

Background: Hepatic glucose metabolism is altered in type 2 diabetes mellitus (T2DM).
Results: Detailed kinetic modeling of hepatic glucose metabolism including its hormonal regulation predicts the hepatic T2DM phenotype and hypoglycemia in insulin treatment.
Conclusion: Rigorous insulin therapy increases the risk of hypoglycemic events due to reduced hepatic counter-regulatory capacity.
Significance: Modeling allows to assess the efficiency and risk of hormone-substitution therapies in T2DM.
Keywords: Diabetes, Glucose Metabolism, Hepatocyte, Hormones, Liver Metabolism, Hypoglycemia, T2DM
Abstract
A major problem in the insulin therapy of patients with diabetes type 2 (T2DM) is the increased occurrence of hypoglycemic events which, if left untreated, may cause confusion or fainting and in severe cases seizures, coma, and even death. To elucidate the potential contribution of the liver to hypoglycemia in T2DM we applied a detailed kinetic model of human hepatic glucose metabolism to simulate changes in glycolysis, gluconeogenesis, and glycogen metabolism induced by deviations of the hormones insulin, glucagon, and epinephrine from their normal plasma profiles. Our simulations reveal in line with experimental and clinical data from a multitude of studies in T2DM, (i) significant changes in the relative contribution of glycolysis, gluconeogenesis, and glycogen metabolism to hepatic glucose production and hepatic glucose utilization; (ii) decreased postprandial glycogen storage as well as increased glycogen depletion in overnight fasting and short term fasting; and (iii) a shift of the set point defining the switch between hepatic glucose production and hepatic glucose utilization to elevated plasma glucose levels, respectively, in T2DM relative to normal, healthy subjects. Intriguingly, our model simulations predict a restricted gluconeogenic response of the liver under impaired hormonal signals observed in T2DM, resulting in an increased risk of hypoglycemia. The inability of hepatic glucose metabolism to effectively counterbalance a decline of the blood glucose level becomes even more pronounced in case of tightly controlled insulin treatment. Given this Janus face mode of action of insulin, our model simulations underline the great potential that normalization of the plasma glucagon profile may have for the treatment of T2DM.
Introduction
The human plasma glucose level is kept in a narrow range between minimum values of ∼3 mm after prolonged fasting or extensive muscle activity and maximum values of ∼9 mm reached postprandially (1, 2). Homoeostasis of plasma glucose is crucial for the organism: hyperglycemia results in nonenzymatic glycosylation (commonly referred to as glycation) and thus loss-of-function of proteins (3), glucose-induced oxidative damage (4, 5), and other adverse effects (6, 7). Hypoglycemia, on the other hand, leads to an undersupply of tissues with glucose and is thereby of particular danger for neuronal cells, erythrocytes, and fibroblasts, using glucose as dominant or even exclusive energy-delivering fuel under normal physiological conditions.
The liver is the central organ for the homeostatic control of plasma glucose responding with either net hepatic glucose utilization (HGU)2 or net hepatic glucose production (HGP) depending on the plasma glucose level exceeding or falling below a critical threshold value (set-point) of ∼6 mm. The hepatic glucose metabolism is controlled by several hormones, with insulin and glucagon being the main counteracting players (1, 8).
In T2DM glucose homeostasis is impaired as the hormonal signals do not match the prevalent plasma glucose concentration. The normally observed increase in insulin with increasing plasma glucose (9, 10) as well as the increase in glucagon and epinephrine with decreasing plasma glucose (11, 12), are altered or nonexistent, resulting in an increased HGP (2), which contributes to the observed hyperglycemia in diabetes (13). In addition to the relative insulin deficiency due to an inadequate insulin secretory response, T2DM is also characterized by a gradually increasing insulin resistance (13).
Supplementation of insulin is required for patients with T2DM if other medications fail to control blood glucose levels adequately. One of the most common and severe problems in the insulin treatment of diabetic patients is the occurrence of transient hypoglycemia (14, 15) leading in extreme cases to coma, seizures, or even death. Insulin supplementation is one of the standard treatments in diabetes, but is associated with a higher risk of hypoglycemic events. Several studies reported on a significant increase of hypoglycemic events under insulin treatment aimed at tight control of plasma glucose (7, 15–17). A conclusive molecular-mechanistic explanation for the occurrence of hypoglycemia in diabetes, the increase of hypoglycemic events under strict insulin treatment, and the role of the liver in these settings is lacking so far.
To address this problem, we applied a detailed kinetic model of hepatic glucose metabolism integrated with the hormonal regulation by insulin, glucagon, and epinephrine (18) to simulate metabolic alterations in T2DM. We demonstrate the reliability of our approach by the good agreement between model predictions and experimental data from a multitude of studies. Simulating the effects of various schemes of insulin and glucagon treatment on hepatic glucose metabolism, we provide evidence that the restricted gluconeogenic capacity of the liver may well account for an increased frequency of hypoglycemic episodes in insulin treatment and demonstrate the great potential of normalizing the blood glucagon profile in the treatment of T2DM.
MATERIALS AND METHODS
The kinetic model of human hepatic glucose metabolism in normal subjects is described in detail in Ref. 18. The model comprises all enzymatic reactions and membrane transport processes involved in hepatic glycolysis, gluconeogenesis from lactate, and glycogen metabolism (Fig. 1). The time-dependent changes of metabolite concentrations are described by ordinary differential equations with the constituting enzymatic rate laws taking into account the specific kinetic properties of enzymes in the hepatocyte. In particular, different rate laws hold for the phosphorylated and dephosphorylated form of interconvertible enzymes. The regulatory impact of hormones (insulin, glucagon and epinephrine) on the glucose metabolism is included in a phenomenological manner in that their plasma levels directly determine the phosphorylation status of interconvertible enzymes, and the plasma level of hormones is related to the plasma glucose level by glucose-hormone response (GHR) profiles observed in healthy subjects.
FIGURE 1.

Model of human hepatocyte glucose metabolism comprising glycolysis, gluconeogenesis, and the glycogen pathway, and the spatial reaction compartments: blood, cytosol, and mitochondrion. The figure is reproduced from Ref. 18. Key enzymes of HGP are depicted in red, key enzymes of HGU in green. Enzymes regulated via allosteric mechanisms are marked with red A, interconvertible enzymes with yellow P. Insulin, glucagon, and epinephrine regulate the glucose metabolism by changing the phosphorylation state (γ) of key interconvertible enzymes, with insulin (γ) decreasing, epinephrine (γ+) and glucagon (γ+) increasing γ. Reactions: ALD, aldolase; AK, adenylate kinase; CS, citrate synthase; EN, enolase; FBP1, fructose-1,6-bisphosphatase; FBP2, fructose-2,6-bisphosphatase; GAPDH, glyceraldehyde-phosphate dehydrogenase; G1PI, glucose-1 phosphate-1,6-phosphomutase; G6P, glucose-6-phosphatase; GK, glucokinase; GLUT2, glucose transporter 2; GP, glycogen phosphorylase; GPI, glucose-6-phosphate isomerase; GS, glycogen synthase; LDH, lactate dehydrogenase; MALT, malate transporter; MDH, malate dehydrogenase; NDK, nucleoside diphosphate kinase; PC, pyruvate carboxylase; PEPCK, phosphoenolpyruvate carboxykinase; PEPT, phosphoenolpyruvate transporter; PDH, pyruvate dehydrogenase; PFK1, phosphofructokinase 1 PFK2, phosphofructokinase 2; PGK, phosphoglycerate kinase; PGM, 3-phosphoglycerate mutase; PK, pyruvate kinase; PPASE, pyrophosphate phosphohydrolase; PYRT, pyruvate transporter; TPI, triose-phosphate isomerase; UGT, UTP:glucose-1 phosphate uridylyltransferase. Metabolites: 13P2G, 1,3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; ACoA, acetyl-CoA; Cit, citrate; CoA, coenzyme A; DHAP, dihydroxyacetone phosphate; Fru16P2, fructose 1,6-bisphosphate; Fru26P2, fructose 2,6-bisphosphate; Fru6P, fructose 6-phosphate; Glc, glucose; Glc1P, glucose 1-phosphate; Glc6P, glucose 6-phosphate; GRAP, glyceraldehyde 3-phosphate; Lac, lactate; OA, oxalacetate; Mal, malate; P, phosphate; PEP, phosphoenolpyruvate; PP, pyrophosphate; Pyr, pyruvate; UDP-Glc, UDP-glucose. For detailed description see ”Materials and Methods“ and Ref. 18.
In this study we assumed that the kinetic properties of metabolic enzymes involved in hepatic glucose metabolism remain unchanged in T2DM. The model simulations for T2DM are solely based on the observed alterations in the GHR of insulin, glucagon, and epinephrine in T2DM. Except these changes in the hormone profiles the model is identical to the model of hepatic glucose metabolism in normal subjects. An annotated SBML of the model including all kinetic equations and parameters is available at BioModels (MODEL1209260000).
Altered GHR in T2DM
The GHR functions (Equations 1–3 and Fig. 2, A–C) describe the change in plasma concentration of insulin (inscond), glucagon (glucond), and epinephrine (epicond) with changing plasma glucose concentration (Glc) for normal subjects (cond = normal) and subjects with T2DM (cond = T2DM).
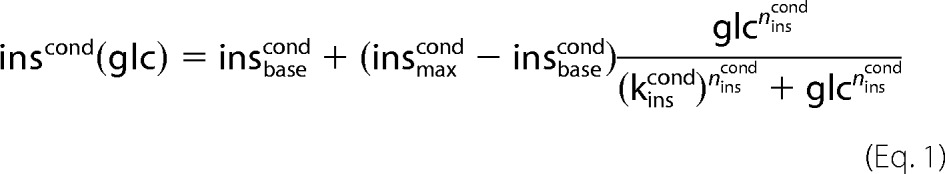 |
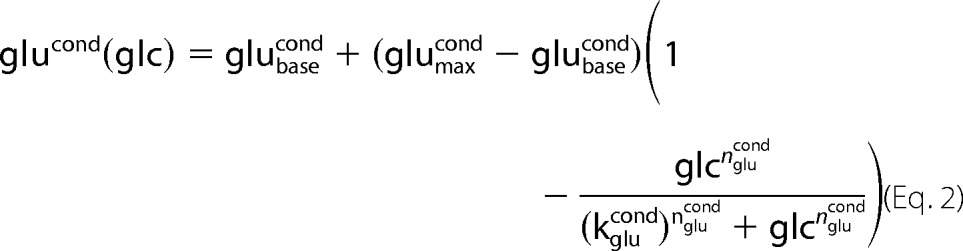 |
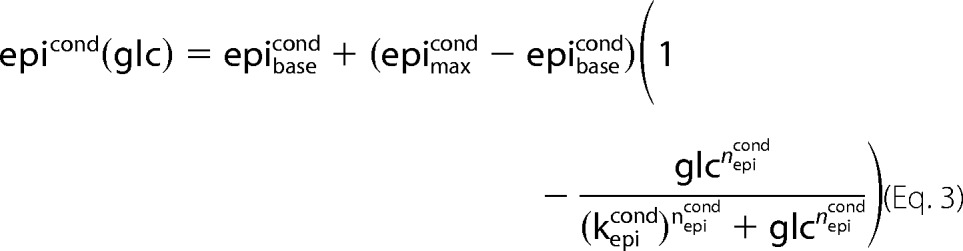 |
All GHR parameters (Table 1) were determined by least-square fit to oral glucose tolerance tests and hypoglycemic, hyperinsulinemic clamp studies in normal subjects (insnormal, glunormal, epinormal) or in subjects with T2DM (insT2DM, gluT2DM, epiT2DM), respectively. The parameters insmaxcond, glumaxcond, epimaxcond denote the maximal, insbasecond, glubasecond, epibasecond the basal hormone concentration. The Hill coefficient ncond determines the steepness of the sigmoid hormone-response curves and the parameter kcond defines the inflection point. Experimental data with standard deviations were extracted from figures, tables, and supplemental information of the references cited in the captions of the corresponding figures. Data points correspond to mean data for multiple subjects from the studies.
FIGURE 2.

GHR, insulin treatments, and phosphorylation state γ. Experimental data and simulated curves for normal subjects are shown in black, for subjects with T2DM in blue. The figure parts regarding normal subjects are reproduced from Ref. 18. All experimental data are from oral glucose tolerance tests and hypoglycemic, hyperinsulinemic clamp studies (1, 9–12, 18, 24, 35–43) (see Table 1) with error bars corresponding to ±S.E. A, glucagon GHR (Equation 2). Glucagon increases with decreasing blood glucose in normal subjects (T2DM subjects) from a basal concentration of 37.9 pmol/liter (50.8 pmol/liter) to a maximal concentration of 190.0 pmol/liter (160 pmol/liter) with the inflection point at 3.01 mm (3.69 mm). The basal glucagon level is increased due to an impaired suppression of glucagon secretion (36, 44, 45). As a consequence of the elevated glucagon level, the counter-regulatory response of the liver to hypoglycemia is impaired (11). In addition, the plasma profile of glucagon is shifted to higher blood glucose concentrations. B, epinephrine GHR (Equation 3). Epinephrine increases with decreasing glucose in normal subjects (T2DM subjects) from a basal concentration of 100 pmol/liter (100 pmol/liter) to a maximal concentration of 6090 pmol/liter (7500 pmol/liter) with the inflection point at 3.1 mm (2.48 mm). The plasma profile of epinephrine is shifted to lower blood glucose concentrations. C, insulin GHR (Equation 1). Insulin increases with increasing glucose in normal subjects (T2DM subjects) from a basal concentration of 0 pmol/liter (0 pmol/liter) to a maximal concentration of 818.9 pmol/liter (280 pmol/liter) with the inflection point at 8.6 mm (10.6 mm). A key characteristic of T2DM is the decrease in relative insulin response (9, 24). D, phosphorylation state (Equation 6) for normal subjects (black) and subjects with T2DM (blue) based on the respective GHRs for insulin, glucagon, and epinephrine. E, insulin concentrations for additive insulin treatments (violet, Equation 4) and linear insulin treatments (green, Equation 5). Insulin GHR for normal subjects (black) and subjects with T2DM (blue) are shown for comparison. F, phosphorylation state (Equation 6) for normal subjects (black), subjects with T2DM (blue), additive insulin treatments (violet), linear insulin treatments (green), the insulin restored case (red), and the glucagon restored case (red dashed).
TABLE 1.
Fitted parameters for insulin, glucagon, and epinephrine GHR with corresponding references for experimental data
| GHR | Max | Base | k | n | Experimental data |
|---|---|---|---|---|---|
| pmol/liter | mm | ||||
| insnormal | insmaxnormal = 818.9 | insbasenormal = 0 | kinsnormal = 8.6 | ninsnormal = 4.2 | 1, 9, 10, 24, 35–39 |
| insT2DM | insmaxT2DM = 280 | insbaseT2DM = 0 | kinsT2DM = 10.6 | ninsT2DM = 6.4 | 1, 9, 10, 24, 35–37 |
| glunormal | glumaxnormal = 190 | glubasenormal = 37.9 | kglunormal = 3.01 | nglunormal = 6.4 | 1, 9–12, 35, 36, 38–42 |
| gluT2DM | glumaxT2DM = 160 | glubaseT2DM = 50.8 | kgluT2DM = 3.69 | ngluT2DM = 5.17 | 1, 9–11, 35, 36, 39, 41 |
| epinormal | epimaxnormal = 6090 | epibasenormal = 100 | kepinormal = 3.1 | nepinormal = 8.4 | 11, 12, 38, 40–43 |
| epiT2DM | epimaxT2DM = 7500 | epibaseT2DM = 100 | kepiT2DM = 2.48 | nepiT2DM = 6.06 | 11, 12, 41 |
Insulin Treatment Schemes
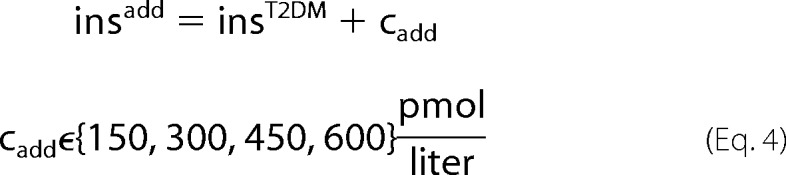
For the simulation of insulin treatment in T2DM two different treatment schemes were applied (see Fig. 2E). Additive treatment (insadd, Equation 4) consists of constant insulin supplementation (cadd) independent of the plasma glucose level to the impaired T2DM insulin response (insT2DM), whereas linear progressive treatment (Equation 5) consists of glucose-dependent insulin supplementation (mlin·glc).
 |
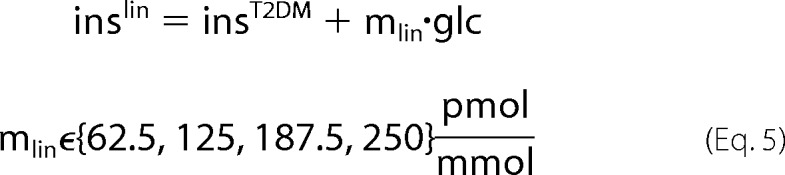 |
Phosphorylation State (γ) of Interconvertible Enzymes
The short term effects of insulin, glucagon, and epinephrine result from changes in the phosphorylation state of key interconvertible enzymes of glucose metabolism, namely GS, GP, PFK2, FBP2, PK, and PDH. Analog to Ref. 18, the relationship between the phosphorylation state γ and the plasma hormone concentrations of insulin (inscond), glucagon (glucond), and epinephrine (epicond) is modeled by the empirical regression function,
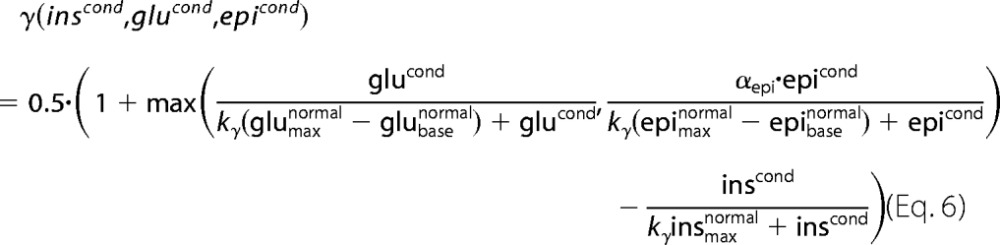 |
with maximal and basal hormone concentrations given in Table 1. To account for the reduced effectiveness of epinephrine compared with glucagon the factor αepi = 0.8 was introduced. The half-saturation parameter of the hormones was set to ky = 0.1. For the given hormone concentrations the phosphorylation state γ was assumed to be equal for all interconvertible enzymes.
The choice of this function is motivated by the measured sigmoid relationship between the phosphorylation state of the interconvertible enzymes like pyruvate kinase and glycogen synthase and the concentration of glucagon and cAMP (19–21). As measurements of the phosphorylation state of other interconvertible enzymes in dependence of plasma hormone levels are lacking so far we applied the regression function (Equation 6) to all interconvertible enzymes of our model, i.e. at given plasma level of insulin, glucagon, and epinephrine all interconvertible enzymes of our model possess the same phosphorylation status. Note that depending on the specific pathological situation considered in our simulations, different values were used for the maximal and basal level of the hormones indicated by the subscripts max base (Table 1).
At given plasma level of glucose, the hormone levels in Equation 6 are determined by the glucose-hormone response functions (Equations 1–3). In case of insulin supplementation therapy these glucose-hormone response functions have to be extended by an additional term describing the specific type of insulin treatment (see above). Depending on the objective of the model simulation, six different combinations of glucose-hormone response profiles were used resulting in the following variants of the phosphorylation function γ: (i) normal glucose-hormone profiles γ(insnormal, glunormal, epinormal) (Fig. 2D, black), (ii) untreated T2DM γ(insT2DM, gluT2DM, epiT2DM) (Fig. 2D, blue), (iii) T2DM with additive insulin supplementation γ(insadd, gluT2DM, epiT2DM) (Fig. 2F, violet), (iv) T2DM with linear insulin supplementation γ(inslin, gluT2DM, epiT2DM) (Fig. 2F, green), (v) T2DM with completely restored normal insulin response γ(insnormal, gluT2DM, epiT2DM) (Fig. 2F, red solid), (vi) T2DM with completely restored normal glucagon response γ(insT2DM, glunormal, epiT2DM) (Fig. 2F, red dashed).
HGP, Gluconeogenesis, and Glycogenolysis in T2DM
Analog to the data for HGP, gluconeogenesis, and glycogenolysis in subjects with normal systemic glucose metabolism (see Table 2 in Ref. 18), data from subjects with T2DM were extracted from the literature (Table 2). Single data points are mean values based on individual values of multiple subjects of one study. The fasting time is measured after administration of specific gluconeogenic substrates by an intravenous infusion, by ingestion, or after ingestion of a mixed meal (2).
TABLE 2.
Experimental data for HGP, GNG, glycogenolysis (GLY) in T2DM after administration of specific gluconeogenic substrates by an intravenous infusion, or by ingestion, or after ingestion of a mixed meal
| Method | Time | HGP | GNG | GLY | GNG/HGP | Ref. |
|---|---|---|---|---|---|---|
| h | μmol/kg/min | % | ||||
| [14C]Acetate | 14 | 22.7 | 12.7 | 10 | 56 | 46 |
| [14C]Bicarbonate | 12–14 | 10.8 | 3.8 | 7 | 35 | 47 |
| 13C NMR | 23 | 11.1 | 9.8 | 1.3 | 88 | 22 |
| 2H2O | 15 | 16.7 | 10.8 | 5.9 | 65 | 48 |
| 2H2O | 17 | 10.4 | 6.9 | 3.1 | 66 | 49 |
| 2H2O | 22 | 7.6 | 5.7 | 1.9 | 69 | 49 |
| 2H2O moderate diabetes mellitus, glucose <10 mm | 16 | 8.6 | 4.8 | 3.8 | 56 | 50 |
| 2H2O severe diabetes mellitus, glucose >10 mm | 16 | 12.6 | 8.5 | 4.1 | 65 | 50 |
| 2H2O mild diabetes | 17.7 | 11.9 | 5.8 | 67 | 51 | |
| 2H2O severe diabetes | 24.1 | 15.7 | 8.3 | 65 | 51 | |
| 2H2O | 15 | 16.7 | 11.2 | 5.5 | 67 | 52 |
| 2H2O | 16 | 15.2 | 9.4 | 5.8 | 62 | 48 |
Glycogenolysis and Glycogen Synthesis in T2DM
In addition to data for normal glucose metabolism (18), data for subjects with T2DM (supplemental Table S1) were extracted from (22).
Hepatic Glucose Response Coefficient (HGRC)
HGRC (Equation 7) measures the change of the stationary net hepatic glucose exchange rate with the blood (represented by the flux through the glucose transporter GLUT2) elicited by a small change in blood glucose concentration.
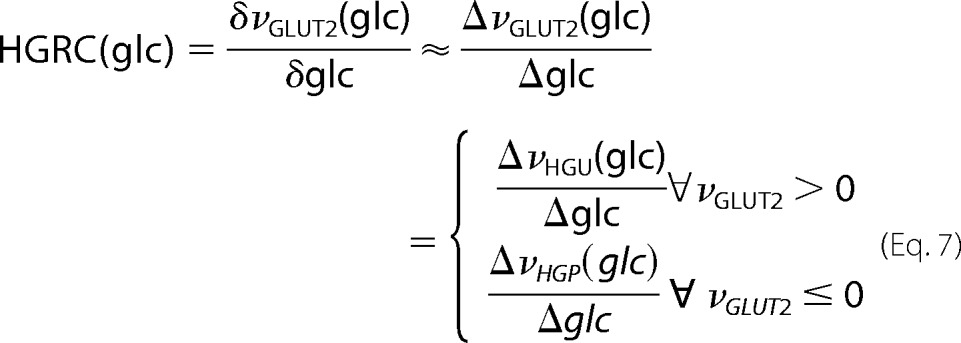 |
The HGRC is a measurement of the counter-regulatory capacity of the liver to react to changes in blood glucose with changes in HGP or HGU, respectively. The HGRC was calculated numerically from the steady state GLUT2 fluxes.
RESULTS
We analyzed the changes in hepatic metabolism in T2DM by using a detailed kinetic model of human hepatic glucose metabolism that includes the hormonal regulation of key regulatory enzymes by reversible phosphorylation/dephosphorylation (Fig. 1, Fig. 2, and “Materials and Methods”). A detailed analysis and validation of the model based on experimental and clinical data for normal subjects is given in Ref. 18. Here we used the model to analyze the alterations in hepatic glucose metabolism in subjects with T2DM.
First, we checked extensively the capability of our model to correctly describe the metabolic disorder in subjects with T2DM. To this end we carried out a series of simulations, where we used the model with identical parameterization as it was initially developed for normal subjects and only altered the glucose-hormone response profiles to those observed in T2DM (see Table 1 and Fig. 2, A–C). As shown below, these simulations recapitulated quantitatively the experimentally determined alterations of the glucose and glycogen metabolism in subjects with T2DM after overnight fasting and short term fasting (Fig. 3, supplemental Fig. S1 and Table S1, Table 3). Next, after having validated the applicability of our model to the hepatic glucose metabolism in T2DM, we performed another series of model simulations to reveal the impact of the altered hormone plasma profiles in T2DM on (i) the set point defining the plasma glucose level at which the liver switches between the glucose producing (HGP) and glucose consuming mode (HGU); (ii) the contributions of glycogenolysis/gluconeogenesis and gluconeogenesis/glycolysis to the diabetes phenotype; (iii) the capability of the liver to respond to changes in blood glucose in untreated T2DM (Fig. 4) and in different modes of insulin treatment of T2DM (Figs. 5, 6, and supplemental Fig. S2–S4).
FIGURE 3.

HGP, GNG, glycogenolysis (GLY), and GNG/HGP in short term fasting over 70 h. Experimental data and simulated curves and simulations for normal subjects are shown in black, for subjects with T2DM in blue. The parts of the figure regarding normal subjects are reproduced from Ref. 18. Experimental data for T2DM subjects were from Refs. 22 and 46–52 (see Table 2), experimental data for normal subjects from Ref. 18, Table 2. Simulated time courses (dashed lines) were started from initially filled glycogen stores (500 mm). Blood glucose levels varied between 3.6 and 5 mm in steps of 0.2 mm with bold curves depicting simulations carried out at blood glucose concentrations of 3.6 and 5 mm, respectively. A, HGP decreases with increasing fasting time and reaches constant levels after ∼25 h. With decreasing blood glucose, HGP increases. HGP is increased in T2DM. B, GNG is almost constant over time in normal subjects and subjects with T2DM with increased GNG in T2DM. C, glycogenolysis (GLY) decreases over time paralleled by a decrease of glycogen. Lower blood glucose levels result in an initially higher GLY rate. GLY rate decreases faster in T2DM and drops to 0 within 30 h. D, GNG/HGP. The relative contribution of gluconeogenesis to the hepatic glucose production increases over time. Whereas in normal subjects gluconeogenesis accounts for 90% of HGP after 40 h fasting, in subjects with T2DM it accounts for 90% of HGP already after 25 h. In T2DM the contribution of GNG to HGP is reduced in the first 20 h.
TABLE 3.
Comparison of model predictions for subjects with T2DM with experimental data
| Description | Model | Experimental data | Refs. | |
|---|---|---|---|---|
| A | HGP/HGU set point at increased plasma glucose concentrations | T2DM set point between 7.1 and 11.1 mm depending on glycogen concentration: 7.1 mm (no glycogen), 9.5 mm (glycogen 250 mm), 11.1 mm (maximal glycogen) | T2DM set point above 8 mm: at 8 mm with insulin concentration of 150 pmol/liter | 26 |
| B | HGP in T2DM | HGP of ∼6 μmol/kg/min of HGP at 8 mm (glycogen 250 mm) | Net splanchnic glucose production (SGP) of ∼4 μmol/min/kg (difference of splanchnic glucose production ∼13 μmol/min/kg and ∼9 μmol/min/kg splanchnic glucose uptake) | 26 |
| HGP > SGP due to the high experimental insulin concentration of 150 pmol/liter (∼50 pmol/liter predicted by the T2DM insulin GHR). | ||||
| C | HGP in normal subjects and T2DM | ∼4 μmol/kg/min of HGP for ∼5 mm blood glucose normal ∼4 μmol/kg/min of HGP for ∼8 mm blood glucose T2DM | Similar basal endogenous glucose production in normal subjects and T2DM, but increased basal glucose levels of ∼8 mm in T2DM compared to normal ∼5 mm | 9, 24 |
| D | HGP for short term starvation and contributions to HGP from gluconeogenesis/glycogenolysis | Half-filled glycogen: ∼20 μmol/kg/min of HGP at ∼3.8 mm blood glucose with ∼9 μmol/min/kg of gluconeogenesis (45%) and ∼11 μmol/kg/min of glycogenolysis (55%) | ∼14.5 μmol/kg/min of HGP with ∼9.5 μmol/kg/min of gluconeogenesis (∼65%) and ∼5 μmol/kg/min of glycogenolysis (∼35%) | 2, 22 |
| One-third glycogen: ∼13 μmol/kg/min of HGP with 7.5 μmol/kg/min of gluconeogenesis (58%) and 5.5 μmol/kg/min of glycogenolysis (42%) | ∼14.5 μmol/kg/min of HGP with ∼9.5 μmol/kg/min of gluconeogenesis (∼65%) and ∼5 μmol/kg/min of glycogenolysis (∼35%) | |||
| E | HGP, gluconeogenesis, and glycogenesis | At ∼8 mm glucose T2DM similar to HGP, gluconeogenesis, and glycogenesis as the normal liver at ∼5 mm | At ∼8 mm glucose T2DM similar to HGP, gluconeogenesis, and glycogenesis as the normal liver at ∼5 mm | 49 |
FIGURE 4.

Stationary glucose exchange rates between liver and blood (HGP/HGU), contributions of gluconeogenesis/glycolysis, contributions of glycogenolysis/glycogenesis and HGRC under constant glycogen, and blood glucose concentration for normal subjects (top row) and subjects with T2DM (bottom row). The parts of the figure regarding normal subjects are reproduced from Ref. 18. Glycogen is varied between 0 and 500 mm in 5 mm steps, blood glucose between 2 and 14 mm in 0.05 mm steps. A, HGP (red)/HGU (green) in μmol/min/kg corresponding to GLUT2 flux. The liver switches between HGP and HGU depending on the blood glucose and glycogen. The set point varies in normal subjects between 5.4 mm for empty and 7.5 mm for completely filled glycogen stores (6.6 mm at a half-filled glycogen pool of 250 mm). B, gluconeogenesis (red)/glycolysis (green) in μmol/min/kg corresponding to glucose-6-phosphate isomerase (GPI) flux. The gluconeogenesis/glycolysis set point varies in normal subjects between 8.8 mm for empty and 7.0 mm for filled glycogen stores. C, glycogenolysis (red)/glycogenesis (green) ([μmol/min/kg]) corresponding to flux through glucose-1-phosphate isomerase (G1PI). D, HGRC in μmol/min/kg/mm (Equation 7). Response of the liver to changes in blood glucose. In normal subjects two main regions of high response exist: a hypoglycemic region below 4 mm, and a hyperglycemic region above 8 mm glucose. E, in T2DM the set point between HGP and HGU is shifted to higher glucose and varies between 7.1 mm (no glycogen) and 11.1 mm (maximal glycogen) (9.5 mm at glycogen concentration of 250 mm). HGP is increased and HGU decreased in T2DM. F, in T2DM the set point between gluconeogenesis and glycolysis is shifted to higher blood glucose and varies between 13.5 mm (no glycogen) and 10.0 mm (maximal glycogen) (12.7 mm at glycogen 250 mm). In T2DM gluconeogenesis is increased and the switch to glycolysis occurs only at very high blood glucose. G, in T2DM the glycogenolysis/glycogenesis set point is shifted to higher blood glucose (5.1 mm normal versus 7.2 mm T2DM at 250 mm glycogen). H, in T2DM the HGRC and hepatic response to changes in blood glucose is considerably decreased. The hepatic response to hypoglycemia is markedly impaired and the hyperglycemic response is shifted to higher glucose (11.1 versus 7.5 mm at 250 mm glycogen).
FIGURE 5.

HPG/HPU (A and B), HGRC (C and D), and shift in set points (E-G) for various treatment regimes in T2DM. Normal subjects (black), T2DM (blue), linear treatments (green), constant insulin treatments (violet), insulin restored (red), and glucagon restored (red dashed). The corresponding simulations for the dependence on glucose and glycogen are given in supplemental Figs. S2–S4. A, HGP/HGU in μmol/min/kg for linear insulin treatments; B, constant insulin treatments in each case at 250 mm hepatic glycogen. T2DM leads to an increased HGP and reduced HGU. Additive and linear insulin treatments further increase HGU and decrease HGP. C, HGRC (given in μmol/min/kg/mm) for linear and D, constant insulin treatments in each case at 250 mm hepatic glycogen. HGRC is markedly reduced in hypoglycemia in T2DM and the hyperglycemic response normally at 8 mm is shifted to ∼11 mm. Set points for HGP/HGU (E), gluconeogenesis/glycolysis (F), and glycogenesis/glycogenolysis (G) for normal subjects, untreated T2DM, and various insulin treatments. The insulin treatments partially normalize the changes in T2DM with glycogen set points being almost normalized in all treatments, whereas HGP/HGU and gluconeogenesis/glycolysis set points are only partially normalized. Glucagon restoration is by far the most effective treatment in normalizing hepatic glucose metabolism (A–F).
FIGURE 6.

Simulated temporal changes of the hepatic production rate and blood glucose level in untreated and insulin-treated T2DM patients elicited by abrupt glucose utilization challenges a hypoglycaemic basal state. A, time course of HGP after an additional glucose utilization challenge of ΔGU = 5 μmol/kg/min for Δt = 30 min at t = 0 min for normal subjects (black) and subjects with T2DM (blue). B, changes in the blood glucose level of normal subjects (black) and subjects with T2DM (blue). C, changes in the blood glucose level of normal subjects (black) and subjects with untreated T2DM (blue) or additive insulin treatment (violet). D, changes in the blood glucose level of normal subjects (black) and subjects with untreated T2DM (blue) or linear insulin treatment (green). E–H, minimum blood glucose level reached after abrupt glucose utilization challenges of various amplitude ΔGU and duration Δt. With increasing ΔGU and Δt the minimal blood glucose level decreases. E, normal subjects; F, untreated T2DM; G, T2DM with linear insulin treatments; H, T2DM with additive insulin treatments.
Alterations in Hormonal Regulation in T2DM
The GHR of insulin, epinephrine, and glucagon to changes in blood glucose are considerably changed in subjects with T2DM compared with normal subjects (Fig. 2, Table 1, and Equations 1–3). These altered glucose-hormone profiles result in an altered relationship between plasma glucose and the phosphorylation state of key enzymes of hepatic glucose metabolism in T2DM (Fig. 2D and Equation 6). Dephosphorylation of key interconvertible enzymes occurs only at highly elevated glucose concentrations in T2DM, half-maximal phosphorylation is achieved at γ0.5T2DM = 9.1 mm compared with γ0.5 = 4.6 mm in the normal case. Moreover, the basal phosphorylation level is significantly increased (34% in T2DM versus 5% in normal case at 20 mm glucose). As the kinetic properties of the interconvertible enzymes are different in the phosphorylated and dephosphorylated state (see Ref. 18) with the phosphorylated states of key enzymes of glucose metabolism supporting HGP, changes in the phosphorylation state in T2DM give rise to a markedly higher HGP, a hallmark of T2DM (2).
HGP, Gluconeogenesis, and Glycogenolysis in Short Term Fasting
The liver is the main glucose supplier in overnight fasting and short term fasting. HGP (Fig. 3A) results either from de novo synthesis via gluconeogenesis (Fig. 3B, GNG) or from degradation of hepatic glycogen via glycogenolysis (Fig. 3C, GLY). The relative contributions of these two pathways to HGP change over the time course of fasting with gluconeogenesis becoming more and more important, whereas the fraction of glycogenolysis to HGP decreases (Fig. 3D, GNG/HGP). Remarkably, by solely taking into account the observed hormonal alterations in T2DM, our model predicts the observed metabolic changes in HGP, as well as the changed contributions of gluconeogenesis and glycogenolysis in T2DM (Fig. 3, blue) in line with experimental data from a multitude of clinical studies (Table 2).
HGP is increased in T2DM (Fig. 3A) contributing to the elevated plasma glucose levels characteristic for diabetes. With on-going fasting HGP decreases to an increased basal rate of 8–9 μmol/kg/min after ∼25 h compared with 7–8 μmol/kg/min at ∼40 h for normal subjects. The contribution of gluconeogenesis to HGP (Fig. 3B) is constant at the given blood glucose concentration for normal subjects as well as for subjects with T2DM (see Ref. 2 for review), whereby the rate of gluconeogenesis increases with decreasing blood glucose level. However, gluconeogenesis is considerably increased in T2DM, being the reason for the increased HGP after longer fasting periods and the increase in HGP in overnight fasting. In contrast, glycogenolysis decreases sharply during fasting due to the fast consumption of glycogen (see also Fig. 4A). In the initial phase of fasting (∼15 h) the rate of glycogenolysis is increased in T2DM but then drops faster compared with the normal case due to the more rapid decrease in glycogen. As a consequence, the contribution of glycogenolysis to HGP ceases in T2DM already after ∼25 h compared with the longer lasting contribution of glycogenolysis to HGP for ∼40 h of the fast in normal subjects. The fractional contributions of glycogenolysis and glycogen synthesis to HGP shift from an approximately equal initial contribution of both pathways to finally complete de novo synthesis. In normal subjects after an overnight fast (∼10 h) half of the HGP is contributed by gluconeogenesis (2), in T2DM only 30–40%. The contributions of glycogenolysis decrease much faster in T2DM and after around 25 h only 10% of HGP result from glycogen, 90% from gluconeogenesis, a state reached in normal subjects only after 40 h of fasting. Remarkably, the experimentally observed alterations in the hormone responses (Fig. 2) are sufficient to predict the observed changes in hepatic glucose metabolism in fasting (Fig. 3).
Glycogenolysis in Short Term Fasting and Glycogen Synthesis Postprandial
Liver glycogen has an important role as a short term glucose buffer enabling homeostasis of the blood glucose level at a short term increase of systemic glucose utilization. At low blood glucose concentrations, like in the fasting state or during extensive muscle activity, glucose is produced from glycogen, whereas during periods of high blood glucose occurring postprandially, glycogen is synthesized from glucose. Our simulations predict an impaired glycogen metabolism in T2DM, which is characterized by an increased glycogenolysis (supplemental Fig. S1A) and decreased glycogen synthesis (supplemental Fig. S1B) in accordance with the experimental data (22, 23).
The Contribution of the Liver to the Homeostasis of the Blood Glucose Level
The central role of the liver for homeostasis of plasma glucose results from its ability to switch between an anabolic glucose producing mode (HGP) and a catabolic glucose utilizing mode (HGU) depending on the blood glucose concentration and associated hormonal signals. In this process the hepatic metabolism is altered from glucose production via gluconeogenesis and glycogenolysis at hypoglycemia to glucose utilization via glycolysis and glycogen synthesis at hyperglycemia, a short term switch between anabolic and catabolic metabolic pathways occurring in the range of minutes. To analyze this switch and its role for plasma glucose homeostasis, we calculated the stationary exchange rate of glucose between liver and blood (=flux through the glucose transporter GLUT2) at varying concentrations of blood glucose and hepatic glycogen for normal subjects and subjects with T2DM (Fig. 4). In normal subjects and at half-filled glycogen stores the hepatic glucose metabolism switches between HGP and HGU at blood glucose concentrations of 6.6 mm (=set point) (Fig. 4A). Intriguingly, the model predicts a clear shift of this HGP/HGU set point to markedly higher glucose levels of about 9.5 mm in T2DM (Fig. 4E and Table 3A). Hence, except for very high plasma levels of glucose the liver works permanently as a glucose producer. The predicted increase in HGP and concomitant decrease in HGU (Fig. 4, E versus A) in T2DM contribute to the increased plasma glucose level in fasting (∼8–10 versus normal ∼5.2 mm) reported in several studies (9, 24) and in line with the experimental finding that hyperglycemia during fasting strongly correlates with increased HGP in T2DM (10, 25).
The switch between gluconeogenesis and glycolysis occurs normally at 8.5 mm glucose for half-filled glycogen stores (Fig. 4B). In T2DM it is shifted to very high blood glucose concentrations of 12–13 mm (Fig. 4F) resulting in increased gluconeogenesis and a switch to hepatic glycolysis only at severe hyperglycemia. Finally, the set point for glycogenolysis/glycogenesis is strongly elevated to 7.2 mm in T2DM (Fig. 4G) compared with 5.1 mm in normal subjects (Fig. 4C).
Taken together, our model predictions are in concordance with numerous experimental findings showing that altered glucose-hormone profiles in T2DM shift the glucose metabolism of the liver toward increased glucose production and thus higher plasma glucose levels, the hallmark of T2DM. The calculated set points of gluconeogenesis/glycolysis as well as glycogenolysis/glycogen synthesis are considerably shifted, both contributing to a hepatic glucose output characterized by increased HGP and decreased HGU leading to an increase in plasma glucose compared with normal, all in good agreement with experimental data (Table 3).
The Liver in T2DM, an Impaired Glucose Homeostate (HGRC)
The capability of the liver to respond to changes in blood glucose concentration was evaluated in normal subjects (Fig. 4D) and subjects with T2DM (Fig. 4H) using as measure the HGRC (Equation 7). This analysis revealed that in T2DM not only the set points of HGP/HGU, gluconeogenesis/glycolysis, and glycogenolysis/glycogen synthesis are shifted, but also the hepatic ability to react to changing glucose concentrations is strongly impaired. Normally, the liver is able to respond over the whole range of physiological blood glucose concentrations with a HGRC ≥ 1.9 ml/kg·min whereby the counter-regulatory response of the liver to hypoglycemia (blood glucose levels <5 mm) is stronger as to hyperglycemia (blood glucose levels >8 mm) (18). Intriguingly, the simulations reveal a strongly impaired counter-regulation in T2DM, especially to hypoglycemia, but also at elevated blood glucose levels. The lower values of HGRC for blood glucose levels below 5 mm provide an explanation for the occurrence and severity of hypoglycemic events in T2DM. As a consequence of the reduced HGP and HGRC in T2DM, the liver is not able to react appropriately to a decrease in blood glucose, resulting in hypoglycemic episodes.
Furthermore, the region of efficient hyperglycemic counter-regulation is shifted from 7.5 mm (normal case) to much higher glucose concentrations of about 11 mm (at glycogen 250 mm), leading to an impaired postprandial response to increased glucose levels. Our computations suggest that the diminished hyperglycemic response of the liver in combination with reduced HGU is one of the main reasons of the much larger postprandial increase in blood glucose in T2DM compared with normal subjects (1, 26).
Insulin Treatment May Increase the Risk of Hypoglycemic Events
Insulin medication is one of the standard treatments in T2DM. However, a corollary of insulin treatment is the increased risk of hypoglycemic events (27). To better understand the molecular mechanism rendering T2DM subjects under insulin treatment more susceptible to hyperglycemic episodes we used our model to predict the impact of different modes of insulin treatment on the hepatic glucose metabolism. We simulated two modes of insulin treatment (Fig. 2E) mimicking administration of short term insulin like, for example, aspart/lispro (insulin dose linearly increasing with blood glucose, Equation 5) or of long term insulin like, for example, glargine (constant insulin dose independent of blood glucose, Equation 4) (28). The effect of these treatments was compared with two reference cases in which either the glucose-insulin response was fully normalized, i.e. was identical with that of healthy subjects and only glucagon and epinephrine levels were impaired (“insulin restoration”) or, alternatively, in which the glucose-glucagon profile was fully normalized and only insulin and epinephrine levels were impaired (“glucagon restoration”).
We found that none of the treatments was able to normalize the altered phosphorylation state of key enzymes in T2DM (Fig. 2, D and F). Compared with the normal case, all treatments result in a decrease in the phosphorylation state of interconvertible enzymes at blood glucose concentrations below 3.8 mm and an increase for glucose concentrations above 5.1 mm. All treatments, with the exception of the glucagon restoration case, show a highly elevated baseline phosphorylation of about 30% at plasma glucose levels above 11 mm. Intriguingly, the glucagon restoration case almost normalizes the altered phosphorylation state in T2DM, and is according to our simulations the only treatment able to considerably decrease the phosphorylation state under hyperglycemia. For low blood glucose levels below 5 mm the insulin restoration and glucagon restoration treatments almost normalize the T2DM changes, whereas the linear and constant insulin therapies result in a decreased phosphorylation state under hypoglycemia. The diminished phosphorylation state in insulin treatment is dose-dependent and increases with an increasing amount of applied insulin.
All simulated therapies as well as insulin restoration and glucagon restoration are effective in shifting the hepatic T2DM phenotype toward the normal phenotype, but to a varying extent. Our model simulations predict that under insulin therapy, the set points of HGP/HGU (Fig. 5E), gluconeogenesis/glycolysis (Fig. 5F), and glycogenolysis/glycogenesis (Fig. 5G) are shifted toward the normal set points, resulting in lowered HGP and gluconeogenesis and consequently blood glucose levels closer to normal. A dose-effect is observed, with the higher insulin treatments being more effective in shifting from the T2DM to the normal phenotype. This is in accordance with the reported correlation between insulin dose and reduction in hemoglobin A1C (29), a retrospective marker of mean blood glucose levels. Interestingly, the glycogenolysis/glycogenesis set point can be almost normalized in all treatments (Fig. 5G), whereas the gluconeogenesis/glycolysis and HGP/HGU set points are still substantially different from the normal value (Fig. 5, E and F). The simulated reference treatment of glucagon restoration provided by far the best normalization of the gluconeogenesis/glycolysis set point, due to the considerably reduced phosphorylation state under hyperglycemia compared with T2DM.
All considered treatments reduce HGP and increase HGU (Fig. 5, A and B). The increase in HGU is only moderate, but a strong reduction in HGP, especially under hypoglycemic glucose levels can be seen. The insulin restoration and glucagon restoration cases also increase HGU and decrease HGP. However, in contrast to the linear and constant insulin treatments, HGP is only marginally decreased for plasma glucose concentrations below 3 mm. The largest increase in HGU was obtained with the glucagon restoration treatment.
The simulated treatments showed contradictory effects on the HGRC (see Equation 7) used to quantify the capacity of the liver to counterbalance variations in the plasma glucose level (Fig. 5, C and D). On the one hand, our model predicts that insulin treatments shift the range of efficient counter-regulation to hyperglycemia toward lower plasma glucose levels, thereby normalizing the T2DM phenotype. Again, glucagon restoration is predicted to have the strongest normalization effect. On the other hand, however, the hepatic counter-regulatory capacity to hypoglycemia, which is already strongly reduced in T2DM, is even further impaired as a consequence of insulin medication. In the additive therapies the hypoglycemic response is nearly abolished, with even low dose insulin treatments almost completely removing the HGRC peak in hypoglycemia (around 4 mm plasma glucose). The linear therapy also reduces the HGRC at 4 mm, but leads to an increase for very low glucose concentrations. By contrast, in the insulin restoration and glucagon restoration cases high HGRC in the hypoglycemic region are observed.
Temporal Response of Hepatic Glucose Metabolism to Glucose Utilization Challenges in Hypoglycemia
The measure of HGRC quantifies the shift of the stationary glucose exchange rate between liver and blood plasma elicited by a small change of the stationary blood glucose level. Because changes in the blood glucose level can be abrupt and quite significant, e.g. in case of an onset of muscle work, it is interesting to study the temporal response of the hepatic glucose metabolism and its impact on homeostasis of the blood glucose level after an abrupt increase of the systemic glucose utilization rate. With respect to the central problem addressed in this work, the increased risk of hypoglycaemic episodes, we focused in these time-dependent simulations on a situation where the patient is already in a hypoglycemic regime (blood glucose level = 4 mm) and experiences a sudden additional glucose utilization challenge.
The simulations started from steady state at a blood glucose concentration of 4 mm were basal systemic glucose utilization of the body, GUbase, and equals the hepatic glucose production rate, HGP. At t = 0 min, the steady state of the blood glucose level was perturbed by additional glucose utilization ΔGU (μmol/kg/min) of duration Δt (min). The resulting temporal changes of the hepatic HGP and blood glucose levels (Fig. 6, A–D) were calculated for a generic patient with a body weight of mbw = 75 kg and a blood volume of Vblood = 5 liters.
HGP in untreated T2DM is decreased (Fig. 6A) and consequently the blood glucose drop is stronger in T2DM than in normal subjects (Fig. 6B). Linear insulin treatments (Fig. 6C) and constant insulin treatments (Fig. 6D) result in a further drop of blood glucose under identical glucose utilization challenges, i.e. the risk of hypoglycemia increases. Normal subjects can cope with this challenge over the whole range of glucose utilization without a drop in blood glucose below 3 mm (Fig. 6E). In T2DM the drop of blood glucose increases considerably (Fig. 6F) resulting in hypoglycemia in untreated T2DM. Insulin treatments further reduce the capacity to react to hypoglycemia and blood glucose drops even further than in untreated T2DM (Fig. 6, G, linear treatment 4, and H, additive treatment 4). Under hypoglycemia, treatment with even low doses of insulin impairs the capacity of the liver to adequately counteract hypoglycemia.
Taken the findings of all simulations together we conclude that the considered insulin treatments are effective in reducing blood glucose levels by an increase in HGU and a decrease in HGP. However, the normal phenotype of hepatic HGP/HGU cannot be restored. Due to the decrease of HGP at plasma glucose levels below 3.8 mm, the capacity of the liver to counteract hypoglycemia is strongly reduced. The probability for hypoglycemic episodes increases in insulin treatment, as a consequence of the further impairment of the already diminished T2DM counter-regulation. Due to the reduced HGP and the reduced reaction to lowered blood glucose (HGRC), the liver as the main organ of gluconeogenesis is not able to react appropriately to a sudden enforcement of hypoglycemia. With increasing insulin doses (additive and linear) HGP and HGRC are further reduced in hypoglycemia. Tight glycemic control, which necessitates higher insulin doses, amplifies the decrease in HGP and HGRC and leads to an increase in hypoglycemic events, explaining the observed increase in hypoglycemic events in tightly controlled T2DM. Our simulations suggest that especially additive treatments by means of long term insulin should have a dramatic effect on the response capacity of the liver. For blood glucose levels already below 3.8 mm, small amounts of insulin above normal increase the risk of hypoglycemia.
DISCUSSION
Our detailed kinetic model of human hepatic glucose metabolism integrated with the hormonal control by insulin, glucagon, and epinephrine to diabetes replicated a large set of experimental data from different studies on metabolic alterations in the hepatic glucose metabolism of patients with T2DM (Fig. 3 and supplemental Fig. S1). Except for changes in the glucose-hormone response profiles, the model used for the study of alterations of the hepatic glucose metabolism in T2DM is identical with the model used for the normal physiological state based on the following premises: (i) usage of empirical mathematical functions to incorporate hormone-induced signal transduction; (ii) a constant cellular redox and energy status of hepatocytes; (iii) modeling of an ”average“ hepatocyte without distinction between periportal and pericentral hepatocytes; and (iv) no inclusion of changes in gene expression of metabolic enzymes and as discussed in detail in Ref. 18.
It is important to note that none of the T2DM data were used for parameterization of the normal model (18) and none of the parameters of the original model were altered in the T2DM simulations. The observed changes in GHR in T2DM are sufficient to change the hepatic glucose metabolism via an altered phosphorylation state to the T2DM phenotype characterized by an increased HGP, mainly due to increased gluconeogenesis, reduced HGU, and a shift of the set point of HGP/HGU to higher blood glucose levels.
The fact that no changes in the expression level of metabolic enzymes were required to fully recapitulate reported experimental findings on the hepatic glucose metabolism in normal conditions and T2DM underlines the importance of short term regulation of the hepatic glucose metabolism by interconvertible enzymes. Hormone-induced changes in the phosphorylation state of key regulatory enzymes occur within a few seconds, thus allowing the rapid adaptation of the hepatic metabolism to varying hormonal signals and glucose levels in the plasma, by changing in a synergistic manner the activity of counteracting anabolic and catabolic pathways.
Impaired hormonal signals have dramatic effects on metabolism and in the case of T2DM may shift the metabolic phenotype from normal glucose homeostasis to a T2DM disease phenotype. This shift in metabolism results in an impaired response of the liver to changes in blood glucose levels. Counter-regulation to hypoglycemia is almost non-existent, leading to hypoglycemic episodes in untreated T2DM.
In addition to the observed changes in glucose-hormone profiles, especially of insulin, T2DM is also characterized by insulin resistance (13), a feature not explicitly taken into account in our model. The altered hormone profiles in T2DM were sufficient to predict the alterations in hepatic glucose metabolism in diabetes. Interestingly, incorporating insulin resistance in our model via a reduced effect of insulin on the phosphorylation state further amplifies the observed T2DM phenotype (simulations not shown). The effect of the two main characteristics of T2DM, reduced relative insulin concentrations and reduced insulin sensitivity, both result in reduced insulin signaling and altered phosphorylation states of key enzymes shifting hepatic metabolism toward a T2DM phenotype.
Our simulations of insulin treatments provided a molecular mechanistic explanation for the contribution of liver metabolism to the occurrence of iatrogenic hypoglycemia observed in insulin therapy and for the increased frequency of hypoglycemic events in tightly controlled T2DM therapy. The presented simulations clearly show how elevated insulin levels during low blood glucose further impair the counter-regulatory capacity of the liver in T2DM and consequently may lead to hypoglycemic episodes. Risk factors of iatrogenic hypoglycemia in insulin therapy are among others: incorrect insulin dose and timing, improper type of insulin due to their pharmacokinetics and pharmacodynamics, bad patterns of food ingestion and exercise, and altered clearance of insulin or sensitivity to insulin, like alcohol and other drugs (28–30), all potentially leading to increased insulin during low blood glucose values and thereby increasing the risk of hypoglycemia.
Furthermore, the model simulations predict a great potential of normalizing the glucagon hormone profile in treatment of T2DM, a theoretical finding that is in concordance with experimental studies (31, 32). Indeed, treatment with glucagon receptor inhibitors and antibodies have been shown to be very effective in blood glucose control in animal models (33) and inhibition of postprandial glucagon considerably reduces blood glucose values in T2DM. Perhaps it is time for a paradigm shift: effective treatment of T2DM should not only aim at the normalization of insulin signaling via insulin supplementation or administration of drugs like sulfonylureas and meglitinides increasing insulin secretion. It should also aim at the normalization of glucagon signaling, which is equally important especially with regard to hypoglycemia. Recent incretin treatments with GLP-1 based on GLP-1 agonists and dipeptidyl peptidase-4 inhibitors are the first steps in this direction by normalizing blood glucose levels not only due to increased insulin secretion, but also by reducing glucagon secretion (34). The next step could be the design of drugs directly targeting glucagon secretion.
This work was supported by the Federal Ministry of Education and Research (BMBF, Germany) under Virtual Liver Network VLN Grant 0315741.

This article contains supplemental Table S1 and Figs. S1–S4.
- HGU
- hepatic glucose utilization
- HGP
- hepatic glucose production
- T2DM
- type 2 diabetes mellitus
- GHR
- glucose-hormone response
- HGRC
- hepatic glucose response coefficient
- GNG
- gluconeogenesis.
REFERENCES
- 1. Gerich J. E. (1993) Control of glycaemia. Baillieres Clin. Endocrinol. Metab. 7, 551–586 [DOI] [PubMed] [Google Scholar]
- 2. Nuttall F. Q., Ngo A., Gannon M. C. (2008) Regulation of hepatic glucose production and the role of gluconeogenesis in humans. Is the rate of gluconeogenesis constant? Diabetes Metab. Res. Rev. 24, 438–458 [DOI] [PubMed] [Google Scholar]
- 3. Degenhardt T. P., Thorpe S. R., Baynes J. W. (1998) Chemical modification of proteins by methylglyoxal. Cell Mol. Biol. 44, 1139–1145 [PubMed] [Google Scholar]
- 4. Baynes J. W., Thorpe S. R. (1999) Role of oxidative stress in diabetic complications. A new perspective on an old paradigm. Diabetes 48, 1–9 [DOI] [PubMed] [Google Scholar]
- 5. Brownlee M. (2005) The pathobiology of diabetic complications. A unifying mechanism. Diabetes 54, 1615–1625 [DOI] [PubMed] [Google Scholar]
- 6. Skyler J. S. (1996) Diabetic complications. The importance of glucose control. Endocrinol. Metab. Clin. North Am. 25, 243–254 [DOI] [PubMed] [Google Scholar]
- 7. Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N. Engl. J. Med. 329, 977–986 [DOI] [PubMed] [Google Scholar]
- 8. Cryer P. E. (1993) Glucose counterregulation. Prevention and correction of hypoglycemia in humans. Am. J. Physiol. 264, E149-E155 [DOI] [PubMed] [Google Scholar]
- 9. Basu A., Dalla Man C., Basu R., Toffolo G., Cobelli C., Rizza R. A. (2009) Effects of type 2 diabetes on insulin secretion, insulin action, glucose effectiveness, and postprandial glucose metabolism. Diabetes Care 32, 866–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Féry F., Melot C., Balasse E. O. (1993) Glucose fluxes and oxidation after an oral glucose load in patients with noninsulin-dependent diabetes mellitus of variable severity. Metabolism 42, 522–530 [DOI] [PubMed] [Google Scholar]
- 11. Israelian Z., Szoke E., Woerle J., Bokhari S., Schorr M., Schwenke D. C., Cryer P. E., Gerich J. E., Meyer C. (2006) Multiple defects in counterregulation of hypoglycemia in modestly advanced type 2 diabetes mellitus. Metabolism 55, 593–598 [DOI] [PubMed] [Google Scholar]
- 12. Segel S. A., Paramore D. S., Cryer P. E. (2002) Hypoglycemia-associated autonomic failure in advanced type 2 diabetes. Diabetes 51, 724–733 [DOI] [PubMed] [Google Scholar]
- 13. American Diabetes Association (2010) Diagnosis and classification of diabetes mellitus. Diabetes Care 33, S62-S69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cryer P. E. (2007) Severe iatrogenic hypoglycemia in type 2 diabetes mellitus. Nat. Clin. Pract. Endocrinol. Metab. 3, 4–5 [DOI] [PubMed] [Google Scholar]
- 15. Diabetes Control and Complications Trial Research Group (1991) Epidemiology of severe hypoglycemia in the diabetes control and complications trial. The DCCT Research Group. Am. J. Med. 90, 450–459 [PubMed] [Google Scholar]
- 16. Action to Control Cardiovascular Risk in Diabetes Study Group, Gerstein H. C., Miller M. E., Byington R. P., Goff D. C., Jr., Bigger J. T., Buse J. B., Cushman W. C., Genuth S., Ismail-Beigi F., Grimm R. H., Jr., Probstfield J. L., Simons-Morton D. G., Friedewald W. T. (2008) Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 358, 2545–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. ADVANCE Collaborative Group, Patel A., MacMahon S., Chalmers J., Neal B., Billot L., Woodward M., Marre M., Cooper M., Glasziou P., Grobbee D., Hamet P., Harrap S., Heller S., Liu L., Mancia G., Mogensen C. E., Pan C., Poulter N., Rodgers A., Williams B., Bompoint S., de Galan B. E., Joshi R., Travert F. (2008) Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 358, 2560–2572 [DOI] [PubMed] [Google Scholar]
- 18. König M., Bulik S., Holzhütter H. G. (2012) Quantifying the contribution of the liver to glucose homeostasis. A detailed kinetic model of human hepatic glucose metabolism. PLoS Comput. Biol. 8, e1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Berkel T. J., Kruijt J. K., Van den Berg G. B., Koster J. F. (1978) Difference in the effect of glucagon and starvation upon L-type pyruvate kinase from rat liver. Eur. J. Biochem. 92, 553–561 [DOI] [PubMed] [Google Scholar]
- 20. Beavo J. A., Bechtel P. J., Krebs E. G. (1974) Activation of protein kinase by physiological concentrations of cyclic AMP. Proc. Natl. Acad. Sci. U.S.A. 71, 3580–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishibashi H., Cottam G. L. (1978) Glucagon-stimulated phosphorylation of pyruvate kinase in hepatocytes. J. Biol. Chem. 253, 8767–8771 [PubMed] [Google Scholar]
- 22. Magnusson I., Rothman D. L., Katz L. D., Shulman R. G., Shulman G. I. (1992) Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C-nuclear magnetic resonance study. J. Clin. Invest. 90, 1323–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krssak M., Brehm A., Bernroider E., Anderwald C., Nowotny P., Dalla Man C., Cobelli C., Cline G. W., Shulman G. I., Waldhäusl W., Roden M. (2004) Alterations in postprandial hepatic glycogen metabolism in type 2 diabetes. Diabetes 53, 3048–3056 [DOI] [PubMed] [Google Scholar]
- 24. Ferrannini E., Simonson D. C., Katz L. D., Reichard G., Jr., Bevilacqua S., Barrett E. J., Olsson M., DeFronzo R. A. (1988) The disposal of an oral glucose load in patients with noninsulin-dependent diabetes. Metabolism 37, 79–85 [DOI] [PubMed] [Google Scholar]
- 25. Bogardus C., Lillioja S., Howard B. V., Reaven G., Mott D. (1984) Relationships between insulin secretion, insulin action, and fasting plasma glucose concentration in nondiabetic and noninsulin-dependent diabetic subjects. J. Clin. Invest. 74, 1238–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rizza R. A. (2010) Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes. Implications for therapy. Diabetes 59, 2697–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akram K., Pedersen-Bjergaard U., Borch-Johnsen K., Thorsteinsson B. (2006) Frequency and risk factors of severe hypoglycemia in insulin-treated type 2 diabetes. A literature survey. J. Diabet. Complications 20, 402–408 [DOI] [PubMed] [Google Scholar]
- 28. Hirsch I. B. (2005) Insulin analogues. N. Engl. J. Med. 352, 174–183 [DOI] [PubMed] [Google Scholar]
- 29. Swinnen S. G., Hoekstra J. B., DeVries J. H. (2009) Insulin therapy for type 2 diabetes. Diabetes Care 32, S253-S259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cryer P. E., Davis S. N., Shamoon H. (2003) Hypoglycemia in diabetes. Diabetes Care 26, 1902–1912 [DOI] [PubMed] [Google Scholar]
- 31. Ali S., Drucker D. J. (2009) Benefits and limitations of reducing glucagon action for the treatment of type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 296, E415-E421 [DOI] [PubMed] [Google Scholar]
- 32. Sloop K. W., Michael M. D., Moyers J. S. (2005) Glucagon as a target for the treatment of type 2 diabetes. Expert. Opin. Ther. Targets 9, 593–600 [DOI] [PubMed] [Google Scholar]
- 33. Yan H., Gu W., Yang J., Bi V., Shen Y., Lee E., Winters K. A., Komorowski R., Zhang C., Patel J. J., Caughey D., Elliott G. S., Lau Y. Y., Wang J., Li Y. S., Boone T., Lindberg R. A., Hu S., Véniant M. M. (2009) Fully human monoclonal antibodies antagonizing the glucagon receptor improve glucose homeostasis in mice and monkeys. J. Pharmacol. Exp. Ther. 329, 102–111 [DOI] [PubMed] [Google Scholar]
- 34. Nauck M. A., Kleine N., Orskov C., Holst J. J., Willms B., Creutzfeldt W. (1993) Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (noninsulin-dependent) diabetic patients. Diabetologia 36, 741–744 [DOI] [PubMed] [Google Scholar]
- 35. Butler P. C., Rizza R. A. (1991) Contribution to postprandial hyperglycemia and effect on initial splanchnic glucose clearance of hepatic glucose cycling in glucose-intolerant or NIDDM patients. Diabetes 40, 73–81 [PubMed] [Google Scholar]
- 36. Henkel E., Menschikowski M., Koehler C., Leonhardt W., Hanefeld M. (2005) Impact of glucagon response on postprandial hyperglycemia in men with impaired glucose tolerance and type 2 diabetes mellitus. Metabolism 54, 1168–1173 [DOI] [PubMed] [Google Scholar]
- 37. Knop F. K., Vilsbøll T., Madsbad S., Holst J. J., Krarup T. (2007) Inappropriate suppression of glucagon during OGTT but not during isoglycaemic intravenous glucose infusion contributes to the reduced incretin effect in type 2 diabetes mellitus. Diabetologia 50, 797–805 [DOI] [PubMed] [Google Scholar]
- 38. Lerche S., Soendergaard L., Rungby J., Moeller N., Holst J. J., Schmitz O. E., Brock B. (2009) No increased risk of hypoglycaemic episodes during 48 h of subcutaneous glucagon-like peptide-1 administration in fasting healthy subjects. Clin. Endocrinol. 71, 500–506 [DOI] [PubMed] [Google Scholar]
- 39. Mitrakou A., Kelley D., Mokan M., Veneman T., Pangburn T., Reilly J., Gerich J. (1992) Role of reduced suppression of glucose production and diminished early insulin release in impaired glucose tolerance. N. Engl. J. Med. 326, 22–29 [DOI] [PubMed] [Google Scholar]
- 40. Degn K. B., Brock B., Juhl C. B., Djurhuus C. B., Grubert J., Kim D., Han J., Taylor K., Fineman M., Schmitz O. (2004) Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and counterregulation during hypoglycemia. Diabetes 53, 2397–2403 [DOI] [PubMed] [Google Scholar]
- 41. Levy C. J., Kinsley B. T., Bajaj M., Simonson D. C. (1998) Effect of glycemic control on glucose counterregulation during hypoglycemia in NIDDM. Diabetes Care 21, 1330–1338 [DOI] [PubMed] [Google Scholar]
- 42. Mitrakou A., Ryan C., Veneman T., Mokan M., Jenssen T., Kiss I., Durrant J., Cryer P., Gerich J. (1991) Hierarchy of glycemic thresholds for counterregulatory hormone secretion, symptoms, and cerebral dysfunction. Am. J. Physiol. 260, E67-E74 [DOI] [PubMed] [Google Scholar]
- 43. Jones T. W., Porter P., Sherwin R. S., Davis E. A., O'Leary P., Frazer F., Byrne G., Stick S., Tamborlane W. V. (1998) Decreased epinephrine responses to hypoglycemia during sleep. N. Engl. J. Med. 338, 1657–1662 [DOI] [PubMed] [Google Scholar]
- 44. Müller W. A., Faloona G. R., Aguilar-Parada E., Unger R. H. (1970) Abnormal α-cell function in diabetes. Response to carbohydrate and protein ingestion. N. Engl. J. Med. 283, 109–115 [DOI] [PubMed] [Google Scholar]
- 45. Spellman C. W. (2010) Pathophysiology of type 2 diabetes: targeting islet cell dysfunction. J. Am. Osteopath. Assoc. 110, S2-S7 [PubMed] [Google Scholar]
- 46. Consoli A., Nurjhan N., Capani F., Gerich J. (1989) Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes 38, 550–557 [DOI] [PubMed] [Google Scholar]
- 47. Woerle H. J., Szoke E., Meyer C., Dostou J. M., Wittlin S. D., Gosmanov N. R., Welle S. L., Gerich J. E. (2006) Mechanisms for abnormal postprandial glucose metabolism in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 290, E67-E77 [DOI] [PubMed] [Google Scholar]
- 48. Gastaldelli A., Toschi E., Pettiti M., Frascerra S., Quiñones-Galvan A., Sironi A. M., Natali A., Ferrannini E. (2001) Effect of physiological hyperinsulinemia on gluconeogenesis in nondiabetic subjects and in type 2 diabetic patients. Diabetes 50, 1807–1812 [DOI] [PubMed] [Google Scholar]
- 49. Wajngot A., Chandramouli V., Schumann W. C., Ekberg K., Jones P. K., Efendic S., Landau B. R. (2001) Quantitative contributions of gluconeogenesis to glucose production during fasting in type 2 diabetes mellitus. Metabolism 50, 47–52 [DOI] [PubMed] [Google Scholar]
- 50. Boden G., Chen X., Stein T. P. (2001) Gluconeogenesis in moderately and severely hyperglycemic patients with type 2 diabetes mellitus. Am. J. Physiol. Endocrinol. Metab. 280, E23-E30 [DOI] [PubMed] [Google Scholar]
- 51. Basu R., Basu A., Johnson C. M., Schwenk W. F., Rizza R. A. (2004) Insulin dose-response curves for stimulation of splanchnic glucose uptake and suppression of endogenous glucose production differ in nondiabetic humans and are abnormal in people with type 2 diabetes. Diabetes 53, 2042–2050 [DOI] [PubMed] [Google Scholar]
- 52. Gastaldelli A., Miyazaki Y., Pettiti M., Santini E., Ciociaro D., Defronzo R. A., Ferrannini E. (2006) The effect of rosiglitazone on the liver. Decreased gluconeogenesis in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 91, 806–812 [DOI] [PubMed] [Google Scholar]