

Background: Catalase activity in catalase-peroxidases occurs through a poorly defined mechanism requiring residues Arg-418, Asp-137, and the amino acid adduct (M255Y229W107).
Results: Along with an adduct radical, Arg-418 stimulates O2 release from dioxyheme, the rapid formation of which requires Asp-137.
Conclusion: The electronics of the Arg-418:MYW-radical interaction enables discharge of O2 during H2O2 turnover.
Significance: An atypical catalase mechanism operates in KatG.
Keywords: Electron paramagnetic resonance (EPR), Heme, Hydrogen Peroxide, Peroxidase, Radicals, DFT, KatG, Catalase
Abstract
Catalase activity of the dual-function heme enzyme catalase-peroxidase (KatG) depends on several structural elements, including a unique adduct formed from covalently linked side chains of three conserved amino acids (Met-255, Tyr-229, and Trp-107, Mycobacterium tuberculosis KatG numbering) (MYW). Mutagenesis, electron paramagnetic resonance, and optical stopped-flow experiments, along with calculations using density functional theory (DFT) methods revealed the basis of the requirement for a radical on the MYW-adduct, for oxyferrous heme, and for conserved residues Arg-418 and Asp-137 in the rapid catalase reaction. The participation of an oxyferrous heme intermediate (dioxyheme) throughout the pH range of catalase activity is suggested from our finding that carbon monoxide inhibits the activity at both acidic and alkaline pH. In the presence of H2O2, the MYW-adduct radical is formed normally in KatG[D137S] but this mutant is defective in forming dioxyheme and lacks catalase activity. KatG[R418L] is also catalase deficient but exhibits normal formation of the adduct radical and dioxyheme. Both mutants exhibit a coincidence between MYW-adduct radical persistence and H2O2 consumption as a function of time, and enhanced subunit oligomerization during turnover, suggesting that the two mutations disrupting catalase turnover allow increased migration of the MYW-adduct radical to protein surface residues. DFT calculations showed that an interaction between the side chain of residue Arg-418 and Tyr-229 in the MYW-adduct radical favors reaction of the radical with the adjacent dioxyheme intermediate present throughout turnover in WT KatG. Release of molecular oxygen and regeneration of resting enzyme are thereby catalyzed in the last step of a proposed catalase reaction.
Introduction
The catalase-peroxidase enzyme, KatG,3 is found in many microorganisms but in the human pathogen Mycobacterium tuberculosis (M. tb) it is the only “catalase” (2 H2O2 → O2 + 2 H2O) and plays an important role in protecting this organism against oxidative stress (1–2). The structural origins of its high catalase activity are therefore of special interest since KatG may fulfill a role different from the ubiquitous monofunctional catalases, especially in organisms that contain genes for both the monofunctional and dual function enzymes (3). KatG in M. tb is also responsible for peroxidative activation of the pro-drug isoniazid (isonicotinic acid hydrazide, INH) used to treat tuberculosis infection, though a genuine peroxidase function for the enzyme in normal bacterial physiology is not known. Mutations in M. tb KatG that confer resistance to INH create ongoing challenges worldwide in treating TB.
The catalase activity of KatG depends upon three conserved amino acids (Met-255, Tyr-229, and Trp-107, M. tb numbering, MYW) with side chains post-translationally linked into a unique adduct on the distal side of the heme pocket (Fig. 1) (4–7). Mutation of any of these residues reduces catalase activity to less than one percent of the rate in wild-type (WT) KatG, but without compromising peroxidase activity (8–12). These findings demonstrate that the MYW-adduct enables formation of a unique intermediate required for efficient turnover of H2O2. Using rapid freeze-quench (RFQ) EPR, we reported the characterization of a narrow doublet radical signal (17-Gauss linewidth, aH1,H2 = 11 and 2.5 Gauss for β-methylene hydrogens) from a modified tyrosine in M. tb KatG (13–14). More recent isotope labeling and mutagenesis experiments (15), allowed assignment of the radical found during catalase turnover to the MYW-adduct and elucidated a function for this unique cofactor first suggested several years ago (16). Explaining the operation of this radical in the catalase reaction particular to KatG is among our present goals.
FIGURE 1.

The active site and nearby amino acids of M. tb catalase-peroxidase KatG (2cca.pdb). The side chains of Met-255, Tyr-229, and Trp-107 are covalently linked forming a unique cofactor required for catalase activity. The side chain of residue Arg-418 is shown in the two conformations, vicinal and remote, found in the crystal structure. Arg-418 and Asp-137 were replaced by mutagenesis for this work.
Residues beyond the MYW-adduct but near the heme are also required for high catalase activity in KatG. For example, replacement of Arg-418 with aliphatic amino acids reduces catalase turnover to less than 2% that of the WT rate (17, 18). This residue, despite its location remote from the heme pocket, was proposed to induce electronic steering effects on the heme through its guanidiniuim group, which interacts with the phenolate oxygen of Tyr-229 (Fig. 1) at alkaline pH. In the low pH range, the Arg side chain is found in a non-interacting conformation (17). The pH dependence of the conformational change was shown crystallographically for Burkholderia pseudomallei KatG, and alternate conformations of the Arg-418 side chain are also seen in the two subunits of the M. tb KatG structure. Another residue, Asp-137, located in a substrate access channel is also essential for KatG catalase activity for reasons that are not completely clear (19).
Investigating the role of Arg-418 (and briefly that of Asp-137) in the context of a potentially novel catalase reaction mechanism seemed warranted and was pursued for this work. According to current evidence, this mechanism in KatG has certain features in common with, and others quite distinct from, those known for the monofunctional catalases. In all cases, the reaction is assumed to proceed through a species at the oxidation level of peroxidase Compound I (ferryl heme:porphyrin-π-cation radical) formed in the first turnover with peroxide. KatGs contain conserved distal side His and Arg residues that participate in this initial reaction (18, 20). In monofunctional catalases, very rapid dismutation steps prevent the accumulation of the intermediate assumed to be solely Compound I. For the monofunctional catalase enzymes and KatG, both atoms of the O2 product come from the same peroxide molecule oxidized in the second phase of turnover (21). One unusual feature of KatG catalysis is that an oxygenated form of heme (dioxyheme) accumulates and persists in the presence of excess H2O2 during catalase turnover (13, 16). (In this work, “dioxyheme” designates the complex in which molecular oxygen is bound to heme iron, formally a ferrous heme-dioxygen/ferric heme-superoxy complex analogous to peroxidase Compound III.) In most peroxidases, Compound III is a catalytically inert species slowly formed in the presence of excess H2O2 and it does not directly dissociate oxygen (22).
A key step proposed for the KatG catalase cycle implicates a radical, possibly on the MYW-adduct, in discharging or reacting with dioxyheme (13, 16). The series of reactions above (Scheme 1) includes such a step and shows: 1) oxidation of resting enzyme by H2O2 to form ferryl heme and the MYW-adduct radical assuming Compound I formation (the latter not seen experimentally); 2) conversion of ferryl heme to dioxyheme by the second peroxide, in enzyme bearing the adduct radical; 3) recombination of unpaired electrons restoring the resting enzyme and releasing O2.
SCHEME 1.
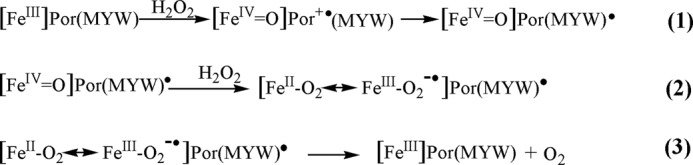
This mechanism or similar ones have been proposed before (16) but detailed verification of the steps is of ongoing interest. Here, we focus on three issues: 1) formation and discharge of dioxyheme in KatG; 2) the role of an MYW-adduct radical specific to the KatG reaction; and 3) the role of key remote residues in the mechanism. Optical stopped-flow spectrophotometric and rapid freeze-quench EPR experiments were used to compare reactions in wild-type (WT) M. tb KatG and in catalase-deficient mutants. Supporting the experimental work is a theoretical analysis using Density Functional Theory (DFT) to model the interaction of Arg-418 with the reactant in Rxn. 3, a dioxyheme:MYW-adduct radical structure. The latter became of obvious importance based on results collected for the catalase-deficient mutant KatG[R418L]. A new perspective emerges on how the rapid KatG catalase reaction depends directly on the interactions between the MYW-adduct radical, dioxyheme, and the conserved residue Arg-418 and on the role of Asp-137 in the reaction pathway.
EXPERIMENTAL PROCEDURES
Mutagenesis, Overexpression, and Purification of M. tuberculosis KatG Enzymes
The WT M. tb katG gene in the pKAT II plasmid was a gift from Dr. Stewart Cole. KatG[R418L] and KatG[D137S] mutants were prepared using the Quickchange (Stratagene) kit for site-directed mutagenesis. Mutagenesis primers used to introduce the desired replacements (mutated codons are in bold) were as follows: (R418L) 5′-CAAGCTGATCCACCTAGACATGGGTCCCG-3′ and 5′-CGGGACCCATGTCTAGGTGGATCAGCTTG-3′; (D137S) 5′-CAGCTGGCCCTCCAACGCCAGCTTG-3′ and 5′-CAAGCTGGCGTTGGAGGGCCAGCTG-3′. Successful mutations were confirmed by sequencing of mutated plasmids (GENEWIZ, South Plainfield, NJ). Overexpression of recombinant WT KatG and its mutant enzymes, and their purification by FPLC were achieved as described in previous reports (23); the heme precursor δ-aminolevulinic acid was added to culture media to promote formation of holoenzyme. KatG concentration is expressed as the heme concentration based on the pyridine hemochromogen assay (ϵ557 nm = 34.5 mm−1·cm−1) (24). Catalase and peroxidase activities were determined spectrophotometrically (NT-14 UV/Vis, AVIV Instruments) in replicate analyses using initial rates of reactions, in which the consumption of hydrogen peroxide was monitored at 240 nm for catalase activity (ϵ240 nm = 43.6 m−1·cm−1), or by color production using t-butyl hydroperoxide and o-dianisidine (peroxidase activity) according to previous reports (23).
Kinetic Measurements
RFQ-EPR samples were prepared using an Update Instrument, Inc. model 1000 chemical-freeze quench apparatus, as described previously (25). Briefly, the enzyme (50 μm final) and H2O2 (50 mm final) were mixed at room temperature in a 1:1 volume ratio, and the mixture was allowed to react for the indicated time periods determined by the length of the reactor tubes or, for longer periods by programming a delay interval; it was then sprayed into a funnel attached to an EPR tube submerged in an isopentane bath held at approximately −130 °C. The frozen powder was packed into precision bore EPR tubes, and samples were transferred to liquid nitrogen for storage.
EPR spectra were recorded at 77 K on a Bruker E500 ElexSys EPR spectrometer operating at X-band (9 GHz). Data acquisition and manipulation were performed using XeprView and WinEPR software (Bruker). Instrumental parameters were as follows: modulation amplitude, 1 G; microwave power, 0.1 milliwatt; modulation frequency, 100 kHz; microwave frequency, 9.445 (± 0.005) GHz; number of scans averaged, 9. For quantitative EPR analysis, signal intensities were evaluated by double integration of first-derivative EPR signals using TEMPOL (4-hydroxy-2,2,6,6- tetramethylpiperidine-N-oxyl) or CuSO4 as standards. The TEMPOL radical concentration was evaluated optically (ϵ428 nm = 13.2 m−1·cm−1) (26). The spins per heme values are corrected for the effect of dilution of frozen samples by trapped isopentane (packing dilution factor ∼0.4). Errors can be significant for later time points, however, because O2 gas dilutes the mixed samples in variable amounts during incubations.
The companion experiments for evaluating the kinetics of H2O2 consumption were performed on a stopped-flow spectrophotometer (HiTech Scientific, UK) using the same enzyme and H2O2 concentrations as for EPR samples. The absorbance of H2O2 at 240 nm was monitored for the desired time period. Due to strong absorbance caused by the high concentrations of enzyme and peroxide, the instrument was adapted with a 1.5 mm light path flow-cell.
CO Inhibition of Catalase Activity of KatG
To check the effect of carbon monoxide on catalase activity, KatG and H2O2 solutions were purged with carbon monoxide for 15 min before being loaded into the syringes of the stopped-flow apparatus. The sample reservoirs containing the CO-saturated solutions were sealed immediately after loading, to avoid gas exchange. The consumption of H2O2 was monitored at 240 nm as a function of time after mixing the enzyme with H2O2 in the airtight stopped-flow system. For time courses at pH 6.0 and 8.5, 20 mm potassium phosphate buffer and 20 mm sodium borate buffer were used respectively.
Oligomerization of KatG during Catalytic Reactions
KatG enzymes (15 μm) were incubated at pH 7.2 for 30 min (37 °C) in the presence of a slow flux of H2O2 generated by aerobic glucose (10 mm) and glucose oxidase. The rate of H2O2 generation was varied from 0–100 μm/min by changing the amount of glucose oxidase. H2O2 flux was separately determined using a horseradish peroxidase-based assay (27). Immediately after incubations, the H2O2-treated KatG (or mutants) was mixed with an equal volume of 2× SDS-PAGE sample loading buffer and incubated at 100 °C for 5 min. Samples were then loaded onto a precast gradient polyacrylamide gel (PhastGel, 4–15%, GE Healthcare) and electrophoresed using a Pharmacia PhastSystem followed by Coomassie Blue staining (23).
DFT Calculations
Additional support for features of the catalase mechanism was achieved through DFT-based calculations by evaluating active site model structures and the relative energies of various electronic states. All computations were performed for gas phase models at the level of the well-documented B3LYP hybrid density functional in combination with moderate 6–31G* basis sets as implemented in the Gaussian 09 program package (28). A starting model was built based upon the crystal structure of the M. tb KatG enzyme (2cca.pdb) using coordinates for all atoms of the MYW-adduct, including the alpha carbons (formulated as methyl groups) as well as the Arg-418 residue with its side chain in vicinal and remote conformations (as in Fig. 1). Incorporation of dioxyheme (with propionates replaced with methyl groups for simplification) and the proximal histidine completes the structure of the species we focused on for evidence of catalytic properties. The Arg-remote model was optimized with constraints applied to freeze the positions of the α-carbon atoms of each residue and one of the meso carbons (between rings A and B) of the heme. Using the optimized geometry of this starting model, a radical form was produced by removing an H atom from the phenolic oxygen of Tyr-229 of the MYW-adduct as in our prior report (13). Then, relative energies were computed for states of the model having total unpaired spin values from S = 1/2 to S = 7/2 in unit increments (doublet to octet multiplicities). Among the Arg-vicinal models, the sextet (S(total) = 5/2) state was of lowest energy after geometry re-optimization, which led to release of triplet state molecular oxygen. Relative energies for other spin multiplicity states were also computed for the oxygen-dissociated model. The lowest energy states are addressed in detail in “Results.” No negative vibrational frequencies were found in any calculation, showing that all optimized geometries were at true energy minima. Spin contamination values were within acceptable limits, showing that the computed energies and spin distributions are reliable. Spin distributions for each electronic state were evaluated using Natural Orbital Analysis as implemented in NBO 5.0 (29). Additional details are given in the text.
RESULTS
Purified mutant enzymes KatG[R418L] and KatG[D137S] exhibited optical spectra similar to that of WT KatG, with purity ratios, A407/A280 ∼0.45–0.5 (not shown), consistent with a normal stoichiometry of one heme per monomer. The spectroscopic features of KatG[R418L] are consistent with a mixture of 5 and 6-coordinate ferric heme in the resting enzyme much like that in WT KatG. The KatG[D137S] mutant contains more 5-coordinate heme than WT KatG, with a Soret peak at 405 nm, similar to the analogous mutant D152S of Synechocystis KatG (19). Small differences in the properties of the resting enzymes are not expected to reduce catalase activity (30). For KatG[R418L], the steady-state peroxidase activity assayed with the artificial substrate o-dianisidine was close to wild type (WT) activity (0.7 versus 0.9 units/mg) but its catalase activity (57 units/mg) was about 2% that of WT enzyme. The peroxidase activity of KatG[D137S] was one-third that of WT (0.3 versus 0.9 units/mg), and its catalase activity (24 units/mg) was much lower (0.8% that of WT enzyme).
1) MYW-adduct Radical in WT KatG, KatG[R418L] and KatG[D137S] Mutants
In an earlier study, we presented evidence that a protein-based radical and dioxyheme were simultaneously present in KatG during catalase turnover at neutral pH (13). The radical, which exhibits a narrow doublet EPR signal, was detected using large molar excesses (8000-fold) of peroxide to accumulate it in samples frozen after manual mixing, as well as in samples frozen within milliseconds of mechanical mixing, for which a smaller molar excess of H2O2 (1000-fold) was sufficient.
Here we first characterize the behavior of the MYW-adduct radical in WT KatG under various conditions, for comparisons with the catalase-deficient mutants KatG[R418L] and KatG[D137S]. The EPR experiments were performed near the pH optimum (7.2) for WT KatG catalase activity. Fig. 2A shows the narrow doublet EPR signal characteristic of the MYW-adduct radical (13, 15), frozen 8 ms after mixing, corresponding to a concentration of 0.3 spin/heme. Spectra are also shown for samples frozen at two or three later time points as indicated, for WT and mutants respectively. A rise in radical concentration was not seen in the accessible time regimes due to the high reaction rates in each case. The radical signal is detected during a time interval corresponding to the trace for H2O2 consumption monitored optically at 240 nm (Fig. 2A, right panel). Increasing peroxide concentration to a 100,000-fold molar excess produced only a small increase in radical yield (up to 0.4 spin/heme), while limiting H2O2 concentration to only a 5-fold molar excess still elicited a detectable yield of radical (∼0.05 spins/heme) at 20 ms (not shown). The yields of radical at time points beyond 1 s are not considered to be accurately observed due to the dilution of the samples by oxygen evolution. We note that the decrease in radical signal intensity illustrated in Fig. 2A is not a reflection of its “lifetime” during a catalytic step but rather represents the decreasing steady-state level of the radical intermediate as the peroxide concentration diminishes.
FIGURE 2.

MYW-radical formation and H2O2 consumption in A) WT KatG; B) KatG[R418L]; C) KatG[D137S] mutants. Left panels: rapid freeze quench X-band EPR spectra (77 K) of enzymes (50 μm) reacted with 1000-fold molar excess of H2O2 at pH 7.2. Samples were frozen at the indicated times after mixing at room temp. Spectra are plotted on the same signal intensity scales. The narrow doublet signal contains one strong and one weak hyperfine coupling interaction (aH1, H2 = 11 and ∼2.5 Gauss) arising from the β-methylene hydrogens of residue Tyr-229 (13). Right panels: stopped-flow spectrophotometric time courses showing H2O2 consumption monitored at 240 nm under the same conditions used for the EPR experiments.
As the loss of catalase activity in certain mutant enzymes might arise from a lack of a required MYW-adduct radical, we examined two catalase-deficient mutant KatGs under the conditions used above. Fig. 2, B and C, show that KatG[R418L] and KatG[D137S] mutants produced the narrow doublet EPR signal in yields similar to WT KatG at the earliest time points. And the persistence of the radical signals coincided with the period required for H2O2 consumption (∼30 s for KatG[R418L] and ∼60 s for KatG[D137S]). These findings suggest that the poor catalase activity of these two mutant enzymes arises from some defect in radical utilization. Interestingly, the initial yield of MYW-adduct radical in both mutants was not significantly higher than for WT protein, nor did the radical persist significantly longer than the substrate H2O2. These observations suggest that an MYW-adduct radical escape pathway may exist in the mutant enzymes, which limits accumulation despite poor utilization of the radical in a catalase reaction step. We later show evidence for such a pathway from the enhanced formation of surface amino acid radicals capable of forming enzyme cross-links.
2) Dioxyheme Formation
Dioxyheme and the MYW-adduct radical are found concurrently during turnover of excess H2O2 by WT KatG (13–14). However, very rapid turnovers prevent evaluation of the kinetics of heme conversion to oxygenated species in the case of wild-type KatG. The KatG[R418L] mutant, which conserves all the catalytically essential heme pocket residues including the intact MYW-adduct (18) and its radical, provides a model for dioxyheme formation thanks to very slow catalase turnover. As shown in Fig. 3A, formation of dioxyheme was complete within 4 ms of mixing (1000-fold excess of H2O2), evidenced by the shift of the Soret peak to 418 nm and the appearance of the bands at 542 nm and 580 nm typical of oxyperoxidase (31). Moreover, dioxyheme could be observed within 4 ms of mixing, using as little as a 2-fold molar excess (Fig. 3A). No evidence for a Compound II-like spectrum appeared, demonstrating the very fast transitions from ferric to dioxyheme. The second-order rate constant for dioxyheme formation determined from a plot of kobs versus H2O2 concentration (not shown) was 2.3 × 104 m−1·s−1 for KatG[R418L]. In WT KatG, the only conditions under which an intermediate can be observed involves the use of very large excesses of H2O2 because dioxyheme, while it should be formed at a rate similar to that seen in the mutant, is rapidly turned over. In contrast to this rapid turnover, dioxyheme prepared without H2O2 and without the MYW-adduct radical (by aerobic photolysis of carbonyl KatG (14)) is stable for minutes. We show later how the lack of radical or the lack of Arg-418 can produce similar effects on the catalase mechanism.
FIGURE 3.

Dioxyheme formation in KatG[R418L] and KatG[D137S]. Spectra were recorded 4 ms after mixing H2O2 with KatG[R418L] (A) or KatG[D137S] (B). Enzyme concentrations were 10 μm (final) and the molar equivalents of H2O2 were as indicated. All reactions were performed in 20 mm potassium phosphate buffer, pH 7.2 at room temperature using a stopped-flow spectrophotometer. The peaks in the visible wavelength region through which lines are drawn closely match those assigned to typical peroxidase Compound III and indicate the dioxyheme component in both reactions.
KatG[D137S] was reported several years ago to lack catalase activity (19) yet formation of the MYW-adduct radical in this mutant is unchanged (shown above). Optical spectra show that only traces of dioxyheme are produced with excess H2O2 (500 or 5000-fold) (Fig. 3B), with the state of heme in the remainder of the enzyme remaining uncharacterized. We suggest that for the small fraction of mutant enzyme that forms dioxyheme, the WT catalase reaction pathway can operate normally, since the MYW-adduct radical and Arg-418 are both available. The lost catalase activity is then suggested to arise simply from the lack of abundant dioxyheme. These observations indicate the importance of residue Asp-137 in formation of dioxyheme and overall, the results are crucial to formulating key steps in the KatG catalase reaction mechanism.
3) CO Inhibition and the pH Dependence of the Catalase Reaction
The origin of the pH dependence of the catalase reaction in KatG is not understood in terms of the participation of specific proton transfer events. One unresolved issue is the absence of a dioxyheme optical signature during catalase turnover in alkaline conditions (pH 8.5) (13, 16), raising the question: is there a change in mechanism with pH or merely a change in the identity of the prevailing steady-state intermediate? If dioxyheme is in fact part of a catalytically competent intermediate, it should occur throughout the pH range of the catalase reaction. Furthermore, if this intermediate dissociates oxygen gas, CO could inhibit the catalase reaction by binding tightly to ferrous heme iron, thereby removing enzyme from catalytic competence by trapping it in the stable carbonyl form.
To explore these ideas, the catalase activity of WT KatG was monitored in CO-saturated buffer. The activity at pH 6.0 was inhibited by 95%, but after a lag of several seconds (Fig. 4A). A similar level of inhibition was found at pH 8.5 after a longer lag phase (Fig. 4B). Thus, under both acidic and basic conditions, either free ferrous heme or a dissociable form of dioxyheme must be present during catalase turnover. Formation of free ferrous heme is difficult to rationalize since there are no reductants available (except possibly for superoxide anion radical that might be present from side reactions; but this species would tend to combine with ferric heme to give dioxyheme in any case, rather than free ferrous heme), while the formation of dioxyheme is readily rationalized by invoking well-known reactions of heme in peroxidases as given in Scheme 1.
FIGURE 4.

Carbon monoxide inhibits catalase activity in KatG at low and high pH. H2O2 concentration was monitored at 240 nm. A, WT KatG (20 nm) was mixed with 25 mm H2O2 in air-saturated (solid lines) or CO-saturated (dashed lines) buffer at pH 6.0 (A) or pH 8.5 (B). C, bovine liver catalase (BLC) activity measured in air (solid line) or CO saturated (dashed line) phosphate buffer, pH 7.0. The conditions were the same as in A except that BLC concentration was 0.5 nm. D, steady-state peroxidase activity assay of KatG using t-butyl hydroperoxide and o-dianisidine performed aerobically or in CO-saturated acetate buffer.
It can be estimated that about 20,000 cycles occur before the maximal inhibition is reached in the CO inhibition process. In contrast to these observations, the activity of the monofunctional enzyme bovine liver catalase (BLC) (Fig. 4C) was not inhibited by CO, confirming that these two enzymes use different catalase mechanisms. The peroxidase activity of KatG assayed with alkyl peroxide is not inhibited by CO (Fig. 4D), which is reasonable as neither ferrous nor dioxyheme is expected to form under such conditions.
4) Radical Migration and Oligomerization of KatG
Insights into the KatG catalase reaction mechanism and defects in the mutants may also be gained by examining oligomerization of the enzymes. We reasoned that the noted inefficient utilization of the MYW-adduct radical in the mutants might allow for increased electron transfers that ultimately produce surface tyrosyl radicals and then dityrosine cross-links. Such radicals are known to form in WT KatG in the presence of a slow flux of H2O2 (32) under conditions that we propose sustain inefficient catalase turnover; therefore, the oligomerization behavior of the catalase-deficient mutants was explored.
Here, while we do not directly detect radical formation in the presence of enzymatically generated H2O2 used as the substrate, we do show enhanced oligomer formation in the catalase-deficient mutants under those conditions. For example, high and moderate yields of oligomers were found for KatG[R418L] and KatG[D137S] respectively, while only traces appeared for WT KatG under the slow-flux conditions (Fig. 5). Oligomerization is an inefficient and/or slow process, as the direct addition of a bolus of H2O2 (1000-fold molar excess) did not produce significant yields (not shown). The enhanced cross-linking of KatG[R418L] is evidence for a higher yield of surface radicals upon decay of the catalytically defective MYW-adduct radical in this mutant. An enhancement in decay processes is more directly evident from the EPR results above, showing that the accumulation of MYW-adduct radical in KatG[R418L] does not exceed that in WT KatG. Decay can be enhanced in this mutant because Arg-418 is required for a rapid Rxn. 3 of Scheme 1 (see DFT results). A slower Rxn. 3 favors escape of the adduct radical through pathways different from its direct reaction with dioxyheme.
FIGURE 5.

SDS-PAGE showing a band for the KatG monomer (80 kDa) and higher molecular weight species appearing in multiples of 80 kDa formed in WT KatG, KatG[R418L] and KatG[D137S] in the presence of a slow flux of H2O2. Enzymes (15 μm) were incubated for 30 min with increasing fluxes of H2O2 (0–100 μm/min) generated by glucose plus glucose oxidase as described under “Experimental Procedures.”
KatG[D137S] forms less oligomer than KatG[R418L], but far more than WT. This result is also consistent with enhanced migration of the MYW-adduct radical, as this mutant lacks the dioxyheme species required for a rapid adduct-radical quenching reaction (Rxn. 3, Scheme 1).
5) DFT Calculations and the Function of Arg-418
A crucial role for residue Arg-418 in the catalase reaction mechanism is evident from the results presented above. The observation that replacing Arg-418 with Leu had the effect of extending the persistence of the MYW-adduct radical suggested that the residue is involved in restoring resting enzyme via Rxn. 3 (Scheme 1). DFT calculations were applied to evaluate energies for a series of models of the MYW-adduct radical:dioxyheme species (the reactant in Rxn. 3) to examine whether Arg-418 influences its properties. Models were built to extend earlier calculations for the MYW-adduct radical alone (13) by incorporating dioxyheme with its proximal imidazole ligand and Arg-418 in two alternate conformations suggested by the M. tb KatG crystal structure.
The new DFT calculations show that Arg-418 can facilitate conversion of the dioxyheme/MYW-adduct radical unit into triplet (molecular) oxygen, ferric heme, and a closed shell (non-radical) MYW-adduct. The most relevant results are illustrated by the energy level diagram and the models shown in Fig. 6. The electronic configurations of models include two isoenergetic lower energy states for the Arg-remote model (doublet and quartet) formally consisting of low spin d6 heme iron (S = 0) and a bound oxygen ligand (S = 0) along with the MYW-adduct radical (S = 1/2) for a total spin = 1/2; or low-spin ferric heme iron (d5, S = 1/2) with a bound superoxide anion radical (S = 1/2), along with the MYW-adduct radical (S = 1/2) for S(total) = 3/2. Higher total spin multiplicities were higher in energy for the Arg-remote case. Most notable is that oxygen remained bound in all four electronic states for Arg-remote structures but was dissociated in all Arg-vicinal models.
FIGURE 6.

DFT-calculated models (in vacuo) of the heme with proximal histidine, MYW-adduct and the Arg-418 side chain in remote and vicinal conformations. Panel A shows the relative energies of three states; the highest corresponds to the structure shown in panel B, which formally contains FeII (S = 0), bound dioxygen (S = 0) and the MYW-adduct radical (S = 1/2) and is isoenergetic with a quartet (not shown) formally containing FeIII (S = 1/2), bound superoxide anion radical (S = 1/2), and MYW-adduct radical (S = 1/2). The middle level in A represents a fictitious state in which the Arg side chain is hydrogen bonded to the Tyr-229 moiety in the MYW-adduct (non-radical), with FeIII S = 1/2 and oxygen dissociated in a singlet state. The lowest energy state is for the model formally containing S = 3/2 heme iron, S = 1 dioxygen and the MYW-adduct in its reduced (non-radical) form as in 6C. The structure used for the model shown in panel B was built from coordinates of Subunit A of the M. tb KatG structure (2cca.pdb), while the structure for 6C was built based on the subunit B coordinates. The structures in B and C represent the energy-minimized models in both cases. The magenta contours depict 98% of calculated unpaired spin densities (isovalue = 0.02). One hydrogen from each terminal nitrogen of the guanidinium group of Arg-418 forms a hydrogen bond to the phenolate oxygen of Tyr-229 in the closed-shell model shown in C. The Arg-vicinal model with S(total) = 7/2 was higher in energy than that with S(total) = 5/2. For KatG in solution, the heme iron exists in 5- and 6-coordinate forms in a quantum-mechanically mixed spin state which includes S = 3/2 and 5/2 iron. Both the sextet and oxtet total spin forms for the Arg-vicinal models were also lower in energy than the lowest energy species of the Arg-remote model.
Panel A shows three energy levels for models selected from the calculated series to illustrate the changes that depend on the Arg-418 configuration. The energy values must be viewed cautiously as the present calculations were performed in vacuo. The overall difference of 44.2 kcal/mol represents the computed energy released upon conversion of the dioxyheme/MYW-adduct radical unit into the resting enzyme species (S = 3/2 iron, non-radical form of MYW-adduct) plus triplet-state molecular oxygen when Arg-418 is vicinal to Tyr-229. The intermediate energy level corresponds to the Arg-vicinal/dissociated 1O2 doublet state model, with FeIII (S = 1/2) and no MYW-adduct radical, useful to estimate the energy gained solely from the Arg-418–Tyr-229 interaction (17.3 kcal/mol). This value compares well with the 22.3 kcal/mol estimated for the Arg-Tyr hydrogen bonding stabilization calculated for the isolated MYW-adduct model (without heme). Dominating the overall energy gain is the 26.9 kcal/mol contribution (lower part, Fig. 6A) closely corresponding to the singlet-triplet gap for free oxygen.
An important feature of the electronic changes caused by the Arg-Tyr interaction was revealed in DFT calculations on smaller models consisting of the MYW-adduct radical without the dioxyheme component, with and without the Arg interaction (not shown). We found that the guanidinium-phenolate interaction causes a shift in unpaired-electron spin density from the Tyr ring into the Trp indole of the MYW-adduct radical. This shift arises because an increase in negative charge can be stabilized on the phenolic oxygen when the positively charged guanidinium group is nearby. Concomitant with this shift is an increase in net positive character of the Trp indole moiety, the nitrogen of which is adjacent to the oxygen ligand in the models incorporating dioxyheme described above. With dioxyheme present, electron transfer from the “superoxide anion radical” iron ligand into the adduct radical could be favored by the charge and spin redistribution. These electronic changes could not be observed in the extended models described above because of oxygen dissociation and reduction of the adduct radical when Arg was vicinal to Tyr-229.
None of the calculated models indicated covalent changes, such as hydrogen or oxygen transfers to or from the MYW-adduct. Thus, the DFT study predicts that a role for Arg-418 in the catalase reaction is to facilitate conversion of the dioxyheme/MYW-adduct radical species into free triplet-state oxygen, ferric heme iron and the closed shell MYW-adduct to complete the cycle in Scheme 1.
DISCUSSION
The new finding, that the MYW-adduct radical is formed in two catalase deficient mutants, demonstrates that residues Asp-137 and Arg-418 are responsible either for formation or discharge of dioxyheme in the proposed catalase reaction mechanism. The very rapid formation of the MYW-adduct radical likely proceeds the same way in all three cases, through a peroxidase Compound I (Scheme 1, Rxn. 1) or possibly in a concerted turnover that bypasses a free Compound I intermediate. Once the initial turnover has occurred, the most likely route to dioxyheme in the proposed scheme (Rxn. 2) is by rapid H2O2-reduction of ferryl heme in a reaction common to other peroxidases (33–34) including KatG[Y229F] (10). Residue Asp-137 seems to be crucial for this conversion and may be responsible for an important proton transfer step that enhances the reactivity of ferryl heme (35). This aspartate residue is absent from Class III peroxidases such as HRP, in which Compound III (dioxyheme) is only slowly formed (20 m−1·s−1) in the presence of excess H2O2 (34). Thus, facile formation of dioxyheme in KatG distinguishes this enzyme from other heme peroxidases.
A strong argument for the catalytic competence of an intermediate comprised of dioxyheme/MYW-adduct radical is based on the fact that at alkaline pH, the catalase rate of WT KatG is decreased nearly 10-fold compared with pH 7, while the steady-state heme species is different from dioxyheme, and the adduct radical is still present at this pH. Additional suggestions come from the observation of CO inhibition, which indicates that dioxyheme is present over the full pH range of catalase activity despite its lack of accumulation at alkaline pH.
CO binds to Fe(II) heme exclusively. It is not acting like an inhibitor of the substrate H2O2, since CO does not bind to the Fe(III) heme or to forms in other oxidation states above Fe(II) heme. The lag period we observed is then a manifestation of inhibition occurring when CO binds to a small fraction of dioxyheme enzyme that spontaneously dissociated molecular oxygen to allow replacement by the CO ligand. Such dissociation is apparently rare and many turnover cycles are required to increase the population of molecules that can be siphoned off the catalytic pathway by CO binding.
The observed lag phase is thus consistent with a mechanism-based process and we estimate that if a very small percentage of dioxyheme (say 1%) were to spontaneously decay by ligand (oxygen) dissociation, several thousand turnover cycles would be needed for inhibition, as was observed. Whether CO is capturing a kinetically competent or some other intermediate species remains to be determined.
The DFT results presented here, combined with results from earlier calculations, provide valuable insights into the behavior of the dioxyheme/MYW-adduct radical intermediate and Rxn. 3 of the proposed catalase pathway. The calculations show that the side chain of residue Arg-418 promotes discharge of the oxygen ligand and quenching of the adduct radical through electronic effects mediated by the guanidinium salt bridge to the phenolic oxygen of Tyr-229 in the adduct radical. This effect will be favored in WT KatG at pH values near neutral and above that are known to favor the Arg-418/Tyr-229 interaction, and will be absent in the KatG[R418L] mutant.
These phenomena and the pH dependence of the KatG catalase reaction can be integrated to provide insights into the origin of the rate increase occurring from pH 5 to 7 and the decrease from pH 7 to 9 (36). One of the key pH-dependent interactions is very likely the salt bridge between Arg-418 and Tyr-229, which will be favored when the phenolic oxygen of Tyr-229 is deprotonated and the guanidinium group is protonated. Interestingly, dioxyheme does not accumulate at alkaline pH, while the MYW-adduct radical is present at normal abundance. In the Scheme 1 mechanism, Rxn. 3 should exhibit a higher rate in the higher pH range with the salt bridge in place. In contrast to this, formation of dioxyheme from ferryl heme is disfavored at high pH, since protonation of the oxo-ligand is required in the typical pathway to the dioxyheme species (34). Thus, during catalase turnover at alkaline pH, dioxyheme is not detected because a slow rate of its formation occurs under conditions of efficient quenching: high pH leads to a decreased reaction rate as Rxn. 2 becomes rate-limiting. In the lower pH range, the rate of formation of dioxyheme will be enhanced but the Arg-418–Tyr-229 interaction is disfavored and quenching of the dioxyheme/MYW-adduct radical unit becomes slower. Then, dioxyheme is the observed steady-state species and the turnover rate decreases due to slowing of Rxn. 3.
CONCLUSIONS
Overall, the current results demonstrate that formation of the MYW-adduct radical is not sufficient for efficient catalase turnover by KatG; dioxyheme and a regulating remote influence (Arg418) on the electronic structure of the MYW-adduct radical must be present for maximal activity. It seems clear that M. tb KatG is structured for very rapid formation of dioxyheme and robust catalase activity under a wide range of H2O2 concentrations, arguably important for the survival of an intracellular pathogen subjected to fluctuating oxidative stress from host cells.
Acknowledgment
We thank Janan Zhu for helping in the preparation of the KatG[D137S] mutant.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-AI060014 and 2R56AI060014-06A1 (NIAID) and NSF Grant CHE-1058116 (to R. S. M.).
- KatG
- catalase-peroxidase
- M. tb
- M. tuberculosis
- Bp
- B. pseudomallei
- KatG[R418L]
- R418L mutant of KatG
- KatG[D137S]
- D137S mutant of KatG
- BLC
- bovine liver catalase
- EPR
- electron paramagnetic resonance
- RFQ
- rapid freeze-quench
- MYW
- Met Tyr Trp.
REFERENCES
- 1. Ng V. H., Cox J. S., Sousa A. O., MacMicking J. D., McKinney J. D. (2004) Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol. Microbiol. 52, 1291–1302 [DOI] [PubMed] [Google Scholar]
- 2. Wilson T. M., de Lisle G. W., Collins D. M. (1995) Effect of inhA and katG on isoniazid resistance and virulence of Mycobacterium bovis. Mol. Microbiol. 15, 1009–1015 [DOI] [PubMed] [Google Scholar]
- 3. Jittawuttipoka T., Buranajitpakorn S., Vattanaviboon P., Mongkolsuk S. (2009) The catalase-peroxidase KatG is required for virulence of Xanthomonas campestris pv. campestris in a host plant by providing protection against low levels of H2O2. J. Bacteriol. 191, 7372–7377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamada Y., Fujiwara T., Sato T., Igarashi N., Tanaka N. (2002) The 2.0 A crystal structure of catalase-peroxidase from Haloarcula marismortui. Nat. Struct. Biol. 9, 691–695 [DOI] [PubMed] [Google Scholar]
- 5. Carpena X., Loprasert S., Mongkolsuk S., Switala J., Loewen P. C., Fita I. (2003) Catalase-peroxidase KatG of Burkholderia pseudomallei at 1.7A resolution. J. Mol. Biol. 327, 475–489 [DOI] [PubMed] [Google Scholar]
- 6. Bertrand T., Eady N. A., Jones J. N., Jesmin, Nagy J. M., Jamart-Grégoire B., Raven E. L., Brown K. A. (2004) Crystal Structure of Mycobacterium tuberculosis Catalase-Peroxidase. J. Biol. Chem. 279, 38991–38999 [DOI] [PubMed] [Google Scholar]
- 7. Zhao X., Yu H., Yu S., Wang F., Sacchettini J. C., Magliozzo R. S. (2006) Hydrogen peroxide-mediated isoniazid activation catalyzed by Mycobacterium tuberculosis catalase-peroxidase (KatG) and its S315T mutant. Biochemistry 45, 4131–4140 [DOI] [PubMed] [Google Scholar]
- 8. Jakopitsch C., Auer M., Ivancich A., Rüker F., Furtmüller P. G., Obinger C. (2003) Total conversion of bifunctional catalase-peroxidase (KatG) to monofunctional peroxidase by exchange of a conserved distal side tyrosine. J. Biol. Chem. 278, 20185–20191 [DOI] [PubMed] [Google Scholar]
- 9. Jakopitsch C., Ivancich A., Schmuckenschlager F., Wanasinghe A., Pöltl G., Furtmüller P. G., Rüker F., Obinger C. (2004) Influence of the unusual covalent adduct on the kinetics and formation of radical intermediates in synechocystis catalase peroxidase: a stopped-flow and EPR characterization of the MET275, TYR249, and ARG439 variants. J. Biol. Chem. 279, 46082–46095 [DOI] [PubMed] [Google Scholar]
- 10. Yu S., Girotto S., Zhao X., Magliozzo R. S. (2003) Rapid formation of compound II and a tyrosyl radical in the Y229F mutant of Mycobacterium tuberculosis catalase-peroxidase disrupts catalase but not peroxidase function. J. Biol. Chem. 278, 44121–44127 [DOI] [PubMed] [Google Scholar]
- 11. Ghiladi R. A., Medzihradszky K. F., Ortiz de Montellano P. R. (2005) Role of the Met-Tyr-Trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG) as revealed by KatG(M255I). Biochemistry 44, 15093–15105 [DOI] [PubMed] [Google Scholar]
- 12. Ghiladi R. A., Knudsen G. M., Medzihradszky K. F., Ortiz de Montellano P. R. (2005) The Met-Tyr-Trp cross-link in Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 280, 22651–22663 [DOI] [PubMed] [Google Scholar]
- 13. Suarez J., Ranguelova K., Jarzecki A. A., Manzerova J., Krymov V., Zhao X., Yu S., Metlitsky L., Gerfen G. J., Magliozzo R. S. (2009) An oxyferrous heme/protein-based radical intermediate is catalytically competent in the catalase reaction of Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 284, 7017–7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao X., Yu S., Ranguelova K., Suarez J., Metlitsky L., Schelvis J. P., Magliozzo R. S. (2009) Role of the oxyferrous heme intermediate and distal side adduct radical in the catalase activity of Mycobacterium tuberculosis KatG revealed by the W107F mutant. J. Biol. Chem. 284, 7030–7037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhao X., Suarez J., Khajo A., Yu S., Metlitsky L., Magliozzo R. S. (2010) A radical on the Met-Tyr-Trp modification required for catalase activity in catalase-peroxidase is established by isotopic labeling and site-directed mutagenesis. J. Am. Chem. Soc. 132, 8268–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jakopitsch C., Vlasits J., Wiseman B., Loewen P. C., Obinger C. (2007) Redox intermediates in the catalase cycle of catalase-peroxidases from Synechocystis PCC 6803, Burkholderia pseudomallei, and Mycobacterium tuberculosis. Biochemistry 46, 1183–1193 [DOI] [PubMed] [Google Scholar]
- 17. Carpena X., Wiseman B., Deemagarn T., Herguedas B., Ivancich A., Singh R., Loewen P. C., Fita I. (2006) Roles for Arg-426 and Trp-111 in the modulation of NADH oxidase activity of the catalase-peroxidase KatG from Burkholderia pseudomallei inferred from pH-induced structural changes. Biochemistry 45, 5171–5179 [DOI] [PubMed] [Google Scholar]
- 18. Cade C. E., Dlouhy A. C., Medzihradszky K. F., Salas-Castillo S. P., Ghiladi R. A. (2010) Isoniazid-resistance conferring mutations in Mycobacterium tuberculosis KatG: catalase, peroxidase, and INH-NADH adduct formation activities. Protein Sci. 19, 458–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jakopitsch C., Auer M., Regelsberger G., Jantschko W., Furtmüller P. G., Rüker F., Obinger C. (2003) Distal site aspartate is essential in the catalase activity of catalase-peroxidases. Biochemistry 42, 5292–5300 [DOI] [PubMed] [Google Scholar]
- 20. Hillar A., Peters B., Pauls R., Loboda A., Zhang H., Mauk A. G., Loewen P. C. (2000) Modulation of the activities of catalase-peroxidase HPI of Escherichia coli by site-directed mutagenesis. Biochemistry 39, 5868–5875 [DOI] [PubMed] [Google Scholar]
- 21. Vlasits J., Jakopitsch C., Schwanninger M., Holubar P., Obinger C. (2007) Hydrogen peroxide oxidation by catalase-peroxidase follows a non-scrambling mechanism. FEBS Lett. 581, 320–324 [DOI] [PubMed] [Google Scholar]
- 22. Wittenberg J. B., Noble R. W., Wittenberg B. A., Antonini E., Brunori M., Wyman J. (1967) Studies on the equilibria and kinetics of the reactions of peroxidase with ligands. II. The reaction of ferroperoxidase with oxygen. J. Biol. Chem. 242, 626–634 [PubMed] [Google Scholar]
- 23. Chouchane S., Lippai I., Magliozzo R. S. (2000) Catalase-peroxidase (Mycobacterium tuberculosis KatG) catalysis and isoniazid activation. Biochemistry 39, 9975–9983 [DOI] [PubMed] [Google Scholar]
- 24. Berry E. A., Trumpower B. L. (1987) Simultaneous determination of hemes a, b, and c from pyridine hemochrome spectra. Anal. Biochem. 161, 1–15 [DOI] [PubMed] [Google Scholar]
- 25. Chouchane S., Girotto S., Yu S., Magliozzo R. S. (2002) Identification and characterization of tyrosyl radical formation in Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 277, 42633–42638 [DOI] [PubMed] [Google Scholar]
- 26. Yordanov N. D., Ranguelova K. (2000) Quantitative electron paramagnetic resonance and spectrophotometric determination of the free radical 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 56A, 373–378 [DOI] [PubMed] [Google Scholar]
- 27. Cotton M. L., Dunford H. B., Raycheba J. M. (1973) Studies on horseradish peroxidase. 13. The kinetic effect of cyanide on the oxidation-reduction cycle. Can. J. Biochem. 51, 627–631 [DOI] [PubMed] [Google Scholar]
- 28. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A. (2009) Gaussian 09, Revision B. 01. Gaussian, Inc, Wallingford CT [Google Scholar]
- 29. Glendening E. D., Badenhoop J. K., Reed A. E., Carpenter J. E., Bohmann J. A., Morales C. M., Weinhold F. (2001) NBO 5.0 Theoretical Chemistry Institute, University of Wisconsin, Madison, WI [Google Scholar]
- 30. Chouchane S., Girotto S., Kapetanaki S., Schelvis J. P., Yu S., Magliozzo R. S. (2003) Analysis of heme structural heterogeneity in Mycobacterium tuberculosis catalase-peroxidase (KatG). J. Biol. Chem. 278, 8154–8162 [DOI] [PubMed] [Google Scholar]
- 31. Keilin D., Hartree E. F. (1951) Purification of horseradish peroxidase and comparison of its properties with those of catalase and methaemoglobin. Biochem. J. 49, 88–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ranguelova K., Suarez J., Magliozzo R. S., Mason R. P. (2008) Spin trapping investigation of peroxide- and isoniazid-induced radicals in Mycobacterium tuberculosis Catalase-Peroxidase. Biochemistry 47, 11377–11385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jakopitsch C., Wanasinghe A., Jantschko W., Furtmüller P. G., Obinger C. (2005) Kinetics of interconversion of ferrous enzymes, compound II and compound III, of wild-type synechocystis catalase-peroxidase and Y249F: proposal for the catalatic mechanism. J. Biol. Chem. 280, 9037–9042 [DOI] [PubMed] [Google Scholar]
- 34. Adediran S. A., Lambeir A.-M. (1989) Kinetics of the reaction of compound II of horseradish peroxidase with hydrogen peroxide to form compound III. Eur. J. Biochem. 186, 571–576 [DOI] [PubMed] [Google Scholar]
- 35. Nakajima R., Yamazaki I. (1987) The mechanism of oxyperoxidase formation from ferryl peroxidase and hydrogen peroxide. J. Biol. Chem. 262, 2576–2581 [PubMed] [Google Scholar]
- 36. Johnsson K., Froland W. A., Schultz P. G. (1997) Overexpression, purification, and characterization of the catalase-peroxidase KatG from Mycobacterium tuberculosis. J. Biol. Chem. 272, 2834–2840 [DOI] [PubMed] [Google Scholar]