

Background: The process of matrix degradation is an integral component of cell invasion.
Results: Actopaxin phosphorylation is required for matrix degradation.
Conclusion: Actopaxin can act upstream of Rho GTPase signaling via an interaction with β-PIX and PAK to control matrix degradation.
Significance: Matrix degradation is a pivotal step in cell invasion and cancer cell metastasis.
Keywords: Cell Adhesion, Cell Invasion, Extracellular Matrix, Matrix Metalloproteinase (MMP), Rho GTPases, Actopaxin, Invadopodia, Matrix Degradation, PAK, β-PIX
Abstract
Dysregulation of cell adhesion and motility is known to be an important factor in the development of tumor malignancy. Actopaxin (α-parvin) is a paxillin, integrin-linked kinase, and F-actin binding focal adhesion protein with several serine phosphorylation sites in the amino terminus that contribute to the regulation of cell spreading and migration. Here, phosphorylation of actopaxin is shown to contribute to the regulation of matrix degradation and cell invasion. Osteosarcoma cells stably expressing wild type (WT), nonphosphorylatable (Quint), and phosphomimetic (S4D/S8D) actopaxin demonstrate that actopaxin phosphorylation is necessary for efficient Src and matrix metalloproteinase-driven degradation of extracellular matrix. Rac1 was found to be required for actopaxin-induced matrix degradation whereas inhibition of myosin contractility promoted degradation in the phosphomutant-expressing Quint cells, indicating that a balance of Rho GTPase signaling and regulation of cellular tension are important for the process. Furthermore, actopaxin forms a complex with the Rac1/Cdc42 GEF β-PIX and Rac1/Cdc42 effector PAK1, to regulate actopaxin-dependent matrix degradation. Actopaxin phosphorylation is elevated in the invasive breast cancer cell line MDA-MB-231 compared with normal breast epithelial MCF10A cells. Expression of the nonphosphorylatable Quint actopaxin in MDA-MB-231 cells inhibits cell invasion whereas overexpression of WT actopaxin promotes invasion in MCF10A cells. Taken together, this study demonstrates a new role for actopaxin phosphorylation in matrix degradation and cell invasion via regulation of Rho GTPase signaling.
Introduction
The mechanism controlling tumor dissemination and metastasis, the major cause of cancer-related fatalities, is poorly understood, although the dysregulation of cell adhesion and motility is known to be an important factor in tumor progression. Interactions between cells and the extracellular matrix (ECM)2 occur via organized assemblies of structural and signaling proteins known as focal complexes and focal adhesions. Focal adhesion proteins serve as a structural and signaling nexus connecting integrins to the dynamic actin cytoskeleton to coordinate cell spreading, motility, and cell invasion (1).
We have previously described actopaxin (α-parvin, CH-ILKBP) (2, 3) as a paxillin- and F-actin-binding protein that localizes to focal adhesions and focal complexes via its interaction with paxillin (4, 5). Disruption of the actopaxin-paxillin interaction inhibits cell adhesion and spreading (4, 5). Additionally, actopaxin forms a complex with the integrin-linked kinase (ILK) and PINCH, commonly referred to as the IPP complex (ILK-Parvin-PINCH), which also links integrin signaling to the actin cytoskeleton (6). The amino terminus of actopaxin contains a series of serine residues, that when phosphorylated in an ERK-dependent manner during cell adhesion can regulate cell spreading and migration (7). Actopaxin is also phosphorylated on these sites during mitosis by cyclin B1/cdc2 kinase to facilitate G2/M transition (8).
The Rho family of GTPases, Cdc42, Rac1, and RhoA, and their effectors are responsible for regulating the actin cytoskeleton during cell spreading, adhesion turnover, migration, and invasion. A large family of guanine nucleotide exchange factors (GEFs) act to positively regulate these GTPases. β-PIX/Cool 1 (PAK-interacting exchange factor/cloned out of library, herein referred to as β-PIX), is a GEF for Rac1 and Cdc42 (9). Knockdown of β-PIX, or expression of a GEF-defective mutant inhibits cell migration by reducing adhesion turnover (10). β-Parvin/affixin, a closely related actopaxin family member, has been reported to bind directly to β-PIX to regulate Rac1 activity (11, 12). Using phosphomimetic and nonphosphorylatable mutants of actopaxin, we have previously determined that actopaxin phosphorylation regulates cell migration in part through the regulation of Rac1 activity (7). Additionally, actopaxin binds CdGAP, a negative regulator of Rac1/Cdc42 activity, and this interaction with actopaxin contributes to CdGAP recruitment to focal adhesions to negatively regulate cell spreading and adhesion turnover (13). Together these data suggest a bimodal role for actopaxin in the regulation of Rho GTPase activity to coordinate cytoskeletal organization and cell migration through the binding and regulation of both GEFs and GAPs.
In addition to the regulation of cell spreading and motility, Rho GTPase signaling has also been shown to play an important role in other aspects of tumor cell invasion including the regulation of matrix degradation associated with the formation of specialized adhesions termed invadopodia that degrade the ECM via elevated matrix metalloproteinase (MMP) activity (14). Herein, we describe a novel function for actopaxin phosphorylation in regulating matrix degradation and invasion. Actopaxin-induced matrix degradation was found to be dependent on Src and MMP activity and also required Rac1 activity. Actopaxin was found to interact with β-PIX in a complex with the p21-activated serine/threonine kinase (PAK1), providing a possible mechanism for actopaxin regulation of Rac1 signaling. In addition, PAK and p38 MAPK, both downstream targets of Rac1, were required for matrix degradation. Actopaxin phosphorylation was elevated in invasive versus normal breast epithelial cells and the introduction of nonphosphorylatable actopaxin into invasive cells inhibited cell invasion, suggesting that actopaxin signaling through its phosphorylation state plays an essential role in the development of tumor cell malignancy.
MATERIALS AND METHODS
Cell Culture and Transfection
U2OS osteosarcoma cells expressing Xpress-tagged wild-type (WT), nonphosphorylatable (S(4,8,14,19)G/T16A) mutant referred to herein as the Quint mutant and phosphomimetic S4D/S8D actopaxin were previously described (7). U2OS (ATCC) and MDA-MB-231 (ATCC) cells were cultured in DMEM with 10% FBS, 1 mm glutamine, 50 units/ml penicillin and 50 μg/ml streptomycin. MCF10A (ATCC) cells were cultured in 50:50 DMEM/Ham's F12 with 5% horse serum, 15 mm HEPES, pH 7.5, 2 mm l-glutamine, 0.5 μg/ml hydrocortisone, 10 μg/ml insulin, 0.02 μg/ml EGF, 50 units/ml penicillin, and 50 μg/ml streptomycin. Cells were maintained at 37 °C in a humidified chamber with 5% CO2. Transfection of U2OS cells was performed with FuGENE 6 (Roche Molecular Biochemicals) according to the manufacturer's instructions. β-PIX and PAK1 constructs were kindly provided by Richard Cerione (Cornell University). Retroviral pLNCX2 Xpress-tagged actopaxin WT and Quint were produced, and MCF10A and MDA-MB-231 cells were infected as previously described (15).
Indirect Immunofluorescence
Cells were fixed and permeabilized in 4% paraformaldehyde/1% Triton-X-100 in PBS, quenched in 0.1 m glycine, and blocked in 3% BSA before staining. Primary antibodies were used at 1:250 for 90 min at 37 °C: Xpress (Invitrogen) and paxillin clone 165 (BD Biosciences). Rhodamine phalloidin (1:1000, Invitrogen) was used to visualize F-actin. Secondary antibodies (Jackson Immunoresearch Laboratories) were used at 1:250 for 1 h at 37 °C. Images were acquired on a Nikon Eclipse TE2000 inverted microscope with a Spot camera using Spot Advance software. Image analysis was performed using National Institutes of Health ImageJ software.
Gelatin Matrix Degradation Assay
Fluorescent 488-gelatin coverslips were prepared as described previously (16). Briefly, glass coverslips were washed overnight in 20% sulfuric acid and then sterilized in ethanol. Coverslips were coated with 50 μg/ml poly-l-lysine (Sigma), washed in PBS, and then incubated in 0.5% glutaraldehyde (Sigma) and coated for 30 min with 488-gelatin (Invitrogen) 1:40 with 0.2% unlabeled gelatin solution (w/v; Sigma) at 37 °C. Cells were plated for 16 h in serum-containing medium, and coverslips were processed as above. Inhibitors were used as follows: 2 μm Src inhibitor PP2, 25 μm MMP inhibitor GM6001, 50 μm Rac1 inhibitor NSC23766, 25 μm myosin inhibitor blebbistatin, 10 μm p38 MAPK inhibitor SB203580 (Calbiochem), and 1 μg/ml Rho activator II (CN03; Cytoskeleton).
Gelatin Zymography
Conditioned medium samples were collected from cells plated overnight on collagen in serum-free medium. Samples were mixed with 2× SDS sample buffer without reducing reagent and run on a 7.5% SDS-PAGE gel co-polymerized with 0.1% gelatin type A (Sigma). Gels were incubated in reaction buffer (50 mm Tris, pH 7.6, 150 mm NaCl, 5 mm CaCl2, 0.05% sodium azide) for 40 h and then fixed with methanol and stained with Coomassie Blue.
Matrigel Invasion Assay
For invasion assays, cells were suspended in serum-free medium and plated in the top well of Matrigel-coated invasion chambers (8-μm pore size; BD BioCoatTM; BD Transduction Laboratories). Serum-containing medium (5% horse serum) was placed in the lower chamber, and cells were allowed to invade for 20 h at 37 °C. Cells on the upper chamber were removed with a cotton swab, and cells on the lower chamber were fixed in 100% methanol and stained with Giemsa. The number of cells invaded was counted for 20 random fields per condition/experiment using a 20× objective.
Immunoprecipitation
Cells were lysed in a co-immunoprecipitation lysis buffer (20 mm Tris, pH 7.6, 0.5% Nonidet P-40, 100 mm NaCl, 10% glycerol, 1 mm MgCl2, 10 μg/ml leupeptin, 2 mm NaF, 200 mm NaVO4). Clarified lystates were incubated end over end with antibodies for 2 h at 4 °C followed by 1 h with protein A/G beads. Beads were washed rapidly with lysis buffer then boiled in 2× SDS sample buffer.
Western Blotting
Cell lysates were resolved on 10% SDS-polyacrylamide gels. Proteins were transferred to nitrocellulose membranes. Primary antibody incubations were done for 2 h at room temperature at 1:1000 dilution: Xpress, actopaxin (Sigma), actopaxin clone 38 (5), phospho-PAK (Ser199/204), phospho-p38 MAPK (Cell Signaling), PAK (Santa Cruz Biotechnology), β-PIX (Millipore), α-actinin (Sigma), 9E10 anti-Myc (Developmental Studies Hybridoma Bank). Secondary HRP-conjugated antibodies (Jackson Immunoresearch Laboratories) were diluted 1:10,000 and incubated for 1 h at room temperature. Immunoblots were developed by chemiluminescence using ECL (GE Healthcare).
Statistical Analysis
Values calculated from at least three independent experiments were compared by Student's t test and p < 0.05 was considered statistically different. Error bars represent the S.E.
RESULTS
Actopaxin Phosphorylation Is Required for Matrix Degradation
Actopaxin phosphorylation of Ser4/8 enhances cell migration whereas a nonphosphorylatable (Quint) mutant suppresses migration in U2OS osteosarcoma cells (7). Invasive tumor cells also develop the ability to degrade ECM to facilitate migration through tissue stroma which can be visualized in vitro by plating cells on fluorescently labeled ECM and monitoring the increase in nonfluorescent areas corresponding to degraded matrix. Interestingly, parental U2OS cell plated on fluorescent 488-gelatin exhibit only modest matrix degradation whereas cells expressing either wild-type (WT) and phosphomimetic (S4D/S8D) actopaxin demonstrate increased matrix degradation (Fig. 1A). The pattern of matrix degradation was both punctate and focal adhesion-like (Fig. 1A, insets). In striking contrast, the Quint actopaxin-expressing cells had diminished ability to degrade matrix (Fig. 1A). Expression levels of the different actopaxin mutants are shown by Western blotting (Fig. 1B). Quantitation of both the percent of cells associated with matrix degradation (Fig. 1C) and the area of matrix degradation/cell area (Fig. 1D) demonstrated that the WT and S4D/S8D actopaxin cells had a significant enhancement in matrix degradation whereas the Quint actopaxin cells significantly inhibited matrix degradation compared with parental cells. Interestingly, overexpression of WT actopaxin induced higher levels of matrix degradation than S4D/S8D actopaxin, suggesting that cycling of phosphorylation/dephosphorylation might be necessary to achieve optimal matrix degradation. These data demonstrate a novel role for actopaxin phosphorylation in the regulation of matrix degradation.
FIGURE 1.

Actopaxin phosphorylation promotes and is necessary for matrix degradation in U2OS cells. A, U2OS parental cells and cells stably expressing Xpress-actopaxin WT, Quint, and S4D/S8D were plated on fluorescent 488-gelatin for 16 h. Expression of WT and S4D/S8D actopaxin promotes degradation of underlying matrix, whereas Quint expression inhibits matrix degradation. B, Western blot shows expression levels of Xpress-tagged WT, Quint, and S4D/S8D actopaxin in U2OS cells. C, quantitation of the percentage of cells associated with matrix degradation demonstrates a significant increase in WT and S4D/S8D-expressing cells whereas Quint cells show a significant decrease compared with parental cells. D, quantitation of the area of matrix degraded/total cell area demonstrates a similar trend to the percent of cells showing matrix degradation. Error bars, S.E.
A role for Src kinase activity in matrix degradation, invadopodia formation, and cell invasion is well established (17). Western blot analysis revealed that the WT and S4D/S8D actopaxin cells have increased levels of phospho-Src Y418 compared with parental and Quint actopaxin-expressing cells (Fig. 2A), consistent with their elevated matrix-degrading capacity (18, 19). Accordingly, treatment of the WT and S4D/S8D actopaxin cells with the Src inhibitor PP2 or the broad MMP inhibitor GM6001 abolished matrix degradation (Fig. 2, B and C), indicating that actopaxin regulates matrix degradation via regulation of Src and MMP activity. In addition, evaluation of MMP activity by in-gel zymography revealed that Quint actopaxin cells produced significantly less secreted MMP2 and MMP9 (Fig. 2, D and E), in accordance with their decreased ability to degrade matrix. Interestingly, the parental U2OS cells secrete MMP9 at levels similar to the WT and S4D/S8D actopaxin cells, but lack MMP2 secretion. This suggests that actopaxin overexpression/phosphorylation is specifically driving increased matrix degradation via elevated secretion and/or activation of MMP2.
FIGURE 2.

Actopaxin-dependent matrix degradation requires Src and MMP activity. A,Western blot analysis shows that levels of phospho-Src Y418 are elevated in cells expressing WT and S4D/S8D actopaxin compared with parental and Quint cells. B, plating WT and S4D/S8D cells in the presence of the Src and MMP inhibitors PP2 and GM6002, respectively, results in the inhibition of matrix degradation. Scale bar, 20 μm. C, quantitation of percentage of cells degrading matrix shows that Src or MMP inhibition results in a highly significant decrease in matrix degradation of WT and S4D/S8D cells. D, gelatin zymography of U2OS actopaxin cells shows the levels of secreted MMP2 and MMP9. E, gelatin zymography is quantified. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Error bars, S.E.
Rac1 and Myosin Contractility Regulate Matrix Degradation
The regulation of matrix degradation involves the formation of invadopodia that have been shown to require a fine balance of signaling between Rho GTPases (17). We have shown previously that expression of S4D/S8D actopaxin results in a Rac1-like phenotype including enhanced lamellipodia formation (supplemental Fig. 1) that can be reversed with the expression of dominant negative Asn17 Rac1 (7). Conversely, Quint actopaxin-expressing cells have a more angular phenotype (supplemental Fig. 1), indicative of high Rho activity and actomyosin contractility.
To address whether phosphorylated actopaxin-driven matrix degradation is dependent on elevated Rac1 activity and/or decreased actomyosin contractility, cells were plated in the presence of the Rac1 inhibitor NSC23766 (20, 21) or the myosin II inhibitor blebbistatin (22) (Fig. 3, A and B). Inhibition of Rac1 greatly reduced the ability of the WT and S4D/S8D actopaxin cells to degrade matrix. In addition, the WT and S4D/S8D actopaxin cells that were treated with the Rac1 inhibitor were smaller in area and more angular, indicative of increased cell contractility and similar to the morphology of the Quint actopaxin-expressing cells. Interestingly, inhibition of myosin activity with blebbistatin was sufficient to induce matrix degradation in the Quint actopaxin-expressing cells. Similar results were observed following treatment of the Quint actopaxin cells with ML-7, a myosin light chain kinase inhibitor (data not shown). Conversely, treatment of the WT actopaxin cells with the Rho activator II inhibited matrix degradation and resulted in a contractile phenotype similar to Quint actopaxin cells (Fig. 3, C and D), Together, these data support previous studies (14, 23) indicating that a tight balance of Rho GTPases signaling and associated cell contractility is required for optimal matrix-degrading capabilities and place actopaxin as a central mediator of these processes.
FIGURE 3.

Rac1 activity and myosin contractility regulate matrix degradation. A, plating WT and S4/8D actopaxin cells in the presence of the Rac1 inhibitor NSC23766 blocks matrix degradation and results in morphological changes similar to that seen in Quint actopaxin-expressing cells. Quint cells plated in the presence of the myosin II inhibitor blebbistatin have increased matrix degradation. B, quantitation of matrix degradation shows a highly significant decrease in WT and S4D/S8D actopaxin-mediated matrix degradation in the presence of the Rac1 inhibitor NCS23766 and a highly significant increase in matrix degradation by Quint cells in the presence of blebbistatin. C, treatment of WT actopaxin cells with Rho activator II inhibits matrix degradation. D, quantitation of matrix degradation shows a highly significant decrease in degradation associated with WT cells treated with Rho activator. Scale bars, 20 μm. Error bars, S.E. *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
Actopaxin Interacts with β-PIX and PAK1, and β-PIX and PAK1 Activity Are Required for Matrix Degradation
β-Parvin can interact directly with the Rac/Cdc42 GEF β-PIX via its CH1 (calponin homology) domain to control Rac1 activity (12). Given the high degree of homology between parvin family members (24), we reasoned that actopaxin may also bind β-PIX in a similar fashion. Accordingly, immunoprecipitation of endogenous actopaxin was found to co-immunoprecipitate β-PIX (Fig. 4A), also via a direct interaction (data not shown). Xpress-tagged WT, Quint, and S4D/S8D actopaxin all interacted equally with β-PIX (Fig. 4A) and co-localized with β-PIX at focal adhesions (supplemental Fig. 2). PAK, a downstream effector of Rac1, binds directly to β-PIX (25), regulating β-PIX activity, Rac1 binding, and PAK signaling to the cytoskeleton (10). PAK1 was also observed to precipitate with WT, Quint, and S4D/S8D actopaxin (Fig. 4A). PAK1 can be recruited to cell and ECM adhesions as part of a paxillin-PKL-β-PIX-PAK complex (26). However, PKL (paxillin kinase linker/GIT2) was not detected in the Xpress-actopaxin immunoprecipitation (Fig. 4A), indicating that actopaxin-β-PIX-PAK form a complex that is independent of the paxillin-PKL-β-PIX-PAK complex. Therefore, actopaxin may provide an alternative mechanism for recruiting β-PIX/PAK to adhesions.
FIGURE 4.

Actopaxin forms a complex with β-PIX and PAK, which are important for actopaxin-mediated matrix degradation. A, endogenous β-PIX co-immunoprecipitated with actopaxin. Xpress-actopaxin co-immunoprecipitates β-PIX and PAK1, but not PKL. B, expression of β-PIX mutants affects matrix degradation. GEF-dead (LL) β-PIX significantly inhibits matrix degradation in WT actopaxin cells, whereas a GEF active (ΔT1) β-PIX mutant promotes matrix degradation in Quint actopaxin-expressing cells. C, GEF-dead (LL) and PAK-binding (WW) mutants of β-PIX inhibit matrix degradation whereas GEF active β-PIX (ΔT1) promotes matrix degradation. The percentage of cells associated with matrix degradation is quantified. Scale bar, 20 μm. *, p < 0.05; **, p < 0.005. Error bars, S.E.
To evaluate whether the GEF activity of β-PIX is required for actopaxin-induced matrix degradation, Myc-tagged WT, GEF-dead L238S/L239S (LL), and active GEF (ΔT1) β-PIX mutants were co-transfected into the WT or Quint actopaxin cells along with Cherry-paxillin, to identify transfected cells (Fig. 4B). The expression of β-PIX WT did not affect matrix degradation in these cells. In contrast, transfection of the WT actopaxin cells with GEF-dead β-PIX LL mutant inhibited matrix degradation, whereas expression of the GEF active β-PIX ΔT1 in the Quint actopaxin cells, which increases Rac1 activity and PAK phosphorylation (10), resulted in a significant increase in matrix degradation (Fig. 4, B and C). In contrast, the expression of β-PIX WW (W43P/W44G), a PAK-binding mutant that blocks PAK activation (9), significantly inhibited matrix degradation in the WT actopaxin cells (Fig. 4C).
PAK1 functions in regulating cytoskeletal organization and cell motility (27). In addition, the expression of active PAK1 can induce matrix degradation and invadopodia formation in smooth muscle cells (28). Furthermore, cells transfected with the autoinhibitory domain of PAK1, which inhibits endogenous PAK1, significantly decreases matrix degradation of melanoma cells (29). Western blot analysis demonstrated that the serine phosphorylation of PAK (Ser199/204) is reduced in the Quint actopaxin-expressing cells compared with the WT and S4D/S8D actopaxin cells (Fig. 5A), indicating less active PAK in Quint actopaxin cells. Introduction of a PAK1 kinase-dead mutant (K299R) (30) into WT actopaxin cells caused a significant reduction of cells degrading matrix, whereas expression of a PAK1 active mutant (T423E) (30) in Quint actopaxin cells resulted in increased ability to degrade matrix (Fig. 5, B and C). Taken together, these data indicate that actopaxin phosphorylation promotes Rac1-dependent matrix degradation through the formation of an active actopaxin-β-PIX-PAK signaling complex.
FIGURE 5.

PAK kinase activity promotes matrix degradation. A, Western blot analysis reveals that the level of PAK phosphorylation is reduced in Quint actopaxin-expressing cells compared with WT and S4D/S8D actopaxin-expressing cells. B, introduction of a PAK1 kinase-dead mutant (K299R) into WT actopaxin cells significantly inhibits matrix degradation, whereas a PAK1 kinase-active mutant (T423E) promotes matrix degradation in Quint cells. C, percent of cells associated with matrix degradation was quantified. Scale bar, 20 μm. ***, p < 0.0005. Error bars, S.E.
p38 MAPK Is Required for Matrix Degradation
Enhanced matrix degradation and cell invasion are frequently associated with an increase in the activity of p38 MAPK (31, 32). p38 MAPK can be activated in a β-PIX/PAK-dependent manner in response to Rac1 signaling and is necessary for cell motility (33). Therefore, we sought to determine whether p38 MAPK is required for actopaxin-induced matrix degradation.
Western blot analysis using an activation-specific phosphoantibody for p38 MAPK showed that phospho-p38 MAPK was reduced in Quint actopaxin cells compared with WT and S4D/S8D actopaxin cells (Fig. 6A). To examine the role p38 MAPK activity in actopaxin-induced matrix degradation, WT, Quint, and S4D/S8D actopaxin-expressing cells were plated in the presence of the p38 MAPK inhibitor SB203580 (Fig. 6, B and C). p38 MAPK inhibition abolished matrix degradation in WT and S4D/S8D actopaxin cells. p38 MAPK has been shown to be regulated in a Rac1-dependent manner (34); therefore, we evaluated whether treatment with the Rac1 inhibitor also reduced p38 MAPK phosphorylation. Treatment of WT actopaxin cells with the Rac1 inhibitor NCS23766 decreased p38 MAPK phosphorylation (Fig. 6D). These data suggest that p38 MAPK is required for matrix degradation and that actopaxin phosphorylation can regulate p38 MAPK activity in a Rac1-dependent manner.
FIGURE 6.

p38 MAPK activity is required for actopaxin-mediated matrix degradation. A, Western blot analysis of WT, Quint, and S4D/S8D actopaxin cells demonstrates that phospho-p38 MAPK is elevated in WT and S4D/S8D actopaxin-expressing cells compared with Quint actopaxin cells. B, WT and S4D,S8D actopaxin cells plated in the presence of the p38 MAPK inhibitor SB203580 show significantly reduced matrix degradation. C, percent of cells displaying matrix degradation in the presence of p38 MAPK inhibitor was quantified. D, WT actopaxin cells plated in the presence of Rac1 inhibitor demonstrate reduced levels of p38 MAPK phosphorylation. Scale bar, 20 μm. *, p < 0.05; **, p < 0.005; ***, p < 0.0005. Error bars, S.E.
Actopaxin Phosphorylation Regulates Cell Invasion
A comparison of normal MCF10A human breast epithelial cells with the highly invasive human breast cancer cell line MDA-MB-231 by Western blot analysis revealed that actopaxin Ser8 phosphorylation is elevated in the MDA-MB-231 cells (Fig. 7A). To determine whether actopaxin phosphorylation contributes to cell invasion, MDA-MB-231 or MCF10A cells were transfected with WT and Quint actopaxin (Fig. 7B). Both actopaxin constructs localize efficiently to focal adhesions in these cells (supplemental Fig. 3) as reported previously for the U2OS cells (7). Parental MCF10A cells do not exhibit significant matrix degradation as measured by the fluorescent 488-gelatin assay. In contrast, overexpression of WT actopaxin in these cells induced matrix degradation whereas Quint actopaxin-expressing cells failed to degrade matrix (Fig. 7C) as observed in the U2OS cells (Fig. 1). MCF10A cells expressing WT actopaxin formed classical invadopodia, comprising a core of actin that co-localized with areas of matrix degradation and surrounded by a ring of adhesion proteins including WT actopaxin (Fig. 7D). This distribution is consistent with localization of endogenous actopaxin to invadopodia formed in parental MCF10A cells following TGF-β-induced epithelial mesenchymal transition (32). In addition, overexpression of WT actopaxin in the normally, noninvasive MCF10A cells resulted in a significant increase in invasion compared with parental and Quint actopaxin-expressing cells (Fig. 7E). Conversely, invasion of the MDA-MB-231 breast cancer cells through Matrigel was significantly decreased in populations expressing the Quint actopaxin compared with parental and WT actopaxin-expressing cells (Fig. 7F). Treatment of the Quint actopaxin MDA-MB-231 cells with the myosin inhibitor blebbistatin was able to restore cell invasion to parental levels (Fig. 7G), similar to the rescue of matrix degradation observed in the Quint actopaxin-expressing U2OS cells following myosin inhibition (Fig. 3). These data suggest that the elevated actopaxin phosphorylation observed in MDA-MB-231 compared with normal breast epithelial cells is contributing directly to the invasive nature of these cells, most likely through promoting both increased matrix degrading activity as well as enhanced motility (7).
FIGURE 7.

Actopaxin phosphorylation is required for invasion through Matrigel of breast tumor cells. A, Western blot analysis demonstrates that endogenous actopaxin pS8 phosphorylation is increased in the invasive breast cancer cell line MDA-MB-231 compared with the normal breast epithelial cell line, MCF10A. B, Western blot shows expression of Xpress-actopaxin WT and Quint constructs in MDA-MB-231 and MCF10A cells. C, WT actopaxin overexpression in MCF10A cells results in increased matrix degradation. Scale bar, 20 μm. D, WT actopaxin localizes to invadopodia in MCF10A cells. Scale bar, 10 μm. E, expression of WT actopaxin in MCF10A cells promotes invasion through Matrigel. F, overexpression of nonphosphorylatable Quint actopaxin significantly inhibits invasion of MDA-MB-231 cells through Matrigel. G, Quint actopaxin MDA-MB-231 cells treated with blebbistatin exhibit increased invasion. *, p < 0.05; **, p < 0.005. Error bars, S.E.
DISCUSSION
In this study we describe a novel role for actopaxin phosphorylation in the regulation of matrix degradation in U2OS osteosarcoma cells. In addition, a novel interaction between actopaxin and β-PIX is described that acts to regulate downstream signaling via Rac1 to PAK1 and p38 MAPK to promote the matrix degradation (Fig. 8). We further show that actopaxin phosphorylation is elevated in breast tumor cells to regulate cell invasion, suggesting that actopaxin phosphorylation may be a key event in the development of an invasive phenotype in cancer cells.
FIGURE 8.
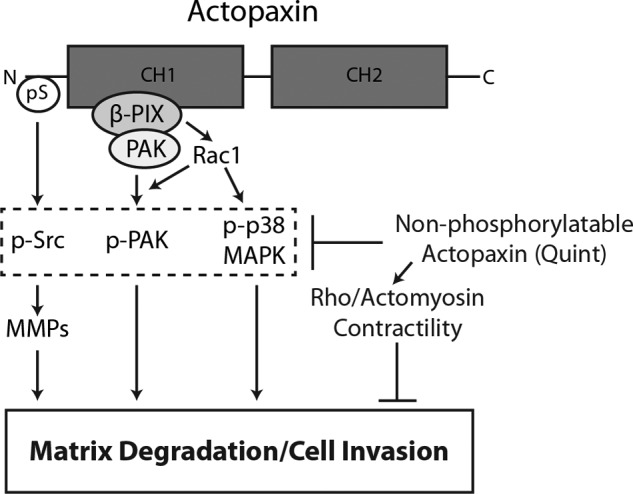
Schematic of phosphoactopaxin-driven matrix degradation and tumor cell invasion. Phosphorylation of the actopaxin amino terminus (pS) or overexpression of wild-type, or the phosphomimetic S4D/S8D actopaxin (data not shown) results in activation of Src (p-Src), elevated MMP secretion and increased matrix degradation. Actopaxin interacts constitutively with the Rac1 GEF, β-PIX in a complex with the Rac1 effector, PAK. Actopaxin phosphorylation promotes PAK activation (p-PAK) in a Rac1/β-PIX-dependent manner, and each is required for actopaxin-mediated matrix degradation. p38 MAPK is also phosphorylated in a Rac1-dependent manner and is necessary for actopaxin-induced matrix degradation. The expression of nonphosphorylatable actopaxin (Quint) mutant results in suppression of phosphorylation of Src, PAK, and p38, leading to decreased MMP secretion, matrix degradation, and tumor cell invasion. This inhibition is due, in part, to enhanced Rho/actomyosin-mediated contractility in the Quint actopaxin-expressing cells. CH, calponin homology domain.
Overexpression of either WT or the phosphomimetic S4D/S8D actopaxin enhanced the ability of U2OS cells to degrade matrix whereas nonphosphorylatable Quint actopaxin abolished matrix degradation. Src and MMP activity were required for matrix degradation to occur, consistent with invadopodia-mediated matrix degradation (17, 35). Invadopodia are highly organized invasive structures, first observed in v-Src-transformed cells, which comprise an actin core surrounded by a ring of adhesion-related proteins (18). However, prototypical invadopodia were not clearly observed in the U2OS cells. Instead, the pattern of matrix degradation was a combination of punctate and focal adhesion-like degradation. A similar type of matrix degradation was recently reported in human fibrosarcoma cells (HT-1080) where a FAK-p130Cas complex was shown to regulate matrix degradation at focal adhesions through binding to MT1-MMP (36). The relatively small percentage of focal adhesions that we observed co-localizing with the sites of matrix degradation in the U2OS cells is most likely due to the highly dynamic nature of these cells. Interestingly, in the MCF10A cells expressing WT actopaxin we observed the formation of more typical invadopodia structures containing the WT actopaxin in their ring structure and a core of actin that coincided with the matrix degradation. This is consistent with our previous study showing the localization of endogenous actopaxin to invadopodia formed in these cells in response to TGF-β-induced epithelial mesenchymal transition (32), suggesting that the mode of degradation is cell type- and possibly context-specific.
Expression of WT and S4D/S8D actopaxin resulted in increased Src activity that was critical for promoting matrix degradation in the U2OS cells. Src kinase phosphorylates many invadopodia components such as cortactin (37) and Tks5 (38) to regulate invadopodia formation and matrix degradation. Interestingly, Src can phosphorylate membrane-associated MT1-MMP (39) and was also shown to be required for the formation of the FAK-p130Cas-MT1-MMP complex (36). Taken together, these data suggest that actopaxin phosphorylation may regulate the focal adhesion pattern of degradation observed in the U2OS cells by promoting the formation of this complex, as well as stimulating the increased secretion of soluble MMPs, in particular MMP2, through increasing Src activity (36).
Expression of the S4D/S8D phosphomimetic mutant of actopaxin produced a highly protrusive phenotype, and phosphoactopaxin-mediated cell spreading, motility, and matrix degradation are all dependent on elevated Rac1 activity (7). Conversely, cells expressing the nonphosphorylatable Quint actopaxin are angular and more contractile, indicative of elevated RhoA and actomyosin activity. In accordance with this phenotype, vascular smooth muscle cells derived from actopaxin-null mice also display a hypercontractile phenotype as a result of increased RhoA activity and elevated myosin light chain phosphorylation (40). A similar phenotype is observed following knockdown of ILK in the same cells (41) or as a result of perturbing the ILK-actopaxin interaction in epithelial cells (42). Thus, a failure to phosphorylate actopaxin phenocopies the actopaxin ablation, suggesting an important role for actopaxin phosphorylation in regulating the ILK-actopaxin signaling axis.
A critical balance of signaling between Rho GTPase family members is necessary for invadopodia formation and matrix degradation, with both Rac1 and RhoA being required (14, 17, 18). Any shift in this balance can result in inhibited formation or maturation of invadopodia (35). Thus, Quint actopaxin cells may inhibit invadopodia formation and matrix degradation through increased actomyosin contractility. Indeed, addition of myosin inhibitors rescued matrix degradation in Quint actopaxin U2OS cells as well as cell invasion of Quint actopaxin MDA-MB-231 cells. Conversely, both the Cdc42/Rac1 GEF, β-PIX, and its effector and binding partner PAK1 have previously been reported to be required for invadopodia formation and function, but little is known about the mechanism regulating their activity in this context (28, 43–45). Actopaxin binding to β-PIX provides a new mechanism by which β-PIX and PAK1 may be recruited to adhesions to coordinate their turnover during migration and/or transformation to invasive structures. The phosphorylation state of actopaxin, which we show impacted PAK1 phosphorylation may in turn regulate the GEF activity of β-PIX to promote Rac1-dependent activation of PAK1 and downstream signaling to the actin cytoskeleton.
It remains to be determined precisely how actopaxin phosphorylation promotes β-PIX GEF activity. One possibility is that actopaxin undergoes a phosphorylation-dependent conformational change, which is then translated to β-PIX. Alternatively, β-PIX has been shown to be activated by Src- and/or ERK-mediated phosphorylation (46, 47). Interestingly, actopaxin phosphorylation is also ERK-dependent (7), and we have shown here that Src is stimulated downstream of actopaxin. Thus, actopaxin phosphorylation may serve to recruit or stabilize these kinases to adhesions and/or modulate their activity toward downstream targets such as β-PIX.
p38 MAPK is also required for matrix degradation and cell invasion (32, 48, 49) at least in part through regulating the expressing and activity of MMPs such as MMP2 and 9 (49). Our data place actopaxin phosphorylation upstream of p38 MAPK phosphorylation. p38 MAPK has previously been shown to be activated in a β-PIX/PAK1- and Rac1-dependent manner (34). Thus, phosphoactopaxin may also regulate p38 MAPK activity via its interaction with β-PIX. Furthermore, actopaxin, by binding both GEFs (β-PIX) and GAPs (CdGAP) (13, 50), is well positioned to serve as a bimodal regulator of Rho GTPase signaling to elicit tight spatial and temporal control of the balance of Rac1 and RhoA signaling. Any perturbation of the phosphorylation state of actopaxin or its protein level may result in a shift in this regulation, thus inhibiting or promoting matrix degradation and cell invasion accordingly.
Despite their similarities in domain structure and binding partners (24), actopaxin (α-parvin) and β-parvin have been shown to have opposing roles on cell proliferation and ILK activation. Actopaxin promotes cell survival, whereas β-parvin promotes apoptosis through their interactions with ILK (51–53). Actopaxin and β-parvin compete for the same binding site on ILK. In breast cancer cell lines and advanced breast tumors mRNA and proteins levels of β-parvin are decreased (54), thus allowing for increased actopaxin-ILK binding and potentially increased actopaxin phosphorylation and signaling. Interestingly, the MDA-MB-231 breast cancer cells have been reported to lack β-parvin expression entirely (55), and we show that these cells have elevated actopaxin phosphorylation compared with normal breast epithelial cells. Forced expression of β-parvin in these cells inhibits proliferation and tumor growth (55), and overexpression of nonphosphorylatable Quint actopaxin inhibits cell invasion; thus, actopaxin phosphorylation may contribute to both tumor growth and metastasis. The analysis of actopaxin levels and phosphorylation in human cancer cells and tumors is incomplete, and further studies will determine the in vivo significance of actopaxin phosphorylation through the use of mouse tumor and metastasis models.
Acknowledgments
We thank members of the Turner laboratory for insightful discussions of the data.
This work was supported, in whole or in part, by National Institutes of Health Grants R01HL070244 and R01GM47607 (to C. E. T.).

This article contains supplemental Figs. 1–3.
- ECM
- extracellular matrix
- GEF
- guanine nucleotide exchange factor
- ILK
- integrin-linked kinase
- MMP
- matrix metalloproteinase
- PAK
- p21-activated kinase
- PIX
- PAK-interacting exchange factor
- PKL
- paxillin kinase linker.
REFERENCES
- 1. Carragher N. O., Frame M. C. (2004) Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14, 241–249 [DOI] [PubMed] [Google Scholar]
- 2. Olski T. M., Noegel A. A., Korenbaum E. (2001) Parvin, a 42-kDa focal adhesion protein, related to the α-actinin superfamily. J. Cell Sci. 114, 525–538 [DOI] [PubMed] [Google Scholar]
- 3. Tu Y., Huang Y., Zhang Y., Hua Y., Wu C. (2001) A new focal adhesion protein that interacts with integrin-linked kinase and regulates cell adhesion and spreading. J. Cell Biol. 153, 585–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nikolopoulos S. N., Turner C. E. (2002) Molecular dissection of actopaxin-integrin-linked kinase-paxillin interactions and their role in subcellular localization. J. Biol. Chem. 277, 1568–1575 [DOI] [PubMed] [Google Scholar]
- 5. Nikolopoulos S. N., Turner C. E. (2000) Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J. Cell Biol. 151, 1435–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Legate K. R., Montañez E., Kudlacek O., Fässler R. (2006) ILK, PINCH and parvin: the tIPP of integrin signaling. Nat. Rev. Mol. Cell Biol. 7, 20–31 [DOI] [PubMed] [Google Scholar]
- 7. Clarke D. M., Brown M. C., LaLonde D. P., Turner C. E. (2004) Phosphorylation of actopaxin regulates cell spreading and migration. J. Cell Biol. 166, 901–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Curtis M., Nikolopoulos S. N., Turner C. E. (2002) Actopaxin is phosphorylated during mitosis and is a substrate for cyclin B1/cdc2 kinase. Biochem. J. 363, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manser E., Loo T. H., Koh C. G., Zhao Z. S., Chen X. Q., Tan L., Tan I., Leung T., Lim L. (1998) PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol. Cell 1, 183–192 [DOI] [PubMed] [Google Scholar]
- 10. Feng Q., Albeck J. G., Cerione R. A., Yang W. (2002) Regulation of the Cool/Pix proteins: key binding partners of the Cdc42/Rac targets, the p21-activated kinases. J. Biol. Chem. 277, 5644–5650 [DOI] [PubMed] [Google Scholar]
- 11. Rosenberger G., Jantke I., Gal A., Kutsche K. (2003) Interaction of α-PIX (ARHGEF6) with β-parvin (PARVB) suggests an involvement of α-PIX in integrin-mediated signaling. Hum. Mol. Genet. 12, 155–167 [DOI] [PubMed] [Google Scholar]
- 12. Matsuda C., Kameyama K., Suzuki A., Mishima W., Yamaji S., Okamoto H., Nishino I., Hayashi Y. K. (2008) Affixin activates Rac1 via β-PIX in C2C12 myoblast. FEBS Lett. 582, 1189–1196 [DOI] [PubMed] [Google Scholar]
- 13. LaLonde D. P., Grubinger M., Lamarche-Vane N., Turner C. E. (2006) CdGAP associates with actopaxin to regulate integrin-dependent changes in cell morphology and motility. Curr. Biol. 16, 1375–1385 [DOI] [PubMed] [Google Scholar]
- 14. Albiges-Rizo C., Destaing O., Fourcade B., Planus E., Block M. R. (2009) Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J. Cell Sci. 122, 3037–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tumbarello D. A., Turner C. E. (2007) Hic-5 contributes to epithelial-mesenchymal transformation through a RhoA/ROCK-dependent pathway. J. Cell. Physiol. 211, 736–747 [DOI] [PubMed] [Google Scholar]
- 16. Bowden E. T., Onikoyi E., Slack R., Myoui A., Yoneda T., Yamada K. M., Mueller S. C. (2006) Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells. Exp. Cell Res. 312, 1240–1253 [DOI] [PubMed] [Google Scholar]
- 17. Destaing O., Block M. R., Planus E., Albiges-Rizo C. (2011) Invadosome regulation by adhesion signaling. Curr. Opin. Cell Biol. 23, 597–606 [DOI] [PubMed] [Google Scholar]
- 18. Buccione R., Caldieri G., Ayala I. (2009) Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer Metastasis Rev. 28, 137–149 [DOI] [PubMed] [Google Scholar]
- 19. Guarino M. (2010) Src signaling in cancer invasion. J. Cell. Physiol. 223, 14–26 [DOI] [PubMed] [Google Scholar]
- 20. Pankov R., Endo Y., Even-Ram S., Araki M., Clark K., Cukierman E., Matsumoto K., Yamada K. M. (2005) A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 170, 793–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao Y., Dickerson J. B., Guo F., Zheng J., Zheng Y. (2004) Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. U.S.A. 101, 7618–7623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kovács M., Tóth J., Hetényi C., Málnási-Csizmadia A., Sellers J. R. (2004) Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 279, 35557–35563 [DOI] [PubMed] [Google Scholar]
- 23. Burgstaller G., Gimona M. (2004) Actin cytoskeleton remodeling via local inhibition of contractility at discrete microdomains. J. Cell Sci. 117, 223–231 [DOI] [PubMed] [Google Scholar]
- 24. Sepulveda J. L., Wu C. (2006) The parvins. Cell. Mol. Life Sci. 63, 25–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bagrodia S., Cerione R. A. (1999) Pak to the future. Trends Cell Biol. 9, 350–355 [DOI] [PubMed] [Google Scholar]
- 26. Brown M. C., West K. A., Turner C. E. (2002) Paxillin-dependent paxillin kinase linker and p21-activated kinase localization to focal adhesions involves a multistep activation pathway. Mol. Biol. Cell 13, 1550–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bokoch G. M. (2003) Biology of the p21-activated kinases. Annu. Rev. Biochem. 72, 743–781 [DOI] [PubMed] [Google Scholar]
- 28. Webb B. A., Eves R., Crawley S. W., Zhou S., Côté G. P., Mak A. S. (2005) PAK1 induces podosome formation in A7r5 vascular smooth muscle cells in a PAK-interacting exchange factor-dependent manner. Am. J. Physiol. Cell Physiol. 289, C898–907 [DOI] [PubMed] [Google Scholar]
- 29. Ayala I., Baldassarre M., Giacchetti G., Caldieri G., Tetè S., Luini A., Buccione R. (2008) Multiple regulatory inputs converge on cortactin to control invadopodia biogenesis and extracellular matrix degradation. J. Cell Sci. 121, 369–378 [DOI] [PubMed] [Google Scholar]
- 30. Sells M. A., Boyd J. T., Chernoff J. (1999) p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 145, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim M. S., Lee E. J., Kim H. R., Moon A. (2003) p38 kinase is a key signaling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 63, 5454–5461 [PubMed] [Google Scholar]
- 32. Pignatelli J., Tumbarello D. A., Schmidt R. P., Turner C. E. (2012) Hic-5 promotes invadopodia formation and invasion during TGF-β-induced epithelial-mesenchymal transition. J. Cell Biol. 197, 421–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee J., Jung I. D., Chang W. K., Park C. G., Cho D. Y., Shin E. Y., Seo D. W., Kim Y. K., Lee H. W., Han J. W., Lee H. Y. (2005) p85 β-PIX is required for cell motility through phosphorylations of focal adhesion kinase and p38 MAP kinase. Exp. Cell Res. 307, 315–328 [DOI] [PubMed] [Google Scholar]
- 34. Bakin A. V., Rinehart C., Tomlinson A. K., Arteaga C. L. (2002) p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 115, 3193–3206 [DOI] [PubMed] [Google Scholar]
- 35. Linder S. (2009) Invadosomes at a glance. J. Cell Sci. 122, 3009–3013 [DOI] [PubMed] [Google Scholar]
- 36. Wang Y., McNiven M. A. (2012) Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J. Cell Biol. 196, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Webb B. A., Jia L., Eves R., Mak A. S. (2007) Dissecting the functional domain requirements of cortactin in invadopodia formation. Eur. J. Cell Biol. 86, 189–206 [DOI] [PubMed] [Google Scholar]
- 38. Courtneidge S. A., Azucena E. F., Pass I., Seals D. F., Tesfay L. (2005) The SRC substrate Tks5, podosomes (invadopodia), and cancer cell invasion. Cold Spring Harbor Symp. Quant. Biol. 70, 167–171 [DOI] [PubMed] [Google Scholar]
- 39. Nyalendo C., Michaud M., Beaulieu E., Roghi C., Murphy G., Gingras D., Béliveau R. (2007) Src-dependent phosphorylation of membrane type I matrix metalloproteinase on cytoplasmic tyrosine 573: role in endothelial and tumor cell migration. J. Biol. Chem. 282, 15690–15699 [DOI] [PubMed] [Google Scholar]
- 40. Montanez E., Wickström S. A., Altstätter J., Chu H., Fässler R. (2009) α-Parvin controls vascular mural cell recruitment to vessel wall by regulating RhoA/ROCK signaling. EMBO J. 28, 3132–3144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kogata N., Tribe R. M., Fässler R., Way M., Adams R. H. (2009) Integrin-linked kinase controls vascular wall formation by negatively regulating Rho/ROCK-mediated vascular smooth muscle cell contraction. Genes Dev. 23, 2278–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lange A., Wickström S. A., Jakobson M., Zent R., Sainio K., Fässler R. (2009) Integrin-linked kinase is an adaptor with essential functions during mouse development. Nature 461, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 43. Furmaniak-Kazmierczak E., Crawley S. W., Carter R. L., Maurice D. H., Côté G. P. (2007) Formation of extracellular matrix-digesting invadopodia by primary aortic smooth muscle cells. Circ. Res. 100, 1328–1336 [DOI] [PubMed] [Google Scholar]
- 44. Webb B. A., Zhou S., Eves R., Shen L., Jia L., Mak A. S. (2006) Phosphorylation of cortactin by p21-activated kinase. Arch. Biochem. Biophys. 456, 183–193 [DOI] [PubMed] [Google Scholar]
- 45. Gringel A., Walz D., Rosenberger G., Minden A., Kutsche K., Kopp P., Linder S. (2006) PAK4 and α-PIX determine podosome size and number in macrophages through localized actin regulation. J. Cell. Physiol. 209, 568–579 [DOI] [PubMed] [Google Scholar]
- 46. Shin E. Y., Shin K. S., Lee C. S., Woo K. N., Quan S. H., Soung N. K., Kim Y. G., Cha C. I., Kim S. R., Park D., Bokoch G. M., Kim E. G. (2002) Phosphorylation of p85 β-PIX, a Rac/Cdc42-specific guanine nucleotide exchange factor, via the Ras/ERK/PAK2 pathway is required for basic fibroblast growth factor-induced neurite outgrowth. J. Biol. Chem. 277, 44417–44430 [DOI] [PubMed] [Google Scholar]
- 47. Feng Q., Baird D., Yoo S., Antonyak M., Cerione R. A. (2010) Phosphorylation of the cool-1/β-Pix protein serves as a regulatory signal for the migration and invasive activity of Src-transformed cells. J. Biol. Chem. 285, 18806–18816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yong H. Y., Kim I. Y., Kim J. S., Moon A. (2010) ErbB2-enhanced invasiveness of H-Ras MCF10A breast cells requires MMP-13 and uPA up-regulation via p38 MAPK signaling. Int. J. Oncol. 36, 501–507 [PubMed] [Google Scholar]
- 49. Kim E. S., Kim M. S., Moon A. (2004) TGF-β-induced up-regulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int. J. Oncol. 25, 1375–1382 [PubMed] [Google Scholar]
- 50. Wormer D., Deakin N. O., Turner C. E. (2012) CdGAP regulates cell migration and adhesion dynamics in two- and three-dimensional matrix environments. Cytoskeleton 69, 644–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Attwell S., Mills J., Troussard A., Wu C., Dedhar S. (2003) Integration of cell attachment, cytoskeletal localization, and signaling by integrin-linked kinase (ILK), CH-ILKBP, and the tumor suppressor PTEN. Mol. Biol. Cell 14, 4813–4825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fukuda T., Chen K., Shi X., Wu C. (2003) PINCH-1 is an obligate partner of integrin-linked kinase (ILK) functioning in cell shape modulation, motility, and survival. J. Biol. Chem. 278, 51324–51333 [DOI] [PubMed] [Google Scholar]
- 53. Zhang Y., Chen K., Tu Y., Wu C. (2004) Distinct roles of two structurally closely related focal adhesion proteins, α-parvins and β-parvins, in regulation of cell morphology and survival. J. Biol. Chem. 279, 41695–41705 [DOI] [PubMed] [Google Scholar]
- 54. Mongroo P. S., Johnstone C. N., Naruszewicz I., Leung-Hagesteijn C., Sung R. K., Carnio L., Rustgi A. K., Hannigan G. E. (2004) β-Parvin inhibits integrin-linked kinase signaling and is down-regulated in breast cancer. Oncogene 23, 8959–8970 [DOI] [PubMed] [Google Scholar]
- 55. Johnstone C. N., Mongroo P. S., Rich A. S., Schupp M., Bowser M. J., Delemos A. S., Tobias J. W., Liu Y., Hannigan G. E., Rustgi A. K. (2008) Parvin-β inhibits breast cancer tumorigenicity and promotes CDK9-mediated peroxisome proliferator-activated receptor γ1 phosphorylation. Mol. Cell. Biol. 28, 687–704 [DOI] [PMC free article] [PubMed] [Google Scholar]