site.
Although we observed CREB phosphorylation and a relatively low level of p19 promoter activity after treatment with TNFα alone, we could not detect CREB binding to the p19 promoter. Because TNFα induces delayed CREB phosphorylation as compared with the combined TNFα/PGE1OH treatment, p19 binding of pCREB induced by TNFα alone might be detectable only at later time points.
The C/EBP family consists of six leucine zipper TF factors, i.e. C/EBP α, β, γ, δ, ϵ, and ζ, which bind to the CCAAT box motifs in various promoters (for review, see Refs. 49). C/EBPβ and -δ are the major factors in inflammation, and TNFα as well as LPS have been reported to modulate C/EBPβ transcriptional activity (50). After phosphorylation by various kinases, C/EBPα, -β, and -δ form heterodimers required for DNA binding. Although the three C/EBP family members bind to a virtually identical DNA sequence, there are some differences in binding site specificity, especially for C/EBPβ. Therefore, cell exposure to various stimuli might lead to the activation of different C/EBP family members, which could result in binding to different C/EBP sites within the same promoter. The IL-23p19 promoter has five C/EBP binding sites, and four of those appear to be essential for promoter activity in response to TNFα and PGE1OH. Interestingly, in vivo C/EBPβ shows binding to different sites for TNFα and PGE1OH, as determined by ChIP assays. This could be due to the formation of different dimers after stimulation with either TNFα or PGE1OH or to epigenetic modifications that could open up various parts of the promoter. Alternatively, TNFα- and EPAC-induced C/EBPβ binding might follow different kinetics.
Our results suggest that PGE2 enhances p19 transcriptional activity through a second pathway initiated by EP4, i.e. EP4 → cAMP → EPAC → PC/EBPβ → binding to the two proximal C/EBP sites. Although C/EBP is not among the classical EPAC targets that include primarily the Ras-like GTPases Rap1 and -2 (51, 52), activation of EPAC has been recently reported to lead to C/EBPβ phosphorylation and subsequent SOCS3 induction in endothelial cells and fibroblasts (38, 53).
In conclusion, we identified CREB and C/EBPβ as positive TFs in the PGE2-induced up-regulation of Il23a gene in TLR- or TNFα-stimulated bone marrow-derived DC. The effect is mediated through EP4-induced activation of adenylate cyclase, resulting in cAMP increases, activation of both PKA and EPAC, and subsequent phosphorylation and DNA binding of CREB and C/EBPβ, respectively, to the p19 promoter. PGE2 is released from immune cells in the early inflammatory phase and can act either as a pro- or an anti-inflammatory agent depending on the target cell type, nature of EP receptors, concentration, and inflammatory environment. Although PGE2 has been reported to inhibit proinflammatory cytokine and chemokine production by macrophages and dendritic cells and to reduce Th1 differentiation primarily through the inhibition of IL-12 and IL-2 (for review, see Refs. 5 and 6), most in vivo data support a proinflammatory role (for review, see Refs. 1, 5, and 7–9). This is particularly relevant in models of autoimmune diseases with a strong Th17 component such as arthritis and EAE. In this respect, our previous and present findings that PGE2 contributes in a significant manner to the up-regulation of IL-23 expression and to the amplification of Th17 response are highly relevant, particularly in view of the recent finding that IL-23 plays the central role in the generation of pathogenic Th17 cells (for review, see Refs. 17).
Acknowledgment
We thank Dr. Youhai Chen (University of Pennsylvania) for the generous gift of the p19-GL3 construct.
This work was supported, in whole or in part, by National Institutes of Health Grants 2RO1AI052306, 2T32AI007101-30, and 3RO1AI084065-03S1 (to D. G.) and by Bridge To Doctorate funding (National Science Foundation; to V. P. K.).

This article contains supplemental Figs. 1–4.
- PGE2
- prostaglandin E2
- 8′-CPT
- 8-CPT-2Me-cAMP
- AKT
- protein kinase B
- C/EBP
- C/AATT enhancer-binding protein
- CREB
- cAMP-responsive element binding protein
- EP
- E-prostanoid receptor
- EPAC
- exchange protein activated by cAMP
- PGE1OH
- prostaglandin E1 alcohol
- PKI-(6–22)
- PKA peptide inhibitor amide
- TLR
- Toll-like receptor
- TF
- transcription factor
- EAE
- experimental autoimmune encephalomyelitis
- DC
- dendritic cells.
REFERENCES
- 1. Hara S., Kamei D., Sasaki Y., Tanemoto A., Nakatani Y., Murakami M. (2010) Prostaglandin E synthases. Understanding their pathophysiological roles through mouse genetic models. Biochimie 92, 651–659 [DOI] [PubMed] [Google Scholar]
- 2. Smith W. L., DeWitt D. L., Garavito R. M. (2000) Cyclooxygenases. Structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182 [DOI] [PubMed] [Google Scholar]
- 3. Matsuoka T., Narumiya S. (2007) Prostaglandin receptor signaling in disease. ScientificWorldJournal 7, 1329–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sugimoto Y., Narumiya S. (2007) Prostaglandin E receptors. J. Biol. Chem. 282, 11613–11617 [DOI] [PubMed] [Google Scholar]
- 5. Tang E. H., Libby P., Vanhoutte P. M., Xu A. (2012) Anti-inflammation therapy by activation of prostaglandin EP4 receptor in cardiovascular and other inflammatory diseases. J. Cardiovasc. Pharmacol. 59, 116–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalinski P. (2012) Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liang X., Lin L., Woodling N. S., Wang Q., Anacker C., Pan T., Merchant M., Andreasson K. (2011) Signaling via the prostaglandin E2 receptor EP4 exerts neuronal and vascular protection in a mouse model of cerebral ischemia. J. Clin. Investig. 121, 4362–4371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Narumiya S. (2009) Prostanoids and inflammation. A new concept arising from receptor knockout mice. J. Mol. Med. 87, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 9. Sakata D., Yao C., Narumiya S. (2010) Emerging roles of prostanoids in T cell-mediated immunity. IUBMB life 62, 591–596 [DOI] [PubMed] [Google Scholar]
- 10. Becher B., Segal B. M. (2011) T(H)17 cytokines in autoimmune neuroinflammation. Curr. Opin. Immunol. 23, 707–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tang C., Chen S., Qian H., Huang W. (2012) Interleukin-23. As a drug target for autoimmune inflammatory diseases. Immunology 135, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu S., Qian Y. (2012) IL-17/IL-17 receptor system in autoimmune disease. Mechanisms and therapeutic potential. Clin. Sci. 122, 487–511 [DOI] [PubMed] [Google Scholar]
- 13. Ahern P. P., Schiering C., Buonocore S., McGeachy M. J., Cua D. J., Maloy K. J., Powrie F. (2010) Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 33, 279–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ghoreschi K., Laurence A., Yang X. P., Tato C. M., McGeachy M. J., Konkel J. E., Ramos H. L., Wei L., Davidson T. S., Bouladoux N., Grainger J. R., Chen Q., Kanno Y., Watford W. T., Sun H. W., Eberl G., Shevach E. M., Belkaid Y., Cua D. J., Chen W., O'Shea J. J. (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-β signaling. Nature 467, 967–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hirota K., Duarte J. H., Veldhoen M., Hornsby E., Li Y., Cua D. J., Ahlfors H., Wilhelm C., Tolaini M., Menzel U., Garefalaki A., Potocnik A. J., Stockinger B. (2011) Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petermann F., Rothhammer V., Claussen M. C., Haas J. D., Blanco L. R., Heink S., Prinz I., Hemmer B., Kuchroo V. K., Oukka M., Korn T. (2010) γδT cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity 33, 351–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duvallet E., Semerano L., Assier E., Falgarone G., Boissier M. C. (2011) Interleukin-23. A key cytokine in inflammatory diseases. Ann. Med. 43, 503–511 [DOI] [PubMed] [Google Scholar]
- 18. Oppmann B., Lesley R., Blom B., Timans J. C., Xu Y., Hunte B., Vega F., Yu N., Wang J., Singh K., Zonin F., Vaisberg E., Churakova T., Liu M., Gorman D., Wagner J., Zurawski S., Liu Y., Abrams J. S., Moore K. W., Rennick D., de Waal-Malefyt R., Hannum C., Bazan J. F., Kastelein R. A. (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725 [DOI] [PubMed] [Google Scholar]
- 19. Li Y., Chu N., Hu A., Gran B., Rostami A., Zhang G. X. (2008) Inducible IL-23p19 expression in human microglia via p38 MAPK and NF-κB signal pathways. Exp. Mol. Pathol. 84, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Migita K., Koga T., Torigoshi T., Motokawa S., Maeda Y., Jiuchi Y., Izumi Y., Miyashita T., Nakamura M., Komori A., Ishibashi H. (2010) Induction of interleukin-23 p19 by serum amyloid A (SAA) in rheumatoid synoviocytes. Clin. Exp. Immunol. 162, 244–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhu L., Wu Y., Wei H., Xing X., Zhan N., Xiong H., Peng B. (2011) IL-17R activation of human periodontal ligament fibroblasts induces IL-23 p19 production. Differential involvement of NF-κB versus JNK/AP-1 pathways. Mol. Immunol. 48, 647–656 [DOI] [PubMed] [Google Scholar]
- 22. Al-Salleeh F., Petro T. M. (2008) Promoter analysis reveals critical roles for SMAD-3 and ATF-2 in expression of IL-23 p19 in macrophages. J. Immunol. 181, 4523–4533 [DOI] [PubMed] [Google Scholar]
- 23. Carmody R. J., Ruan Q., Liou H. C., Chen Y. H. (2007) Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J. Immunol. 178, 186–191 [DOI] [PubMed] [Google Scholar]
- 24. Liu W., Ouyang X., Yang J., Liu J., Li Q., Gu Y., Fukata M., Lin T., He J. C., Abreu M., Unkeless J. C., Mayer L., Xiong H. (2009) AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J. Biol. Chem. 284, 24006–24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sheikh S. Z., Kobayashi T., Matsuoka K., Onyiah J. C., Plevy S. E. (2011) Characterization of an interferon-stimulated response element (ISRE) in the Il23a promoter. J. Biol. Chem. 286, 1174–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khayrullina T., Yen J. H., Jing H., Ganea D. (2008) In vitro differentiation of dendritic cells in the presence of prostaglandin E2 alters the IL-12/IL-23 balance and promotes differentiation of Th17 cells. J. Immunol. 181, 721–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sheibanie A. F., Khayrullina T., Safadi F. F., Ganea D. (2007) Prostaglandin E2 exacerbates collagen-induced arthritis in mice through the inflammatory interleukin-23/interleukin-17 axis. Arthritis Rheum. 56, 2608–2619 [DOI] [PubMed] [Google Scholar]
- 28. Sheibanie A. F., Tadmori I., Jing H., Vassiliou E., Ganea D. (2004) Prostaglandin E2 induces IL-23 production in bone marrow-derived dendritic cells. FASEB J. 18, 1318–1320 [DOI] [PubMed] [Google Scholar]
- 29. Sheibanie A. F., Yen J. H., Khayrullina T., Emig F., Zhang M., Tuma R., Ganea D. (2007) The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23 → IL-17 axis. J. Immunol. 178, 8138–8147 [DOI] [PubMed] [Google Scholar]
- 30. Chizzolini C., Chicheportiche R., Alvarez M., de Rham C., Roux-Lombard P., Ferrari-Lacraz S., Dayer J. M. (2008) Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood 112, 3696–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Myer R. G., El Mezayen R., High K. P. (2010) Prostaglandin E2-dependent IL-23 production in aged murine dendritic cells. Exp. Gerontol. 45, 834–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stefanelli P., Teloni R., Carannante A., Mariotti S., Nisini R., Gagliardi M. C. (2012) Neisseria gonorrhoeae triggers the PGE2/IL-23 pathway and promotes IL-17 production by human memory T cells. Prostaglandins Other Lipid Mediat. 99, 24–29 [DOI] [PubMed] [Google Scholar]
- 33. Yao C., Sakata D., Esaki Y., Li Y., Matsuoka T., Kuroiwa K., Sugimoto Y., Narumiya S. (2009) Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 15, 633–640 [DOI] [PubMed] [Google Scholar]
- 34. Yen J. H., Khayrullina T., Ganea D. (2008) PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood 111, 260–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yen J. H., Kocieda V. P., Jing H., Ganea D. (2011) Prostaglandin E2 induces matrix metalloproteinase 9 expression in dendritic cells through two independent signaling pathways leading to activator protein 1 (AP-1) activation. J. Biol. Chem. 286, 38913–38923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jing H., Vassiliou E., Ganea D. (2003) Prostaglandin E2 inhibits production of the inflammatory chemokines CCL3 and CCL4 in dendritic cells. J. Leukoc. Biol. 74, 868–879 [DOI] [PubMed] [Google Scholar]
- 37. Jonuleit H., Kühn U., Müller G., Steinbrink K., Paragnik L., Schmitt E., Knop J., Enk A. H. (1997) Proinflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur. J. Immunol. 27, 3135–3142 [DOI] [PubMed] [Google Scholar]
- 38. Sands W. A., Woolson H. D., Yarwood S. J., Palmer T. M. (2012) Exchange protein directly activated by cyclic AMP-1-regulated recruitment of CCAAT/enhancer-binding proteins to the suppressor of cytokine signaling-3 promoter. Methods Mol. Biol. 809, 201–214 [DOI] [PubMed] [Google Scholar]
- 39. Shaywitz A. J., Greenberg M. E. (1999) CREB. A stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem. 68, 821–861 [DOI] [PubMed] [Google Scholar]
- 40. Kim H. R., Cho M. L., Kim K. W., Juhn J. Y., Hwang S. Y., Yoon C. H., Park S. H., Lee S. H., Kim H. Y. (2007) Up-regulation of IL-23p19 expression in rheumatoid arthritis synovial fibroblasts by IL-17 through PI 3-kinase-, NF-κB-, and p38 MAPK-dependent signaling pathways. Rheumatology 46, 57–64 [DOI] [PubMed] [Google Scholar]
- 41. Yang L., Du C., Chen T., Li S., Nie W., Zhu W., Fan F., Zhu J., Yan H. (2012) Distinct MAPK pathways are involved in IL-23 production in dendritic cells cocultured with NK cells in the absence or presence of angiotensin II. Mol. Immunol. 51, 51–56 [DOI] [PubMed] [Google Scholar]
- 42. Minami M., Shimizu K., Okamoto Y., Folco E., Ilasaca M. L., Feinberg M. W., Aikawa M., Libby P. (2008) Prostaglandin E receptor type 4-associated protein interacts directly with NF-κB1 and attenuates macrophage activation. J. Biol. Chem. 283, 9692–9703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Takayama K., García-Cardena G., Sukhova G. K., Comander J., Gimbrone M. A., Jr., Libby P. (2002) Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J. Biol. Chem. 277, 44147–44154 [DOI] [PubMed] [Google Scholar]
- 44. McCoy J. M., Wicks J. R., Audoly L. P. (2002) The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J. Clin. Investig. 110, 651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Q., Muramoto K., Masaaki N., Ding Y., Yang H., Mackey M., Li W., Inoue Y., Ackermann K., Shirota H., Matsumoto I., Spyvee M., Schiller S., Sumida T., Gusovsky F., Lamphier M. (2010) A novel antagonist of the prostaglandin E(2) EP(4) receptor inhibits Th1 differentiation and Th17 expansion and is orally active in arthritis models. Br. J. Pharmacol. 160, 292–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mise-Omata S., Kuroda E., Niikura J., Yamashita U., Obata Y., Doi T. S. (2007) A proximal κB site in the IL-23 p19 promoter is responsible for RelA- and c-Rel-dependent transcription. J. Immunol. 179, 6596–6603 [DOI] [PubMed] [Google Scholar]
- 47. Ruan Q., Kameswaran V., Tone Y., Li L., Liou H. C., Greene M. I., Tone M., Chen Y. H. (2009) Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 31, 932–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wen A. Y., Sakamoto K. M., Miller L. S. (2010) The role of the transcription factor CREB in immune function. J. Immunol. 185, 6413–6419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roos A. B., Nord M. (2012) The emerging role of C/EBPs in glucocorticoid signaling. Lessons from the lung. J. Endocrinol. 212, 291–305 [DOI] [PubMed] [Google Scholar]
- 50. Poli V. (1998) The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 273, 29279–29282 [DOI] [PubMed] [Google Scholar]
- 51. Billington C. K., Hall I. P. (2012) Novel cAMP signaling paradigms. Therapeutic implications for airway disease. Br. J. Pharmacol. 166, 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Breckler M., Berthouze M., Laurent A. C., Crozatier B., Morel E., Lezoualc'h F. (2011) Rap-linked cAMP signaling Epac proteins. Compartmentation, functioning, and disease implications. Cell. Signal. 23, 1257–1266 [DOI] [PubMed] [Google Scholar]
- 53. Yarwood S. J., Borland G., Sands W. A., Palmer T. M. (2008) Identification of CCAAT/enhancer-binding proteins as exchange protein activated by cAMP-activated transcription factors that mediate the induction of the SOCS-3 gene. J. Biol. Chem. 283, 6843–6853 [DOI] [PubMed] [Google Scholar]