

Background: Wnt/β-catenin signaling is a therapeutic target in cancer and other diseases but also has important physiological functions.
Results: CS-E can inhibit Wnt/β-catenin signaling and interfere with selective transcriptional and biological outcomes.
Conclusion: Wnt/β-catenin signaling thresholds control distinct downstream transcriptional and biological outcomes.
Significance: The development of pharmacological strategies to dissociate disease-related from physiological functions of Wnt/β-catenin signaling is critical for clinical applications.
Keywords: Carbohydrate Function, Chondroitin Sulfate, Drug Discovery, Signaling, Wnt Pathway
Abstract
Aberrant activation of the Wnt/β-catenin signaling pathway is frequently associated with human disease, including cancer, and thus represents a key therapeutic target. However, Wnt/β-catenin signaling also plays critical roles in many aspects of normal adult tissue homeostasis. The identification of mechanisms and strategies to selectively inhibit the disease-related functions of Wnt signaling, while preserving normal physiological functions, is in its infancy. Here, we report the identification of exogenous chondroitin sulfate-E (CS-E) as an inhibitor of specific molecular and biological outcomes of Wnt3a signaling in NIH3T3 fibroblasts. We demonstrate that CS-E can decrease Wnt3a signaling through the negative regulation of LRP6 receptor activation. However, this inhibitory effect of CS-E only affected Wnt3a-mediated induction, but not repression, of target gene expression. We went on to identify a critical Wnt3a signaling threshold that differentially affects target gene induction versus repression. This signaling threshold also controlled the effects of Wnt3a on proliferation and serum starvation-induced apoptosis. Limiting Wnt3a signaling to this critical threshold, either by CS-E treatment or by ligand dilution, interfered with Wnt3a-mediated stimulation of proliferation but did not impair Wnt3a-mediated reduction of serum starvation-induced apoptosis. Treatment with pharmacological inhibitors demonstrated that both induction and repression of Wnt3a target genes in NIH3T3 cells require the canonical Wnt/β-catenin signaling cascade. Our data establish the feasibility of selective inhibition of Wnt/β-catenin transcriptional programs and biological outcomes through the exploitation of intrinsic signaling thresholds.
Introduction
Wnt proteins are secreted glycoproteins that activate signaling pathways controlling a variety of developmental processes, including cell fate determination, morphogenesis, and differentiation, as well as tissue homeostasis (1–7). Aberrantly activated Wnt/β-catenin signaling has also been implicated in a number of human diseases and malignancies, including cancer, fibrosis, and cardiovascular disease (8–12). The involvement of activated Wnt/β-catenin signaling in human disease has sparked intense interest in the identification and characterization of inhibitory molecules that could be utilized to interfere with aberrant Wnt signaling (3, 13). However, although pharmacological inhibition of activated Wnt/β-catenin signaling is desirable for the treatment of many cancers, concomitant impairment of Wnt/β-catenin function in normal tissue physiology and homeostasis, for example in intestinal epithelium and liver, could have negative consequences (2, 14, 15). Thus, the ability to target disease-related biological outcomes of Wnt signaling while preserving the outcomes necessary for normal physiology would be critical for future clinical applications.
Several studies have demonstrated a correlation of Wnt/β-catenin signaling levels with embryonic stem cell differentiation, anterior specification during mouse embryogenesis, adult hepatic homeostasis, phenotypic severity of intestinal tumorigenesis, and lineage determination during hematopoiesis (16–21). However, it is not known whether Wnt signaling thresholds play a role in dissociating global Wnt transcriptional programs or the associated biological outcomes of Wnt/β-catenin signaling.
The glycosaminoglycan chondroitin sulfate (CS)2 consists of linear chains of repeating disaccharide units covalently linked to cell surface and secreted proteins to form chondroitin sulfate proteoglycans (22, 23), which have been shown to control multiple aspects of cellular behavior and communication (22, 24–27). Differentially sulfated CS forms include the monosulfated chondroitin 4-sulfate (C4S) and chondroitin 6-sulfate (C6S) units, as well as the disulfated units chondroitin sulfate D (CS-D) and chondroitin sulfate E (CS-E). Differential sulfation of chondroitin can drastically affect its function (22, 28–31). Interestingly, exogenous CS-E recently has been shown to bind Wnt3a ligand with high affinity in vitro, and treatment of L-cells with CS-E, or alterations in endogenous levels of CS-E by modulations of expression of chondroitin-4-sulfotransferase-1 (C4ST-1), could affect the overall cellular levels of β-catenin (32, 33).
Here, we initially set out to determine the effects of exogenous CS-E treatment on Wnt3a signaling output in order to establish CS-E as a bona fide Wnt pathway inhibitor. We demonstrate that treatment with CS-E decreased Wnt3a signaling output by ∼75%. Accordingly, treatment with CS-E was able to interfere with the phosphorylation of cell surface LRP6, a component of the Wnt receptor complex, suggesting that CS-E treatment interferes with Wnt3a-mediated receptor activation. Surprisingly, genome-wide gene expression profiling experiments demonstrated an inhibitory effect of CS-E on Wnt3a-mediated induction of target gene expression but not target gene repression. We go on to show that these effects of CS-E were due to differential requirements of Wnt3a signaling thresholds for induced versus repressed target genes and that different signaling thresholds also control specific biological outcomes of Wnt3a signaling. Consistent with our observations, CS-E is able to impair the Wnt3a-mediated stimulation of proliferation but cannot interfere with Wnt3a-mediated reduction of apoptosis. By utilizing Wnt3a ligand dilutions in the absence of CS-E, we demonstrate that the identified signaling threshold levels are an intrinsic property of the Wnt3a signaling cascade. Treatment with pharmacological inhibitors established that both induction and repression of Wnt3a target genes in NIH3T3 cells are mediated by the canonical Wnt/β-catenin signaling cascade. Thus, our data establish the feasibility of selective inhibition of Wnt3a transcriptional programs and biological outcomes through the exploitation of intrinsic signaling thresholds. We believe that these data will have an important impact on future functional evaluations of differential biological impacts of Wnt pathway inhibitors. This work is of substantial clinical interest, as it is a first step toward a better understanding of pathway inhibitors that interfere with specific disease-related outcomes of Wnt signaling while sparing physiological functions. Moreover, we provide support for a potential use of CS-E as a therapeutic inhibitor of specific biological outcomes of canonical Wnt signaling.
EXPERIMENTAL PROCEDURES
Cell Lines, Reagents, and Treatments
NIH3T3, L-cells, and L-Wnt3a-cells (34) were obtained from ATCC. C4S, C6S, CS-D, and CS-E were obtained from Seikagaku/The Associates of Cape Cod. Wnt3a recombinant protein (Wnt3a-RP) was purchased from R&D Systems. Wnt antagonist I (IWR-1-endo; catalogue No. 681669) was purchased from Calbiochem and quercetin hydrate from Acros Organics (catalogue No. 174070250).
Immunofluorescence Studies
For β-catenin immunofluorescence studies, nonconfluent NIH3T3 cells were incubated in L-CM or Wnt3a-CM with or without chondroitin sulfates (concentrations as indicated below) for 24 h. Alternatively, cells were incubated with Wnt3a-RP (500 ng/ml) and CS-E (concentrations indicated in text). Cells were fixed in 4% paraformaldehyde (PFA) and permeabilized, and immunofluorescence was performed by standard methods. The primary antibody was mouse anti-β-catenin (Santa Cruz Biotechnology, Inc.), followed by Alexa-488-conjugated secondary antibody (Invitrogen). Nuclei were counterstained with DAPI. For EdU proliferation studies, the Click-iT EdU kit (Invitrogen) was used according to the manufacturer's instructions. For TUNEL labeling of cells undergoing apoptosis, an in situ cell death detection kit, fluorescein (Roche Applied Science) was used according to the manufacturer's instructions. For nuclear luminescence quantification of the β-catenin signal in Wnt3a-stimulated cells, the average and standard deviation of the nuclear luminescence of 40 nuclei for each condition were measured in immunofluorescence images in Adobe Photoshop.
TOPFLASH Reporter Assays
NIH3T3 cells were transiently transfected with firefly TOPFLASH (35) and Renilla luciferase transfection control reporter constructs using linear PEI (Mr 25,000, Polysciences, Inc.; PEI/DNA ratio of 5:1) as the transfection reagent. Cells were stimulated with Wnt3a-CM or Wnt3a-RP for 24 h. Dual-Luciferase assays were performed as per the manufacturer's instructions (Promega or Biotium, Inc.).
Genome-wide Microarray Gene Expression Profiling
Subconfluent NIH3T3 cells were treated with L-CM or Wnt3a-CM in the presence or absence of CS-E at 100 μg/ml for 36 h followed by RNA preparation using TRI Reagent or TRIzol (Fisher Scientific) and the RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Subsequently, gene expression profiling was performed at the Northwestern University Center for Genetic Medicine Genomics Core Facility utilizing Illumina Mouse-8 gene expression chips on an Illumina iScan platform.
Quantitative Real-time PCR (qRT-PCR)
RNA was prepared using TRI Reagent or TRIzol according to the manufacturer's instructions. Subsequently, 1–4 μg of RNA was reverse-transcribed using reverse transcriptase (Promega) followed by real-time amplification using Power-SYBR Green PCR Master Mix (Applied Biosystems) on an Applied Biosystems 7500 real-time PCR platform in 20-μl reactions with an annealing temperature of 60 °C.
The following primer pairs were used (5′ to 3′): HPRT, ATGCCGACCCGCAGTCCCAGC and CGAGCAAGTCTTTCAGTCCTGTCC; Calr, GCATAGGCCTCATCATTGGT and AATACTCCCCCGATGCAAAT; Tnfsf9, GGCTGTGCCAGTTCAGAGTT and AGACCAGGTCACCCCTGTTT; Mrgprf, GCGTAGCAATCTGCTCAATG and GGAAACTGTTCATGGGAAGC; Slpi, TGTGGCTCCTGTTACTCAGC and GCCAAACCCCTACCTAACCA; Txnip, AGGCCTCATGATCACCATCT and GATGCAAGGGTCTCAGCAGT; Prickle1, TGCTCAGGAGATCCAAGTCC and CTCTCTTCAAAGTGATACGC; Arhgap18, GGAACTTTGGTGGAGCCTTC and GGACAGCCATGTAAAGGGAG; Arrdc4, AACGATGTTGCCAAAGGTTC and TTGAACCTGCGGTTGAGTCT; Cyp2f2, TAATGTTGGACACAGAGCGG and ATCCTGGAAGAAGGCAGCTT; and Lcn2, TGGCCACTTGCACATTGTAG and ATGTCCCTCCATCCTGGTC.
Western Blots
NIH3T3 cells were stimulated with L-CM or Wnt3a-CM for 1 h in the presence or absence of CS-E at 100 μg/ml. Alternatively, NIH3T3 cells were stimulated with Wnt3a-RP (500 ng/ml) in the presence or absence of CS-E at 200 μg/ml. Whole cell lysates were separated on 6% SDS-polyacrylamide gels. Antibodies used included rabbit anti-LRP6, rabbit anti-pLRP6 (both from Cell Signaling Inc.), and mouse anti-α-tubulin (Santa Cruz Biotechnology).
RESULTS
CS-E Interferes with Wnt3a Signaling
In initial experiments, we set out to evaluate whether treatment with exogenous CS could interfere with Wnt3a signaling output. We stimulated canonical Wnt signaling in NIH3T3 cells with Wnt3a conditioned medium (Wnt3a-CM) from L-cells stably overexpressing a Wnt3a cDNA; and as a control, we used conditioned medium from the parental nontransfected L-cells (L-CM). To test the effects of differentially sulfated chondroitin forms on canonical Wnt signaling, we treated these cultures concomitantly with C4S, C6S, CS-D, or CS-E, all at 100 μg/ml, or with no CS as a control. Immunofluorescence staining for β-catenin showed that in L-CM-treated control cultures, low levels of β-catenin were located mainly in cell membrane and cytoplasmic regions (Fig. 1A, white arrow). Treatment with C4S (Fig. 1B), C6S (Fig. 1C), CS-D (Fig. 1D), or CS-E (Fig. 1E) did not alter β-catenin localization or levels. When control NIH3T3 cultures where treated with Wnt3a-CM, we observed a drastic accumulation and nuclear localization of β-catenin (Fig. 1F, yellow arrow) as shown previously. Concomitant treatment with C4S (Fig. 1G), C6S (Fig. 1H), or CS-D (Fig. 1I) did not alter this Wnt3a-mediated effect. However, CS-E was able to severely reduce the Wnt3a-mediated accumulation of β-catenin (Fig. 1J); but CS-E did not completely abolish the effect of Wnt3a, as weak nuclear staining for β-catenin was still visible (Fig. 1J, yellow arrow) indicating that residual Wnt3a signaling was still present. Quantification of β-catenin nuclear luminosity from Wnt3a-CM-treated cultures confirmed that treatment with C4S, C6S, and CS-D did not interfere with the Wnt3a-mediated nuclear accumulation of β-catenin (Fig. 1K). However, treatment with CS-E decreased the nuclear luminosity of β-catenin to ∼27% (Fig. 1K).
FIGURE 1.

CS-E treatment interferes with Wnt3a signaling in NIH3T3 cells. A–J, immunofluorescence detection of β-catenin in NIH3T3 cells treated with L-CM or Wnt3a-CM in the presence or absence of exogenous CS forms at 100 μg/ml. Treatment with L-CM results in low levels of β-catenin expression, localized mainly in the cytoplasm and the cell membrane (A, white arrow). This pattern of β-catenin expression was identical in cultures treated with C4S (B), C6S (C), CS-D (D), and CS-E (E). Treatment with Wnt3a-CM caused a drastic increase in β-catenin levels and nuclear accumulation (F, yellow arrow). This outcome of Wnt3a-CM was unaffected by treatment with C4S (G), C6S (H), and CS-D (I). CS-E treatment caused a severe reduction in β-catenin expression levels (J) while still maintaining nuclear localization (F–J, yellow arrows). Scale bar = 10 μm. K, quantification of nuclear luminosity of Wnt3a-induced nuclear β-catenin signal from experiment depicted in F–J. Treatment with CS-E, but not C4S, C6S, or CS-D, reduced nuclear luminosity to ∼27% when compared with Wnt3a treatment alone. (L, treatment with Wnt3a-CM for 24 h caused a significant increase in TOPFLASH luciferase activity when compared with treatment with L-CM. Concomitant treatment with CS-E, but not C4S, C6S, or CS-D (all at 100 μg/ml), significantly reduced TOPFLASH reporter activity. (*, p < 0.01). M, dose response of the effects of CS-E on Wnt3a-induced TOPFLASH activity. CS-E at 5 μg/ml reduced Wnt3a-induced TOPFLASH activity slightly, whereas CS-E at 20, 100, and 200 μg/ml reduced the effects of Wnt3a-CM stimulation significantly (*, p < 0.01). No significant differences among CS-E concentrations of 20, 100, and 200 μg/ml were observed. N, treatment of NIH3T3 cells with Wnt3a-CM (W3a) but not L-CM (L) for 1 h led to phosphorylation of LRP6 (pLRP6). Concomitant treatment with CS-E (E) (100 μg/ml) significantly (*, p < 0.05) interfered with phosphorylation of the LRP6 protein. Levels of LRP6 and α-tubulin proteins are shown as controls. Note that only the upper band of the two specific LRP6 bands is phosphorylated in response to Wnt3a.
We next examined whether CS-E could also interfere with the nuclear function of β-catenin. To this end, we employed the widely used TOPFLASH reporter assay, in which a luciferase reporter is driven by multiple T-cell factor/lymphoid enhancer factor binding sites and serves as a functional nuclear output of β-catenin activity. Stimulation with Wnt3a-CM led to a strong increase in TOPFLASH activity when compared with stimulation with L-CM (Fig. 1L). This was not altered by treatment with C4S, C6S, or CS-D; however, CS-E reduced Wnt3a-CM-stimulated TOPFLASH activity significantly (Fig. 1L). Quantitation of this effect showed that CS-E could reduce Wnt3a-CM-mediated TOPFLASH activity to ∼20–25% (Fig. 1L), corroborating our quantitative findings above. Dose-response experiments established that CS-E at 5 μg/ml had a moderate effect on Wnt3a-CM-induced TOPFLASH activity that did not reach statistical significance, whereas CS-E at 20 μg/ml reduced TOPFLASH activity significantly, and CS-E at 100 as well as 200 μg/ml reduced TOPFLASH even further, with an apparent plateau reached at 100 μg/ml and no statistical difference between 20, 100, and 200 μg/ml (Fig. 1M). These data demonstrated that CS-E at 100 μg/ml achieved a maximum inhibitory effect on Wnt3a signaling when stimulating with Wnt3a-CM.
CS-E Interferes with LRP6 Phosphorylation
Previous Biacore experiments showed that CS-E can bind Wnt3a ligand with high affinity (32). Thus, we hypothesized that CS-E treatment could interfere with the activation of the cell surface receptor complex by Wnt3a ligand. To this end, we analyzed phosphorylation of LRP6, an integral component of the Wnt receptor complex (Fig. 1N). Western blot analysis of NIH3T3 total cell lysates showed that treatment with Wnt3a-CM for 60 min led to phosphorylation of LRP6 (pLRP6), whereas concomitant treatment with CS-E abolished this phosphorylation (Fig. 1N). The levels of total LRP6 protein were not significantly altered by our treatments, and CS-E alone had no effect on pLRP6 levels. Quantitation of these effects showed a significant reduction in the levels of pLRP6 when cells were co-treated with Wnt3a-CM and CS-E, whereas CS-E alone did not alter pLRP6 levels (Fig. 1N). These results demonstrated that CS-E could interfere with Wnt3a-mediated activation of its cell surface receptor complex.
CS-E Interferes with Pathway Stimulation by Both Wnt3a-CM and Wnt3a-RP
To corroborate that this effect of CS-E on Wnt3a signaling is not caused by nonspecific interactions of CS-E with components of the conditioned medium other than Wnt3a, we also analyzed the effects of CS-E on β-catenin accumulation and localization, TOPFLASH activity, and LRP6 phosphorylation in response to recombinant Wnt3a protein (Wnt3a-RP) (supplemental Fig. S1). First, NIH3T3 cells were treated with Wnt3a-RP in the presence or absence of CS-E at 50, 100, 200, or 300 μg/ml. CS-E was able to significantly inhibit Wnt3a-RP-induced TOPFLASH activity at all concentrations, with a plateau effect at 200 μg/ml (Fig. supplemental S1A). We next analyzed the effect of CS-E treatment on Wnt3a-RP-stimulated LRP6 phosphorylation. Western blot analysis demonstrated that treatment with CS-E could significantly reduce Wnt3a-RP-mediated phosphorylation of LRP6 (supplemental Fig. S1B). Finally, we analyzed the effects of CS-E on Wnt3a-RP-stimulated accumulation and nuclear localization of β-catenin in immunofluorescence staining (supplemental Fig. S1, C–F). As shown for Wnt3a-CM above, CS-E treatment drastically reduced Wnt3a-RP-stimulated β-catenin staining (supplemental Fig. S1F) when compared with cultures treated with Wnt3a-RP alone (supplemental Fig. S1E). Measurement and quantification of β-catenin nuclear luminosity from supplemental Fig. S1, E and F showed that CS-E treatment reduced Wnt3a-RP-stimulated β-catenin staining to ∼29% (supplemental Fig. S1G). Together, these data demonstrated that treatment with exogenous CS-E could interfere in a basically identical manner with signaling initiated by either Wnt3a-CM or Wnt3a-RP, thus corroborating our conclusions above that CS-E treatment negatively regulates Wnt3a signaling.
CS-E Interferes with Wnt3a-mediated Induction, but Not Repression, of Target Genes
As a next step, we wanted to extend our analysis to the effects of CS-E treatment on Wnt3a-controlled global transcriptional programs. NIH3T3 cells were treated with L-CM, L-CM+CS-E, Wnt3a-CM, or Wnt3a-CM+CS-E. After 36 h, RNA was prepared, and gene expression profiles were analyzed by microarray analysis on Illumina Mouse BeadChips. We chose a time frame of 36 h to capture not only immediate early target genes but also a more complete set of target genes affected by Wnt3a signaling. This analysis revealed 96 genes that were induced more than 2-fold by Wnt3a-CM treatment (supplemental Table S1), whereas 22 genes were repressed more than 2-fold by Wnt3a-CM treatment (supplemental Table S2). Several genes in both the induced and repressed groups were identified previously as Wnt3a target genes in NIH3T3 cells after 6 h of treatment (supplemental Tables S1 and S2) (36). Identification of additional target genes suggested that our 36-h treatment might include direct target genes that require treatment with Wnt3a for more than 6 h and/or indirect target genes. When we examined the effects of CS-E treatment on Wnt3a-CM target gene induction, we observed a uniform reduction in the Wnt3a-induced expression of all positively regulated target genes (supplemental Table S1 and Fig. 2A). Quantification showed that on average, CS-E treatment reduced Wnt3a-CM-stimulated gene expression to ∼18%, when compared with Wnt3a treatment in the absence of CS-E (Fig. 2B). This result correlated with the CS-E-mediated reduction in Wnt3a signaling described above. Notably, CS-E alone did not significantly affect expression of these Wnt3a target genes (Fig. 2, A and B). We then analyzed the effect of CS-E on expression of target genes negatively regulated by Wnt3a. Surprisingly, treatment with CS-E did not interfere with Wnt3a-mediated repression of any of the target genes in this group (supplemental Table S2 and Fig. 2C). Quantification of this effect demonstrated no interference of CS-E with the negative regulation of Wnt3a of these target genes (Fig. 2D). CS-E alone had a modest overall repressive function on these genes (supplemental Table S2 and Fig. 2, C and D). Thus, these results suggested a surprising differential effect of CS-E on Wnt3a-mediated gene induction versus repression. To confirm these results, we selected five positively regulated Wnt target genes (calreticulin (Calr), Rho GTPase-activating protein 18 (Arhgap18), Prickle homolog 1 (Prickle1), Tumor necrosis factor ligand superfamily 9 (Tnfsf9), MAS-related GPR, member F (Mrgprf)) and five negatively regulated target genes (cytochrome P450 polypeptide 2 (Cyp2f2), secretory leukocyte peptidase inhibitor (Slpi), arrestin domain-containing protein 4 (Arrdc4), lipocalin 2 (Lcn2), SRY-box-containing gene 9 (Sox9)) and quantified the effect of CS-E treatment on Wnt3a stimulation of target gene mRNA expression by qRT-PCR (Fig. 3). These experiments confirmed the ability of Wnt3a-CM to induce expression of all five positive target genes when compared with treatment with L-CM (Fig. 3A). Concomitant treatment with CS-E caused a significant reduction in the expression of these positively regulated target genes, (Fig. 3A), confirming that CS-E could interfere with the Wnt3a-mediated stimulation of target gene expression. All genes in the group of negatively regulated target genes were drastically repressed by Wnt3a-CM (Fig. 3B); importantly, these experiments confirmed that concomitant treatment with CS-E did not interfere with Wnt3a repression and therefore could not reverse the Wnt3a-mediated reduction in target gene expression (Fig. 3B). This effect was observed independently of the effect of CS-E alone on target gene expression (Fig. 3B), suggesting that the absence of any effect of CS-E on Wnt3a-mediated target gene repression was not due to strong independent effects of CS-E on expression of these genes. These results confirmed our microarray data and showed that CS-E could interfere with Wnt3a-mediated induction, but not repression, of target gene mRNA expression. Because we showed above that even in the presence of CS-E a residual Wnt3a signaling output of ∼25% was maintained, we hypothesized that the differential effects of CS-E on Wnt3a target gene induction versus repression might have identified a critical Wnt3a signaling threshold that allows selective control of global Wnt3a transcriptional programs.
FIGURE 2.

Microarray analysis: CS-E differentially affects positively and negatively regulated Wnt3a transcriptional programs. A, graphic presentation of the -fold change in mRNA expression of the 96 genes induced 2-fold or more by Wnt3a-CM in NIH3T3 cells. Wnt3a-CM (W3a, marked in blue) increased mRNA expression of these target genes from ∼7- to 2-fold over treatment with L-CM. Concomitant treatment with CS-E at 100 μg/ml (W3a+CS-E, marked in red) reduced mRNA expression of all Wnt3a-induced genes. Treatment with CS-E alone in L-CM (CS-E, marked in yellow) did not significantly affect target gene expression over L-CM alone. B, quantification of results shown in A. In the presence of Wnt3a-CM, CS-E reduced target gene expression to ∼18%. CS-E did not have a significant effect on gene expression in L-CM alone. C, graphic presentation of the fold-mRNA expression of the 22 genes repressed 2-fold or more by Wnt3a-CM in NIH3T3 cells. Wnt3a-CM (blue) repressed mRNA expression of these target genes to levels of ∼0.2–0.5 over treatment with L-CM. Concomitant treatment with CS-E at 100 μg/ml (red) was not able to interfere with this repression and in many cases slightly increased repression of mRNA expression. Treatment with CS-E alone in L-CM (yellow) also slightly decreased target gene expression over L-CM alone. D, quantification of results shown in C. CS-E treatment could not interfere with Wnt3a-CM-mediated target gene repression and instead increased repression slightly. A similar effect was observed when cells were treated with CS-E in L-CM alone.
FIGURE 3.

Confirmation of the effects of CS-E on Wnt3a target gene expression by qRT-PCR. NIH3T3 cells were treated with L-CM (L) or Wnt3a-CM (W3a) in the presence or absence of CS-E at 100 μg/ml for 36 h followed by RNA preparation and qRT-PCR. A, evaluation of effects of CS-E treatment on expression of the positively regulated target genes Calr, Arhgap18, Prickle1, Tnfsf9, and Mrgprf in the presence of L-CM or Wnt3a-CM. All five target genes were confirmed to be induced by treatment with Wnt3a-CM. Concomitant treatment with CS-E resulted in a significant decrease in expression of Calr, Arhgap18, Prickle1, Tnfsf9, and Mrgprf (*, p < 0.05). B, evaluation of effects of CS-E treatment on expression of the negatively regulated target genes Cyp2f2, Slpi, Arrdc4, Lcn2, and Sox9 in the presence of L-CM or Wnt3a-CM. All five target genes were confirmed to be repressed by treatment with Wnt3a-CM, and concomitant treatment with CS-E did not result in any significant change in Wnt3a-mediated repression in any of the target genes.
Differential Wnt3a Signaling Thresholds for Induced Versus Repressed Target Genes
To test our hypothesis that differential Wnt3a signaling thresholds govern target gene induction versus repression, and to ensure that the effects we observed were not due to nonspecific effects of CS-E, we modulated Wnt3a signaling levels in the absence of CS-E. We generated a Wnt3a signaling gradient by dilution of CM with 0, 16, 30, 55, and 100% TOPFLASH activity in conditions with 0, 10, 25, 50, and 100% Wnt3a-CM, respectively (Fig. 4, A and C). We then asked whether expression of positive and negative Wnt3a target genes in NIH3T3 cells was responsive to different Wnt3a signaling levels. Analysis of expression of the positively regulated target genes Mrgprf, Tnfsf9, Arhgap18, and Prickle1 by qRT-PCR showed low levels of ∼8–18% induction at Wnt3a signaling levels of 16% (Fig. 4B, W10%), whereas a 17–42% induction of expression was observed at a Wnt3a signaling level of 30% (W25%). Even at 55% signaling activity (Fig. 4B, W50%) expression levels of induced target genes reached only 10–38% of the levels observed at 100% signaling activity (W100%). In contrast, the negatively regulated Wnt3a target genes Cyp2f2, Lcn2, Arrdc4, and Txnip showed 35–75% repression even at the lowest signaling levels tested (16%) (Fig. 4D, W10%). At 30% Wnt3a signaling activity (W25%), we observed 68–89% repression, and at 55% signaling activity (W50%) repression reached 40–94% of the levels observed at 100% signaling activity (W100%). These results support our hypothesis that differential Wnt3a signaling thresholds control target gene induction versus repression. Whereas only minimal induction of target gene expression was achieved at low levels of Wnt3a signaling activity, gene repression was much more sensitive at these low signaling levels and allowed significant repression. These results also supported our notion that inhibition of Wnt3a signaling to ∼25% by treatment with CS-E could interfere with target gene induction but not with target gene repression.
FIGURE 4.

Differential Wnt3a thresholds control target gene induction versus repression. A and C, TOPFLASH reporter assays in NIH3T3 cells in the presence of Wnt3a-CM at 0 (noW), 10 (W10%), 25 (W25%), 50 (W50%), or 100% (W100%) led to a gradient of Wnt3a signaling activity with 0, 16, 30, 55, and 100% TOPFLASH activity, respectively. B, expression of the positively regulated Wnt3a target genes Mrgprf, Tnfsf9, Arhgap18, and Prickle1 at different Wnt3a signaling levels. Low levels of ∼8–18% induction were observed at Wnt3a signaling levels of 16% (W10%), whereas a 17–42% induction of expression was observed at a Wnt3a signaling level of 30% (W25% (B)). Even at 55% signaling activity (W50%) expression levels of induced target genes only reached 10–38% of the levels observed at 100% signaling activity (W100%). D, the negatively regulated Wnt3a target genes Cyp2f2, Lcn2, Arrdc4, and Txnip showed repression of 35–75% even at the lowest signaling levels tested (16% = W10%). At 30% Wnt3a signaling activity (W25%), we observed 68–89% repression, and at 55% signaling activity (W50%) repression reached 40–94% of the levels observed at 100% signaling activity (W100%). The dashed line box indicates signaling levels that bracket the levels we observed with CS-E treatment (∼25%; see Fig. 1).
Differential Wnt3a Signaling Thresholds Regulate Proliferation Versus Apoptosis
Wnt/β-catenin signaling has been shown in many studies to regulate proliferation positively through the induction of genes involved in the positive regulation of the cell cycle and proliferation (37, 38). An analysis of Gene Ontology (GO) Biological Processes of the positively regulated genes in our gene profiling experiments indeed showed an enrichment in genes involved in the positive regulation of cell proliferation (supplemental Table S1). In contrast, canonical Wnt signaling has also been shown to interfere with serum starvation-induced apoptosis (38–40), which has been correlated with the repression of pro-apoptotic genes (36, 39). Accordingly, analysis of GO Biological Processes, as well as literature searches, of the genes repressed by Wnt3a in our gene profiling experiments demonstrated an enrichment of pro-apoptotic genes (supplemental Table S2). Because we had demonstrated that Wnt3a could regulate positive and negative target genes at different threshold signaling levels, we hypothesized that these Wnt3a thresholds might also affect proliferation and apoptosis in a similar fashion. To test our hypothesis, we first examined at which signaling levels Wnt3a could efficiently induce proliferation of NIH3T3 cells. For this, we established Wnt3a-RP signaling gradients in TOPFLASH assays; concentrations of Wnt3a-RP of 0, 50, 62.5, 100, 167, and 500 ng/ml resulted in TOPFLASH activities of 0, 9, 27, 46, 76, and 100%, respectively (Fig. 5A). We then measured proliferation of NIH3T3 cells at these Wnt3a signaling levels by EdU incorporation. Cells were treated with Wnt3a-RP for 24 h, with the nucleotide analog EdU added for the last 60 min followed by the analysis of detection and quantification of EdU by immunofluorescence methods as an output for proliferation (Fig. 5B). Wnt3a signaling levels of 9% (50 ng/ml Wnt3a-RP), 27% (62.5 ng/ml Wnt3a-RP), 46% (100 ng/ml Wnt3a-RP), and 76% (167 ng/ml Wnt3a-RP) increased EdU incorporation by 1–10% when compared with unstimulated cells (Fig. 5B). At 100% signaling activity (500 ng/ml Wnt3a-RP), a dramatic increase of EdU-positive cells to 28% was observed (Fig. 5B). These data suggested that high levels of Wnt3a signaling, approaching 100% activity, are required in NIH3T3 cells to elicit a maximum effect on cell proliferation.
FIGURE 5.
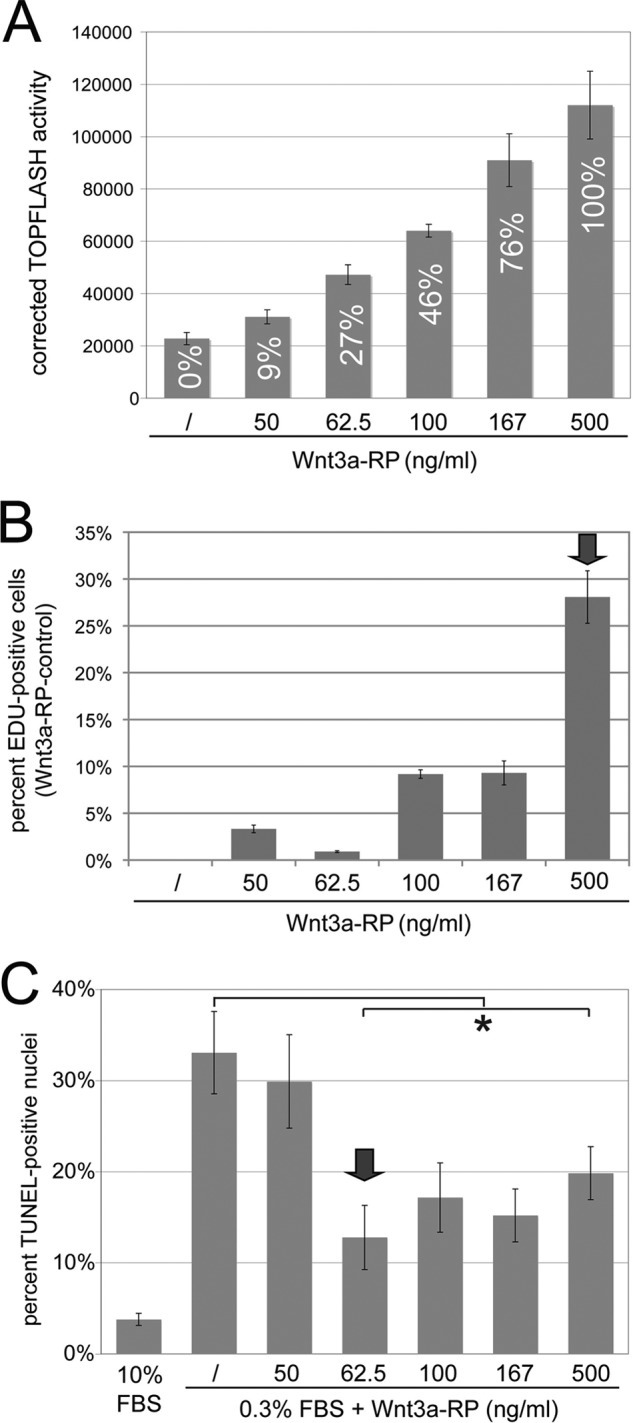
Differential Wnt3a thresholds control proliferation versus apoptosis. A, establishment of a TOPFLASH Wnt3a signaling gradient with Wnt3a-RP. Wnt3a-RP at 0, 50, 62.5, 100, 167 and 500 ng/ml resulted in TOPFLASH activities (Wnt3a-RP/control) of 0, 9, 27, 46, 76, and 100%, respectively. The concentration of 500 ng/ml is above the concentrations frequently used for Wnt3a-RP, and TOPFLASH activity at this concentration was set arbitrarily to 100%. B, quantification of EdU incorporation to measure proliferation of NIH3T3 cells. Cells were treated with Wnt3a-RP for 24 h at 0% (/), 9% (50 ng/ml Wnt3a-RP), 27% (62.5 ng/ml Wnt3a-RP), 46% (100 ng/ml Wnt3a-RP), 76% (167 ng/ml Wnt3a-RP), or 100% (500 ng/ml Wnt3a-RP). The percentage of EdU-positive nuclei in relation to total DAPI-stained nuclei was established for all conditions; subsequently, the value for control conditions (/) was set to zero and subtracted from experimental condition to show the percent increase in EdU-positive cells obtained with Wnt3a treatment over control conditions. Wnt3a signaling levels of 9% (50 ng/ml Wnt3a-RP), 27% (62.5 ng/ml Wnt3a-Rp), 46% (100 ng/ml Wnt3a-RP), and 76% (167 ng/ml Wnt3a-RP) increased EdU incorporation by 1–10% when compared with unstimulated cells (/). At 100% signaling activity (500 ng/ml Wnt3a-RP), a dramatic increase of EdU-positive cells to 28% was observed. The arrow marks the signaling level with maximum biological response. C, quantification of serum starvation-induced apoptosis by TUNEL assay in the presence of Wnt3a-RP gradient signaling levels. Cells were treated in medium containing 0.3% serum and with the indicated concentrations of Wnt3a-RP for 24 h. The percentage of TUNEL-positive cells was quantified by co-staining with DAPI. Control cells in 10% serum showed ∼3% TUNEL-positive cells; this increased to 33% when cell were incubated in 0.3% serum. Low Wnt3a signaling activity of 9% (50 ng/ml Wnt3a-RP) was not able to significantly interfere with apoptosis. However, a Wnt3a signaling activity of 27% (62.5 ng/ml Wnt3a-RP) was able to significantly reduce apoptosis to 13% (*, p < 0.05). Higher Wnt3a signaling activities of 46, 76, or 100% reduced TUNEL staining to a degree similar to that seen with 27% Wnt3a activity. The arrow marks the lowest signaling level with maximum biological response.
Next, we analyzed the effects of the different Wnt3a signaling levels established in Fig. 5A on serum starvation-induced apoptosis by TUNEL assays (Fig. 5C). Cells were starved for 24 h in medium containing 0.3% serum and were treated concomitantly with Wnt3a-RP at the indicated concentrations. As a control, cells were incubated in 10% serum. After treatments, cells were processed for TUNEL staining, and the percentage of positively stained nuclei was quantified (Fig. 5C). Cells in 10% serum showed ∼3% TUNEL-positive cells; this increased to 33% when cell were incubated instead in 0.3% serum, thus demonstrating that serum starvation could lead to increased apoptosis in these cells. A low Wnt3a signaling activity of 9% (50 ng/ml Wnt3a-RP) did not interfere with apoptosis, but signaling activity of 27% (62.5 ng/ml Wnt3a-RP) significantly reduced apoptosis to ∼13%. Strikingly, higher Wnt3a signaling activities of 46, 76, or 100% did not increase this anti-apoptotic effect any further (Fig. 5C), suggesting that a Wnt3a signaling threshold of ∼27% is sufficient to mediate the full anti-apoptotic effects of Wnt3a. In summary, these data supported our hypothesis that Wnt3a can regulate proliferation and apoptosis at different threshold levels. Wnt3a signaling activity of close to 100% was required to efficiently promote proliferation, whereas a signaling activity of only 27% was sufficient for a maximum anti-apoptotic effect.
CS-E Treatment Can Interfere with Wnt3a-induced Proliferation but Not with Wnt3a-mediated Repression of Apoptosis
The data given above led us to hypothesize that reduction of Wnt3a signaling to ∼25% by treatment with CS-E will interfere with the effects of Wnt3a signaling on proliferation but will not affect the functional role of Wnt3a signaling in the prevention of apoptosis in NIH3T3 cells. To investigate proliferative responses, cells were incubated in DMEM, 10% FBS in the presence or absence of Wnt3a-RP for 24 h, either alone (Fig. 6, A and D) or in the presence or absence of CS-D (Fig. 6, B and E) or CS-E (Fig. 6, C and F), followed by EdU incorporation. The percentage of EdU-positive cells was quantified by co-staining with DAPI. Few EdU-positive cells were seen in control conditions (Fig. 6A), and similar numbers of cells were observed when treated with CS-D (Fig. 6B) or CS-E (Fig. 6C). Stimulation with Wnt3a-RP caused an increase in EdU-positive cells (Fig. 6D), which was not altered by concomitant treatment with CS-D (Fig. 6E). However, concomitant treatment with CS-E led to a reduction in the number of EdU-positive cells (Fig. 6F). Quantification of these data (Fig. 6G) showed that the proliferation rate of ∼42% in control conditions was increased to 54% with the addition of Wnt3a-RP. Treatment with CS-D did not change the pro-proliferative effect of Wnt3a, whereas CS-E completely abolished the ability of Wnt3a to stimulate proliferation (Fig. 6G). To corroborate these findings, we also sought to determine the effects of CS-E on Wnt3a-mediated cell growth over a longer time frame. Equal numbers of NIH3T3 cells were incubated for 24 h in the presence or absence of Wnt3a-RP, with or without CS-D or CS-E (Fig. 6H), followed by quantification of cell numbers. Wnt3a-RP treatment resulted in a 28% increase in cell growth; this was not significantly altered by concomitant CS-D treatment. However, the addition of CS-E completely abolished Wnt3a-stimulated cell growth and in fact led to a 2% decrease in cell numbers (Fig. 6H).
FIGURE 6.

CS-E interferes with Wnt3a-mediated stimulation of proliferation but not with the anti-apoptotic effects of Wnt3a. A–H, effect of CS-E on Wnt3a-induced proliferation. NIH3T3 cells were treated under control conditions (DMEM/10% FBS) or with DMEM/10% FBS plus Wnt3a-RP (500 ng/ml) for 24 h in the presence or absence of CS-D or CS-E at 200 μg/ml followed by incorporation of EdU. The percentage of EdU-positive cells was quantified by co-staining with DAPI. Few EdU-positive cells were seen in control conditions (A). Treatment with CS-D (B) or CS-E (C) showed similar results. Treatment with Wnt3a-RP caused an increase in EdU-positive cells (D). This was not altered by treatment with CS-D (E) but was reduced by treatment with CS-E (F). G, quantification of these data showed that the proliferation rate of ∼42% in control conditions was increased to 54% with Wnt3a-RP. Treatment with CS-D did not change this proliferation-promoting effect of Wnt3a, whereas treatment with CS-E completely abolished the effect of Wnt3a (arrow; *, p < 0.01). H, Wnt3a-induced cell growth was quantified by cell counts 24 h after NIH3T3 cells were treated under control conditions (DMEM/10% FBS) or with DMEM/10% FBS plus Wnt3a-RP (500 ng/ml) in the presence or absence of CS-D or CS-E at 200 μg/ml. Stimulation of NIH3T3 cells with Wnt3a-RP (no CS), Wnt3a-RP+CS-D (CS-D), or Wnt3a-RP+CS-E (CS-E) treatments is shown as a percentage of increase in cell growth over the control conditions (*, p < 0.01). Wnt3a-RP treatment resulted in a 28% increase in cell growth, whereas Wnt3a-RP+CS-E treatment led to a 2% decrease in cell growth. I–N, effect of CS-E on Wnt3a-mediated repression of serum starvation-induced apoptosis. NIH3T3 cells were incubated in medium containing 0.3% serum with 100 ng/ml Wnt3a-RP in the presence or absence of CS-D or CS-E at 100 μg/ml for 24 h. Subsequently, apoptotic cells containing DNA double strand break cells were visualized using a TUNEL fluorescence staining procedure. Nuclei were counterstained with DAPI. Control cultures incubated in 10% serum showed almost no positively stained cells (I). Serum starvation led to a significant increase in TUNEL-positive cells (J), which was significantly reduced by treatment with Wnt3a-RP (K). Concomitant treatment with CS-D did not alter this effect of Wnt3a (L) and neither did treatment with CS-E (M). N, quantification of these data showed ∼1% TUNEL-positive cells at 10% serum conditions, which increased to 47% TUNEL-positive cells under serum starvation conditions. Treatment with Wnt3a-RP reduced the number of cells undergoing apoptosis to 12%, which was not significantly altered by CS-D or by CS-E treatment (arrow; p < 0.01).
We next analyzed the effect of treatment with CS-D and CS-E treatment on the rescue of serum starvation-induced apoptosis by Wnt3a (Fig. 6, I–N). NIH3T3 cells were serum-starved in medium containing 0.3% serum, with Wnt3a-RP at 500 ng/ml in the presence or absence of CS-D or CS-E at 100 μg/ml, for 24 h. Subsequently, TUNEL staining was performed to visualize cells undergoing apoptosis. Cells incubated in 10% serum showed very little positive staining (Fig. 6I). Serum starvation led to a significant increase in TUNEL-positive cells (Fig. 6J), which was significantly reduced by treatment with Wnt3a-RP (Fig. 6K). Concomitant treatment with CS-D did not alter this effect of Wnt3a (Fig. 6L). Importantly, treatment with CS-E also did not interfere with Wnt3a function in this assay (Fig. 6M). Quantification of these data (Fig. 6N) showed that almost no TUNEL-positive cells were present in 10% serum conditions, which increase to 47% TUNEL-positive cells in 0.3% serum. Treatment with Wnt3a-RP reduced the number of cells undergoing apoptosis to 12%, which was not significantly altered by CS-D (as we showed that CS-D did not alter Wnt3a signaling) or by CS-E (Fig. 6N). In summary, these data supported our hypothesis that a reduction of Wnt3a signaling levels to ∼25% by treatment with exogenous CS-E interfered with Wnt3a-stimulated proliferation but not with the anti-apoptotic effects of Wnt3a in NIH3T3 cells.
Wnt3a Signaling Thresholds Achieved by Pharmacological Inhibition Differentially Affect Induced Versus Repressed Target Genes
Thus far, we have described novel Wnt3a signaling thresholds that differentially control target gene induction and repression as well as the biological consequences of a Wnt3a signal. We hypothesized that establishment of Wnt3a signaling thresholds by treatment with previously established pharmacological inhibitors should differentially affect Wnt3a-mediated target gene expression in a manner similar to inhibition with CS-E or to dilution of Wnt3a ligand in the absence of any inhibitors. For this purpose, we utilized two known pharmacological inhibitors of the Wnt/β-catenin pathway: the tankyrase inhibitor IWR-1 (inhibitor of Wnt response-1), which stabilizes axin, a scaffold for the intracellular β-catenin destruction complex, and therefore inhibits canonical β-catenin signaling (41), and the flavonoid quercetin, which has been utilized to interfere with the transcriptional activity of β-catenin/TCF complexes in the nucleus (42, 43). We first utilized different concentrations of IWR-1 and quercetin in TOPFLASH assays to establish a Wnt3a signaling gradient (Fig. 7A). NIH3T3 cells were treated with DMSO (control) and IWR-1 (5, 10, 25, and 50 μm) and quercetin (50, 100, 150, and 500 μm) concomitantly with L-CM or Wnt3a-CM for 24 h. IWR-1 and quercetin inhibited Wnt3a signaling activity in a concentration-dependent manner. Importantly, IWR-1 at 5 μm and quercetin at 150 μm reduced Wnt3a/β-catenin-induced TOPFLASH activity to the critical threshold levels of 27 and 29%, respectively.
FIGURE 7.
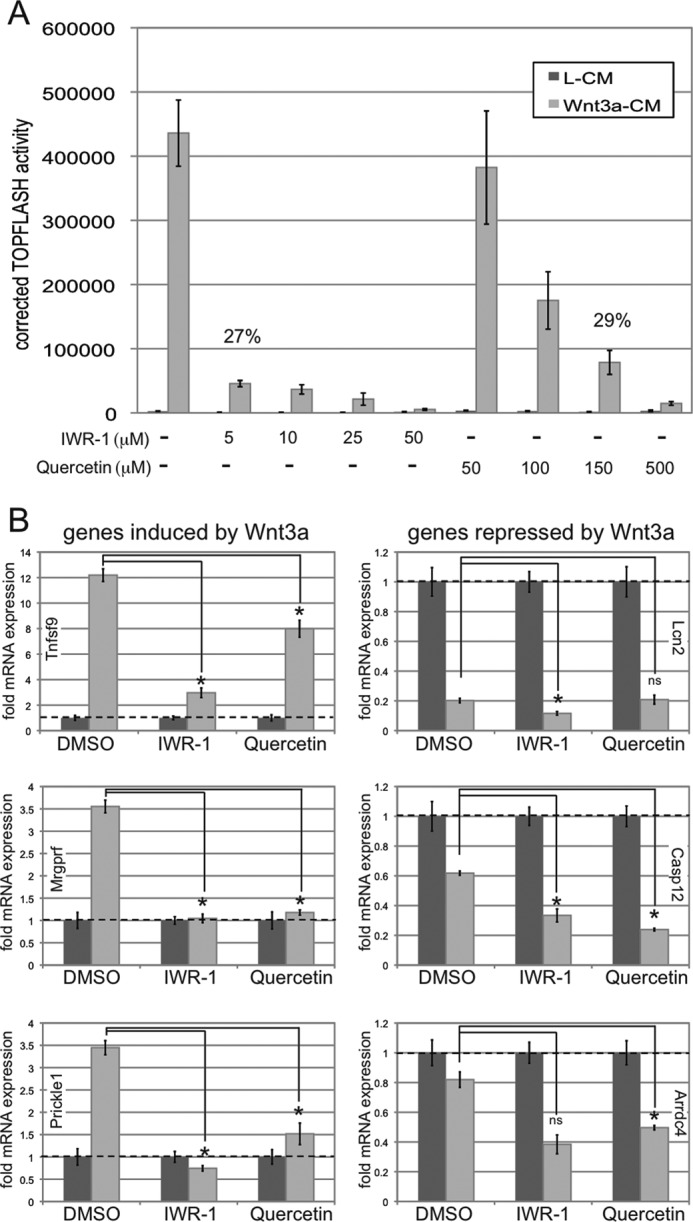
Wnt3a signaling thresholds achieved by pharmacological inhibition differentially affect induced versus repressed target genes. A, the pharmacological Wnt/β-catenin inhibitors IWR-1 and quercetin inhibit Wnt3a signaling activity in a concentration-dependent manner in TOPFLASH assays. NIH3T3 cells were treated with DMSO (control), IWR-1 (5, 10, 25, and 50 μm), or quercetin (50, 100, 150, and 500 μm) concomitantly with L-CM or Wnt3a-CM for 24 h. IWR-1 at 5 μm and quercetin at 150 μm reduced Wnt3a/β-catenin-induced TOPFLASH activity to 27 and 29%, respectively, in NIH3T3 cells. B, NIH3T3 cells were treated with L-CM or Wnt3a-CM for 36 h in the presence or absence of IWR-1 and quercetin at 5 and 150 μm, respectively, followed by RNA preparation and qRT-PCR. The expression levels of three positively regulated (Tnfsf9, Mrgprf, and Prickle1) and three negatively regulated (Lcn2, Casp12, and Arrdc4) Wnt3a target genes were analyzed as described in the legend for Fig. 3. IWR-1 and quercetin at these concentrations interfered with Wnt3a-mediated target gene induction, whereas target gene repression was not impaired (*, p < 0.01). ns, not significant.
We then analyzed the effects of these inhibitor concentrations on Wnt3a-mediated target gene expression. NIH3T3 cells were treated with L-CM or Wnt3a-CM for 36 h in the presence or absence of IWR-1 and quercetin at 5 and 150 μm, respectively, followed by RNA preparation and qRT-PCR (Fig. 7B). The expression levels of three positively regulated (Tnfsf9, Mrgprf, and Prickle1) and three negatively regulated (Lcn2, Casp12, and Arrdc4) Wnt3a target genes were analyzed. These experiments demonstrated that IWR-1 and quercetin, at concentrations that reduced Wnt/β-catenin signaling activity to ∼25%, significantly reduced Wnt3a-mediated gene induction but could not interfere with Wnt3a-mediated target gene repression. Overall, these studies confirmed that a critical intrinsic Wnt3a signaling threshold, established by either Wnt3a ligand dilution or by pharmacological inhibition using CS-E, IWR-1, or quercetin, leads to differential effects on target gene induction versus repression.
Both Induction and Repression of Target Genes by Wnt3a Require the Canonical β-Catenin Pathway
It was reported recently that in articular chondrocytes, Wnt3a could simultaneously activate the canonical β-catenin and the noncanonical calcium/calmodulin kinase II (Ca2+/CaMKII) pathways (44). Therefore, we sought to determine whether target gene induction and repression by Wnt3a in our system was solely dependent on the canonical β-catenin pathway. For this purpose, we again utilized the pharmacological inhibitors IWR-1 and quercetin, We hypothesized that if either induction or repression of target genes was dependent on a noncanonical pathway, a complete inhibition of canonical β-catenin signaling by IWR-1 or quercetin should leave the noncanonical transcriptional response intact. We first established that IWR-1 at 100 μm and quercetin at 500 μm completely eliminated Wnt3a-mediated canonical β-catenin signaling in TOPFLASH assays (Fig. 8A). We then asked how this complete inhibition of β-catenin signaling by IWR-1 and quercetin would affect target gene induction and repression by Wnt3a. NIH3T3 cells were treated with Wnt3a-RP for 36 h in the presence or absence of IWR-1 (100 μm) or quercetin (500 μm). Subsequently, we prepared RNA and analyzed the effects of IWR-1 and quercetin on the expression levels of three positively regulated (Tnfsf9, Mrgprf, and Prickle1) and three negatively regulated (Lcn2, Casp12, and Arrdc4) Wnt3a target genes (Fig. 8B). Inhibition of canonical β-catenin signaling by IWR-1 and quercetin completely eliminated both the induction and repression of target genes by Wnt3a-RP (Fig. 8B), suggesting that both Wnt3a-induced and -repressed transcriptional programs require a functional canonical β-catenin pathway.
FIGURE 8.

Induction and repression of Wnt3a target genes require the canonical β-catenin pathway. A, TOPFLASH assay to establish complete inhibition of Wnt/β-catenin signaling with IWR-1 and quercetin. NIH3T3 cells were treated for 24 h with DMSO (Control), IWR-1 at 100 μm, or quercetin at 500 μm either in control medium (DMEM/10% FBS) or in DMEM/10% FBS plus 500 ng/ml Wnt3a-RP. IWR-1 and quercetin at these concentrations completely blocked Wnt3a/β-catenin-induced TOPFLASH activity in NIH3T3 cells. B, NIH3T3 cells were treated with Wnt3a-RP (500 ng/ml) for 36 h in the presence or absence of IWR-1 and quercetin at 100 and 500 μm, respectively, followed by RNA preparation and qRT-PCR. The expression levels of three positively regulated (Tnfsf9, Mrgprf, and Prickle1) and three negatively regulated (Lcn2, Casp12, and Arrdc4) Wnt3a target genes were analyzed as described in the legend for Fig. 3. Complete inhibition of Wnt/β-catenin signaling eliminated both the induction and repression of target genes by Wnt3a-RP (*, p < 0.05).
DISCUSSION
Here, we have demonstrated that the glycosaminoglycan CS-E can function as an inhibitor of Wnt/β-catenin signaling by interfering with Wnt3a-mediated phosphorylation of LRP6 and activation of the Wnt receptor complex. Surprisingly, genome-wide gene expression profiling experiments showed a selective inhibitory effect of CS-E on Wnt3a-mediated target gene induction but not target gene repression. These effects of CS-E were due to differential Wnt3a signaling thresholds for induced versus repressed target genes. Moreover, these signaling thresholds also control the effects of Wnt3a on proliferation and apoptosis. Specifically, at a critical threshold level of ∼25% signaling activity, Wnt3a could not efficiently induce target gene expression or stimulate cell proliferation. In contrast, 25% Wnt3a signaling activity was sufficient to efficiently repress negatively regulated target genes and interfere with serum starvation-induced apoptosis. Consistent with these observations, CS-E, which reduced Wnt3a signaling to ∼25%, was able to impair Wnt3a-mediated stimulation of proliferation but could not interfere with Wnt3a-mediated reduction of apoptosis (summarized in Fig. 9). These biologically important signaling threshold levels were also observed when utilizing Wnt3a ligand dilutions in the absence of CS-E, thus demonstrating that the identified signaling thresholds are an intrinsic property of the Wnt3a signaling cascade. We also demonstrated that both positively and negatively regulated transcriptional programs downstream of Wnt3a required a functional canonical β-catenin signaling cascade. Of note, one negatively regulated target gene, Txnip, consistently showed stronger repression at 25% Wnt3a signaling activity when compared with 50% activity. This was the only negatively regulated gene that displayed this response; we hypothesize that so far undefined thresholds in the midrange of Wnt3a signaling might fine-tune the repression of a small subset of negatively regulated target genes. Future studies looking specifically into the regulation of specific target genes might expand the principal mechanism of Wnt3a signaling thresholds we first described here. Our data establish the feasibility of selective inhibition of Wnt3a transcriptional programs and biological outcomes through the exploitation of intrinsic signaling thresholds. Specifically, these data support the notion that targeted pharmacological inhibition of Wnt/β-catenin to a specific signaling threshold might interfere with disease-related biological outcomes, for example proliferation, but maintain aspects of Wnt/β-catenin function in normal physiology through differential effects on positively and negatively regulated transcriptional programs.
FIGURE 9.

Model of selective interference with Wnt3a transcriptional programs and biological outcomes through differential Wnt3a signaling thresholds. Transcriptional programs negatively regulated by Wnt3a signaling are repressed at low levels of Wnt3a signaling activity (red response curve). As repressed transcriptional programs include pro-apoptotic genes, Wnt3a can also interfere with serum starvation-induced apoptosis at these low signaling levels. In contrast, transcriptional programs positively regulated by Wnt3a require high levels of Wnt3a signaling activity for efficient induction (blue response curve). As induced transcriptional programs include positive regulators of proliferation, the maximal effects of Wnt3a on proliferation require high signaling levels. Treatment with CS-E reduced Wnt3a signaling activity to a threshold level of ∼25%, which elicited a differential response of molecular and biological outcomes of Wnt3a signaling; positively regulated target genes and proliferation cannot be efficiently induced, whereas negatively regulated target genes and apoptosis are efficiently repressed.
Several previous studies have suggested the importance of Wnt signaling dosage and ligand gradients in embryonic development and disease (16–21). Our data presented here suggest that Wnt gradients might elicit differential transcriptional and biological responses depending on the distance of the Wnt ligand source and signaling activity levels. In areas close to the source of the Wnt signal, Wnt ligand concentrations would be high and therefore signaling could function at a sufficiently high level of close to 100% to allow target gene induction, as well as the promotion of biological programs that rely on induced target genes, including cell proliferation. In areas further removed from the Wnt source, with signaling levels of ∼25%, the signaling strength would not suffice to induce target genes efficiently or to promote proliferation. In contrast, target gene repression and interference with serum starvation-induced apoptosis would still be possible at these positions in the gradient. Even further along the gradient, at very low signaling levels of ∼9% and below, Wnt3a-mediated target gene repression and interference with apoptosis would also diminish.
We have shown here that the canonical β-catenin-dependent signaling cascade is critical for both positive and negative regulation of target gene expression by Wnt3a. The mechanism by which Wnt3a/β-catenin signaling differentially regulates transcriptional programs and biological function remains to be elucidated. In the absence of Wnt3a signaling, LEF and TCF function as transcriptional repressors by recruiting co-repressors such as Groucho to the classic Wnt response element (WRE) (45–47). When Wnt3a signaling is initiated, stabilized β-catenin can translocate to the nucleus and displace Groucho from the TCF/LEF complex, which switches the activity of the complex from a repressor to a transcriptional activator (47). However, recently published evidence suggests that β-catenin/TCF/LEF could also function as a transcriptional repressor through the binding of an alternative DNA regulatory sequence (48). In the context of our data, this could suggest that the interactions of a β-catenin/TCF/LEF protein complex with the classic Wnt response element to stimulate target gene expression might exhibit different kinetics from the interaction of the β-catenin/TCF/LEF complex with an alternative repressor DNA element.
A recent publication demonstrates that Wnt3a stimulation of articular chondrocytes could simultaneously activate β-catenin-dependent as well as Ca2+/CaMKII-dependent signaling pathways (44). These two pathways were activated at different Wnt3a signaling thresholds; at high concentrations, Wnt3a activates the β-catenin pathway, whereas lower signaling levels are sufficient to activate the Ca2+/CaMKII-dependent pathway (44). In contrast, our data presented here suggest that in NIH3T3 cells, both target gene induction and repression require a functional β-catenin cascade. It is intriguing to speculate that Wnt3a stimulation of different downstream pathways might be coordinated and regulated in a cell type- or context-dependent manner.
This work has important pharmacological implications. Wnt/β-catenin signaling is a critical therapeutic target in a number of human diseases including cancer, fibrosis, heart disease, and osteoarthritis (11, 49, 50). Thus, much effort has gone into the identification and characterization of Wnt pathway inhibitors, although translation of this research into clinical applications has not been successful thus far. Moreover, targeting Wnt signaling harbors the risk of interfering with the normal physiological functions of Wnt/β-catenin signaling (2, 14, 15). A detailed understanding of the effects of Wnt inhibitors on downstream disease-related versus physiological biological events is crucial for any translational efforts. We show here that CS-E could reduce Wnt3a signaling and interfere with target gene induction and proliferative responses but not with Wnt3a-mediated target gene repression and anti-apoptotic responses. Moreover, we demonstrate that the pharmacological Wnt inhibitors IWR-1 and quercetin also differentially affect target gene expression when utilized at concentrations that specify the critical canonical Wnt signaling threshold. Indeed, Shi et al. (51) report that inhibition of Wnt/β-catenin signaling impairs proliferation and expression of MYC and CCND1, genes associated with proliferation, but did not affect apoptosis or expression of Survivin, an apoptosis-related gene. It remains to be seen whether the critical Wnt signaling threshold that we identified here is functional in different cell types or whether it is modulated in a cell type-specific or temporal manner in development and disease. Our work represents an important step toward targeting individual biological outcomes of Wnt signaling, for example increased proliferation in cancer progression, while leaving other outcomes, for example tissue homeostasis and repair, unaffected.
CS-E has been shown to interfere with tissue colonization of mouse osteosarcoma and lung cancer cells injected into the tail veins of recipient mice (52, 53). Our preliminary data indicate that CS-E could also interfere with breast cancer progression.3 We are currently investigating whether localized or systemic treatment with CS-E in clinically relevant mouse cancer models could indeed differentially affect specific downstream biological consequences of Wnt signaling and whether CS-E treatment is a promising therapeutic avenue in these models. From a clinical point of view, it is interesting to note that oral administration of CS, registered as a drug on the European market for the treatment of osteoarthritis symptoms, has been studied extensively and shown to be safe, well tolerated, and effective (54, 55).
Acknowledgment
We thank Dr. Cara Gottardi for the gift of the Super 8x TOPFlash luciferase reporter plasmid.
This work was supported by the Children's Hospital of Chicago Research Center.

This article contains supplemental Fig. S1 and Tables S1 and S2.
C. M. Willis and M. Klüppel, unpublished results.
- CS
- chondroitin sulfate
- RP
- recombinant protein
- qRT-PCR
- quantitative real-time RT-PCR
- DMSO
- dimethyl sulfoxide
- EdU
- 5-ethynyl-2′-deoxyuridine
- TCF/LEF
- T-cell factor/lymphoid enhancer factor.
REFERENCES
- 1. Cadigan K. M., Peifer M. (2009) Wnt signaling from development to disease: insights from model systems. Cold Spring Harb. Perspect. Biol. 1, a002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 3. Clevers H., Nusse R. (2012) Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 [DOI] [PubMed] [Google Scholar]
- 4. Moon R. T. (2005) Wnt/β-catenin pathway. Sci. STKE 2005, cm1. [DOI] [PubMed] [Google Scholar]
- 5. Nusse R. (2005) Wnt signaling in disease and in development. Cell Res. 15, 28–32 [DOI] [PubMed] [Google Scholar]
- 6. Nusse R. (2012) Wnt signaling. Cold Spring Harb. Perspect. Biol. 4, a011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Valenta T., Hausmann G., Basler K. (2012) The many faces and functions of β-catenin. EMBO J. 31, 2714–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Behrens J., Lustig B. (2004) The Wnt connection to tumorigenesis. Int. J. Dev. Biol. 48, 477–487 [DOI] [PubMed] [Google Scholar]
- 9. Polakis P. (2007) The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 17, 45–51 [DOI] [PubMed] [Google Scholar]
- 10. Moon R. T., Kohn A. D., De Ferrari G. V., Kaykas A. (2004) WNT and β-catenin signalling: diseases and therapies. Nat. Rev. Genet. 5, 691–701 [DOI] [PubMed] [Google Scholar]
- 11. Blankesteijn W. M., van de Schans V. A., ter Horst P., Smits J. F. (2008) The Wnt/frizzled/GSK-3β pathway: a novel therapeutic target for cardiac hypertrophy. Trends Pharmacol. Sci. 29, 175–180 [DOI] [PubMed] [Google Scholar]
- 12. Wei J., Melichian D., Komura K., Hinchcliff M., Lam A. P., Lafyatis R., Gottardi C. J., MacDougald O. A., Varga J. (2011) Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma? Arthritis Rheum. 63, 1707–1717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polakis P. (2012) Drugging Wnt signalling in cancer. EMBO J. 31, 2737–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fevr T., Robine S., Louvard D., Huelsken J. (2007) Wnt/β-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell. Biol. 27, 7551–7559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huelsken J., Held W. (2009) Canonical Wnt signalling plays essential roles. Eur. J. Immunol. 39, 3582–3584 [DOI] [PubMed] [Google Scholar]
- 16. Albuquerque C., Breukel C., van der Luijt R., Fidalgo P., Lage P., Slors F. J., Leitão C. N., Fodde R., Smits R. (2002) The “just-right” signaling model: APC somatic mutations are selected based on a specific level of activation of the β-catenin signaling cascade. Hum. Mol. Genet. 11, 1549–1560 [DOI] [PubMed] [Google Scholar]
- 17. Buchert M., Athineos D., Abud H. E., Burke Z. D., Faux M. C., Samuel M. S., Jarnicki A. G., Winbanks C. E., Newton I. P., Meniel V. S., Suzuki H., Stacker S. A., Näthke I. S., Tosh D., Huelsken J., Clarke A. R., Heath J. K., Sansom O. J., Ernst M. (2010) Genetic dissection of differential signaling threshold requirements for the Wnt/β-catenin pathway in vivo. PLoS Genet. 6, e1000816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kielman M. F., Rindapää M., Gaspar C., van Poppel N., Breukel C., van Leeuwen S., Taketo M. M., Roberts S., Smits R., Fodde R. (2002) Apc modulates embryonic stem-cell differentiation by controlling the dosage of β-catenin signaling. Nat. Genet. 32, 594–605 [DOI] [PubMed] [Google Scholar]
- 19. Li Q., Ishikawa T. O., Oshima M., Taketo M. M. (2005) The threshold level of adenomatous polyposis coli protein for mouse intestinal tumorigenesis. Cancer Res. 65, 8622–8627 [DOI] [PubMed] [Google Scholar]
- 20. Luis T. C., Ichii M., Brugman M. H., Kincade P., Staal F. J. (2012) Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia 26, 414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luis T. C., Naber B. A., Roozen P. P., Brugman M. H., de Haas E. F., Ghazvini M., Fibbe W. E., van Dongen J. J., Fodde R., Staal F. J. (2011) Canonical Wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell 9, 345–356 [DOI] [PubMed] [Google Scholar]
- 22. Klüppel M. (2010) The roles of chondroitin-4-sulfotransferase-1 in development and disease. Prog. Mol. Biol. Transl. Sci. 93, 113–132 [DOI] [PubMed] [Google Scholar]
- 23. Klüppel M. (2011) Efficient secretion of biologically active chondroitinase ABC from mammalian cells in the absence of an N-terminal signal peptide. Mol. Cell. Biochem. 351, 1–11 [DOI] [PubMed] [Google Scholar]
- 24. Prinz R. D., Willis C. M., Viloria-Petit A., Klüppel M. (2011) Elimination of breast tumor-associated chondroitin sulfate promotes metastasis. Genet. Mol. Res. 10, 3901–3913 [DOI] [PubMed] [Google Scholar]
- 25. Hinek A., Teitell M. A., Schoyer L., Allen W., Gripp K. W., Hamilton R., Weksberg R., Klüppel M., Lin A. E. (2005) Myocardial storage of chondroitin sulfate-containing moieties in Costello syndrome patients with severe hypertrophic cardiomyopathy. Am. J. Med. Genet. A 133A, 1–12 [DOI] [PubMed] [Google Scholar]
- 26. Maeda N., Fukazawa N., Ishii M. (2010) Chondroitin sulfate proteoglycans in neural development and plasticity. Front. Biosci. 15, 626–644 [DOI] [PubMed] [Google Scholar]
- 27. Theocharis A. D., Skandalis S. S., Tzanakakis G. N., Karamanos N. K. (2010) Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 277, 3904–3923 [DOI] [PubMed] [Google Scholar]
- 28. Karangelis D. E., Kanakis I., Asimakopoulou A. P., Karousou E., Passi A., Theocharis A. D., Triposkiadis F., Tsilimingas N. B., Karamanos N. K. (2010) Glycosaminoglycans as key molecules in atherosclerosis: the role of versican and hyaluronan. Curr. Med. Chem. 17, 4018–4026 [DOI] [PubMed] [Google Scholar]
- 29. Klüppel M., Wight T. N., Chan C., Hinek A., Wrana J. L. (2005) Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development 132, 3989–4003 [DOI] [PubMed] [Google Scholar]
- 30. Klüppel M., Vallis K. A., Wrana J. L. (2002) A high-throughput induction gene trap approach defines C4ST as a target of BMP signaling. Mech. Dev. 118, 77–89 [DOI] [PubMed] [Google Scholar]
- 31. Klüppel M., Samavarchi-Tehrani P., Liu K., Wrana J. L., Hinek A. (2012) C4ST-1/CHST11-controlled chondroitin sulfation interferes with oncogenic HRAS signaling in Costello syndrome. Eur. J. Hum. Genet. 20, 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nadanaka S., Ishida M., Ikegami M., Kitagawa H. (2008) Chondroitin 4-O-sulfotransferase-1 modulates Wnt-3a signaling through control of E disaccharide expression of chondroitin sulfate. J. Biol. Chem. 283, 27333–27343 [DOI] [PubMed] [Google Scholar]
- 33. Nadanaka S., Kinouchi H., Taniguchi-Morita K., Tamura J., Kitagawa H. (2011) Down-regulation of chondroitin 4-O-sulfotransferase-1 by Wnt signaling triggers diffusion of Wnt-3a. J. Biol. Chem. 286, 4199–4208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Willert K., Brown J. D., Danenberg E., Duncan A. W., Weissman I. L., Reya T., Yates J. R., 3rd, Nusse R. (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423, 448–452 [DOI] [PubMed] [Google Scholar]
- 35. Molenaar M., van de Wetering M., Oosterwegel M., Peterson-Maduro J., Godsave S., Korinek V., Roose J., Destrée O., Clevers H. (1996) XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell 86, 391–399 [DOI] [PubMed] [Google Scholar]
- 36. Chen S., McLean S., Carter D. E., Leask A. (2007) The gene expression profile induced by Wnt3a in NIH 3T3 fibroblasts. J. Cell Commun. Signal. 1, 175–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chakraborty P. K., Lee W. K., Molitor M., Wolff N. A., Thévenod F. (2010) Cadmium induces Wnt signaling to up-regulate proliferation and survival genes in subconfluent kidney proximal tubule cells. Mol. Cancer 9, 102–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jia L., Miao C., Cao Y., Duan E. K. (2008) Effects of Wnt proteins on cell proliferation and apoptosis in HEK293 cells. Cell Biol. Int. 32, 807–813 [DOI] [PubMed] [Google Scholar]
- 39. Dehner M., Hadjihannas M., Weiske J., Huber O., Behrens J. (2008) Wnt signaling inhibits Forkhead box O3a-induced transcription and apoptosis through up-regulation of serum- and glucocorticoid-inducible kinase 1. J. Biol. Chem. 283, 19201–19210 [DOI] [PubMed] [Google Scholar]
- 40. Mirkovic I., Charish K., Gorski S. M., McKnight K., Verheyen E. M. (2002) Drosophila Nemo is an essential gene involved in the regulation of programmed cell death. Mech. Dev. 119, 9–20 [DOI] [PubMed] [Google Scholar]
- 41. Chen B., Dodge M. E., Tang W., Lu J., Ma Z., Fan C. W., Wei S., Hao W., Kilgore J., Williams N. S., Roth M. G., Amatruda J. F., Chen C., Lum L. (2009) Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat. Chem. Biol. 5, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pahlke G., Ngiewih Y., Kern M., Jakobs S., Marko D., Eisenbrand G. (2006) Impact of quercetin and EGCG on key elements of the Wnt pathway in human colon carcinoma cells. J. Agric. Food. Chem. 54, 7075–7082 [DOI] [PubMed] [Google Scholar]
- 43. Park C. H., Chang J. Y., Hahm E. R., Park S., Kim H. K., Yang C. H. (2005) Quercetin, a potent inhibitor against β-catenin/Tcf signaling in SW480 colon cancer cells. Biochem. Biophys. Res. Commun. 328, 227–234 [DOI] [PubMed] [Google Scholar]
- 44. Nalesso G., Sherwood J., Bertrand J., Pap T., Ramachandran M., De Bari C., Pitzalis C., Dell'accio F. (2011) WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell Biol. 193, 551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Atcha F. A., Syed A., Wu B., Hoverter N. P., Yokoyama N. N., Ting J. H., Munguia J. E., Mangalam H. J., Marsh J. L., Waterman M. L. (2007) A unique DNA binding domain converts T-cell factors into strong Wnt effectors. Mol. Cell. Biol. 27, 8352–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J., Sutter C., Parker D. S., Blauwkamp T., Fang M., Cadigan K. M. (2007) CBP/p300 are bimodal regulators of Wnt signaling. EMBO J. 26, 2284–2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cadigan K. M. (2008) Wnt-β-catenin signaling. Curr. Biol. 18, R943–R947 [DOI] [PubMed] [Google Scholar]
- 48. Blauwkamp T. A., Chang M. V., Cadigan K. M. (2008) Novel TCF-binding sites specify transcriptional repression by Wnt signalling. EMBO J. 27, 1436–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barker N., Clevers H. (2006) Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 5, 997–1014 [DOI] [PubMed] [Google Scholar]
- 50. Chien A. J., Moon R. T. (2007) WNTS and WNT receptors as therapeutic tools and targets in human disease processes. Front. Biosci. 12, 448–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shi Y. L., Hsieh C. B., Lai H. C., Yan M. D., Hsieh T. Y., Chao Y. C., Lin Y. W. (2007) SFRP1 suppressed hepatoma cells growth through Wnt canonical signaling pathway. Int. J. Cancer 121, 1028–1035 [DOI] [PubMed] [Google Scholar]
- 52. Basappa, Murugan S., Sugahara K. N., Lee C. M., ten Dam G. B., van Kuppevelt T. H., Miyasaka M., Yamada S., Sugahara K. (2009) Involvement of chondroitin sulfate E in the liver tumor focal formation of murine osteosarcoma cells. Glycobiology 19, 735–742 [DOI] [PubMed] [Google Scholar]
- 53. Li F., Ten Dam G. B., Murugan S., Yamada S., Hashiguchi T., Mizumoto S., Oguri K., Okayama M., van Kuppevelt T. H., Sugahara K. (2008) Involvement of highly sulfated chondroitin sulfate in the metastasis of the Lewis lung carcinoma cells. J. Biol. Chem. 283, 34294–34304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Uebelhart D. (2008) Clinical review of chondroitin sulfate in osteoarthritis. Osteoarthr. Cartil. 16, Suppl. 3, S19–S21 [DOI] [PubMed] [Google Scholar]
- 55. Vangsness C. T., Jr., Spiker W., Erickson J. (2009) A review of evidence-based medicine for glucosamine and chondroitin sulfate use in knee osteoarthritis. Arthroscopy 25, 86–94 [DOI] [PubMed] [Google Scholar]