

Background: Growth hormone (GH) is involved in hepatic glucose metabolism.
Results: GH increased hepatic gluconeogenesis through STAT5 transactivation, which was abolished by the metformin-ATM-AMPK-SHP pathway.
Conclusion: The metformin-ATM-AMPK-SHP network prevents the increase of hepatic gluconeogenesis by the GH-dependent pathway.
Significance: The ATM-AMPK-SHP pathway may provide a novel mechanism for regulating hepatic glucose homeostasis via a GH-dependent pathway.
Keywords: AMP-activated Kinase (AMPK), Gluconeogenesis, Growth Hormone, Nuclear Receptors, STAT Transcription Factor, Ataxia Telangiectasia Mutated, Small Heterodimer Partner
Abstract
Growth hormone (GH) is a key metabolic regulator mediating glucose and lipid metabolism. Ataxia telangiectasia mutated (ATM) is a member of the phosphatidylinositol 3-kinase superfamily and regulates cell cycle progression. The orphan nuclear receptor small heterodimer partner (SHP: NR0B2) plays a pivotal role in regulating metabolic processes. Here, we studied the role of ATM on GH-dependent regulation of hepatic gluconeogenesis in the liver. GH induced phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6-phosphatase gene expression in primary hepatocytes. GH treatment and adenovirus-mediated STAT5 overexpression in hepatocytes increased glucose production, which was blocked by a JAK2 inhibitor, AG490, dominant negative STAT5, and STAT5 knockdown. We identified a STAT5 binding site on the PEPCK gene promoter using reporter assays and point mutation analysis. Up-regulation of SHP by metformin-mediated activation of the ATM-AMP-activated protein kinase pathway led to inhibition of GH-mediated induction of hepatic gluconeogenesis, which was abolished by an ATM inhibitor, KU-55933. Immunoprecipitation studies showed that SHP physically interacted with STAT5 and inhibited STAT5 recruitment on the PEPCK gene promoter. GH-induced hepatic gluconeogenesis was decreased by either metformin or Ad-SHP, whereas the inhibition by metformin was abolished by SHP knockdown. Finally, the increase of hepatic gluconeogenesis following GH treatment was significantly higher in the liver of SHP null mice compared with that of wild-type mice. Overall, our results suggest that the ATM-AMP-activated protein kinase-SHP network, as a novel mechanism for regulating hepatic glucose homeostasis via a GH-dependent pathway, may be a potential therapeutic target for insulin resistance.
Introduction
Growth hormone (GH)2 binds to its cognate receptor and regulates a number of transcription factors, including STAT5 through recruitment of intracellular activation of JAK2 (1, 2). GH causes STAT5 phosphorylation and nuclear translocation, leading to transcriptional activation of a wide range of target genes, including cell cycle regulators, anti-apoptotic genes, insulin-like growth factor-I, suppressor of cytokine signaling 2, and several hepatocyte nuclear factors (HNFs) (3, 4). GH is known to play a role in the regulation of body growth, differentiation, aging, and metabolism (5). Moreover, GH antagonizes insulin action on glucose and lipid homeostasis in diverse tissues (6, 7). In this respect, elevated GH may promote insulin resistance and diabetes by increasing hepatic glucose production, triglyceride storage, and antagonizing insulin action in human and animal models (8). The molecular mechanism of GH regulation of hepatic gluconeogenesis has not been fully elucidated.
Metformin is an anti-diabetic agent that ameliorates hyperglycemia by decreasing hepatic glucose production (9, 10). Liver kinase B1 is a downstream effector of metformin and activates AMP-activated protein kinase (AMPK) in the liver, thereby regulating glucose homeostasis (10, 11). AMPK is a conserved serine/threonine protein kinase that functions as a major intracellular energy sensor and a master regulator of metabolic homeostasis, including glucose and lipid metabolism (11, 12). AMPK is activated in response to a variety of physiological stimuli such as exercise, hypoxia, and the action of several pharmacological agents and hormones, including metformin, leptin, and adiponectin (13). Previous studies from our group have shown that metformin inhibits hepatic gluconeogenesis by up-regulating the AMPK-dependent pathway in vitro and in vivo (14, 15). The ataxia telangiectasia mutated (ATM) gene encodes a 350-kDa protein that is a serine/threonine protein kinase, which belongs to the superfamily of phosphatidylinositol 3-kinase-related kinase (16). The ATM gene is well characterized in autosomal recessive diseases (e.g., cerebellar ataxia), immunodeficiency, radiation sensitivity, and growth retardation (17). ATM is a regulator of cell cycle progression by activating p53, Brca1, Chk2, and Smc1 in response to general toxic agents, ionizing radiation, DNA damage, and cell cycle arrest (16, 18). Previous studies have demonstrated that 5-aminoimidazole-4-carboxamide ribotide (AICAR), an AMPK activator, induces AMPK phosphorylation in an ATM-dependent manner (19, 20). ATM null mice displayed glucose intolerance and hyperglycemia, leading to deteriorating insulin secretion and elevated blood glucose levels (21). Although ATM activates AMPK in vitro and the loss of ATM leads to diabetes, the possible role for ATM in hepatic gluconeogenesis has yet to be fully elucidated.
The small heterodimer partner (SHP; NR0B2) is an atypical orphan nuclear receptor that functions as a co-repressor of transcription factors, including HNF-3β, HNF-4α, and FoxO1 (22). SHP plays a critical role in the maintenance of metabolic homeostasis, such as glucose, lipid, and bile acid metabolism (23). Previous reports from our group have demonstrated that pharmacological agents that activate AMPK, such as metformin, sodium arsenite, and hepatocyte growth factor, induce SHP to hepatic gluconeogenesis (14, 24, 25). However, a critical role for the ATM-AMPK pathway in SHP gene expression and its subsequent role in mediating GH-mediated up-regulation of hepatic gluconeogenesis have not been fully elucidated.
In this study, we demonstrated that GH is a key regulator of hepatic gluconeogenic gene expression and glucose production in hepatocytes. GH induced STAT5 occupancy of the gluconeogenic gene promoter, and this stimulatory effect of GH was abolished by either a JAK2 inhibitor or DN-STAT5. Induction of SHP by ATM effectively blocked GH-mediated induction of hepatic gluconeogenic genes and glucose production, which was abolished by KU-55933 (an ATM inhibitor) treatment. Overall, these observations suggest that the ATM-AMPK-SHP pathway may confer a novel mechanism for regulating the pathological processes of hepatic glucose homeostasis via a GH-dependent pathway and provide a potential therapeutic strategy for modulating hepatic gluconeogenesis.
EXPERIMENTAL PROCEDURES
Chemicals
Metformin (1,1-dimethylbiguanide hydrochloride; Sigma), recombinant human growth hormone (ProSpec), forskolin (Calbiochem), the JAK2 inhibitor AG490 (Sigma), and the ATM inhibitor KU-55933 (Calbiochem) were purchased from the indicated companies and dissolved in the recommended solvents.
Plasmids
The reporter plasmids encoding PEPCK-Luc and G6Pase-Luc were prepared as described previously (14). Plasmids encoding the constitutively active form of AMPK (CA-AMPK) and the dominant negative mutant of AMPK (DN-AMPK) were prepared as described previously (14). SHP cDNA and siRNAs were prepared as described previously (14, 24). CA-STAT5a and DN-STAT5a/b were generously provided by Dr. Toshio Kitamura (26) and Dr. Michael J. Waters (27), respectively. The point mutant form of PEPCK-Luc was generated using a site-directed mutagenesis kit (Stratagene, La Jolla, CA) and the following primers: forward, 5′-CAATTAAGG-GTTGAGCCTATA-3′, and reverse, 5′-TATAG-GCTCAACCCTTAATTG-3′. All of the plasmids were confirmed by sequencing analysis.
Preparation of Recombinant Adenovirus
Adenoviruses encoding full-length human SHP, siRNA SHP, GFP, c-Myc-tagged DN-AMPK, and CA-AMPK have been described previously (14, 15, 24). Briefly, the cDNA encoding CA-STAT5 and DN-STAT5 was inserted into pAdTrack-CMV shuttle vector. This vector was electroporated into the AdEasy adenoviral vector to generate the recombinant adenoviral plasmid. The viruses were amplified in HEK293 cells and titrated using Adeno-XTM Rapid titer according to the manufacturer's instructions.
Cell Culture and Transient Transfection Assays
HepG2 (human hepatoma) cells were cultured in DMEM (Invitrogen) supplemented with 10% FBS (Hyclone, Logan, UT) and antibiotics in a humidified atmosphere containing 5% CO2 at 37 °C. AML-12 cells (immortalized mouse hepatocytes) were cultured in DMEM/F-12 medium (Invitrogen) supplemented with 10% FBS, insulin-transferrin-selenium (Invitrogen), dexamethasone (40 ng/ml; Sigma), and antibiotics in a humidified atmosphere containing 5% CO2 at 37 °C. Transient transfection assays were carried out as described previously (14).
siRNA Experiments
The siRNAs for STAT5 were chemically synthesized (Cell Signaling Technology, Danvers, MA) and transfected according to the manufacturer's instructions. HepG2 cells were transfected with siRNA using Oligofectamine reagent (Invitrogen). Efficiency of knockdown was performed through Western blot analyses.
Isolation and Culture of Primary Rat Hepatocytes
Rat primary hepatocytes (RPHs) were isolated from the livers of 7-week-old male Sprague-Dawley rats. The hepatocytes were isolated by the collagenase perfusion method, as described previously (14).
Primary Human Hepatocyte Culture
Human primary hepatocytes (HPHs) were obtained through the Liver Tissue and Cell Distribution System of the National Institutes of Health (S. Strom, University of Pittsburgh, PA). HPHs were cultured as described previously (25).
Animal Experiments
Male wild-type C57BL/6J (WT) and SHP null mice (SHP KO) were used for this experiment. As for GH stimulation experiment, both mice were injected intraperitoneally with or without GH for 3 h. The mice were sacrificed, and their liver tissues were harvested. All animal studies and protocols were approved and carried out by the institutional animal use and care committee of Korea Research Institute of Bioscience and Biotechnology.
RNA Isolation and Analysis
Total RNA was extracted from either RPH or HPH under various conditions with TRIzol reagent (Invitrogen) according to the manufacturer's protocol. qPCR analysis using a SYBR Green PCR kit was conducted using PEPCK, G6Pase, SHP, and β-actin primers, as described previously (15, 25).
Western Blot Analysis
RPHs were isolated and processed according to a method described previously (14, 25). The membranes were probed with antibody against phospho-ATM, ATM, phospho-AMPK, AMPK, phospho-STAT5 (Tyr-694; Cell Signaling Technology), STAT5, SHP (H-160), PEPCK (H-300), G6Pase (C-14), and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) and then developed using an ECL Western blot detection kit (Amersham Biosciences).
Co-immunoprecipitation Assay
Total protein was extracted from RPH treated with either GH for 1 h or metformin for 6 h. The cells were immunoprecipitated using STAT5 antibody (Santa Cruz Biotechnology) and then blotted with SHP antibody (Santa Cruz Biotechnology). Signals were developed using the ECL Western blot Detection kit.
Chromatin Immunoprecipitation Assay
36 h after infection with adenoviral siRNA SHP (Ad-si SHP) and control siRNA (Ad-Scram), RPHs were treated with metformin (2 mm) for 6 h and with GH (500 ng/ml) for 1 h. The cells were subsequently harvested, and ChIP assays were conducted using STAT5 and SHP antibodies as described previously (16, 24). After purification, PCRs were performed using two pairs of primers encompassing proximal (−472/−265 bp) or distal (−1245/−1026 bp) regions of the PEPCK promoter. The specific primers used for PCR were: proximal, forward 5′-AGAGCTGAATTCCCTTCTCA-3′ and reverse 5′-TTTGACCGT-GACTGTTGCTG-3′; distal, forward 5′-CCTCGAATCTGTCACACGTC-3′ and reverse 5′-ACACAGACCTTCACGCCAA-T-3′.
Electrophoretic Mobility Shift Analysis
For EMSA, nuclear extracts were prepared from HepG2 as previously described (28). Nuclear extracts were prepared from HepG2 cells treated with GH. For supershift analysis, protein was preincubated with indicated antibody and then incubated with labeled oligonucleotide from −316 to −296 of the PEPCK gene promoter. The protein-DNA complexes were analyzed by EMSA. The sequence of the oligonucleotide used was 5′-TGACAATTAAGGCAAGAG-CCT-3′.
Glucose Output Assay
Glucose production in RPH was measured using a colorimetric glucose oxidase assay (Sigma) according to the manufacturer's protocol. Briefly, the cells were washed three times with PBS and then incubated for 3 h at 37 °C in 5% CO2 in glucose production buffer (glucose-free RPMI 1640, pH 7.4, containing 20 mm sodium lactate, 1 mm sodium pyruvate, and 15 mm HEPES, without phenol red). The glucose assays were performed in triplicate, and the intra-assay coefficient of variation was <5%, as described previously (25).
Statistical Analysis
Data calculation and statistical analysis were performed using GraphPad Prism 3–5.0 software. The statistical significance of differences between groups was determined with Student's t test, and multiple comparisons were analyzed using one-way ANOVA under treatment and experiment as factors. All of the data are presented as the means ± S.E. Differences were considered statistically significant at p < 0.05.
RESULTS
Growth Hormone Increases Hepatic Gluconeogenic Gene Expression in Primary Hepatocytes
First, we examined whether GH shows any potential role in hepatic gluconeogenic gene expression in hepatic cell line and primary hepatocytes. Treatment of immortalized mouse hepatocyte AML-12 cells and RPHs with GH significantly increased the mRNA levels of two key hepatic gluconeogenic genes, PEPCK and G6Pase, in a time-dependent manner (Fig. 1, A and B). In addition, the mRNA levels of PEPCK and G6Pase were effectively increased by GH treatment in HPHs, in a fashion similar to that shown in AML-12 cells and RPH (Fig. 1C). Importantly, induction of PEPCK and G6Pase mRNA expression by 3-h treatment of GH reached the maximum levels in the HPH, which is consistent with the increase in AML-12 and RPH. Next, we examined the effect of GH on the PEPCK and G6Pase protein levels in primary hepatocytes. GH significantly increased PEPCK protein level through STAT5 activation in primary hepatocytes (Fig. 1D). Interestingly, treatment with GH continuously increased G6Pase protein level within 3–24 h in a time-dependent manner (Fig. 1D). Overall, these findings strongly suggest that GH plays an important role in hepatic gluconeogenic gene expression in primary hepatocytes and a hepatic cell lines.
FIGURE 1.

Growth hormone induces hepatic gluconeogenic gene expression in rat and human primary hepatocytes. A–C, AML-12 cells (A), RPHs (B), and HPHs (C) were cultured for 24 h under serum-free conditions. The cells were treated with GH (500 ng/ml) for various time periods up to 24 h. Hepatic gluconeogenic gene expression was measured by qPCR and then normalized to β-actin level. All of the data are representative of at least three independent experiments. D, as mentioned above, RPHs were treated with GH for various time periods up to 24 h. Whole cell extracts were isolated and analyzed using Western blot analysis with the indicated antibodies. *, p < 0.05 compared with untreated control (Student's t test).
GH Increases Hepatic Gluconeogenesis through STAT5 Activation
To determine whether GH-mediated STAT5 activation is involved in the induction of hepatic gluconeogenesis, we examined the effect of STAT5 activation on hepatic gluconeogenesis after treatment of primary hepatocytes with GH and AG490, a specific JAK2 inhibitor. AG490 treatment markedly repressed GH-mediated induction of PEPCK and G6Pase protein level through STAT5 inhibition, in a dose-dependent manner (Fig. 2A). GH treatment significantly increased glucose production in primary hepatocytes, whereas this stimulatory effect of GH was dramatically reduced by AG-490 (Fig. 2B). To determine whether the regulation of hepatic gluconeogenesis by GH is mediated by STAT5, we evaluated the effect of STAT5 on the regulation of gluconeogenic gene expression and glucose production in primary hepatocytes using adenovirus-mediated overexpression of a constitutively active form of STAT5 (Ad-CA-STAT5) or a dominant negative form of STAT5 (Ad-DN-STAT5). Our Western blot analysis demonstrated that both CA-STAT5 and DN-STAT5 were successfully transduced in primary hepatocytes (Fig. 2, C and D). Next, STAT5 overexpression using Ad-CA-STAT5 significantly increased PEPCK and G6Pase protein levels in primary hepatocytes, whereas this stimulatory effect of GH decreased markedly following Ad-DN-STAT5 treatment (Fig. 2E). Consistent with previous data, glucose production in primary hepatocytes was significantly increased by Ad-CA-STAT5 when compared with that of the Ad-GFP control. However, the increase in hepatic glucose production by the GH treatment was markedly reduced by Ad-DN-STAT5, which was comparable with control groups. As expected, the increase of glucose output by both GH and Ad-CA-STAT5 reached the maximum levels relative to control and/or alone treatment (Fig. 2F). Next, we further confirm whether GH-induced hepatic gluconeogenic gene expression is mediated by STAT5 in hepatocytes. GH significantly increased PEPCK and G6Pase protein level, whereas this effect was abolished by endogenous STAT5 knockdown with siRNA STAT5 (Fig. 2G), in a fashion similar to that observed for gluconeogenesis (Fig. 2, E and F). Collectively, these results indicate that GH-mediated up-regulation of hepatic gluconeogenesis was altered by the JAK-STAT5 pathway in primary hepatocytes.
FIGURE 2.

STAT5 mediates GH-mediated induction of hepatic gluconeogenesis. A, RPHs were pretreated with AG490 for 6 h and then exposed to GH (500 ng/ml) for 3 h at the various concentrations. Whole cell extracts were isolated and analyzed using Western blot analysis with the indicated antibodies. Protein levels were normalized to β-actin level and/or total form. B, glucose output assay in RPHs was performed with GH and AG490 at the indicated concentrations in glucose-free medium supplemented with gluconeogenic substrate sodium lactate (20 mm) and sodium pyruvate (1 mm). C, RPHs were infected with Ad-GFP and Ad-CA-STAT5 for 36 h. D, RPHs were infected with Ad-DN-STAT5 for 36 h and then treated with GH for 3 h. Whole cell extracts were isolated and analyzed using Western blot analysis with the indicated antibodies. E, RPHs were infected with Ad-GFP, Ad-CA-STAT5, and Ad-DN-STAT5 for 36 h and then treated with or without GH for 3 h. Whole cell extracts were isolated and analyzed using Western blot analysis with the indicated antibodies. F, glucose output assay in RPH was conducted as described for B, using glucose-free medium supplemented with the gluconeogenic substrate sodium lactate (20 mm) and sodium pyruvate (1 mm). All of the data are representative of at least three independent experiments. G, HepG2 cell lines were transfected with siRNA control and siRNA STAT5 for 36 h and then treated with or without GH for 3 h. Whole cell extracts were isolated and analyzed using Western blot analysis with various antibodies. Protein levels were normalized to β-actin level and/or total forms. *, p < 0.05 (Student's t test); #, p < 0.05 compared with untreated control and/or GH-treated cells (Student's t test or ANOVA).
GH Specifically Mediates STAT5 Occupancy on the Gluconeogenic Gene Promoter
To determine whether GH regulates hepatic gluconeogenic gene expression at a transcriptional level, we carried out transient transfection assays using reporter luciferase constructs containing the PEPCK and G6Pase gene promoter in HepG2 cells. As expected, reporter assays in HepG2 cells showed that GH treatment significantly increased the activity of the PEPCK and G6Pase gene promoters in a dose-dependent manner (Fig. 3A), which is consistent with the increase in gluconeogenic gene expression (Fig. 1). HNF-4α was used as a positive control for activation of both the PEPCK and G6Pase gene promoters. Next, we confirmed whether STAT5 also regulates transcriptional activity of gluconeogenic gene using these promoters. Co-transfection of CA-STAT5, but not DN-STAT5, significantly increased the activity of the PEPCK and G6Pase promoters in a dose-dependent manner (Fig. 3B). Overall, these findings strongly suggest that GH positively regulates the transcriptional activity of hepatic gluconeogenic genes through STAT5 activation in hepatocytes.
FIGURE 3.

Hepatic gluconeogenic gene promoter is activated by STAT5. A–D, HepG2 cell lines (A) were transiently co-transfected with the indicated reporter genes for 36 h and then treated with GH (500 ng/ml) for 3 h in serum-free medium. HNF-4α (100 ng) was used as a positive control. HepG2 cells (B) were transiently co-transfected with CA-STAT5, DN-STAT5, HNF-4α, and the observed reporter genes and then treated with GH (500 ng/ml) for 3 h. HNF-4α (100 ng) was used as a positive control. HepG2 cell were transfected with deletion constructs of the PEPCK reporter gene and CA-STAT5 (D). The cells were treated with GH after transfection (C). E, schematic diagrams of wild-type and mutant forms of the PEPCK promoter encompassing constructs from −310 to −302 bp under the indicated conditions. HepG2 cells were co-transfected with wild type (wt), the mutant form of the PEPCK gene promoter (mt), and CA-STAT5. The cells were treated with GH at the indicated conditions after transfection. Luciferase activity was normalized to β-galactosidase activity to correct for transfection efficiency. F, ChIP assay shows the recruitment of STAT5 on the PEPCK gene promoter. RPHs were treated with GH, and soluble chromatin was immunoprecipitated with anti-STAT5 antibody or IgG as indicated. Purified DNA samples were employed for PCR with primers binding to the specific proximal (top panel) and nonspecific distal (bottom panel) regions on the PEPCK gene promoter. 10% of the soluble chromatin was used as an input. All of the data are representative of at least three independent experiments. *, p < 0.05 (Student's t test); #, p < 0.05 compared with untreated control and/or GH-treated cells (Student's t test or ANOVA).
To identify the molecular mechanism by which GH mediates up-regulation of hepatic gluconeogenic gene transcription, serial deletion constructs of the PEPCK promoter were used to perform a transient transfection assay. As shown in Fig. 3 (C and D), the activity of PEPCK promoter by GH and CA-STAT5 was continuously retained with a deletion up to −355 bp, but it did not maintain completely with the −208-bp construct. These observations show that the STAT5-binding element required for the GH response is located within the region between −355 and −208 bp on the PEPCK gene promoter. Next, site-directed mutagenesis was carried out to introduce CAA to GTT substitution on the PEPCK gene promoter and to further evaluate the functional significance of the STAT5-binding region on the PEPCK gene promoter (Fig. 3E). Wild type (PEPCK wt) and the mutant reporter plasmid (PEPCK mt) were transiently transfected with CA-STAT5 into HepG2 cells. STAT5-dependent activity of the PEPCK gene promoter was increased by either GH treatment or CA-STAT5, and this phenomenon was abolished at a mutant form of the STAT5 site (Fig. 3E). Overall, these results suggest that the STAT5-binding site may be sufficient to mediate activation of the PEPCK gene promoter by GH. We next performed ChIP assays in primary hepatocytes to study STAT5 binding to PEPCK gene at the chromatin level. GH strongly induced STAT5 occupancy on the proximal region (−472/−265) of the PEPCK promoter (Fig. 3F, upper panel) but not on the distal region (−1245/−1026) of the PEPCK promoter (Fig. 3F, lower panel). These findings strongly suggest that STAT5 physically binds to PEPCK gene promoter and mediates GH induction of PEPCK gene transcription.
ATM Represses Hepatic Gluconeogenesis via Induction of SHP
Several studies have demonstrated a correlation between ATM and metabolic dysfunction (21, 29). A previous report has indicated that ATM directly activates AMPK and is required for a response to metformin in humans (30). Therefore, we hypothesized that activation of ATM by metformin might inhibit GH-mediated hepatic gluconeogenesis by inducing SHP. First, we elucidated a critical role of ATM in the AMPK-SHP signal pathway in primary hepatocytes. As expected, metformin increased ATM and AMPK phosphorylation, as well as SHP protein level in primary hepatocytes (Fig. 4A), whereas this stimulatory effect of metformin was repressed by a specific ATM kinase inhibitor KU-55933 (Fig. 4B). Next, we determined how ATM regulates GH-mediated hepatic gluconeogenesis. Up-regulation of SHP by metformin-mediated activation of the ATM-AMPK pathway markedly decreased GH-mediated induction of hepatic gluconeogenic gene expression in primary hepatocytes, which was restored by KU-55933 (Fig. 4, C and D). The increase in glucose production by GH was markedly reduced by metformin (Fig. 4E). However, the inhibitory effects of metformin were reversed by KU-55933 treatment (Fig. 4E) in a pattern similar to that indicated by hepatic gluconeogenic gene expression. Overall, these results strongly suggest that ATM plays a role in the regulation of GH-stimulated hepatic gluconeogenesis by SHP induction.
FIGURE 4.

ATM-AMPK-SHP pathway inhibits GH-mediated hepatic gluconeogenesis. A and B, RPHs were treated with either metformin for 6 h (A) or the ATM inhibitor KU-55933 for 3 h (B) at the indicated concentrations. Whole cell extracts were isolated and analyzed using Western blot analysis with various antibodies. The protein level was normalized to β-actin level and/or total forms. C–E, RPHs were pretreated with metformin for 6 h and then treated with either GH or KU-55933 for 3 h with the indicated conditions. Whole cell extracts were isolated and analyzed by Western blot analysis (C) with various antibodies. Total RNAs were extracted from hepatocytes and utilized for qPCR analysis (D). SHP, PEPCK, and G6Pase mRNA level were normalized to β-actin expression. A glucose output assay (E) was performed using glucose-free medium supplemented with gluconeogenic substrate sodium lactate (20 mm) and sodium pyruvate (1 mm). All of the data are representative of at least three independent experiments. *, p < 0.05 (Student's t test); #, p < 0.05; &, p < 0.05 compared with untreated control, GH-treated cells, metformin (Met)-treated cells, and GH- and metformin-treated cells (ANOVA).
SHP Physically Interacts with STAT5 and Inhibits DNA Binding of STAT5 on PEPCK Gene Promoter
We elucidated the endogenous interaction between SHP and STAT5 using a co-immunoprecipitation assay in primary hepatocytes to identify the molecular mechanism regarding GH-mediated induction of hepatic gluconeogenesis. Endogenous SHP and STAT5 proteins strongly interacted with each other upon treatment of primary hepatocytes with both GH and metformin compared with either controls or GH alone (Fig. 5A). Next, to further confirm a role for SHP in STAT5-binding on the PEPCK gene promoter, we performed a transient transfection assay using wild-type and mutant forms of the PEPCK gene reporter, CA-STAT5, and SHP, as described in Fig. 3E. As anticipated, the increase in PEPCK gene promoter activity by GH treatment was dramatically reduced by SHP in wild-type reporter (PEPCK wt), and this phenomenon was abolished in the mutant form of the promoter (Fig. 5B). Moreover, to further confirm a link between SHP and the DNA binding activity of endogenous STAT5 on the PEPCK gene promoter, we performed ChIP assays using anti-STAT5 in primary hepatocytes. Endogenous STAT5 physically bound to the proximal region (−472/−265) upon GH treatment, and this activity was completely abolished by metformin treatment. However, SHP knockdown using Ad-si SHP reversed STAT5 occupancy on the proximal PEPCK promoter compared with control Ad-Scram (Fig. 5C, left panel). Moreover, the nonspecific distal region (−1245/−1026) of the PEPCK promoter was unable to recruit this protein under all conditions (Fig. 5C, right panel). Neither endogenous SHP nor control IgG bound to the PEPCK gene promoter under both regions (Fig. 5C, bottom panel). Next, we further confirmed the specific binding of STAT5 on the PEPCK gene promoter using an electrophoretic mobility shift assay. As expected, the binding of STAT5 to the PEPCK gene promoter was demonstrated by the protein-DNA complex induced by GH treatment, whereas this complex disappeared with mutant form of STAT5 and cold competitor (Fig. 5D). Moreover, GH showed the strong and specific binding of STAT5 to the PEPCK gene promoter and weakly supershifted with a STAT5 antibody (Fig. 5E), as consistent with the ChIP assay. Overall, these findings demonstrate that SHP negatively regulates GH-mediated induction of hepatic gluconeogenesis via a physical interaction leading to blockage of STAT5 occupancy.
FIGURE 5.

SHP physically interacts with STAT5. A, co-immunoprecipitation (IP) assays with RPHs indicated an association between SHP and STAT5. Protein extracts from primary hepatocytes were immunoprecipitated using anti-STAT5 antibody and then blotted with SHP antibody. Expression of phospho-STAT5, STAT5, and SHP from 10% of lysates were analyzed by Western blotting (WB) with specific antibodies. B, HepG2 cells were co-transfected with the wild type, mutant form of the PEPCK gene promoter, CA-STAT5, and SHP, as described in Fig. 3E. Luciferase activity was normalized to β-galactosidase activity to correct for transfection efficiency. C, RPHs were infected for 36 h with Ad-si SHP (multiplicity of infection of 60) and Ad-Scram (multiplicity of infection of 60) and then treated with either GH (500 ng/ml) or metformin (Met, 2 mm) treatment at the indicated concentration for the ChIP assay. Input represents 10% of purified DNA in each sample. Cell extracts were immunoprecipitated with STAT5 (nonphosphorylated STAT5) and SHP antibodies, and purified DNA samples were used for PCR with primers binding to the specific proximal (left panel) and nonspecific distal (right panel) regions on the PEPCK gene promoter. All of the data are representative of at least three independent experiments. D, nuclear extracts were prepared from HepG2 cells treated with GH (500 ng/ml). For EMSA, protein-DNA complexes (arrow) were analyzed by 4% PAGE, followed by autoradiography. Successive cold competitor (Cold Comp) was performed with unlabeled oligonucleotide. E, nuclear extracts were prepared from HepG2 cells treated with GH (500 ng/ml). For supershift (S.S) analysis, protein was preincubated with the indicated antibody and then incubated with labeled oligonucleotides from −316 to −296 of the PEPCK gene promoter. The protein-DNA complexes (arrows) were analyzed by EMSA. *, p < 0.05 (Student's t test); #, p < 0.05 compared with untreated control and GH-treated cells (Student's t test or ANOVA).
Induction of SHP by Metformin Inhibits GH-mediated Hepatic Gluconeogenesis
As reported previously, several AMPK activators (metformin, sodium arsenite, and hepatocyte growth factor) have a significant effect on the increase in SHP gene expression both in vivo and in vitro (14, 24, 25). Next, we examined whether the induction of SHP by metformin inhibits GH-mediated hepatic gluconeogenesis. The increase in PEPCK and G6Pase mRNA level following GH treatment was markedly inhibited by metformin-induced SHP mRNA expression in both RPH and HPH (Fig. 6, A and B). This inhibition was recapitulated by adenoviral overexpression of SHP (Ad-SHP) relative to controls (Fig. 6C). Moreover, GH induced glucose production was markedly repressed by either metformin treatment or Ad-SHP transduction (Fig. 6D) in primary hepatocytes. To further confirm whether GH-mediated hepatic gluconeogenesis is altered by SHP, we evaluated the potential effect of SHP on GH-induced PEPCK and G6Pase protein expression in primary hepatocytes. Metformin markedly decreased GH-mediated induction of PEPCK and G6Pase protein level through SHP induction, whereas this effect was abolished by endogenous SHP knockdown with Ad-si SHP (Fig. 6E). Furthermore, metformin effectively reduced GH-stimulated hepatic glucose production, in a fashion similar to that observed for gluconeogenic gene expression, and this phenomenon occurred by SHP knockdown (Fig. 6F). Forskolin was used as a positive control to confirm the efficiency of the glucose output assay. A previous study from another group demonstrated that the loss of SHP exacerbates hepatic insulin resistance by increasing glucose intolerance (31). Finally, we confirmed the role of SHP on GH-mediated induction of hepatic gluconeogenesis in the liver of SHP null mice. Induction of PEPCK and G6Pase protein levels by GH was significantly higher in the liver of SHP null mice relative to the wild-type mice (Fig. 6G). Overall, these findings suggest that SHP has an important role in the negative regulation of GH-mediated hepatic gluconeogenesis.
FIGURE 6.

Metformin inhibits GH-mediated induction of hepatic gluconeogenesis through induction of SHP. A and B, RPHs and HPHs were pretreated with metformin (Met) for 6 h and then exposed to GH for 3 h at the indicated concentration. Total RNA was isolated and analyzed using qPCR with the observed primers. C, RPHs were infected with a multiplicity of infection of 60 for Ad-SHP for 36 h and then treated with GH for 3 h. Total RNA was isolated and analyzed using qPCR analysis with the observed primers. D, a glucose output assay in RPH was performed as described in A and/or C, using glucose-free medium supplemented with the gluconeogenic substrates sodium lactate (20 mm) and sodium pyruvate (1 mm). Forskoloin (FSK) was used as a positive control. E and F, RPHs were infected with a multiplicity of infection of 60 for Ad-si SHP and Ad-Scram for 36 h. Then cells were treated with either GH or metformin. Whole cell extracts were isolated and analyzed by Western blot analysis with various antibodies. The experiments were conducted as described in E, and glucose output assay was measured (F). Forskolin was used as a positive control. G, wild-type and SHP null mice were injected intraperitoneally with GH (2 μg/g of body weight) for 3 h. Tissue extracts were isolated from livers of the indicated groups and assessed by Western blot analysis with various antibodies. The protein level was normalized to the β-actin level. All of the mice were separated into experimental groups (n = 5 mice/group). *, p < 0.05 (Student's t test); #, p < 0.05; &, p < 0.05 compared with untreated control, GH-treated cells, and GH- and metformin-treated cells (Student's t test or ANOVA).
DISCUSSION
In this study, we have demonstrated that GH positively regulated hepatic gluconeogenesis by up-regulating STAT5-mediated induction of PEPCK and G6Pase gene expression and glucose production in primary hepatocytes, and this phenomenon was abolished when cells were treated with DN-STAT5 or AG490. However, the inhibitory effects of ATM on hepatic gluconeogenesis were blocked by SHP knockdown and KU-55933 treatment. Based on these findings, we propose that the metformin-ATM-AMPK-SHP network may prevent hepatic metabolic disorders related to insulin resistance by down-regulating the GH-dependent signaling pathway.
Several studies have shown that GH elevates hepatic glucose production by glycogenolysis in rodents and human (6, 32), whereas another group reported no effect of GH on gluconeogenesis (33). Therefore, the role of GH in hepatic gluconeogenesis is still controversial. Based on these findings, we elucidated the critical role of GH on hepatic gluconeogenic gene expression and glucose production in primary hepatocytes. As expected, GH exposure significantly increased gluconeogenic gene expression and glucose production through STAT5 in primary hepatocytes but not in AG490 and DN-STAT5-treated hepatocytes (Figs. 1–3). Overall, these findings strongly suggest that GH is a potent modulator of hepatic gluconeogenesis through STAT5. Lahuna et al. (34) demonstrated that GH stimulates HNF-6 gene transcription by activating STAT5 and HNF-4, and our group reported that SHP down-regulates PEPCK and G6Pase gene transcription via a direct interaction and blockage of HNF-6 recruitment in hepatocytes (35). Our current study strongly suggests that GH up-regulates hepatic gluconeogenesis by STAT5 transactivation, which is consistent with a previous report and our previous study. Moreover, we could not find known transcription factors on the specific binding region of the PEPCK gene promoter, excluding STAT5 in this gene promoter region. However, we cannot exclude the possibility that GH may also depend on other unknown transcription factors to regulate hepatic gluconeogenesis.
Several previous studies have shown that the phorbol ester PMA decreases G6Pase gene expression by the activation of MEK and ERK, and stimulation of G6Pase gene expression by glucose occurs by transcriptional mechanism through transcriptional activation of the G6Pase gene promoter and post-transcriptional mechanism through a decrease in the degradation of G6Pase mRNA (36, 37). Interestingly, our experiments provide the different induction pattern of G6Pase protein and mRNA compared with GH and glucagon signal. Our results demonstrate that forskolin significantly increased the G6Pase protein level within 3–6 h, whereas this induction was gradually decreased from 12 to 24 h. However, treatment with GH continuously increased G6Pase protein level within 3–24 h in a time-dependent manner (data not shown), as previously described (Fig. 1). Based on these findings, our current study suggests a novel insight regarding how GH affects hepatic gluconeogenesis following the different pattern of G6Pase mRNA and protein expression. However, we cannot rule out the possibility that GH may also depend on unknown mechanisms of protein stability, degradation, and other signal pathways to regulate the induction pattern of G6Pase mRNA and protein. Therefore, a further detailed investigation is required to elucidate the detail molecular network of G6Pase gene expression in the future.
Our findings indicate that metformin induced SHP gene expression via the ATM-AMPK signal pathway and that GH-mediated hepatic gluconeogenesis was markedly reduced in an ATM-dependent manner. Moreover, this inhibitory effect of the ATM signaling pathway was abolished by KU-55933 treatment in primary hepatocytes (Fig. 4). Therefore, the current study provides evidence for the first time, to the best of our knowledge, that ATM is a positive regulator of SHP gene expression through the ATM-AMPK signal pathway, and this pathway reduced GH-mediated up-regulation of hepatic glucose metabolism, consistent with a previous report showing exacerbated hyperglycemia in ATM-deficient mice (21). However, a more detailed study on the ATM-dependent pathway and glucose homeostasis is required in an animal model such as ATM-deficient mice and an insulin resistance mice model.
SHP plays an important role regulating diverse metabolic processes and maintaining metabolic homeostasis (22, 23). Therefore, our results indicate that metformin-stimulated SHP gene expression through activation of ATM-AMPK pathway in primary hepatocytes improved hepatic gluconeogenesis by decreasing GH-dependent pathway (Figs. 4 and 6). Moreover, a number of pharmacological agents, including metformin, resveratrol, and A-769662, activate AMPK directly or indirectly both in vivo and in vitro (10, 13). Accordingly, activators of AMPK, such as sodium arsenite, hepatocyte growth factor, and other natural products may suggest the beneficial effect of inducing SHP through the ATM-AMPK pathway and improving hepatic gluconeogenesis by down-regulating the GH-dependent pathway.
A previous study from another group demonstrated that deletion of SHP function significantly protects the mice from diet-induced obesity and hepatic steatosis but accelerates hepatic insulin resistance or development of type 2 diabetes via the disruption of glucose tolerance in animal models of diet-induced obesity (31). On the other hand, our current results demonstrate that GH-mediated hepatic gluconeogenesis in primary hepatocytes and liver is mediated by SHP (Fig. 6), which is consistent with the previous results and the previous study. Therefore, we suggest the possibility that SHP may also rely on complex mechanisms under other physiological conditions to regulate the control fashion of hepatic gluconeogenesis.
GH down-regulates STAT5 occupancy on the fatty acid synthase gene promoter and up-regulates STAT5 recruitment on the pyruvate dehydrogenase kinase 4 promoter in adipocytes (38, 39). Based on our current findings, we propose a molecular mechanism by which metformin induces SHP gene expression by activating AMPK and regulating GH-mediated hepatic gluconeogenesis through STAT5 transactivation in primary hepatocytes. First, GH-induced STAT5 physically interacted with SHP in primary hepatocytes (Fig. 5A). Second, GH-induced STAT5 occupancy on the gluconeogenic gene promoter was reduced by metformin, and this phenomenon was abolished by SHP knockdown in comparison with controls (Fig. 5C). Our results strongly provide a detailed molecular mechanism between the metformin-ATM-AMPK-SHP network and GH-induced hepatic gluconeogenesis. A further detailed explanation is required to elucidate the molecular network in the future. Finally, our findings demonstrate that the GH-dependent pathway represents a major component of hepatic gluconeogenesis, whereas the increase of SHP by the metformin-ATM-AMPK network prevented hepatic gluconeogenesis by down-regulating the GH-STAT5-dependent pathway (Fig. 7).
FIGURE 7.
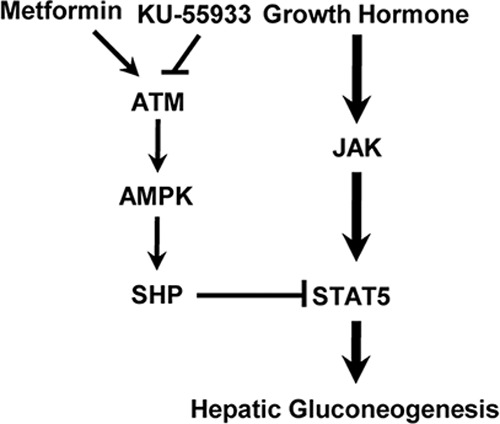
Schematic diagram of the regulation of hepatic gluconeogenesis by the ATM-AMPK-SHP network. Growth hormone induces up-regulation of hepatic gluconeogenesis through STAT5 transactivation, whereas ATM-AMPK-SHP pathway represses GH-induced hepatic gluconeogenesis by inhibiting STAT5-mediated hepatic gluconeogenesis. However, this inhibitory effect of metformin is restored by KU-55933, a specific inhibitor of ATM.
In conclusion, our present study suggests that SHP up-regulation through the ATM-AMPK signaling pathway regulates GH-induced hepatic gluconeogenesis in both primary hepatocytes and liver. Moreover, we also speculate that the metformin-ATM-AMPK-SHP network may prevent hepatic metabolic dysfunction by down-regulating GH-STAT5-mediated hepatic gluconeogenesis. Therefore, as in Fig. 7, a novel molecular mechanism involved in STAT5 inhibition by SHP may provide new insights into the beneficial effects of GH-mediated hepatic metabolic disorders and may be of help to develop novel therapeutic agents to treat hepatic diseases.
Acknowledgments
We thank Dr. Lothar Hennighausen (National Institutes of Health) for helpful discussion. We also thank Dr. Seok-Yong Choi (Chonnam National University Medical School, Gwangju, Republic of Korea) for critical reading of the manuscript.
This work was supported by National Creative Research Initiatives Center for Nuclear Receptor Signals Grant 20110018305 from the Korean Ministry of Education, Science and Technology and the Future-based Technology Development Program (BIO Fields) through the National Research Foundation of Korea funded by Ministry of Education, Science and Technology Grant 20100019512 (to H. S. C.) and a grant of the Korea Health Technology R & D Project, Ministry of Health & Welfare Grant A111345, World Class University Program through the National Research Foundation of Korea funded by Ministry of Education, Science and Technology Grant R32-10064 (to I.-K. L.).
- GH
- growth hormone
- AMPK
- AMP-activated protein kinase
- ATM
- ataxia telangiectasia mutated
- CA
- constitutively active form
- DN
- dominant negative form
- G6Pase
- glucose 6-phosphatase
- PEPCK
- phosphoenolpyruvate carboxykinase
- SHP
- small heterodimer partner
- HNF
- hepatocyte nuclear factor
- RPH
- rat primary hepatocyte
- HPH
- human primary hepatocyte
- qPCR
- quantitative PCR
- Ad
- adenoviral
- si
- siRNA
- ANOVA
- analysis of variance.
REFERENCES
- 1. Carter-Su C., Schwartz J., Smit L. S. (1996) Molecular mechanism of growth hormone action. Annu. Rev. Physiol. 58, 187–207 [DOI] [PubMed] [Google Scholar]
- 2. Lanning N. J., Carter-Su C. (2006) Recent advances in growth hormone signaling. Rev. Endocr. Metab. Disord. 7, 225–235 [DOI] [PubMed] [Google Scholar]
- 3. Buitenhuis M., Coffer P. J., Koenderman L. (2004) Signal transducer and activator of transcription 5 (STAT5). Int. J. Biochem. Cell Biol. 36, 2120–2124 [DOI] [PubMed] [Google Scholar]
- 4. Hennighausen L., Robinson G. W. (2008) Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 22, 711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lichanska A. M., Waters M. J. (2008) How growth hormone controls growth, obesity and sexual dimorphism. Trends Genet. 24, 41–47 [DOI] [PubMed] [Google Scholar]
- 6. Vijayakumar A., Novosyadlyy R., Wu Y., Yakar S., LeRoith D. (2010) Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm. IGF Res. 20, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davidson M. B., Shen D. C., Venkatesan N., Sladen G. (1987) In vivo insulin antagonism but evanescent in vitro tissue effect in rats with growth hormone-secreting tumors. J. Endocrinol. Invest. 10, 569–574 [DOI] [PubMed] [Google Scholar]
- 8. Dominici F. P., Turyn D. (2002) Growth hormone-induced alterations in the insulin-signaling system. Exp. Biol. Med. 227, 149–157 [DOI] [PubMed] [Google Scholar]
- 9. Rotella C. M., Monami M., Mannucci E. (2006) Metformin beyond diabetes. New life for an old drug. Curr. Diabetes Rev. 2, 307–315 [DOI] [PubMed] [Google Scholar]
- 10. Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-Melody J., Wu M., Ventre J., Doebber T., Fujii N., Musi N., Hirshman M. F., Goodyear L. J., Moller D. E. (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shaw R. J., Lamia K. A., Vasquez D., Koo S. H., Bardeesy N., Depinho R. A., Montminy M., Cantley L. C. (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310, 1642–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kahn B. B., Alquier T., Carling D., Hardie D. G. (2005) AMP-activated protein kinase. Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25 [DOI] [PubMed] [Google Scholar]
- 13. Zhang B. B., Zhou G., Li C. (2009) AMPK. An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 9, 407–416 [DOI] [PubMed] [Google Scholar]
- 14. Kim Y. D., Park K. G., Lee Y. S., Park Y. Y., Kim D. K., Nedumaran B., Jang W. G., Cho W. J., Ha J., Lee I. K., Lee C. H., Choi H. S. (2008) Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes 57, 306–314 [DOI] [PubMed] [Google Scholar]
- 15. Lee J. M., Seo W. Y., Song K. H., Chanda D., Kim Y. D., Kim D. K., Lee M. W., Ryu D., Kim Y. H., Noh J. R., Lee C. H., Chiang J. Y., Koo S. H., Choi H. S. (2010) AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 285, 32182–32191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McKinnon P. J. (2004) ATM and ataxia telangiectasia. EMBO Rep. 5, 772–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lavin M. F. (2008) Ataxia-telangiectasia. From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 9, 759–769 [DOI] [PubMed] [Google Scholar]
- 18. Kim S. T., Xu B., Kastan M. B. (2002) Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 16, 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suzuki A., Kusakai G., Kishimoto A., Shimojo Y., Ogura T., Lavin M. F., Esumi H. (2004) IGF-1 phosphorylates AMPK-α subunit in ATM-dependent and LKB1-independent manner. Biochem. Biophys. Res. Commun. 19, 986–992 [DOI] [PubMed] [Google Scholar]
- 20. Sun Y., Connors K. E., Yang D. Q. (2007) AICAR induces phosphorylation of AMPK in an ATM-dependent, LKB1-independent manner. Mol. Cell Biochem. 306, 239–245 [DOI] [PubMed] [Google Scholar]
- 21. Miles P. D., Treuner K., Latronica M., Olefsky J. M., Barlow C. (2007) Impaired insulin secretion in a mouse model of ataxia telangiectasia. Am. J. Physiol. Endocrinol. Metab. 293, E70–E74 [DOI] [PubMed] [Google Scholar]
- 22. Lee Y. S., Chanda D., Sim J., Park Y. Y., Choi H. S. (2007) Structure and function of the atypical orphan nuclear receptor small heterodimer partner. Int. Rev. Cytol. 261, 117–158 [DOI] [PubMed] [Google Scholar]
- 23. Chanda D., Park J. H., Choi H. S. (2008) Molecular basis of endocrine regulation by orphan nuclear receptor small heterodimer partner. Endocr. J. 55, 253–268 [DOI] [PubMed] [Google Scholar]
- 24. Chanda D., Kim S. J., Lee I. K., Shong M., Choi H. S. (2008) Sodium arsenite induces orphan nuclear receptor SHP gene expression via AMP-activated protein kinase to inhibit gluconeogenic enzyme gene expression. Am. J. Physiol. Endocrinol. Metab. 295, E368–E379 [DOI] [PubMed] [Google Scholar]
- 25. Chanda D., Li T., Song K. H., Kim Y. H., Sim J., Lee C. H., Chiang J. Y., Choi H. S. (2009) Hepatocyte growth factor family negatively regulates hepatic gluconeogenesis via induction of orphan nuclear receptor small heterodimer partner in primary hepatocytes. J. Biol. Chem. 284, 28510–28521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Onishi M., Nosaka T., Misawa K., Mui A. L., Gorman D., McMahon M., Miyajima A., Kitamura T. (1998) Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol. Cell Biol. 18, 3871–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barclay J. L., Nelson C. N., Ishikawa M., Murray L. A., Kerr L. M., McPhee T. R., Powell E. E., Waters M. J. (2011) GH-dependent STAT5 signaling plays an important role in hepatic lipid metabolism. Enocrinology 152, 181–192 [DOI] [PubMed] [Google Scholar]
- 28. Kim J. Y., Kim H. J., Kim K. T., Park Y. Y., Seong H. A., Park K. C., Lee I. K., Ha H., Shong M., Park S. C., Choi H. S. (2004) Orphan nuclear receptor small heterodimer partner represses hepatocyte nuclear factor 3/Foxa transactivation via inhibition of its DNA binding. Mol. Endocrinol. 18, 2880–2894 [DOI] [PubMed] [Google Scholar]
- 29. Schalch D. S., McFarlin D. E., Barlow M. H. (1970) An unusual form of diabetes mellitus in ataxia telangiectasia. N. Engl. J. Med. 282, 1396–1402 [DOI] [PubMed] [Google Scholar]
- 30. GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group, Wellcome Trust Case Control Consortium 2, Zhou K., Bellenguez C., Spencer C. C., Bennett A. J., Coleman R. L., Tavendale R., Hawley S. A., Donnelly L. A., Schofield C., Groves C. J., Burch L., Carr F., Strange A., Freeman C., Blackwell J. M., Bramon E., Brown M. A., Casas J. P., Corvin A., Craddock N., Deloukas P., Dronov S., Duncanson A., Edkins S., Gray E., Hunt S., Jankowski J., Langford C., Markus H. S., Mathew C. G., Plomin R., Rautanen A., Sawcer S. J., Samani N. J., Trembath R., Viswanathan A. C., Wood N. W., Harries L. W., Hattersley A. T., Doney A. S., Colhoun H., Morris A. D., Sutherland C., Hardie D. G., Peltonen L., McCarthy M. I., Holman R. R., Palmer C. N., Donnelly P., Pearson E. R. (2011) Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat. Genet. 43, 117–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Park Y. J., Kim S. C., Kim J., Anakk S., Lee J. M., Tseng H. T., Yechoor V., Park J., Choi J. S., Jang H. C., Lee K. U., Novak C. M., Moore D. D., Lee Y. K. (2011) Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J. Lipid Res. 52, 2234–2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ghanaat F., Tayek J. A. (2005) Growth hormone administration increases glucose production by preventing the expected decrease in glycogenolysis seen with fasting in healthy volunteers. Metabolism 54, 604–609 [DOI] [PubMed] [Google Scholar]
- 33. Cho Y., Ariga M., Uchijima Y., Kimura K., Rho J. Y., Furuhata Y., Hakuno F., Yamanouchi K., Nishihara M., Takahashi S. (2006) The novel roles of liver for compensation of insulin resistance in human growth hormone transgenic rats. Endocrinology 147, 5374–5384 [DOI] [PubMed] [Google Scholar]
- 34. Lahuna O., Rastegar M., Maiter D., Thissen J. P., Lemaigre F. P., Rousseau G. G. (2000) Involvement of STAT5 (signal transducer and activator of transcription 5) and HNF-4 (hepatocyte nuclear factor 4) in the transcriptional control of the hnf6 gene by growth hormone. Mol. Endocrinol. 14, 285–294 [DOI] [PubMed] [Google Scholar]
- 35. Lee Y. S., Kim D. K., Kim Y. D., Park K. C., Shong M., Seong H. A., Ha H. J., Choi H. S. (2008) Orphan nuclear receptor SHP interacts with and represses hepatocyte nuclear factor-6 (HNF-6) transactivation. Biochem. J. 413, 559–569 [DOI] [PubMed] [Google Scholar]
- 36. Schmoll D., Grempler R., Barthel A., Joost H. G., Walther R. (2001) Phorbol ester-induced activation of mitogen-activated protein kinase/extracellular-signal-regulated kinase kinase and extracellular-signal-regulated protein kinase decreases glucose-6-phosphatase gene expression. Biochem. J. 357, 867–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Massillon D. (2001) Regulation of the glucose-6-phosphatase gene by glucose occurs by transcriptional and post-transcriptional mechanisms. Differential effect of glucose and xylitol. J. Biol. Chem. 276, 4055–4062 [DOI] [PubMed] [Google Scholar]
- 38. Hogan J. C., Stephens J. M. (2005) The regulation of fatty acid synthase by STAT5A. Diabetes 54, 1968–1975 [DOI] [PubMed] [Google Scholar]
- 39. White U. A., Coulter A. A., Miles T. K., Stephens J. M. (2007) The STAT5A-mediated induction of pyruvate dehydrogenase kinase 4 expression by prolactin or growth hormone in adipocytes. Diabetes 56, 1623–1629 [DOI] [PubMed] [Google Scholar]